

Abstract
Bacterial serine dipeptide lipids are known to promote inflammatory processes and are detected in human tissues associated with periodontal disease or atherosclerosis. Accurate quantification of bacterial serine lipid, specifically lipid 654 [((S)-15-methyl-3-((13-methyltetradecanoyl)oxy)hexadecanoyl)glycyl-l-serine, (3S)-l-serine] isolated from Porphyromonas gingivalis,1 in biological samples requires the preparation of a stable isotope internal standard for sample supplementation and subsequent mass spectrometric analysis. This report describes the convergent synthesis of a deuterium-substituted serine dipeptide lipid, which is an isotopically labeled homologue that represents a dominant form of serine dipeptide lipid recovered in bacteria.
Introduction
The anaerobic Gram-negative organism, Porphyromonas gingivalis, is thought to be a major periodontal pathogen2 associated with destructive periodontal disease in adults. These agonists are also produced by common oral and intestinal Bacteroidetes,3 and they are recovered in chronically inflamed human tissues including destructive periodontal disease and atherosclerosis tissues.2 The most commonly observed serine dipeptide mono-fatty acid species is composed of 3-OH iso-C17:0 fatty acid moiety may form a 3-OH ester linkage to a saturated fatty acid, most frequently to iso-C15:0 (see 1).4 Members of the Bacteroidetes phylum comprise a group of commensal organisms recovered in the human oral and intestinal microbiomes. Oral and intestinal Bacteroidetes organisms have recently been shown to produce novel serine dipeptide lipids identical to those described for specific Flavobacteria. The dominant serine lipid species isolated from Porphyromonas gingivalis, called Lipid 654 (1, ((S)-15-methyl-3-((13-methyltetradecanoyl)oxy)hexadecanoyl)glycyl-l-serine, (3S)-l-serine), acts as a bacterial virulence factor by engaging the innate immune receptor, Toll-like receptor 2 (TLR2). We have identified Lipid 654 in oral tissues from periodontal disease sites as well as in human blood and atherosclerotic lesions. In order to implicate this lipid class in human disease, accurate quantification of Lipid 654 requires supplementing biological samples with a stable isotope internal standard for subsequent quantification using conventional mass spectrometric approaches. The present report describes the preparation of a deuterated serine dipeptide internal standard (2) for use in quantifying 1 in biological samples. In addition to quantifying the bacterial lipid 1 in biological samples, the deuterated lipid standard 2 can also be used for the evaluation of enzymatic hydrolysis of the parent lipid by known esterases as well as peptidases. In addition, the deuterated lipid 2 can be used for evaluation of metabolic breakdown of 1 by cells important in chronic inflammatory disease processes associated with the accumulation of bacterial serine dipeptide lipids. Indeed, 13B will be used as a standard for the lipid 430 isolated from Porphyromonas gingivalis and 2B will be used as a standard for 654 isolated from P. gingivalis. The isotopically labeled homologue 2B, rather than the isotopologue of 1, was chosen because of the cost effectiveness in the synthetic scheme, the requirement that the standard not co-elute with the substrates, and with the approval of our colleagues who plan to use the standards in their research. The racemic compounds are sufficient for mass spectral analysis and their planned use as standards. The racemic targets are deemed sufficient for the planned analyses, at present, but future studies may require the synthesis of enantioenriched or enantiopure standards.
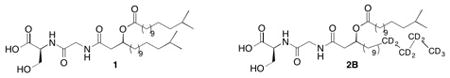
Discussion
Deuterium labeled serine dipeptide lipid was successfully prepared for use in the analysis of biological samples. The first step in the synthesis, however, was to prepare the non-deuterated fatty ester dipeptide via procedures that mimic the convergent synthesis of lipids 430 and 654 in our recent article.1 In order to mitigate the reactivity of the Grignard reagent with the acidic hydroxyl proton, we first protected the commercially available 11-bromoundecanol as the dimethyl(t-butyl)silyl derivative, in 61% yield. The Grignard reagent derived from 1-bromobutane was coupled with the protected alcohol, facilitated by lithium tetrachlorocuprate and NMP (N-methylpyrrolidone) to give the protected alcohol product, 4A, in 50% overall yield from 11-bromoundecan-1-ol.6c Deprotection gave 5A in 87% yield and oxidation with the oxoammonium salt known as Bobbitt’s reagent5 provided pentadecanal, 6A, in 91% yield, without oxidation of the initially formed aldehyde to the carboxylic acid. Coupling with ethyl diazoacetate in the presence of a tin (II) chloride catalyst, using the protocol developed by Roskamp et. al.,6a,b gave 7A (90%), our standard methodology.6c Subsequent reduction with NaBH4, followed by saponification with LiOH, afforded β-hydroxy acid 9A in 65% yield. Coupling of 9A and the previously prepared protected dipeptide 101 using N-hydroxy succinimide for the conversion to the activated carboxylic acid derivative and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) to facilitate the coupling.1 Removal of the N-Cbz group by catalytic hydrogenation gave 11A in 67% yield. Deprotection of the ester of the protected serine afforded 12A in 81% yield. Treatment of 12A with LiOH gave 13A in 84% yield. This sequence completed the first part of our synthesis.
