

Abstract
Recently, protein kinase M ζ (PKMζ) has emerged as an important player for maintaining memory. It has been reported that PKMζ regulates the trafficking of GluA2 in postsynaptic membranes to maintain memory. However, there has been no study on PKMζ outside the synaptic region regarding memory maintenance. Here, we found that PKMζ is transported to the nucleus in a neural activity-dependent manner. Moreover, we found that PKMζ phosphorylates CREB-binding protein (CBP) at serine residues and that PKMζ inhibition reduces the acetylation of histone H2B and H3. Finally, we showed that the amnesic effect of PKMζ inhibition can be rescued by enhancing histone acetylation level. These results suggest the possibility that nuclear PKMζ has a crucial role in memory maintenance.
Keywords: PKMζ, Memory maintenance, Histone acetylation, CREB-binding protein, Translocation into nucleus
1. Introduction
Learning and memory are unique abilities of higher organisms that aid in their survival. Information about the external world is internally transformed and stored in the neural circuits and can be used to guide future behavior. To date, extensive studies have revealed how memory is formed, consolidated, and extinguished in terms of signaling molecules and neural circuits (Kandel, 2001; Kandel, Dudai, & Mayford, 2014). The mechanism of memory maintenance, however, has not been studied to the extent of other stages of memory.
Recently, PKMζ, an atypical protein kinase C (PKC) isoform, has emerged as a key molecule in memory maintenance (Ling et al., 2002; Pastalkova et al., 2006; Sacktor, 2011; Tsokas et al., 2016). PKMζ is a constitutively active form of PKC because it lacks a regulatory subunit. Active PKMζ regulates N-ethylmaleimide-sensitive factor (NSF)-dependent GluA2 trafficking that ultimately maintains the synaptic strength of specific synaptic connections involved in memory (Migues et al., 2010; Yao et al., 2008). However, synapses are not in a static state, and synaptic molecules, including GluA2, are basally degraded and synthesized. If the synaptic configuration is not properly maintained in condition of continuous protein turnover, memories will not be maintained and will eventually collapse. Sustaining specific receptors such as AMPARs, including GluA2, on the synaptic membrane is effective for a relatively short time window; however, this mechanism cannot explain how some memories are maintained permanently. Thus, it is plausible that a transcriptional and translational mechanism may be involved in maintaining memories such as lifelong memories (Kandel et al., 2014).
To date, studies on PKMζ have focused on the function of PKMζ in synaptic regions. However, it has been recently revealed that PKMζ is located in the nucleus as well as postsynaptic sites (Hernandez, Oxberry, Crary, Mirra, & Sacktor, 2014). The presence of PKMζ in the nucleus raises the possibility that it can regulate the gene expression required for memory maintenance. In this study, we examined how PKMζ moves from cytosol into the nucleus and whether nuclear PKMζ can affect transcriptional and epigenetic regulation. Finally, we determined if epigenetic changes can rescue amnesia induced by PKMζ inhibition in the amygdala.
2. Materials and methods
2.1. Animals
Male C57BL/6NCrljBgi mice aged between 6 and 8 weeks were purchased from Orient Bio (Korea). Animals were housed in standard laboratory cages on a 12-h light-dark cycle and provided with access to food and water ad libitum. Mice were used for all experiments 1–2 weeks after being housed in laboratory cages. All experiments were approved by the Institute of Laboratory Animal Resources of Seoul National University.
2.2. Primary neuronal cultures
Embryonic hippocampal neurons were prepared as previously described (Cho et al., 2015). Briefly, rat hippocampi were dissected from E17 embryos and dissociated mechanically after trypsin treatment. Approximately 1,200,000 cells/plate or 100,000 cells/coverslip were plated onto poly-D-lysine-coated 100 mm plastic culture dishes (for Fig. 2D) or cover slips (for Fig. 1C and D). After a 3–4 h recovery in MEM-EBBS with 2 mM glutamine, 10% FBS, 0.45% glucose, 0.11 mg/mL sodium pyruvate, and penicillin/streptomycin, cells were maintained in Neurobasal medium supplemented with B27, GlutaMAX, and penicillin/streptomycin.
Fig. 2.
