

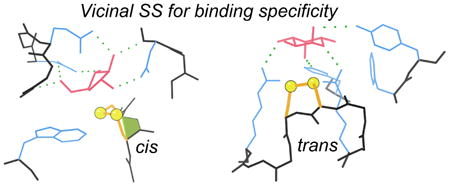
Abstract
Vicinal disulfides between sequence-adjacent cysteine residues are very rare and rather startling structural features which play a variety of functional roles. Typically discussed as an isolated curiosity, they have never received a general treatment covering both cis and trans forms. Enabled by the growing database of high-resolution structures, required deposition of diffraction data, and improved methods for discriminating reliable from dubious cases, we here identify and describe distinct protein families with reliably genuine examples of cis or trans vicinal disulfides and discuss their conformations, conservation, and functions. No cis-trans interconversions and only one case of catalytic redox function are seen. Some vicinal disulfides are essential to large, functionally coupled motions, whereas most form the centers of tightly packed internal regions. Their most widespread biological role is providing a rigid hydrophobic contact surface under the undecorated side of a sugar or multi-ring ligand, contributing an important aspect of binding specificity.
Keywords: structural bioinformatics, sequence-adjacent disulfide, cis non-proline, SS bond, carbohydrate binding
Graphical abstract
Introduction
A disulfide bond between sequence-adjacent cysteines, known as a vicinal disulfide, is a very distinctive and interesting, but extremely rare, conformation. Adjacent Cys residues occur about as often as expected from single-Cys frequency 1, but are usually in the reduced form. Those SH groups typically point away from each other, and in general are not both seen to ligand the same metal ion 2, with an exception for mercury 3. When adjacent Cys do form disulfide bonds, the two bonds are nearly always to different partners 4, such as in the cystine knot motif of transcription factors 5 and toxins 6. The vicinal disulfides studied here are the rare exceptions to all the above rules. Figure 1 shows the highest-resolution example, the conserved Cys 121-122 of the 1.06Å PDB ID: 3CU9 arabinanase structure 7 located at the active site, here occupied by a disordered glycerol.
Figure 1.
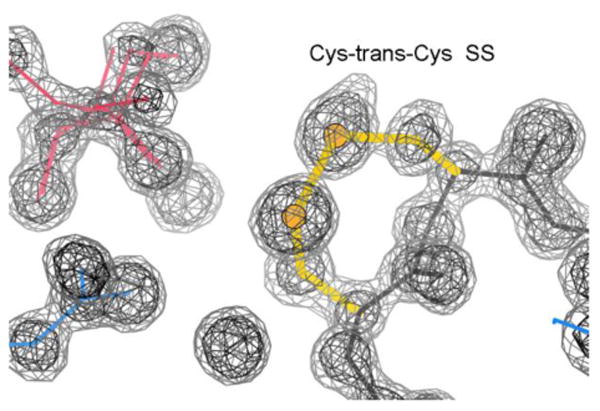
The highest-resolution vicinal SS. Cys121-Cys122 (yellow), in the PDB ID: 3CU9 arabinanase structure at 1.06Å resolution 7 is next to a disordered glycerol (pink) at the active site. 2mFo-DFc electron density contours at 1.2σ (gray) and 3.0σ(black).
For the most part, each vicinal disulfide has been reported as an isolated curiosity, discussed for biological function within the context of its own protein family. Sometimes they are not noticed or not commented upon, even when in a suggestive position. Early calculations implied that only a cis peptide conformation is feasible for an i to i+1 disulfide 8-9, which led some studies to assume that all vicinal SS in proteins would be cis (Kao 10 on acetylcholine receptor; Blake 11 on methanol dehydrogenase). After both those cases were determined as actually trans 12-14, some studies then assumed that all vicinal SS in proteins would be trans although somewhat twisted from 180° 15-16 Vicinal SS are briefly mentioned in several overall reviews of disulfides, with only trans examples given 17-18. The only protein-structure survey we can find about vicinal disulfides 16 was early enough to identify just 9 distinct examples, all then thought to be trans; it categorizes the two different trans backbone conformations as types of β turns. However, computational and NMR work on small-molecule and short-peptide vicinal SS structures describes both cis and trans forms 19-21, and high-resolution protein examples of both types have slowly accumulated. We feel, therefore, that an updated survey and analysis of both cis and trans vicinal disulfides seems useful and timely.
The work presented here takes advantage of the greatly increased resource of high-resolution crystal structures in the worldwide Protein Data Bank (PDB) 22 to identify more numerous and varied examples of this extremely rare feature. Deposited diffraction data, required since 2008, enables evaluation of electron-density quality for robust separation of reliable from dubious instances. We thus can provide a comprehensive and critical structural-bioinformatic analysis of both cis and trans vicinal disulfides, describing their observed conformational variety in well-verified examples along with many of their most notable and interesting functional roles.
Results
Cis vicinal SS examples and functions
We encountered these intriguing sequence-adjacent disulfides in separate work on cis-nonPro peptides 23, which included only two vicinal SS among the 451 cis-nonPro examples in our low-homology reference datasets at ≤1.99Å resolution. To expand the sample, we did a broader X-ray PDB search for Cys-cis-Cys peptides (see Methods). From over 180 Cys-cis-Cys peptides in 51 PDB files, manual examination of those results identified distinct groups of reliably genuine Cys-cis-Cys SS in 13 unrelated protein families. In addition, the search results included three Cys-cis-Cys without the connecting disulfide at 2.6 to 3.9Å resolution (PDB ID: 2HTH, 4H65, and 4Q5Z), but for none of them was there close to adequate evidence that the peptide is actually cis. All three had very poor or missing local density and very high B factors; two are next to chain ends, and the third has one of the Cys assigned as zero occupancy. Also, when a structure is known for a Cys-cis-Cys SS in its open, reduced form (PDB ID: 1KV3, 3SR3), the peptide has changed to trans. We conclude, therefore, that adjacent Cys are unlikely to have a cis peptide conformation unless they are connected in a vicinal SS.
The highest-resolution cis examples for each of the 13 protein families are listed in Table 1 with brief descriptions. They include a wide variety of hydrolase enzymes, receptors, and binding or transport proteins, spread across vertebrates, invertebrates, bacteria, archaea, and viruses.
Table 1. Distinct, validated examples of Cis vicinal disulfides.
| PDBid ch | resa | resol confb | Protein | reference |
|---|---|---|---|---|
| 1WD3 A | 177 | 1.75Å C- | arabinofuranosidase | Miyanaga 2004 36 |
| 1YM0 A | 151 | 2.06Å C- | worm 2-chain trypsin | Wang 2005 15 |
| 2Q3Z A | 371 | 2.00Å C+ | transglutaminase 2 | Pinkas 2007 33 |
| 2WYR A | 232 | 2.24Å C+ | PhTET1 12-mer peptidase | Vellieux unpublished |
| 3EDH A | 65 | 1.25Å C+ | bone morphogenic protein | MacSweeney 2008 37 |
| 3HOL A | 352 | 1.98Å C+ | transferrin-binding protein | Moraes 2009 24 |
| 3SR3 B | 302 | 1.50Å C+ | microcin C peptidase | Nocek 2012 38 |
| 3T6Q A | 388 | 1.90Å C+ | Toll-like Leu-rich repeat | Ohto 2011 31 |
| 4AMQ A | 358 | 2.17Å C+ | nucleotidyl transferase | Abergel unpublished |
| 4DVK A | 71 | 2.21Å C+ | viral env ribonuclease | Krey 2012 28 |
| 4MGE A | 7 | 1.85Å C+ | cellobiose transporter | Anderson unpublished |
| 4NN5 C | 169 | 1.90Å C+ | cytokine/receptor TSLP | Verstraete 2014 26 |
| 4TVP E | 28 | 3.10Å C- | human anti-HIV-1 env FAB | Pancera 2014 32 |
Residue number of second Cys.
C means cis. + or - is handedness of SS χ3 (see Conformations section).
Figure 2a shows Cys-cis-Cys352 of the PDB ID: 3HOL structure 24, in its clear electron density at 1.98Å resolution. This cis peptide example, and seven others in related proteins, occur in bacterial transferrin-binding proteins used to co-opt iron from the host organism 24. The disulfide loop probably plays a structural role here, since it is beautifully packed on a 6-stranded β sheet in a tight domain-domain interface (Figure 2b), affecting and probably stabilizing local conformation and interactions. A similarly well-packed case with a probable structure-stabilizing role is Cys-cis-Cys 169 of the 1.9Å PDB ID: 4NN5 cytokine receptor 26. That vicinal SS turns and caps the top of a Greek key β arch, tightly contacting the curving backbone and a Trp ring on the adjacent-strand β arch. Its role is analogous to the support and specificity provided by conserved Tyr-Gly H-bonded tyrosine corners that turn the β arch in other families of Greek key proteins 27. A similar structural role is played by a Cys-cis-Cys in the PDB ID: 4DVK viral-envelope ribonuclease 28 and by a Cys-trans-Cys in the PDB ID: 3E2V amidohydrolase (Table 2), where the vicinal disulfides form peculiar helix N-caps 29. Those definitely provide the N-cap function of specificity for preventing an earlier helix start, but it is unclear whether they hurt or enhance local stability. A vicinal SS is quite unfavorable in itself, but in 4dvk it creates good N-cap-type interactions in an unprecedented way: the Gln sidechain at the N-cap+3 position makes a cap-box H-bond 30 to the backbone CO of residue N-cap-2 and extensive van der Waals interaction with the disulfide (see the 4dvk_SS_N-cap 3D graphics). In 3e2v there are no van der Waals contacts within the local sequence, and the only H-bond across the vicinal SS is water mediated.