Esterification of 9A with 13-methyltetradecanoic acid gave 14A in 63% yield and deprotection of the OTBDPS group with TBAF gave 15A in 76% yield. Deprotection of the methyl ester group with potassium trimethlsilanolate gave 2A in 65% yield. The unusual reagent used to convert the methyl ester to the carboxylic acid was necessitated by the sensitivity of the fatty ester linkage to the usual conditions used to cleave the methyl ester. The overall conversion for the linear sequence of 10 steps to 13A was 13% overall yield, whereas the linear sequence of 11 steps gave 2A in 9% overall yield.
Following a similar synthetic methodology, the synthesis initially focused on the fatty acid portion of the lipid and the preparation of 9B.1 To avoid undesired reactivity of expensive deuterated Grignard reagent with the acidic hydroxy proton, commercially available 11-bromoundecan-1-ol was protected with tert-butyl dimethyl silyl chloride to form the silyl ether. Coupling with the Grignard derivative of 3B, prepared by bromination of the commercially available [2H9]-butan-1-ol, required a lithium tetrachlorocuprate complex with N-methylpyrrolidone (NMP) as an additive.6c The use of NMP as well as lithium tetrachlorocuprate insured that a minimum amount of 1-bromo-[2H9]-butane was used to produce deuterated silyl ether 4B in 55% overall yield from 11-bromoundecanol.7 Deprotection using TBAF generated alcohol 5B. Oxidation of the alcohol with the oxoammonium reagent Bobbitt’s salt5 yielded aldehyde 6B in 76% yield, with no over-oxidation to the acid. β–Keto ester 7B was prepared in 61% yield by the reaction of the aldehyde 6B with ethyl diazoacetate in the presence of catalytic tin (II) chloride, using Roskamp’s protocol.6a,b Selective reduction of the ketone moiety with sodium borohydride generated racemic β–hydroxy ester 8B in 80% yield Saponification of this ester using lithium hydroxide yielded racemic β-hydroxy acid 9B. The racemic compounds are sufficient for mass spectral analysis and their planned use as standards. The racemic targets are deemed sufficient for the planned analyses, at present, but future studies may require the synthesis of enantioenriched or enantiopure standards.
With nonadeuterated β-hydroxy acid 9B in hand, the convergent synthesis proceeded by coupling with the previously prepared protected dipeptide 10 to give 11B in 78% yield.1 Activation of the acid with N-hydroxysuccinimide combined with the hydrogenolysis product from the dipeptide led to the protected lipid 11B using EDC as the coupling agent.8 Subsequent deprotection with TBAF gave 12B in 85% yield, followed by LiOH saponification gave the nonadeuterated lipid 13B in 73% yield from the C17 fatty acid.
The other targeted lipid utilized esterification of the alcohol moiety 11B with 13-methylpentadecanoic acid. An EDC/DMAP mediated coupling with 10 gave nonadeuterated-protected lipid 14B in 80% yield. Deprotection proceeded via removal of the silyl ether using TBAF to liberate the serine hydroxyl group in 15B in 85% yield The sensitivity of the ester linkage in the lipid required liberation of the methyl ester using the potassium trimethylsilanolate, which led to 2B in 58% yield9
Conclusions
We have successfully incorporated nine deuterium atoms into 2B from the commercially available 11-bromoundecan-1-ol using commercially available [2H9]-butan-1-ol as the deuterium source. The convergent synthesis prepared 13A in 10 linear steps and 13% overall yield from 11-bromoundecan-1-ol and 2A in 11 linear steps and 9% overall yield. Using an identical route we have prepared 13B in 10 linear steps and 12% overall yield and 2B in 10 linear steps and 8% overall yield. The 13B will be used as a standard for the lipid 430 isolated from Porphyromonas gingivalis and 2B will be used as a standard for 654 isolated from P. gingivalis.1
Experimental Section
All glassware was oven-dried, and all reactions were performed under a nitrogen atmosphere. All chemicals were purchased from the Sigma-Aldrich Chemical Co., and used without further purification unless otherwise noted. Tetrahydrofuran (THF) was distilled from sodium benzophenone ketyl, methylene chloride (dichloromethane, DCM) was distilled from calcium hydride, and dimethylformamide (DMF) was distilled in vacuo from calcium hydride. Ethyl acetate (EtOAc), methanol (MeOH), and diethyl ether were used as obtained from the vendor. Bobbitt’s salt5 was provided as a gift from Dr. James Bobbitt (Department of Chemistry, The University of Connecticut)), but it is commercially available from Sigma-Aldrich as 4- acetamido-2,2,6,6-tetramethylpiperidine 1-oxyl. [2H9]-Butan-1-ol was purchased from Sigma-Aldrich and used without further purification.?