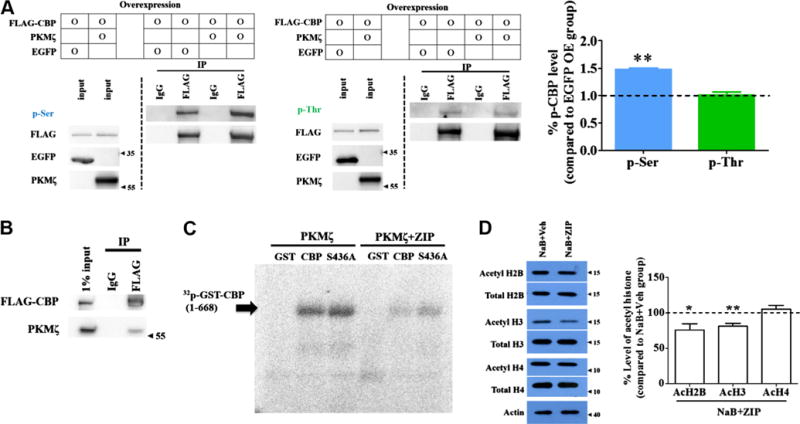
Phosphorylation of CBP induced by PKMζ. (A) Overexpressed FLAG-CBPs in HEK293T cells were immunoprecipitated with 3XFLAG beads and immunoblotted with anti-p-Ser (left panel) or anti-p-Thr (middle panel) antibody. EGFP was overexpressed as a negative control. Phosphorylation of CBP at serine residues was increased by PKMζ but not at threonine residues (right panel). ** p < 0.01. (B) Overexpressed FLAG-CBPs were co-immunoprecipitated with PKMζ in HEK293T cells. (C) Purified FLAG-CBPs were incubated with purified GST-CBP(1-668) or GST-CBP(1-668)S436A fragments in vitro. GST-CBP(1-668) and GST-CBP(1-668)S436A were phosphorylated by PKMζ, and these phosphorylations were inhibited by ZIP co-treatment. GST was used as a negative control. (D) NaB-induced histone acetylation was inhibited by ZIP treatment in cultured neurons. AcH2B and AcH3 levels, but not AcH4 levels, were decreased by ZIP treatment compared to the Veh group. Acetyl histone proteins were normalized with total histone protein. Veh; vehicle. * p < 0.05, **p < 0.01.
Fig. 1.
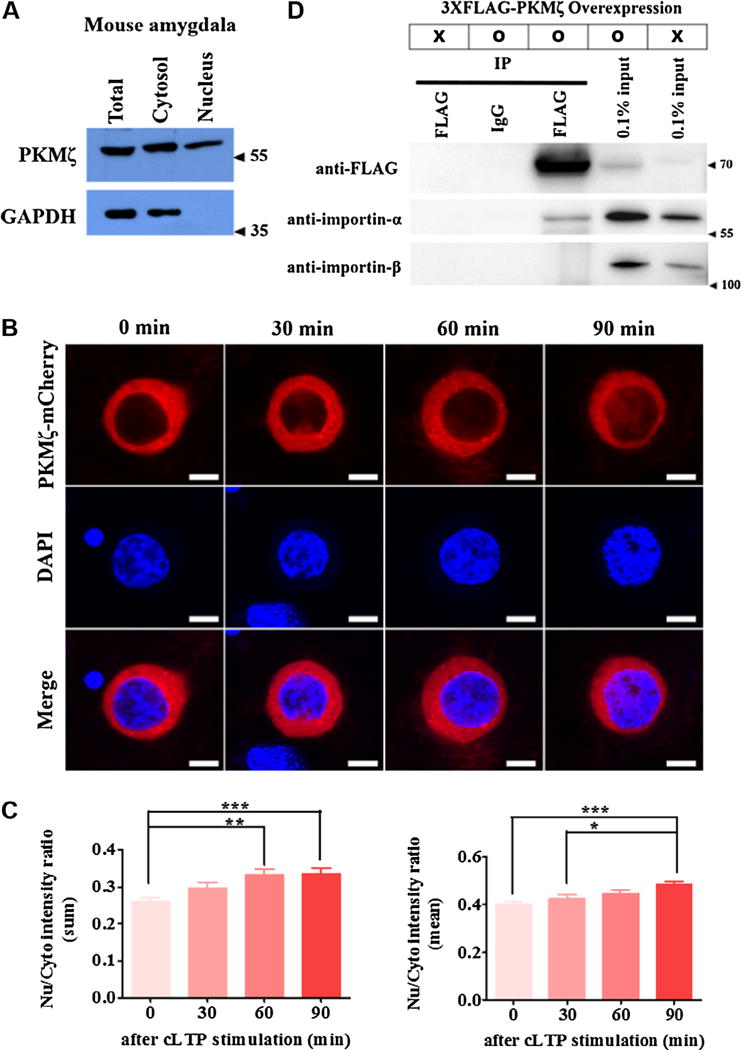
Activity-dependent transport of PKMζ into the nucleus. (A) PKMζ expression in the total, cytosol, and nuclear fraction of the adult mouse amygdala. PKMζ is expressed in the nucleus including the cytosol. (B) and (C) Ectopically expressed PKMζ-mCherry fusion proteins move into the nucleus during cLTP stimulation. The Nu/Cyto intensity ratio of fluorescent intensity was increased in a time-dependent manner after cLTP stimulation (left panel; sum ratio, right panel; mean ratio (Nu/Cyto intensity ratio of fluorescent intensity normalized by each area)). *p < 0.05, ** p < 0.01, ***p < 0.001. Nu; Nucleus, Cyto; Cytosol. Scale bar; 10 μm. (D) Overexpressed 3XFLAG-PKMζ in HEK293T cells was co-immunoprecipitated with importin-α, but not importin-β.