Figure 2.
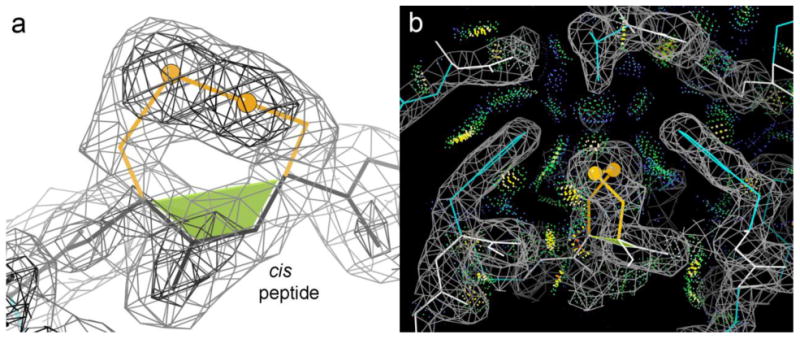
The buried, tightly packed vicinal SS in the PDB ID: 3HOL transferrin-binding protein 24. (a) Closeup of the cis vicinal SS in its clear electron density. The cis-nonPro peptide is marked by the green trapezoid. (b) Extensive, favorable, all-atom contacts 25 (dot-surface patches) with the surrounding Phe rings and β strands.
Table 2. Distinct, validated examples of Trans vicinal disulfides.
| PDBid ch | resa | resol confb | Protein | reference |
|---|---|---|---|---|
| 1GYH A | 242 | 1.89Å Tz | arabinanase | Nurizzo 2002 53 |
| 1KB0 A | 117 | 1.44Å Tx | PQQ alcohol DH | Oubrie 2002 50 |
| 1QAZ A | 189 | 1.78Å Tx | alginate lyase | Yoon 1999 57 |
| 1QLW A | 72 | 1.09Å Tz | bacterial esterase | Bourne 2000 58 |
| 1UV4 A | 206 | 1.50Å Tx | arabinanase | Proctor 2005 54 |
| 1ZK7 A | 465 | 1.60Å outc | mercuric reductase | Ledwidge 2005 3 |
| 2AD6 A | 104 | 1.50Å Tx | PQQ methanol DH | Li 2011 52 |
| 2FHF A | 644 | 1.65Å Tx | pullulanase | Mikami 2006 56 |
| 2PGZ A | 191 | 1.76Å Tx | acetylcholine BP | Hansen 2007 44 |
| 3BWS B | 347 | 1.99Å Tx | leptospiral antigen | Guiseppe 2008 59 |
| 3E2V A | 117 | 1.50Å Tx | amidohydrolase | Almo unpublished |
| 3GXB A | 1670 | 1.90Å Tx | von Willebrand F | Zhang 2009 47 |
Residue number of second Cys.
T means trans. x or z describes SS vs backbone shape (see Conformations section).
Has dihedral outliers, is not either Tx or Tz (see Conformations section).
Besides the transferrin-binding proteins related to 3HOL, another large set of related Cys-cis-Cys SS structures comes from the Leu-rich repeats (LRR) in a group of Tolllike receptor complexes that recognize bacterial lipidA, such as the RP105/MD-1 complex of PDB ID: 3T6Q31. Here the Cys-cis-Cys SS may affect receptor function, because it greatly changes the shape of its LRR repeat (see Figure 3), which includes Asn 402 (circled) whose attached 9-unit carbohydrate provides much of the interaction with the lipidA-binding MD-1 chain (green).
Figure 3.
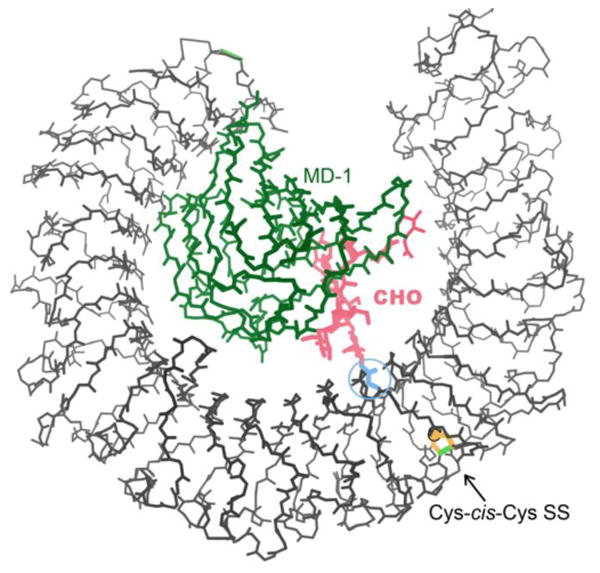
Cys-cis-Cys388 in the Leu-rich repeat Toll-like receptor of the PDB ID: 3T6Q structure31. The vicinal SS (gold) distorts its repeat, which contains Asn402 (blue), which links the branched carbohydrate (pink) that helps bind MD-1 lipidA-binding protein (green).
One example is found in a mature antibody: Cys27c-cis-Cys28 in a light-chain hypervariable loop of the 35o22 human-anti-HIV-1FAB, bound to pre-fusion HIV-1 env trimer in the PDB ID: 4TVP structure32. At 3.1Å resolution its conformational details are not at all reliable, but it is almost certainly both SS-bonded and cis. The vicinal SS seems to help form a stable and unusual local structure which closely backs up the other loops that directly bind to the protein epitope and especially its N-linked glycans.
Human transglutaminase 2 (PDB ID: 2Q3Z) 33 forms Gln-Lys crosslinks in tissue, is implicated in the pathogenesis of celiac disease, and has catalysis-independent functions in signaling and in cell adhesion. Although not at the active site, oxidation of its Cys-cis-Cys SS 371 is a major factor in enzymatic inactivation and in a large conformational rearrangement, probably promoted by disulfide-exchange with the nearby Cys230 34. The (F/Y) CCGP sequence is conserved in five different forms of transglutaminase 33. As usual, the reduced form, seen in PDB ID: 1KV3 35 has a trans peptide conformation.
In the PDB ID: 1WD3/1WD4 structures of a yeast arabinofuranosidase 36, the Cys-cis-Cys 177 is at the active site, and its sulfurs make hydrophobic contact with the arabinofuranose bound in 1WD4. The Ala double mutant of the vicinal SS has a lower Kcat as well as lower binding affinity, and the CC sequence is conserved within the Family 54 (see Methods for the CAZy family definitions) glycosyl hydrolases 36. Family GH43 arabinanases (such as the 3CU9 shown in Figure 1) and the eel agglutinin of PDB ID: 1K12 have vicinal disulfides that bind their substrate sugars in a very similar manner to 1WD4 36. Those Cys-Cys peptides are trans rather than cis, have a different overall fold, and are treated in the next section.
Trans vicinal SS examples and functions
Cys-trans-Cys vicinal disulfides occur more often than cis ones. A search for i to i+1 SS bonds within our non-homologous quality-filtered reference dataset (see Methods) identified 12 distinct trans examples (Table 2), compared with just two cis examples. A full-PDB x-ray search at <2Å resolution found many more, including relatives of the original 12 and 9 new distinct, interesting cases. Most trans vicinal SS are also functionally important conformational arrangements.
Perhaps the best-known example of a biologically critical vicinal SS is in nicotinic acetylcholine receptor (nAChR), where a Cys-trans-Cys vicinal disulfide is necessary both for agonist/antagonist binding and for coupling that binding to ion-channel opening 10. The nAChR homo- or hetero-pentameric ligand-gated ion channels mediate rapid post-synaptic neurotransmission by opening the ion channel when acetylcholine or another agonist binds between pairs of the extracellular receptor domains. nAChRs are implicated in many neurological disorders and are important drug targets 39. It has long been known that the nAChR binding site contains a vicinal disulfide bond (C192-C193) whose redox sensitivity is strongly decreased by the conformational change that occurs on agonist binding, and that this SS occurs only in the lowest molecular-weight isoform, called α 10. The naming convention was subsequently adopted such that by definition the α subunits are those with the vicinal SS. Indeed it has remained true that all functional pentamers in nAChR, and in many similar receptors across a wide variety of organisms, contain at least one α subunit which forms the “principal” face of the intersubunit binding site 40-42, opposite the “complementary” binding face of the adjacent subunit. [Note: the vicinal SS is in “Loop C”, not in the 13-residue “Cys loop” that is also an important feature of the principal binding face.]
Crystal structures of the extracellular domain of nAChR-α1 (PDB ID: 2QC1) 43 and of AChBP a closely related soluble acetylcholine-binding protein (PDB ID: 1I9B, 2PGZ, 3T4M) 12, 44, have shown that the vicinal SS is definitively trans, that its peptide NH H-bonds to a backbone CO of bungarotoxin 43 and that in essentially all bound states the disulfide makes van der Waals contact with the ligand (PDB ID: 1UV6, 1UW6) 44, 45. As shown in Figure 4, the SS and its Loop C moves the farthest (about 10Å) in the allosteric conformational change underlying channel activation 39, 45. Thermodynamic cycles for mutations which include modifying the vicinal SS peptide backbone 46 have recently shown that involvement of a cis/trans conversion in receptor function is highly unlikely, but have strongly implicated an intersubunit H-bond network between the vicinal SS NH and an Asp on the complementary face as critical to nAChR ion-channel function.
Figure 4.
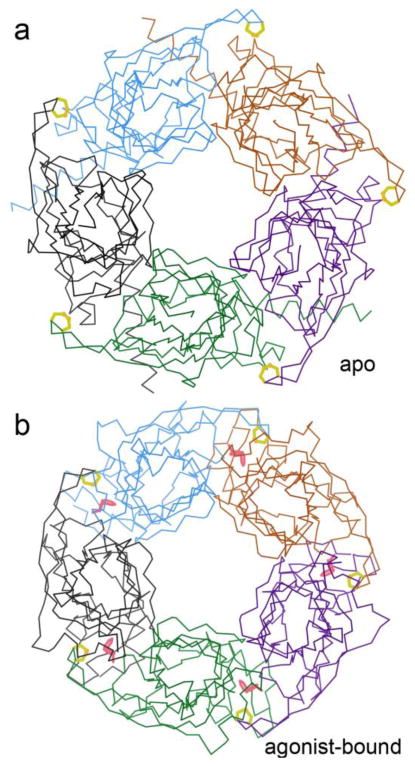
Comparing conformations in different forms of AChBP. (a) Apo pentamer, with the vicinal SS (yellow) and the C loop in their open form (PDB ID: 3T4M 44). (b) Agonist-bound pentamer, with the vicinal SS and C loop closed inward to help bind the nicotine (pink) (PDB ID: 1UW6 45).
In contrast with the many highly ordered, tightly packed vicinal disulfides, several other examples besides nAChR function on mobile loops or arms. Von Willebrand factor is an extremely long blood-clotting factor of disulfide-linked, glycosylated multimers. Each monomer contains over a dozen domains of four quite different types whose various protein-binding and platelet-binding activities depend on its multimer size. That size is tightly regulated by shear flow in the blood, by calcium binding, and by partial unfolding and specific cleavage of the A2 domain. Structures of the A2 domain (PDB ID: 3GXB, 3ZQK,) 47-48 show that its Cys-trans-Cys 1670 vicinal SS is exposed at the end of the C-terminal β strand of the doubly-wound α/β fold, from which the partial unfolding starts. Mutation or deletion of those half-cystines changes the response to shear stress and promotes disease states by accelerating A2 unfolding and cleavage 49. The tight lump of the vicinal SS is proposed to act like a “shear-bolt”, stabilizing the folded form only until suddenly unfolding at the appropriate level of shear stress 47.
An even more mobile example is found in the MerA bacterial mercuric reductase. In the inactive oxidized-form PDB ID: 1ZK7 crystals, the vicinal SS is very close above the active-site disulfide and flavin cofactor in the other chain of the biological unit, across a deep cleft in the dimer interface (Figure 5). In the active reduced-form PDB ID: 1ZK9 crystals, 11 residues at the protein C-terminus are disordered 3. That mobile tail, highlighted in red, allows the reduced SH pair to access and bind incoming mercuric ion substrate, either protein-bound or in solution, and deliver it to the active site for reduction to the uncharged nontoxic Hg0 3.
Figure 5.
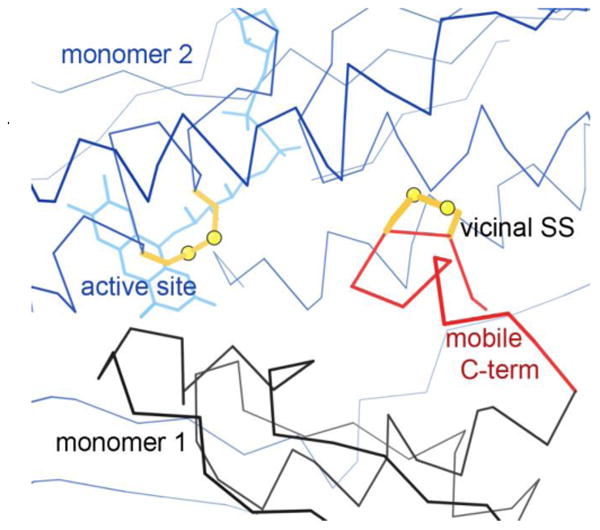
The Cys-trans-Cys465 vicinal SS (gold, at right) in the 1ZK7 inactive oxidized-form structure of mercuric reductase 3. In the active reduced form, the C-terminal region (red) is highly mobile, enabling the 464-465 SHs to deliver Hg ion substrate. Here, the vicinal SS on one chain of the biological dimer (black) sits quite close to the active-site SS and flavin of the neighboring chain (blue, at left).
One large group of Cys-trans-Cys SS is the bacterial quino-heme alcohol dehydrogenases such as PDB ID: 1KB0, 1W6S, and 2AD6 50-52. The Cys-trans-Cys SS forms part of the central cavity in these 8-blade β-propeller structures, and helps bind the pyrroloquinoline quinone (PQQ) cofactor. The vicinal SS lies between the PQQ and the cytochrome C-type heme, and is reported to be essential for the long-range electron transfer 50. As shown in Figure 6, both sulfurs and a Cβ of the vicinal SS make extensive contact with all 3 rings of the PQQ, including carbon C5 which becomes non-planar after the enzymatic reaction 52.
Figure 6.
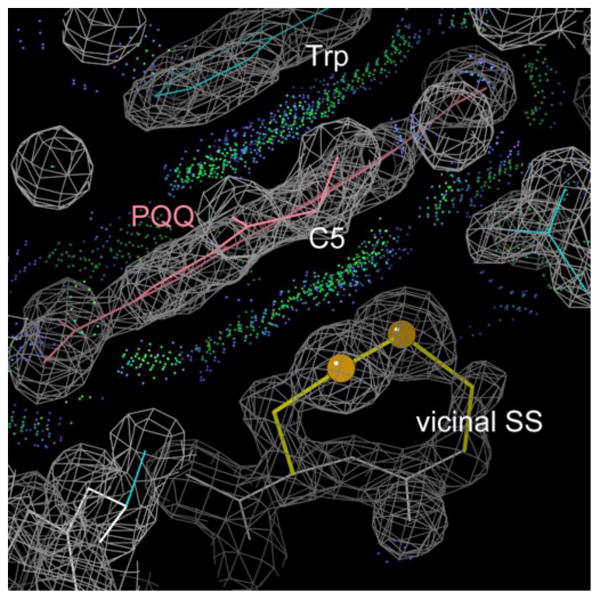
The PQQ quinone (pink) of the 1KB0 quino-heme alcohol dehydrogenase 50 sandwiched between a Trp ring and the hydrophobic platform provided by the vicinal SS. Green dots show favorable van der Waals all-atom contacts.
The trans vicinal SS set contains two family GH43 arabinanases, PDB ID: 1GYH from Cellvibrio japonicus 53 and PDB ID: 1UV4 from Bacillus subtilis 54. In each of these 5-blade β-propeller structures, the Cys-trans-Cys sulfurs provide a hydrophobic platform to bind the undecorated side of the arabinose ring, complementing the sugar oxygen interactions made by polar side chains. The equivalent vicinal SS is also present in the Geobacillus stearothermophilus family GH43 arabinanase of PDB ID: 3CU9 (Figure 1) and PDB ID: 3D61 7. In the fucose lectin of eel agglutinin (PDB ID: 1K12) 55, a small trimeric Greek-key β protein, the Cys-trans-Cys SS sits just below the bound fucose, interacting with its C1 and C2 ring atoms. See interactive graphics of 1k12_overall, and of 1k12_SS-fucose to view this arrangement in 3D. The RGDCCGER sequence is conserved in a number of related proteins 55. The vicinal SS of the pullulanase TIM-barrel domain (PDB ID: 2FHF) 56 also helps bind the carbohydrate substrate, but instead of lying directly below a specific sugar ring the disulfide makes multiple, somewhat looser interactions with both SS and backbone, which appear to help curve the branched oligo-glucose chain around it. Given the similar vicinal-SS-to-ligand interactions in the quino-heme alcohol dehydrogenases above and in the arabinofuranosidase cis example, there are five cases of such ligand-binding in five quite different protein folds. See Discussion section for further comparisons and comments.
Conformations of backbone and SS
A disulfide conformation is described by the succession of χ1, χ2, χ3, χ2′, χ1′ side-chain dihedral angles, where t means near 180° and + or - are near ±60° (±90° for χ3). All trans vicinal SS cases we checked adopt a ++-+- SS conformation, which is nearly compatible with the 3.8Å Cα-Cα distance of a trans peptide (the examples average 3.74Å). The dominant conformation for cis examples is t-+--, with 3 cases of t+-+-, each of which can provide the shorter 2.95Å Cα-Cα separation across a cis peptide (the examples average 2.92Å). Figure 7 panels a vs b compare the χ3+(cis plus) χ3- (cis minus) conformations (χ3 is the central of the 5 dihedral values that describe the SS conformation), where the backbone stays the same and the SS changes handedness. Panels c and d compare the more common trans Tx conformation versus the rarer Tz with a flipped-over peptide. Each example is very clearly one conformation or the other, with no evidence in the electron density for conformational heterogeneity or exchange in either SS or backbone. However, both cis and trans examples often show evidence of an alternate conformation with the SS bond broken, which is a common result of synchrotron radiation damage 60.
Figure 7.