2.3. Immunoprecipitation
HEK293T cells in 100-mm culture dishes were transfected with 16 μg of FLAG-CBP and 4 μg of pCMV-PKMζ expression plasmid (or 4 μg of EGFP-N1 expression plasmid for control groups). Two days later, transfected cells were harvested and washed with cold PBS. The collected cells were lysed with a buffer containing 1% Triton X-100, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM EDTA, benzonase (Sigma), phosphatase-inhibitor cocktail (Roche), and protease-inhibitor cocktail (Roche). After centrifuging at 13,300 rpm for 15 min, the supernatant was diluted 1/10 with a buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM EDTA, and protease-inhibitor cocktail (Roche). After BCA assay for protein quantification, 1 mg of protein was incubated with 3-times washed 50 μl (bead volume) of mouse anti-FLAG M2 antibody-conjugated beads (Sigma) (or mouse IgG-Agarose (Sigma) for control groups) at 4 °C for 2 h. Subsequently, the beads were washed twice with a buffer containing 0.1% Triton X-100, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 2 mM EDTA and then twice with 1X TBS. Finally, the immunoprecipitate was eluted by adding 3XFLAG peptides (Sigma).
Western blotting was performed as previously described (Li et al., 2010). The samples were separated on 4–12% Bis-Tris Plus Gels (Invitrogen) and then transferred to nitrocellulose blotting membranes (GE healthcare) overnight. After blocking with 5% nonfat milk, the membrane was incubated with the primary antibody (rabbit anti-phosphoserine (Millipore) 1:500 in 5% BSA, rabbit anti-phosphothreonine (Millipore) 1:500 in 5% BSA, mouse anti-PKCζ (Santa Cruz) 1:5000 in 5% milk, mouse anti-EGFP (NeuroMab) 1:5000 in 5% milk, and mouse anti-FLAG (Sigma) 1:10,000 in 5% milk) and horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody. After washing, the membrane was treated with Immobilon western chemiluminescent HRP substrate (Millipore) according to the manufacturer’s instructions. Imaging was performed using the ChemiDoc MP system (Bio-Rad).
2.4. Co-immunoprecipitation
Co-immunoprecipitation and western blotting were performed as previously described (Jun et al., 2015; Kim et al., 2014). Briefly, HEK293T cells were transfected with a 3XFLAG-PKMζ (interaction between PKMζ and importin) or 3XFLAG-CBP and pCMV-PKMζ (interaction between PKMζ and CBP) expression plasmid. Immunoprecipitation was conducted as described above. The transferred membranes were incubated with the primary antibody (for PKMζ-importin: mouse anti-importin-α (Sigma) 1:1000, mouse anti-importin-β (Abcam) 1:1000, and mouse anti-FLAG (Sigma) 1:10,000, for PKMζ-CBP: mouse anti-PKC/Mζ (Santa Cruz) 1:5000, and mouse anti-FLAG (Sigma) 1:10,000) and horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody. After washing, the membrane was treated with Immobilon western chemiluminescent HRP substrate (Millipore) according to the manufacturer’s instructions. Imaging was performed using the ChemiDoc MP system (Bio-Rad).
2.5. Kinase assay
Protein purification and in vitro kinase assay were performed as previously described (Lee et al., 2006). Briefly, GST, GST-CBP (1-668), and GST-CBP(1-668)S436A were overexpressed in the BL21 cell line and purified with glutathione Sepharose 4 Fast Flow (GE Healthcare). PKMζ-3XFLAG was overexpressed in the HEK293T cell line and purified with anti-FLAG M2 affinity gels (Sigma). For the in vitro kinase assay, 3.5 μg of substrate was incubated for 30 min at 37 °C in 25 μl of reaction solution (0.2 mM ATP, 1 mCi [γ-32P]ATP, 50 ng of purified PKMζ protein, 50 mM Tris [pH 7.5], 10 mM MgCl2). The zeta inhibitory peptide (ZIP, 10 μM) (Invitrogen) was added to verify the specificity of PKMζ kinase activity. Reactions were stopped by adding SDS sample buffer and heating to 95 °C for 10 min. Samples were separated by SDS-PAGE and analyzed with a Bio-Imaging Analyzer (BAS-2500, Fuji).
2.6. Purification of the nuclear fraction
At 15–17 days in vitro (DIV), cultured neurons were treated with 1 mM sodium butyrate (NaB; Sigma-Aldrich) or/and 10 μM ZIP (Invitrogen) for 1 h, followed by washing with PBS. After harvesting using a scrapper, neurons were lysed with TX buffer (50 mM Tris-Cl, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100) containing a protease inhibitor cocktail (Roche). The homogenate was incubated on ice for 15 min, followed by centrifugation at 500g at 4 °C for 10 min to purify the nuclear fraction. The supernatant was removed, and the pellet was lysed with TX buffer containing 0.2 N HC1 and protease inhibitor cocktail. After incubation on ice for 30 min, the nuclear lysate was subjected to centrifugation at 9300g at 4 °C for 10 min. The supernatant was used for western blot analysis.