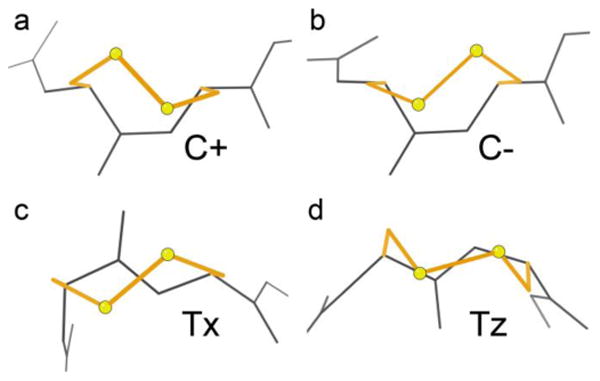
Typical examples of the two cis and two trans conformations of vicinal disulfides. (a) C+, in PDB ID: 3HOL; (b) C-, in PDB ID: 1YM0; (c) Tx, in PDB ID: 2QC1; (d) Tz, in PDB ID: 3CU9.
An early NMR study showed these same 4 conformations for a vicinal SS dipeptide in solution, as assessed by chirality of the SS (χ3) and dipolar couplings around the angles at Cα 20. Those 3 NMR-measurable dihedral angles turn out to distinguish all accessible conformations of the 7 variable parameters in a vicinal SS. Overall, trans was seen as preferred to cis by 1.5:1. Within folded proteins with crystal structures, we see a much stronger preference for trans over cis, on the order of 4:1. A similar imbalance between solution peptides and folded proteins is also seen for the more statistically significant cis-Pro case (15-20% vs 5%) 61, presumably because most proteins have evolved to avoid the kinetic folding problems of requiring a slowly converting conformation disfavored in solution 62.
Within cis, NMR showed the plus χ3 conformer preferred to the minus χ3 by 3:1. The crystal structures completely define the 8-membered ring conformations, and we see plus χ3 (C+) preferred to minus (C-) in the disulfide portion (i.e., more t-+-- than t+-+-) by 10:3 examples in our small dataset, with essentially identical backbone conformations, matching the 3:1 NMR ratio of C+ to C-.
Within trans, Creighton finds all χ3 minus, but detects two distinct but incompletely defined conformer states for ψ1, ϕ2, at a ratio of 3:1, with the acetylcholine receptor example in the commoner form. Out of 12 unrelated examples of Cys-trans-Cys-SS we find 9 that match acetylcholine receptor (Tx), and 2 in the other conformation (Tz). One case (PDB ID: 1ZK7) differs from both; it is quite mobile, has fairly strong but ambiguous electron density and shows dihedral outliers, so it is omitted from our conformational analyses.
We find that in trans, the SS stays about the same for conformations with different peptide directionality. The commoner type (as in acetylcholine receptor) looks like an X in projection from above the SS, while the less common form configures SS and backbone in nearly aligned Z shapes, so the forms are very easy to tell apart (Figure 6 c vs d). As labeled in Figure 6, we suggest calling the four conformations C+, C-, Tx, and Tz. This agreement between solution peptide populations and crystal-structure protein populations is satisfyingly close, except for the above-noted difference that the crystal structures show a considerably higher preference for trans over cis.
As noted in the Introduction, the 2003 review of trans vicinal disulfides by Carugo et al. 16 categorizes their examples as having either type VIII or type II β-turn conformation. In our dataset, the trans Tx examples have an average i to i+4 Cα-Cα distance of 6.5±0.6Å, indeed under the 7Å definitional cutoff for a beta turn, although the Tz average is 7.9±0.8Å. The Tx acetylcholine-binding vicinal SS case has ϕ,ψ(1), ϕ,ψ(2) values within the 60° ranges defined for type VIII turns, but no other examples match either type VIII or type II. Additionally, no case has a β-turn H-bond, so that “turn” no longer seems the best description. Somewhat unintuitively, the cis vicinal SS have even more extended backbones than the trans, with a mean i to i+4 Cα-Cα distance of 8.3±0.6Å for C+ and 8.8±1.2Å for C-. They often occur in or at the end of β strands.
Table 3 lists the above and other parameters for each of the four conformational types of vicinal SS, flagging those that deviate from ideal with p-values <0.01 (see Methods). All of these vicinal disulfide conformations are shown to be possible by their occurrence in clear, high-resolution electron density for peptides in non-homologous structures at better than 2Å resolution. However, their extreme rarity and distorted geometries imply they are probably quite strained. For cis examples the conformation is already less favorable, but they have ω quite close to planar (averaging +4° for C+ and -1° for C-). However, the trans peptides are twisted by an average of +23° in Tx and -17° in Tz with no example within 10° of 180°; this is the largest departure from ideality that we find. To understand this effect, we constructed ideal-covalent-geometry models of trans and cis Cys-Cys peptides, with rotatable φ, ψ, and χ1 conformational angles set to the average of each of the 4 observed forms. For trans the S atoms are much too far apart to bond (3.4Å). Adding the observed 23° twist in ω brings the sulfurs much closer (2.4Å), because that twist swings the two Cα-Cβ bond vectors toward one another. The final adjustment to allow an SS bond is achieved by a consistent pattern of tightening both covalent bond angles across the peptide by an average of -2.2° and both angles internal to the ring at Cα by an average of -2.4°. For the two cis conformational forms, the Cα-Cα 1-2 distance across the peptide more closely matches its ideal of that typical for cis-nonPro, but when they are built in ideal geometry either the S atoms are much too close together or the χ angles are very unfavorable. The necessary adjustments in bond angles and dihedrals seem to follow a less consistent pattern than for trans, and the lower resolution and fewer examples in cis prevent a clear analysis. The near-planarity of ω in the C+ and C- conformations can, however, be rationalized by the fact that an ω twist from cis happens to be much less effective in swinging the Cα-Cβ bond vectors than is a twist from trans.
Table 3. Characterization of the 4 conformational classes for vicinal disulfides.
| Tx | Tz | C+ | C- | ideal | |
|---|---|---|---|---|---|
| # examples | 17 | 9 | 13 | 3 | |
| Distances | |||||
| Cα-Cα 1-2 | 3.73 | 3.74 | 2.93 | 2.89 | 3.80/2.95±0.1Å |
| Cα-Cα 0-4 | 6.51 | 7.85 | 8.26 | 8.80 | ≤7Å for β-turn |
| Angles, SS | |||||
| C-Cα-Cβ 1 | 105.8° | 106.1° | 110.8° | 110.4° | 110.1 ±1.9° |
| Cα-Cβ-S 1 | 115.5° | 113.7° | 114.7° | 114.9° | 114.4 ±2.3° |
| Cβ-S-S 1 | 108.5° | 109.7° | 108.3° | 107.8° | 104.2 ±2.1° |
| Cβ-S-S 2 | 107.6° | 110.1° | 101.3° | 106.2° | 104.2 ±2.1° |
| Cα-Cβ-S 2 | 113.7° | 112.9° | 114.0° | 116.0° | 114.4 ±2.3° |
| N-Cα-Cβ 2 | 106.2° | 106.4° | 109.7° | 110.8° | 110.5 ±1.7° |
| Angles, main-chain | |||||
| tau 1 | 113.6° | 111.4° | 107.2° | 109.9° | 111.0 ±2.8° |
| Cα-C-N | 115.5° | 114.0° | 119.4° | 106.2° | 118.2/119.3 ±2° |
| C-N-Cα | 121.0° | 122.1° | 125.3° | 118.1° | 122.6/125.9±3° |
| tau 2 | 116.0° | 117.1° | 111.2° | 109.3° | 111.0 ±2.8° |
| Dihedrals, SS | |||||
| χ1 1 | 68.2° | 82.5° | -162.3° | 168.8° | 65/-178° ±9° |
| χ2 1 | 79.6° | 89.9° | -71.0° | 55.9° | 79/-73° ±17° |
| χ3 | -103.3° | -86.4° | 93.2° | -92.2° | -86/-93° ±9/11° |
| χ2 2 | 76.0° | 82.8° | -53.2° | -71.8° | 79/-73° ±17° |
| χ1 2 | -50.7° | -41.7° | -49.9° | -84.3° | -65 ±9° |
| Dihedrals, main-chain | |||||
| ψ 1 | -34.3° | 177.4° | 147.7° | 132.9° | |
| Ω | -157.4° | 163.6° | 3.9° | -0.7° | 180/0° ±5° |
| φ2 | -135.0° | 59.6° | -133.9° | -146.4° | |
Notes: For sources of ideal values, extra examples included, and statistical calculations, see Methods. 1ZK7 is not included because it does not fit any of the four conformations and has ambiguous density. For each conformation and parameter, vicinal-SS means that have p-values <0.01 are red if higher than ideal and blue if lower than ideal. Entries with p-values <0.001 are also italicized and underlined. C-, with only 3 examples, is not statistically evaluated.
Like many other rare and energetically disfavored conformations 63-64, these genuine and conserved examples of vicinal disulfides are very likely to be functionally important in some way, as we have seen for both cis and trans examples described above.