2.7. Activity-dependent translocation of PKMζ
At 7 DIV, embryonic hippocampal cultured neurons were transduced with PKMζ-mCherry expressing adeno-associated virus (AAV) (2 × 109/well, coverslip in 12-well plate). PKMζ was tagged with mCherry at the C-terminus. The viral vector expressing the PKMζ-mCherry fusion protein under the CaMKIIα promoter was packaged into AAV (serotype 2/1) as previously reported (Choi et al., 2014). At 18 DIV, a chemical long-term potentiation (cLTP) stimulation protocol was used for inducing neural activity. Briefly, cultured neurons were incubated with cLTP solution (200 μM glycine, 20 μM bicuculline, 124 mM NaCl, 3 mM KCl, 2 mM CaCl2, 10 mM HEPES (pH 7.3), 10 mM glucose) for 5 min, and then the medium was exchanged with cLTP solution without glycine for 30, 60, or 90 min. After completing the cLTP stimulation protocol, cultured neurons were briefly washed with cold PBS and then fixed with 4% paraformaldehyde/4% sucrose in PBS for 15 min on ice. After fixation, cultured neurons onto the coverslip were again briefly washed with PBS and mounted on the slide glass with mounting medium (VECTASHIELD containing DAPI). A confocal laser scanning microscope (LSM700, Zeiss) was used for obtaining images of PKMζ-mCherry signals. To obtain PKMζ-mCherry signals only in the nucleus, the focal plane was set to the region showing the largest DAPI signal. The ImageJ program was used for image analysis. Fluorescence intensity of the nucleus was divided by that of the cytosol (sum ratio, Fig. 1D left panel). To overcome the cytosol and nucleus size differences among neurons, we calculated the mean ratio which was calculated by dividing the normalized value of the nucleus by that of the cytosol. Normalization was done by dividing fluorescence intensity of the nucleus or cytosol by its area (mean ratio, Fig. 1D right panel). Sum ratio and mean ratio were calculated from all neurons. Experimenters blinded to identity of the groups performed the image analysis.
2.8. Cannulation and microinjection
Cannulation and microinjection were performed as described previously (Li et al., 2010). Briefly, mice were anesthetized by an intraperitoneal injection of a mixture of saline, ketamine (0.16 mg/kg), and xylazine (0.01 mg/kg). The scalp was shaved and then cleaned with povidone-iodine and alcohol. The head of the mouse was fixed into an adapter mounted on a stereotaxic frame, and the eyes were protected from drying with saline. An incision was made over the skull, and the surface was exposed. Two small holes were drilled above the amygdala, and the dura was gently reflected. The tips of guide cannulas (22 gauge) were placed at 1.4 mm posterior, ±3.25 mm lateral, and 3.7 mm ventral to the bregma. A screw was inserted into the skull above the cortex to fix the dental cement. Cannulas and screw were fixed with dental cement. Mice were subjected to behavioral experiments at least 1 week after cannulation. For microinjection, the mice were anesthetized by isoflurane. A 30-gauge injector 1.0 mm below the guide was inserted into the guide cannula, and drugs (10 nmol/μl ZIP, 1 or 10 μg/μl NaB, in saline, 0.5 μl/side) were microinjected using a manual syringe pump. An infusion needle was left in place for 1 min after the end of the injection to prevent overflow of drug. After microinjection, mice were immediately returned to their home cage.
2.9. Behavioral procedures
For cued fear conditioning, mice were handled for 4 days. On the training day (Day 1), mice were placed in the conditioning chamber with a stainless grid floor (Med Associates). The first tone was delivered for 30 s, 180 s after entry to the chamber, and the tone was coterminated with electrical foot shock (0.7 mA, 2 s). The second tone and foot shock were delivered 90 s after the first pairing between the tone and shock. The mice were returned to the home cage 30 s after the second pairing. On Day 2, ZIP (10 nmol/μl, 0.5 μl/side, Invitrogen) and/or NaB (2 μg/μl, 0.5 μl/side, Sigma-Aldrich) were bilaterally infused into the amygdala. On Day 3, mice were exposed in the different context (Coulbourn Instruments), and the tone was delivered for 30 s 2 min after entry into the chamber. The behavior of the mice was automatically recorded with a digital camera. Freezing behavior was analyzed by Freezeview software (Coulbourn Instruments). The setting values for measuring freezing were 0.5 s for bout duration and 11 for threshold.