Conformational switching
Given the possibilities both of disulfide oxidation/reduction and of cis-trans isomerization, there could be scope for functionally relevant switching in vicinal disulfides of the sort that has been shown for cis-prolines, where isomerization produces a slow step in protein folding 62 and can control biological clock functions 65. However, we found no authentic cases of a dynamic switch between cis and trans vicinal SS conformations, as alternates or between equivalent structures or substantiated in the literature. Presumably that barrier would be extremely high with the 8-membered ring in place. There were, however, cases where the backbone conformation was originally mis-assigned and subsequently corrected, as described in the Introduction. There is also a dodecamer structure (PDB ID: 2WYR) where 10 of the chains have a well-built cis vicinal SS and the other 2 are incorrectly fit as highly twisted trans, in spite of CO density clearly indicating cis. For the less dramatic peptide flip between Tx and Tz conformations within trans, there is a switch on the evolutionary time scale among family GH43 5-bladed-propeller arabinanase enzymes, between Tx in 1UV4 and Tz in 1GYH and 3CU9.
Oxidation/reduction of vicinal disulfides does happen quite frequently, but the only case in our set that is definitely part of a dynamic, directly catalytic redox cycle is in the 1ZK7 mercuric reductase of Figure 4. In the 2Q3Z transglutaminase, the vicinal SS does not directly take part in catalysis, but its oxidation is coupled in a complex manner with inactivation and conformational change. For other examples (e.g., acetylcholine receptor), the protein is inactive if the disulfide is reduced, but reduction is not a normal part of the functional cycle. An interesting case has been described, which could be more general, of a non-native vicinal disulfide as the major intermediate (accumulating to as much as 60%) during folding of a small cystine-knot protein 66. In contrast, for transglutaminase, a separate disulfide is apparently an intermediate in forming the vicinal one 33. In the bone morphogenetic Zn protease (BMP-1) of PDB ID: 3EDH at 1.27Å 37, the Cys-cis-Cys 65 SS and another residue on each side show clean electron density for each atom at half of full height (some modeled at 50% occupancy), but there is no indication of an alternate conformation. The SS is very close to the Zn site but with a bound DMSO in between. This loop is completely disordered in all other structures of BMP-1, so it is not known whether the disulfide is broken in those forms. At a more trivial level, many vicinal SS examples show difference density peaks indicating an alternate conformation with the SS bond broken; as noted above, that is a very common result of synchrotron radiation damage and may or may not indicate instability in vivo.
There is one protein in our dataset for which the vicinal disulfide is genuinely formed in one chain of a crystal structure and broken in other chains. That is the Cys-cis-Cys 302 SS seen in chain B of the 1.5Å PDB ID: 3SR3 MccF microcin C peptidase, which is reduced and trans in chain A, and is also reduced in all 4 other crystal structures of MccF 38. The electron density (2mFo-DFc) is very clear for the conformation modeled in each chain of 3SR3, but difference peaks (mFo-DFc) show a small component of the other form in each case, indicating that this change is a dynamic equilibrium. The two different states in chains A and B of 3SR3 are illustrated in Figure 8, in stereo. The Nocek et al. paper thoroughly discusses the catalytic and evolutionary aspects, but does not mention the vicinal disulfide.
Figure 8.
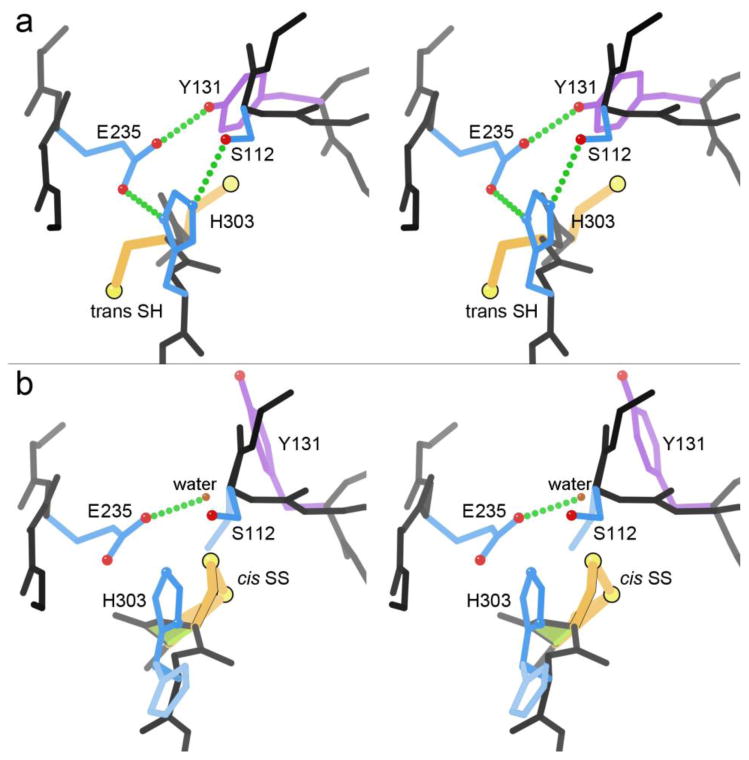
Microcin C peptidase MccF in the 3SR3 structure at 1.5Å 38. (a) The intact Ser-His-Glu-Tyr active site of chain A, with Cys 301 and 302 as reduced SH. (b) The disrupted active site in chain B, where the vicinal Cys-cis-Cys302 is SS-bonded.
Microcin C is a bacterial heptapeptide-adenylate, Trojan-horse antibiotic. The peptide allows uptake into other bacterial cells, where it is processed down to a C-N linked aspartyl-adenylate which inhibits Asp tRNA synthetase. MccF is a serine peptidase specialized to cleave the amino-acid to nucleic-acid linkage and detoxify either intact or processed microcin C, thus protecting the bacterium that produces the antibiotic, or providing resistance in a target cell (in this case, Bacillus anthracis). MccF has a Ser-His-Glu catalytic triad backed up by a Tyr OH that H-bonds with the Glu to help orient it and spread its buried charge, analogous to the classic trypsin-like Ser-His-Asp plus Ser 67-68.
The adjacent Cys residues in MccF immediately precede the catalytic His 303, and are normally in the trans SH form shown in Figure 7a, with an intact and functional catalytic triad. When the Cys-cis-Cys SS forms (Figure 7b), it greatly changes the backbone conformation, pushes away the Tyr (its OH is replaced by a water), disrupts most of the charge-relay H-bonding, and disorders both the His and the Ser. This form would surely be almost totally inactive.
The vicinal disulfide formation in 3SR3 chain B is presumably an unintended crystallographic artifact in this case; the apo W180A 3SR3 crystals grew from slightly different precipitant (although not obviously any more oxidative), and in a different space group, than the other 4 MccF structures. However, the SS-bonded form in 3SR3 chain B suggests a strategy of introducing adjacent Cys to design brute-force, redox-sensitive inactivation of enzyme active sites. A similar idea, but without prior evidence for the extent of disruption, has been used to modestly inhibit ribonuclease A on changing redox state, for a Cys-Cys mutant on the N-terminal helix 69.
Discussion
Although extremely rare, vicinal disulfides are very distinctive and easy to identify. They can be located either in very tightly packed domain interiors or on relatively mobile loops. It should by now be completely uncontroversial that they can occur in proteins with either a cis or a trans peptide, more frequently trans. Each peptide form apparently has just two possible conformations, differing in peptide orientation for trans and in SS-link handedness for cis.
In crystal structures the assignment of whether adjacent Cys are SS-bonded or not is very reliable at any resolution unless there is local disorder. By NMR, such assignment can be made reliably for an isolated pair (e.g., in Wang et al. 15 for the PDB ID: 1DL0 J-atracotoxin), but is very difficult where 3 or 4 Cys are very close together as often true in small SS-rich toxins and hormones. For example, in the 4-Cys cluster of PDB ID: 1M4F hepcidin 70 Cys13 and Cys14 were assigned as SS-bonded. However, the 1.9Å PDB ID: 3H0T FAB-complex crystal structure, along with painstaking chemical, NMR, and mass spectrometry analyses 71, showed that the hepcidin connectivity is actually 13-10 and 14-22. In contrast, NMR assignment of the detailed stereochemical conformation is probably more reliable than in crystal structures with resolution worse than about 2.5Å. We have not evaluated SS connectivity for all NMR structures, but to our surprise there are no demonstrably genuine vicinal SS examples in crystal structures of small SS-rich proteins, so presumably they are quite unusual in that context.
Each vicinal SS found is worth noting and its possible functional role considered, especially if the sequence of two adjacent Cys is conserved in related proteins. Many known examples have been demonstrated essential for function in important systems, by a very wide variety of mechanisms, as described above. Others are apparently structural elements, or are suggestively near a functional site but without a demonstrated mechanistic involvement. We found no cases of peptide cis-trans conformational switching. There are two cases of a verified redox-active functional coupling – catalytic for mercuric reductase and conformational for tissue transglutaminase. In contrast, for acetylcholine receptor and for von Willebrand factor the essential vicinal SS remains bonded during conformational change of the protein. Unexpectedly, the most widespread functional role we find for vicinal disulfides is in the specific binding of sugars or other ring ligands.
Although binding of saccharides is often described only in terms of the polar hydrogen bonds, hydrophobic interactions are also important components of specificity. A sugar ring can be thought of as “bar coded” by which positions have attached OH groups and whether each OH is up or down. An interaction such as these seen for vicinal disulfides (Table 4, Figure 9) can prohibit binding for an axial OH rather than an H substituent at one or more of the contacted ring positions. Thus, in combination with H-bonding groups to recognize positions that do have an OH, the vicinal SS can require non-OH positions.
Table 4. The five unrelated families that show vicinal SS binding to sugar or other rings.
| Protein | Fold | confa | PDB | organism | 50sfb | ligand |
|---|---|---|---|---|---|---|
| Arabinofuranosidase | ConA lectin | C- | 1WD3 | Aspergillus kawa. | √ | |
| C- | 1WD4 | Aspergillus kawa. | arabinose | |||
| GH43 arabinanase | β-propeller 5 | Tz | 1GYH | Cellvibrio japonicus | √ | |
| Tx | 1UV4 | Bacillus subtilis | √ | |||
| Tz | 3D61 | Geobacillus stearo. | arabinobiose | |||
| Tz | 3D5Z | Geobacillus stearo. | arabinotriose | |||
| Fucose lectin | Greek-key β | Tz | 1K12 | Anguilla anguilla | fucose | |
| PQQ methanol DH | β-propeller 8 | Tx | 2AD6 | Methylophilus meth. | √ | PQQ |
| Tx | 1KB0 | Comamonas testost. | √ | PQQ | ||
| Tx | 1W6S | Methylophilus extorq. | PQQ | |||
| Pullulanase | TIM barrel | Tx | 2FHF | Klebsiella pneumon. | √ | oligo-glucose |
See Conformations section.
Checkmarked PDBs are in the Top8000_SFbest_hom50 dataset.
Figure 9.
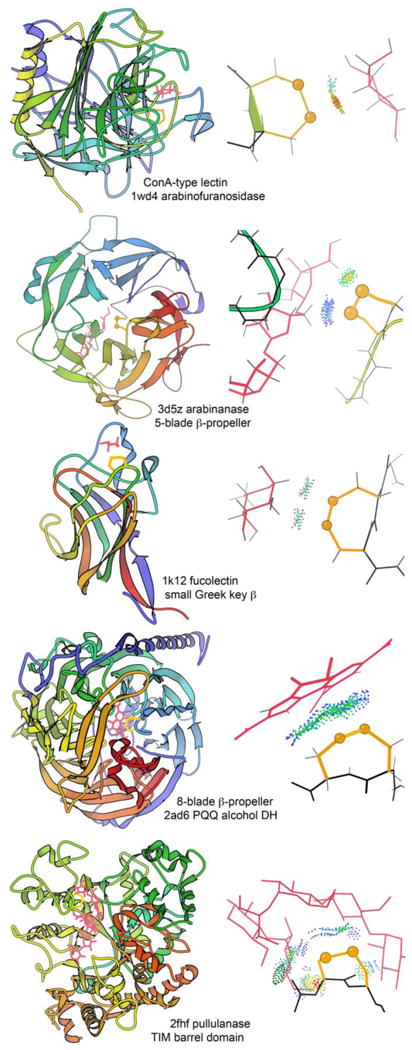
For the five unrelated families of proteins where a vicinal SS provides saccharide or other ring binding specificity, an example ribbon schematic of the chain or domain fold is provided in the lefthand column, and a closeup of the contact interactions between vicinal SS and ring ligand is provided in the righthand column. Ribbons are N-to-C colored, disulfides are in gold, ligands in pink, and all-atom contact dots in blue, green, and yellow. View the 1k12_SS-fucose 3D graphics or the graphical abstract to see the fucolectin site in its local context.
This same negative but specific recognition function is often provided by a tryptophan or other aromatic side chain. However, a vicinal SS is perhaps even harder to move out of the way if the wrong ligand tries to bind, pushing the sugar out of position for the specificity H-bonds to ring OHs. A vicinal disulfide acts conformationally rather like a super-sized proline ring that can rigidly organize a connected region of side chains and backbone.
Methods
Our starting reference dataset of 6,765 protein chains is the Top8000_SFbest_hom50 list 64, available on GitHub at http://github.com/rlabduke/reference_data. It is quality-filtered at the chain level by requiring deposited diffraction data, resolution <2.0Å, MolProbity score 72 (which balances all-atom clashes, Ramachandran, and rotamer criteria) <2.0, ≤5% of residues with bond-length outliers >4σ, ≤5% with bond-angle outliers >4σ, ≤5% with Cβ deviation outliers >0.25Å, and ≥38 residues. To control redundancy, within each PDB sequence-homology cluster at the 50% level as of March 29 2011, only the best otherwise-acceptable chain is included, based on the average of resolution and MolProbity score. Since local quality varies greatly within each structure, individual residues are filtered by requiring no all-atom steric clashes and no atom with: a B-factor ≥40, a 2mFo-DFc map value ≤1.1σ, or a real-space correlation coefficient ≤0.7.
The Top8000_SFbest_hom50 list included only 2 cis and 12 trans vicinal disulfides. To expand the examples, two further searches were done. For cis cases, the search was across the entire x-ray PDB for Cys-cis-Cys peptides, starting from our previously defined cis-nonPro peptides23, and resulting in the 13 separate Cys-cis-Cys protein families of Table 1. For trans cases, the original 12 of Table 2 were augmented by a search for i to i+1 SSBOND records in the headers of current x-ray PDB files at <2.0Å resolution, resulting in a further 9 examples with quite distinct functions (PDB ID: 1K12, 1TG7, 1US4, 2ZWS, 3RPW, 3W15, 3ZZY, 4IHM, 4LQK). To improve coverage in the geometry statistics of Table 3, three further related but non-identical structures were included for cis (2Z63, 3V8U, 4DW5) and 6 further for trans (1W6S, 2QC1, 2VWS, 2WN9, 3CU9, 3ZQK).
For the conformational comparisons in Table 3, ideal values of amino-acid bond angles are taken from Engh and Huber 73 (1991). Ideal angles and dihedrals across the SS bond are from Sobolev et al. 74 (2015), and χ1 values for Cys are from Hintze et al. 64. For cis peptides, ideal backbone bond angles and Cα-Cα 1-2 distance were determined from mean values in 400 cis-nonPro quality-filtered examples. Geometrical parameters for the vicinal SS examples were provided from Phenix 75. Within each conformation, significance of mean deviation from ideal values was determined by one-sample t-tests done with functions in the NumPy library 76. In Table 3, deviations up or down are flagged in color for p-values <0.01. We use a 1% cutoff rather than the usual 5% because some of these data show quite non-normal distributions. Entries with p-values <0.001 are italicized and underlined as well as colored.
The vicinal SS cases were examined and verified in the KiNG molecular graphics and modeling program 77, with display of validation markup from MolProbity 72 (http://molprobity.biochem.duke.edu), electron density contours (usually at 1.2 and 3α) and difference density contours (at ±3.5 σ), and all-atom contacts 25 for which explicit hydrogens were added and optimized in Reduce 78. The rotatable ideal-geometry dipeptides were produced and studied in the Mage interactive display program 79. Figures were produced as KiNG output, somewhat modified in Adobe Photoshop.
To describe Carbohydrate-Active enZymes and binding proteins, we use the CAZy website 80, which provides the accepted classification of these proteins into families based on sequence, structure, and function relationships (http://www.cazy.org).
Supplementary Material
Highlights.