3. Results
To date, studies on PKMζ function have focusedon its role within synaptic regions. Here, we first determined whether PKMζ is also located in the nucleus. Endogenous PKMζ in the mouse amygdala was found in the nucleus as well as cytosol (Fig. 1A). This result is consistent with the recent report that PKMζ was found in the nucleus using post-embedding immuno-electron microscopy (Hernandez et al., 2014). Moreover, when we analyzed a predicted nuclear localization signal (NLS), we found that PKMζ has 3 predicted bipartite NLSs (Kosugi, Hasebe, Tomita, & Yanagawa, 2009). Next, we tested if neuronal activity can facilitate the movement of PKMζ to the nucleus. mCherry-fused PKMζ was overexpressed in cultured hippocampal neurons using AAV. At 18 DIV, after chemical LTP stimulation for 5 min, images were taken by confocal laser scanning microscopy. Consistent with Fig. 1A, mCherry-fused PKMζ was observed in the nucleus, and the ratio of nucleus/cytosol of fluorescent intensity increased over time after chemical LTP stimulation (n = 55–65 for each group, one-way ANOVA followed by Tukey’s multiple comparison test; F3,246 = 6.410 (sum ratio, left panel); F3, 246 = 6.139 (mean ratio, right panel; Nu/Cyto intensity ratio normalized by each area), *p < 0.05, **p < 0.01, ***p < 0.001; Fig. 1B and C). If PKMζ is transported from the cytosol, including synaptic regions, into the nucleus, it might bind to a member of the importin family to enter the nucleus. Thus, we tested if PKMζ can bind to importin using co-immunoprecipitation. As shown in Fig. 1D, PKMζ was co-immunoprecipitated with importin-α but not importin-β (Fig. 1D). These results indicate that PKMζ is transported into the nucleus in a neural activity-dependent manner.
Next, we decided to determine the nuclear function of PKMζ. If nuclear PKMζ also functions in memory maintenance, it is plausible that PKMζ acts on a molecule that can modulate the expression of memory-related genes. Thus, we first examined if PKMζ can interact with CBP because CBP is known to be involved in memory formation and LTP (Korzus, Rosenfeld, & Mayford, 2004; Valor et al., 2011; Wei et al., 2012). In addition, PKMζ has the same catalytic domain as PKCζ that regulates CBP function through phosphorylation (He et al., 2009; Wang et al., 2010). When we examined whether PKMζ interacts with CBP, overexpressed FLAG-tagged CBP was co-immunoprecipitated with PKMζ (Fig. 2B). This result raises the possibility that nuclear PKMζ can regulate gene expression required for memory maintenance via CBP.
Next, we determined whether PKMζ phosphorylates CBP. After PKMζ and CBP were overexpressed in HEK293T cells, CBPs were immunoprecipitated followed by immunoblotting with anti-p-Ser or p-Thr antibodies. PKMζ overexpression increased the phosphorylation of CBP at serine residues but not threonine residues (n = 3; one sample t-test, t2 = 20.67, p < 0.01 (p-Ser); t2 < 1 (p-Thr), **p < 0.01; Fig. 2A). To further examine whether PKMζ can phosphorylate CBP directly, we used an in vitro kinase assay. N-terminal fragments of CBP(1-668) were used as a substrate because PKCζ can phosphorylate CBP at S436 and PKMζ has an identical catalytic subunit as PKCζ (He et al., 2009; Wang et al., 2010). As a result, PKMζ phosphorylated CBP(1-668), and ZIP, a PKMζ-inhibiting peptide, reduced the level of p-CBP(1-668) (Fig. 2C). Notably, PKMζ also phosphorylated CBP(1-668)S436A (Fig. 2C), indicating that there might be additional phosphorylation sites on CBP besides S436.
CBP can regulate gene expression by acting as a transcription coactivator with histone acetyltransferase (HAT) activity (Vo & Goodman, 2001). Thus, we wanted to determine whether PKMζ can regulate the HAT activity of CBP because histone acetylation is involved in metaplasticity, which has long-lasting effects on synaptic function (Zovkic, Guzman-Karlsson, & Sweatt, 2013). To this end, we used a HDAC inhibitor (NaB) to enhance global histone acetylation level and ZIP to inhibit PKMζ activity. In cultured neurons, PKMζ inhibition through ZIP treatment reduced acetylated H2B and H3 levels but not the acetylated H4 level (n = 8, one sample t-test, t7 = 2.68, p < 0.05 (Ac-H2B); t7 = 4.46, p < 0.01 (Ac-H3); t7 < 1 (Ac-H4), *p < 0.05, **p < 0.01; Fig. 2D). Acetylated H2B and H3 levels were also shown to be reduced in the cortex of conditional Cbp knockout mice (Chen, Zou, Watanabe, van Deursen, & Shen, 2010). These results indicate that, in the nucleus, PKMζ phosphorylates CBP at serine residues and may affect global histone acetylation levels regulated by CBP.