The vicinal SS-bond plays highly varied functional roles, but lacks an overall study
Despite earlier claims, protein vicinal SS can use either cis- or trans-peptide forms
Four reliably valid conformations are seen, which do not interconvert dynamically
Functions include rigidity, mobility, or binding specificity, a few with redox change
Broad, validated analysis clarifies conformations, changes & a new sugar-binding role
Acknowledgments
Funding for this work was from National Institutes of Health grants R01-GM073919 and P01-GM063210 project IV. We thank Michael Prisant for database computer system management, Bradley Hintze for running the t-tests, and Steven Lewis for the i to i+1 SS-bond search.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Richardson JS. The Anatomy and Taxonomy of Protein Structure. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 2.Der BS, Jha RK, Lewis SM, Thompson PM, Guntas G, Kuhlman B. Combined computational design of a zinc-binding site and a protein-protein interaction: One open zinc coordination site was not a robust hotspot for de novo ubiquitin binding. Proteins Struc Func Bioinf. 2013;81:1245–1255. doi: 10.1002/prot.24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ledwidge R, Patel B, Dong A, Fiedler D, Falkowski M, Zelikova J, Summers AO, Pai EF, Miller SM. Nmer A, the metal binding domain of mercuric ion reductase, removes Hg2+ from proteins, delivers it to the catalytic core, and protects cells under glutathione-depleted conditions. Biochemistry. 2005;44:11402–11416. doi: 10.1021/bi050519d. [DOI] [PubMed] [Google Scholar]
- 4.Thornton JM. Disulphide bridges in globular proteins. J Mol Biol. 1981;151:261–287. doi: 10.1016/0022-2836(81)90515-5. [DOI] [PubMed] [Google Scholar]
- 5.McDonald NQ, Hendrickson WA. A structural superfamily of growth factors containing a cystine knot motif. Cell. 1993;73:421–424. doi: 10.1016/0092-8674(93)90127-c. [DOI] [PubMed] [Google Scholar]
- 6.Craik DJ, Daly NL, Waine C. The cystine knot motif in toxins and implications for drug design. Toxicon. 2001;39:43–60. doi: 10.1016/s0041-0101(00)00160-4. [DOI] [PubMed] [Google Scholar]
- 7.Alhassid A, Ben-David A, Tabachnikov O, Libster D, Naveh E, Zolotnitsky G, Shoham Y, Shoham G. Crystal structure of an inverting GH 43 1,5-α-L-arabinanase from Geobacillus stearothermophilus complexed with its substrate. Biochem J. 2009;422:73–82. doi: 10.1042/BJ20090180. [DOI] [PubMed] [Google Scholar]
- 8.Ramachandran GN, Sasisekharan V. Conformation of polypeptides and proteins. Adv Protein Chem. 1968;23:283–438. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 9.Chandrasekharan R, Balasubramanian R. Stereochemical studies of cyclic peptides. VI Energy calculations of the cyclic disulphide cysteinyl-cysteine Biochim Biophys Acta. 1969;188:1–9. doi: 10.1016/0005-2795(69)90039-7. [DOI] [PubMed] [Google Scholar]
- 10.Kao PN, Karlin A. Acetylcholine receptor binding site contains a disulfide cross-link between adjacent half-cystinyl residues. J Biol Chem. 1986;261:8085–8088. [PubMed] [Google Scholar]
- 11.Blake CCF, Ghosh M, Harlos K, Avezoux A, Anthony C. The active site of methanol dehydrogenase contains a disulphide bridge between adjacent cysteine residues. Struct Biol. 1994;1:102–105. doi: 10.1038/nsb0294-102. [DOI] [PubMed] [Google Scholar]
- 12.Brejc K, van Dijk WJ, Klaasen RV, Schuurmans M, van der Oost J, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh M, Anthony C, Harlos K, Goodwin MG, Blake C. The refined structure of the quinoprotein methanol dehydrogenase from Methylobacterium extorquens at 1.94Å. Structure. 1995;3:177–187. doi: 10.1016/s0969-2126(01)00148-4. [DOI] [PubMed] [Google Scholar]
- 14.Xia Z-x, Dai W-w, Zhang Y-f, White SA, Boyd GD, Mathews FS. Determination of the gene sequence and the three-dimensional structure at 2.4Å resolution of methanol dehydrogenase from Methylophilus W3A1. J Mol Biol. 1996;259:480–501. doi: 10.1006/jmbi.1996.0334. [DOI] [PubMed] [Google Scholar]
- 15.Wang X-h, Connor M, Smith R, Maciejewski MW, Howden MEH, Nicholson GM, Christie MJ, King GF. Discovery and characterization of a family of insecticidal neurotoxins with a rare vicinal disulfide bridge. Nature Struct Biol. 2000;7:505–513. doi: 10.1038/75921. [DOI] [PubMed] [Google Scholar]
- 16.Carugo O, Cemazar M, Zahariev S, Hudaky I, Gaspari Z, Perczel A, Pongor S. Vicinal disulfide turns. Protein Engin. 2003;16:637–639. doi: 10.1093/protein/gzg088. [DOI] [PubMed] [Google Scholar]
- 17.Wouters MA, George RA, Haworth NL. ‘Forbidden’ disulfides: Their role as redox switches. Curr Protein Peptide Sci. 2007;8:484–495. doi: 10.2174/138920307782411464. [DOI] [PubMed] [Google Scholar]
- 18.Fass D. Disulfide bonding in protein biophysics. Annu Rev Biophys. 2012;41:63–79. doi: 10.1146/annurev-biophys-050511-102321. [DOI] [PubMed] [Google Scholar]
- 19.Mez HC. Cyclo-L-cystine acetic acid. Cryst Struct Comm. 1974;3:657–660. [Google Scholar]
- 20.Creighton CJ, Reynolds CH, Lee DHS, Leo GC, Reitz AB. Conformational analysis of the eight-membered ring of the oxidized cysteinyl-cysteine unit implicated in nicotinic acetylcholine receptor ligand recognition. J Am Chem Soc. 2001;123:12664–12669. doi: 10.1021/ja016505m. [DOI] [PubMed] [Google Scholar]
- 21.Chung BKW, Yudin AK. Disulfide-bridged peptide macrobicycles from nature. Org Biomol Chem. 2015;13:8768–8779. doi: 10.1039/c5ob01115a. [DOI] [PubMed] [Google Scholar]
- 22.Berman HM, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 2003;10:980. doi: 10.1038/nsb1203-980. http://www.wwpdb.org. [DOI] [PubMed] [Google Scholar]
- 23.Williams CJ, Richardson JS. Fitting Tips #9: Avoid excess cis peptides at low resolution or high B. Comput. Cryst Newsletter. 2015;6:2–6. http://www.phenix-online.org/newsletter/CCN2015 01.pdf. [Google Scholar]
- 24.Moraes TF, Yu R-h, Strynadka NCJ, Schryvers AB. Insights into the bacterial transferrin receptor: the structure of transferrin-binding protein B from Actinobacillus pleuropneumoniae. Mol Cell. 2009;35:523–533. doi: 10.1016/j.molcel.2009.06.029. [DOI] [PubMed] [Google Scholar]
- 25.Word JM, Lovell SC, LaBean TH, Taylor HC, Zalis ME, Presley BK, Richardson JS, Richardson DC. Visualizing and quantifying molecular goodness-of-fit: Small-probe contact dots with explicit hydrogen atoms. J Mol Biol. 1999;285:1711–1733. doi: 10.1006/jmbi.1998.2400. [DOI] [PubMed] [Google Scholar]
- 26.Verstraete K, van Schie L, Vyncke L, Bloch Y, Tavernier J, Pauwels E, Peelman F, Savvides SN. Structural basis of the proinflammatory signaling complex mediated by TSLP. Nature Struct Mol Biol. 2014;21:375–382. doi: 10.1038/nsmb.2794. [DOI] [PubMed] [Google Scholar]
- 27.Hemmingsen JM, Gernert KM, Richardson JS, Richardson DC. The tyrosine corner: a feature of most Greek key beta-barrel proteins. Protein Sci. 1994;3:1927–1937. doi: 10.1002/pro.5560031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krey T, Bontems F, Vonrhein C, Vaney M-C, Bricogne G, Rumenapf T, Rey FA. Crystal structure of the pestivirus envelope glycoprotein Erns and mechanistic analysis of its ribonuclease activity. Structure. 2012;20:862–873. doi: 10.1016/j.str.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 29.Richardson JS, Richardson DC. Amino Acid Preferences for Specific Locations at the Ends of α Helices. Science. 1988;240:1648–1652. doi: 10.1126/science.3381086. [DOI] [PubMed] [Google Scholar]
- 30.Harper ET, Rose GD. Helix Stop Signals in Proteins and Peptides: The Capping Box. Biochemistry. 1993;32:7605–7609. doi: 10.1021/bi00081a001. [DOI] [PubMed] [Google Scholar]
- 31.Ohto U, Miyake K, Shimizu T. Crystal structures of mouse and human RP105/MD-1 complexes reveal unique dimer organization of the Toll-like receptor family. J Mol Biol. 2011;413:815–825. doi: 10.1016/j.jmb.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 32.Pancera M, Zhou T, Druz A, Georgiev IS, Soto C, Gorman J, Huang J, Acharya P, Chuang GY, Ofek G, Stewart-Jones GBE, Stuckey J, Bailer RT, Joyce MG, Louder MK, Tumba N, Yang Y, Zhang B, Cohen MS, Haynes BF, Mascola JR, Morris L, Munro JB, Blanchard SC, Mothes W, Connors M, Kwong PD. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature. 2014;514:455–461. doi: 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pinkas DM, Strop P, Brunger AT, Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007;5:e327. doi: 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stamnaes J, Pinkas DM, Fleckenstein B, Khosla C, Sollid LM. Redox regulation of transglutaminase 2 activity. J Biol Chem. 2010;285:25402–25409. doi: 10.1074/jbc.M109.097162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu S, Cerione RA, Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci USA. 2002;99:2743–2747. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyanaga A, Koseki T, Matsuzawa H, Wakagi T, Shoun H, Fushinobu S. Crystal structure of a family 54 alpha-L-arabinofuranosidase reveals a novel carbohydrate-binding module that can bind arabinose. J Biol Chem. 2004;279:44907–44914. doi: 10.1074/jbc.M405390200. [DOI] [PubMed] [Google Scholar]
- 37.Mac Sweeney A, Gil-Parrado S, Vinzenz D, Bernardi A, Hein A, Bodendorf U, Erbel P, Logel C, Gerhartz B. Structural basis for the substrate specificity of bone morphogenetic protein 1/Tolloid-like metalloproteases. J Mol Biol. 2008;384:228–239. doi: 10.1016/j.jmb.2008.09.029. [DOI] [PubMed] [Google Scholar]