It has been reported that inhibition of PKMζ in the hippocampus, insular cortex, and amygdala erases several types of memories such as place memory, conditioned taste aversion, and fear memory, respectively (Kwapis, Jarome, Lonergan, & Helmstetter, 2009; Li et al., 2010; Migues et al., 2010; Pastalkova et al., 2006; Shema, Sacktor, & Dudai, 2007; von Kraus, Sacktor, & Francis, 2010). PKMζ maintains memories by regulating the trafficking of synaptic GluA2 (Migues et al., 2010; Yao et al., 2008). Apart from this mechanism, we examined whether enhanced histone acetylation levels can rescue memory erasure induced by PKMζ inhibition. If PKMζ is involved in regulating histone acetylation levels through CBP, PKMζ inhibition would lead to reduction of histone acetylation level. Thus, we assumed that memory erasure induced by ZIP treatment could be rescued by a HDAC inhibitor (NaB). As shown in Fig 3A and C, ZIP infusion into the amygdala reduced freezing level. However, when NaB was co-infused with ZIP, reduction of freezing level was not observed (n = 15–17 per group, one-way ANOVA followed by Tukey’s multiple comparison test, F2, 44 = 5.716, p < 0.01, *p < 0.05; Fig. 3A and C). HDAC inhibitors such as NaB can enhance memories. Thus, to exclude the possibility that NaB infusion simply enhanced cued fear memory regardless of the action of ZIP, we tested if NaB alone enhances cued fear memory in our experimental scheme. However, 1 μg of NaB infusion into the amygdala did not increase freezing level (n = 9 per group, unpaired t-test, t16 < 1; Fig. 3B and D). These results indicate that there is another memory maintenance mechanism probably mediated by nuclear PKMζ that is independent of the regulation of the surface GluA2 level.
Fig. 3.
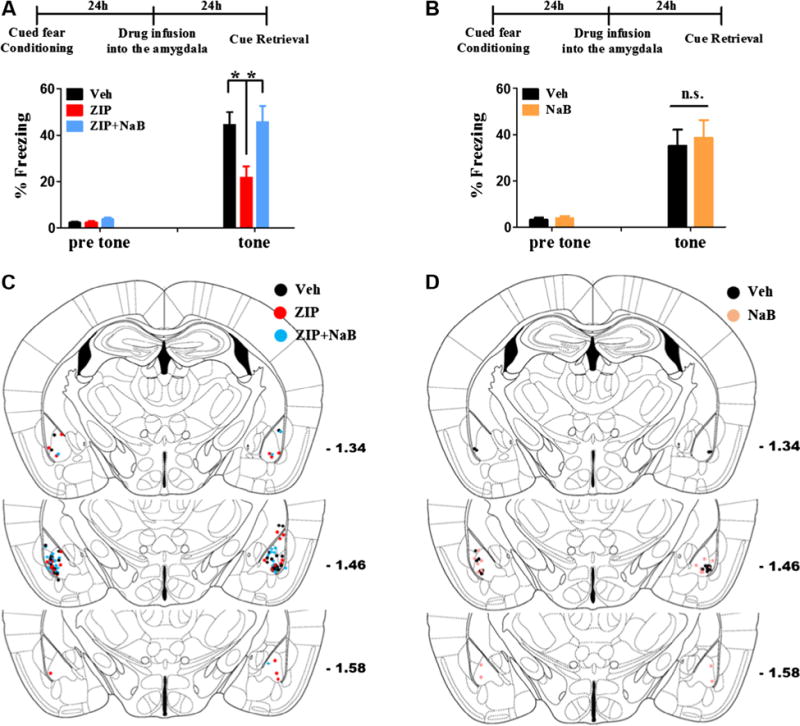
Cued fear memory is decreased by PKMζ inhibition in the amygdala and NaB co-application blocks the amnesic effect of PKMζ inhibition. (A) One day after cued fear conditioning, ZIP was microinjected into the amygdala. Cued fear memory was tested 1 day after drug microinjection. ZIP microinjection reduced the freezing level. However, NaB co-application blocked the amnesic effect of ZIP. Veh; vehicle, * p < 0.05. (B) In same experimental scheme as (A), microinjection of NaB alone did not increased the freezing level. n.s.; not significant. (C) and (D) The location of the microinjection sites in (A) and (B).
4. Discussion
To date, most studies on PKMζ have focused on the function of PKMζ in the synaptic region (Sacktor, 2011). Here, we examined the role of PKMζ in the nucleus. Consistent with a previous study on hippocampal neurons (Hernandez et al., 2014), we revealed that PKMζ is located in the nucleus of amygdalar neurons. In addition, we found that PKMζ is transported to the nucleus in a neural activity-dependent manner. Moreover, PKMζ can bind to the adaptor protein importin-α for nuclear translocation. These results suggest that PKMζ can function in the nucleus to maintain memory in addition to its role in the synaptic regions. Indeed, PKMζ can bind to and phosphorylate CBP at serine residues. Moreover, PKMζ is involved in the regulation of histone acetylation levels based on our finding that PKMζ inhibition reduced acetylated H2B and H3 levels. Finally, we demonstrated that memory erasure through PKMζ inhibition can be rescued by enhancing histone acetylation levels.
Although we found that PKMζ can phosphorylate CBP on serine resides, we did not identify the exact phosphorylation site on CBP. Thus, further studies are needed to identify which sites on CBP are phosphorylated by PKMζ and determine how phosphorylated CBP affects memory maintenance. Whether phospho-CBP induced by PKMζ actually increases its HAT activity also requires clarification. We found that the reduction of freezing after ZIP infusion into the amygdala 1 day after fear conditioning was rescued by NaB co-application with ZIP. This suggests that PKMζ acts to maintain histone acetylation levels during memory maintenance, and that the PKMζ inhibitor erases long-term memory in part by reducing histone acetylation level. These results were supported by Shanping Chen et al.’s report that chelerythrine, a PKC inhibitor selective for PKM at low concentrations, blocked memory maintenance and trichostatin A, HDAC inhibitor, abolished chelerythrine’s effect in Aplysia (Chen et al., 2014). However, this result might appear to conflict with the observation that ZIP treatment could still reduce histone acetylation levels in NaB-treated neuronal culture. This apparent discrepancy, however, might result from the difference between model systems (in vitro vs. in vivo) or concentration of drugs used. For example, in the in vitro experiments, we used NaB-treatment to raise global histone acetylation levels and found that ZIP counteracted this action, indicating that basal PKMζ in the neuronal cultures can act to increase histone acetylation levels when deacetylation is blocked. In contrast, we speculate that in the amygdala of the previously trained animals, PKMζ may already have been increased above basal levels and thus already increased the histone acetylation levels. In these in vivo experiments, ZIP may be normally sufficient to return the increased histone acetylation levels to the basal levels, but not if histone deacetyltransferase are inactivated by the HDAC inhibitor.
Although there have been many studies that examined whether CBP is involved in memory formation, no study has tested whether CBP is important for maintaining memory after the consolidation phase (Chen et al., 2010; Korzus et al., 2004; Valor et al., 2011; Vecsey et al., 2007; Wei et al., 2012). If PKMζ functions through CBP to maintain memory, CBP inhibition might break down memories. Thus, it is worth examining in future experiments whether CBP is required for maintaining memory as a downstream factor of PKMζ. It is well established that after the consolidation phase, memory and LTP become independent of protein synthesis (Abraham & Williams, 2008). After the consolidation phase of a memory, application of a protein synthesis inhibitor (PSI) does not disrupt the original memory. Although many studies have examined the effect of a single injection of a PSI on memory, the effect of the PSI usually lasts for around 6 h (Wanisch & Wotjak, 2008). In contrast, the lifetimes of many proteins in the brain are over 1 day in vivo (Price, Guan, Burlingame, Prusiner, & Ghaemmaghami, 2010). Thus, it is possible that inhibition of protein synthesis for longer periods of time can break down the synaptic configuration bearing information for memory and eventually erases memory. In this case, enhanced ongoing protein synthesis mediated by epigenetic mechanisms protecting against protein turnover may be required for maintaining memory (Zovkic et al., 2013). Thus, it would be valuable to examine whether activation of CBP induced by PKMζ can supply proteins that are necessary to maintain memory. This hypothesis is also important because many synaptic memory-related genes such as AMPA and NMDA receptors, postsynaptic density proteins, and their interacting proteins were found to be reduced in the conditional Cbp knockout mice brain (Chen et al., 2010).
In conclusion, we suggest the hypothesis that nuclear PKMζ is involved in memory maintenance apart from synaptic PKMζ. Once neurons are activated by external stimulation during memory formation, PKMζ expression is increased and it enters the nucleus. Nuclear PKMζ might phosphorylate and activate CBP, and the activated CBP in turn stimulates gene expression which is involved in memory maintenance. Additional studies should be performed to determine whether CBP is actually involved in memory maintenance as a downstream factor of PKMζ and if so to determine how CBP affects memory maintenance.
Acknowledgments
Funding
This work was supported by two National Research Foundation (NRF) of Korea grants funded by the Korean government (MSIP) [NRF-2012R1A3A1050385 to B-KK and 35B-2011-1-C00034 to H-GK]. J-IK was supported by the POSCO TJ Park Foundation. TCS was supported by NIH funding 2R37 MH057068, RO1 MH53576, and RO1 DA034979.
Footnotes
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Authors’ contributions
H-GK and J-IK designed the studies and carried out the molecular experiments and outlined the manuscript and wrote the manuscript. S-ES, W-JY, THK and S-LC carried out molecular experiments. JY carried out the behavioral experiments. SHB, J-BY, TCS and B-KK supervised the experiments, participated in the interpretation of the data, and wrote the manuscript. All authors read and approved the final manuscript.