- 38.Nocek B, Tikhonov A, Babnigg G, Gu M, Zhou M, Makarova KS, Vondenhoff G, Van Aerschot A, Kwon K, Anderson WF, Severinov K, Joachimiak A. Structural and functional characterization of microcin C resistance peptidase MccF from Bacillus anthracis. J Mol Biol. 2012;420:366–383. doi: 10.1016/j.jmb.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bouzat C. New insights into the structural bases of activation of Cys-loop receptors. J Physiol - Paris. 2012;106:23–33. doi: 10.1016/j.jphysparis.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 40.Li SX, Huang S, Bren N, Noridomi K, Dellisanti CD, Sine SM, Chen L. Ligand-binding domain of an α7-nicotinic receptor chimera and its complex with agonist. Nat Neurosci. 2011;14:1253–1259. doi: 10.1038/nn.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Millar NS, Gotti C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology. 2009;56:237–246. doi: 10.1016/j.neuropharm.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 42.Wijckmans E, Nys M, Debaveye S, Brams M, Pardon E, Willegems K, Bertrand D, Steyaert J, Efremov R, Ulens C. Functional and biochemical characterization of Alvinella pompejana Cys-loop receptor homologues. PLoS One. 2016;11:e0151183. doi: 10.1371/journal.pone.0151183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dellisanti CD, Yao Y, Stroud JC, Wang ZZ, Chen L. Crystal structure of the extracellular domain of nAChR α1 bound to α-bungarotoxin at 1.94Å resolution. Nature Neurosci. 2007;10:953–962. doi: 10.1038/nn1942. [DOI] [PubMed] [Google Scholar]
- 44.Nemecz A, Taylor P. Creating an alpha-7 nicotinic acetylcholine recognition domain from the acetylcholine binding protein: crystallographic and ligand selectivity analysis. J Biol Chem. 2011;286:42555–42565. doi: 10.1074/jbc.M111.286583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Celie PHN, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 2004;41:907–914. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 46.Blum AP, Gleitsman KR, Lester HA, Dougherty DA. Evidence for an extended hydrogen bond network in the binding site of the nicotinic receptor: role of the vicinal disulfide of the α1 subunit. J Biol Chem. 2011;286:32251–32258. doi: 10.1074/jbc.M111.254235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Q, Zhou YF, Zhang CZ, Zhang X, Lu C, Springer TA. Structural specializations of A2, a force-sensing domain in the ultralarge vascular protein von Willebrand factor. Proc Natl Acad Sci USA. 2009;106:9226–9231. doi: 10.1073/pnas.0903679106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jakobi AJ, Mashaghi A, Tans SJ, Huizinga EG. Calcium modulates force sensing by the von Willebrand factor A2 domain. Nature Commun. 2011;2:385. doi: 10.1038/ncomms1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luken BM, Winn LYN, Emsley J, Lane DA, Crawley JTB. The importance of vicinal cysteines, C1669 and C1670, for von Willebrand factor A2 domain function. Blood. 2010;115:4910–4913. doi: 10.1182/blood-2009-12-257949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oubrie A, Rozeboom HJ, Kalk KH, Huizinga EG, Dijkstra BW. Crystal structure of quinohemoprotein alcohol dehydrogenase from Comamonas testosteroni: Structural basis for substrate oxidation and electron transfer. J Biol Chem. 2002;277:3727–3732. doi: 10.1074/jbc.M109403200. [DOI] [PubMed] [Google Scholar]
- 51.Williams PA, Coates L, Mohammed F, Gill R, Erskine PT, Coker A, Wood SP, Anthony C, Cooper JB. The atomic resolution structure of methanol dehydrogenase from Methylobacterium extorquens. Acta Crystallogr. 2005;D61:75–79. doi: 10.1107/S0907444904026964. [DOI] [PubMed] [Google Scholar]
- 52.Li J, Gan JH, Mathews FS, Xia ZX. The enzymatic reaction-induced configuration change of the prosthetic group PQQ of methanol dehydrogenase. Biochem Biophys Res Commun. 2011;406:621–626. doi: 10.1016/j.bbrc.2011.02.107. [DOI] [PubMed] [Google Scholar]
- 53.Nurizzo D, Turkenburg JP, Charnock SJ, Roberts SM, Dodson EJ, McKie VA, Taylor EJ, Gilbert HJ, Davies GJ. Cellvibrio japonicus α-L-arabinanase 43A has a novel five-blade β-propeller fold. Nat Struct Biol. 2002;9:665–668. doi: 10.1038/nsb835. [DOI] [PubMed] [Google Scholar]
- 54.Proctor MR, Taylor EJ, Nurizzo D, Turkenburg JP, Lloyd RM, Vardakou M, Davies GJ, Gilbert HJ. Tailored catalysts for plant cell-wall degradation: Redesigning the exo/endo preference of Cellvibrio japonicus arabinanase 43A. Proc. Natl Acad Sci USA. 2005;102:2697–2702. doi: 10.1073/pnas.0500051102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bianchet MA, Odom EW, Vasta GR, Amzel LM. A novel fucose recognition fold involved in innate immunity. Nat Struct Biol. 2002;9:628–634. doi: 10.1038/nsb817. [DOI] [PubMed] [Google Scholar]
- 56.Mikami B, Iwamoto H, Malle D, Yoon HJ, Demirkan-Sarikaya E, Mezaki Y, Katsuya Y. Crystal structure of pullulanase: Evidence for parallel binding of oligosaccharides in the active site. J Mol Biol. 2006;359:690–707. doi: 10.1016/j.jmb.2006.03.058. [DOI] [PubMed] [Google Scholar]
- 57.Yoon HJ, Mikami B, Hashimoto W, Murata K. Crystal structure of alginate lyase A1-III from Sphingomonas species A1 at 1.78Å resolution. J Mol Biol. 1999;290:505–514. doi: 10.1006/jmbi.1999.2883. [DOI] [PubMed] [Google Scholar]
- 58.Bourne PC, Isupov MN, Littlechild JA. The atomic-resolution structure of a novel bacterial esterase. Structure. 2000;8:143–151. doi: 10.1016/s0969-2126(00)00090-3. [DOI] [PubMed] [Google Scholar]
- 59.Guiseppe PO, Neves FO, Nascimento ALTO, Guimaraes BG. The leptospiral antigen Lp49 is a two-domain protein with putative protein-binding function. J Struct Biol. 2008;163:53–60. doi: 10.1016/j.jsb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 60.Ravelli RBG, McSweeney SM. The ‘fingerprint’ that X-rays can leave on structures. Structure. 2000;8:315–328. doi: 10.1016/s0969-2126(00)00109-x. [DOI] [PubMed] [Google Scholar]
- 61.Grathwohl C, Wuthrich K. NMR studies of the rates of proline cis-trans isomerization in oligopeptides. Biopolymers. 1981;20:2623–2633. [Google Scholar]
- 62.Schmid FX. Kinetics of unfolding and refolding of single-domain proteins. In: Creighton TE, editor. Protein Folding. W.H. Freeman & Co.; New York: 1992. pp. 197–241. [Google Scholar]
- 63.Videau LL, Arendall WB, III, Richardson JS. The cis-Pro touch-turn: a rare motif preferred at functional sites. Proteins: Struct Funct Bioinf. 2004;56:298–309. doi: 10.1002/prot.20101. [DOI] [PubMed] [Google Scholar]
- 64.Hintze BJ, Lewis SM, Richardson JS, Richardson DC. MolProbity's ultimate rotamer-library distributions for model validation. Proteins: Struc Func Bioinf. 2016;84:1177–1189. doi: 10.1002/prot.25039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. 2007;3:619–629. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 66.Cemazar M, Zahariev S, Lopez JJ, Carugo O, Jones JA, Hore PJ, Pongor S. Oxidative folding intermediates with nonnative disulfide bridges between adjacent cysteine residues. Proc Natl Acad Sci USA. 2003;100:5754–5759. doi: 10.1073/pnas.2225470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carter P, Wells JA. Dissecting the catalytic triad of a serine protease. Nature. 1988;332:564–568. doi: 10.1038/332564a0. [DOI] [PubMed] [Google Scholar]
- 68.Page MJ, Di Cera E. Serine peptidases: Classification, structure, and function. Cell Mol Life Sci. 2008;65:1220–1236. doi: 10.1007/s00018-008-7565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park C, Raines RT. Adjacent cysteine residues as a redox switch. Protein Engin. 2001;14:939–942. doi: 10.1093/protein/14.11.939. [DOI] [PubMed] [Google Scholar]
- 70.Hunter HN, Fulton DB, Ganz T, Vogel HJ. The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. J Biol Chem. 2002;277:37597–37603. doi: 10.1074/jbc.M205305200. [DOI] [PubMed] [Google Scholar]
- 71.Jordan JB, Poppe L, Haniu M, Arvedson T, Syed R, Li V, Kohno H, Kim H, Schnier PD, Harvey TS, Miranda LP, Cheetham J, Sasu BJ. Hepcidin revisited, disulfide connectivity, dynamics, and structure. J Biol Chem. 2009;284:24155–24167. doi: 10.1074/jbc.M109.017764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. 2010;D66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Engh RA, Huber R. Accurate bond and angle parameters for x-ray protein structure refinement. Acta Crystallogr. 1991;A47:392–400. [Google Scholar]
- 74.Sobolev OV, Moriarty NW, Afonine PV, Hintze BJ, Richardson DC, Richardson JS, Adams PD. Disulfide bond restraints. Comput Crystallogr Newsletter. 2015;6:13. http://www.phenix-online.org/newsletter/CCN_2015_01.pdf. [Google Scholar]
- 75.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bressert E. In: SciPy and NumPy: An Overview for Developers. Roumeliotis R, Blanchette M, editors. O'Reilly Media, Inc.; Sebastopol: 2012. [Google Scholar]
- 77.Chen VB, Davis IW, Richardson DC. KiNG (Kinemage, Next Generation): a versatile interactive molecular and scientific visualization program. Protein Sci. 2009;18:2403–2409. doi: 10.1002/pro.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Word JM, Lovell SC, Richardson JS, Richardson DC. Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol. 1999;285:1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 79.Richardson DC, Richardson JS. MAGE, PROBE, and Kinemages. In: Rossmann MG, Arnold E, editors. International Tables for Crystallography Vol F. Kluwer Academic Publishers; The Netherlands, Dordrecht: 2001. pp. 727–30. [Google Scholar]
- 80.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active enZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2008;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.