References
- Abraham WC, Williams JM. LTP maintenance and its protein synthesis-dependence. Neurobiology of Learning and Memory. 2008;89:260–268. doi: 10.1016/j.nlm.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chen S, et al. Reinstatement of long-term memory following erasure of its behavioral and synaptic expression in Aplysia. Elife. 2014;3:e03896. doi: 10.7554/eLife.03896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Zou X, Watanabe H, van Deursen JM, Shen J. CREB binding protein is required for both short-term and long-term memory formation. Journal of Neuroscience. 2010;30:13066–13077. doi: 10.1523/JNEUROSCI.2378-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, et al. Multiple repressive mechanisms in the hippocampus during memory formation. Science. 2015;350:82–87. doi: 10.1126/science.aac7368. [DOI] [PubMed] [Google Scholar]
- Choi JH, et al. Optimization of AAV expression cassettes to improve packaging capacity and transgene expression in neurons. Molecular Brain. 2014;7:17. doi: 10.1186/1756-6606-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, et al. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell. 2009;137:635–646. doi: 10.1016/j.cell.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez AI, Oxberry WC, Crary JF, Mirra SS, Sacktor TC. Cellular and subcellular localization of PKMzeta. Philosophical Transactions of the Royal Society of London, Series B: Biological sciences. 2014;369:20130140. doi: 10.1098/rstb.2013.0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun MH, et al. TMEM106B, a frontotemporal lobar dementia (FTLD) modifier, associates with FTD-3-linked CHMP2B, a complex of ESCRT-III. Molecular Brain. 2015;8:85. doi: 10.1186/s13041-015-0177-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: A dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Dudai Y, Mayford MR. The molecular and systems biology of memory. Cell. 2014;157:163–186. doi: 10.1016/j.cell.2014.03.001. [DOI] [PubMed] [Google Scholar]
- Kim KH, et al. Intracellular membrane association of the Aplysia cAMP phosphodiesterase long and short forms via different targeting mechanisms. Journal of Biological Chemistry. 2014;289:25797–25811. doi: 10.1074/jbc.M114.572222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:10171–10176. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwapis JL, Jarome TJ, Lonergan ME, Helmstetter FJ. Protein kinase Mzeta maintains fear memory in the amygdala but not in the hippocampus. Behavioral Neuroscience. 2009;123:844–850. doi: 10.1037/a0016343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, et al. PKA-activated ApAF-ApC/EBP heterodimer is a key downstream effector of ApCREB and is necessary and sufficient for the consolidation of long-term facilitation. Journal of Cell Biology. 2006;174:827–838. doi: 10.1083/jcb.200512066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XY, et al. Alleviating neuropathic pain hypersensitivity by inhibiting PKMzeta in the anterior cingulate cortex. Science. 2010;330:1400–1404. doi: 10.1126/science.1191792. [DOI] [PubMed] [Google Scholar]
- Ling DS, et al. Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nature Neuroscience. 2002;5:295–296. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- Migues PV, et al. PKMzeta maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nature Neuroscience. 2010;13:630–634. doi: 10.1038/nn.2531. [DOI] [PubMed] [Google Scholar]
- Pastalkova E, et al. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313:1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- Price JC, Guan S, Burlingame A, Prusiner SB, Ghaemmaghami S. Analysis of proteome dynamics in the mouse brain. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14508–14513. doi: 10.1073/pnas.1006551107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor TC. How does PKMzeta maintain long-term memory? Nature Reviews Neuroscience. 2011;12:9–15. doi: 10.1038/nrn2949. [DOI] [PubMed] [Google Scholar]
- Shema R, Sacktor TC, Dudai Y. Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKM zeta. Science. 2007;317:951–953. doi: 10.1126/science.1144334. [DOI] [PubMed] [Google Scholar]
- Tsokas P, et al. Compensation for PKMzeta in long-term potentiation and spatial long-term memory in mutant mice. Elife. 2016;5 doi: 10.7554/eLife.14846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valor LM, et al. Ablation of CBP in forebrain principal neurons causes modest memory and transcriptional defects and a dramatic reduction of histone acetylation but does not affect cell viability. Journal of Neuroscience. 2011;31:1652–1663. doi: 10.1523/JNEUROSCI.4737-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecsey CG, et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. Journal of Neuroscience. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. Journal of Biological Chemistry. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- von Kraus LM, Sacktor TC, Francis JT. Erasing sensorimotor memories via PKMzeta inhibition. PLoS ONE. 2010;5:e11125. doi: 10.1371/journal.pone.0011125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, et al. CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Developmental Cell. 2010;18:114–125. doi: 10.1016/j.devcel.2009.10.023. [DOI] [PubMed] [Google Scholar]
- Wanisch K, Wotjak CT. Time course and efficiency of protein synthesis inhibition following intracerebral and systemic anisomycin treatment. Neurobiology of Learning and Memory. 2008;90:485–494. doi: 10.1016/j.nlm.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Wei W, et al. P300/CBP-associated factor selectively regulates the extinction of conditioned fear. Journal of Neuroscience. 2012;32:11930–11941. doi: 10.1523/JNEUROSCI.0178-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, et al. PKM zeta maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. Journal of Neuroscience. 2008;28:7820–7827. doi: 10.1523/JNEUROSCI.0223-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovkic IB, Guzman-Karlsson MC, Sweatt JD. Epigenetic regulation of memory formation and maintenance. Learning & Memory. 2013;20:61–74. doi: 10.1101/lm.026575.112. [DOI] [PMC free article] [PubMed] [Google Scholar]