Thin-layer chromatography was done on Sorbent Technologies aluminum-backed TLC plates with fluorescent indicator and 0.2 mm silica gel layer thickness, and p-anisaldehyde or phosphomolybdic acid were used as developing agents. Column chromatography was done using 60 Å porosity, 32-63 μm silica gel. 1H and 13C NMR were collected on a Bruker Avance 300 (300.13 MHz 1H, 75.48 MHz 13C), Bruker DRX-400 (400.144 MHz 1H, 100.65 MHz 13C) or a Bruker Avance 500 (500.13 MHz 1H, 125.65 MHz 13C). Chemical shifts are reported in ppm downfield from tetramethylsilane (TMS) in the following format chemical shift, multiplicity (s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet). Coupling constants are reported in Hz. Mass spectrometry data was collected on a HP 5870B GC/MSD mass spectrometer with an HP-1 column, and high resolution MS and MS/MS spectra were obtained by direct infusion of the target lipids into a QTOF mass spectrometer, QSTAR Elite from Sciex (Foster City, CA) or directly by using AccuTOF™ DART, JEOL (Peabody, MA). Multiple reaction monitoring (MRM) transitions were selected based the MS/MS spectra of 1. Lipid samples were injected into a triple quadrupole mass spectrometer, 4000 QTrap from Sciex (Foster City, CA). IR spectra were taken on an FT/IR-410/C031560585 JASCO or a Nexus 670 FT-IR E.S.P, neat, unless otherwise stated. All melting points to an upper limit of 270 °C were obtained using a Unimelt capillary melting point apparatus or Digimelt MPA160. For products described as waxy solid or semi-solids, melting points could not be obtained. The term brsm is ‘based on recovered starting material.’
((11-Bromoundecyl)oxy)(tert-butyl)dimethylsilane
A flame-dried 100 mL round bottom flask was charged with 11-bromoundecan-1-ol (2.00 g, 7.96 mmol). The starting material was dissolved in CH2Cl2 (50 mL) and cooled on an ice bath. tert-Butyldimethylsilyl chloride (1.40 g, 9.15 mmol) was added and stirred for 15 min before the addition of imidazole (0.705 g, 10.4 mmol). The reaction was stirred overnight at room temperature. Upon completion the suspension was gravity filtered and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (5% EtOAc:hexane) to yield ((11-bromoundecyl)oxy)(tert-butyl)dimethylsilane,10 as a clear liquid (2.85 g, 7.78 mmol, 97.7%). 1H NMR (400 MHz, CDCl3) δ 3.60 (t, J = 6.6 Hz, 2H), 3.41 (t, J = 6.9 Hz, 2H), 1.85 (p, J = 7.0 Hz, 2H), 1.51 (t, J = 6.8 Hz, 2H), 1.42 (t, J = 7.4 Hz, 2H), 1.28 (s, 12H), 0.90 (s, 9H), 0.05 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 63.38, 34.09, 32.94, 32.91, 29.63, 29.53, 29.47, 28.82, 28.24, 26.05, 25.85, 18.44, -5.19. ppm HRMS (AccuTOF): [MH]+ Calc’d for C17H37BrOSi m/z 365.1875. Found: m/z 365.1869.
1-Bromobutane, 3A
A solution of 48% HBr (2.50 g) was added to a flame dried 25 mL round bottomed flask. Dropwise addition of H2SO4 (0.40 mL) was followed by stirring until the flask was cool to the touch. Dropwise addition of butan-1-ol (1.00 g, 13.5 mmol) was followed by treatment with H2SO4 (0.35 mL). A reflux condenser was attached and the solution was heated for 3 h. A vacuum distillation apparatus was set up through the top of the original reflux condenser. The product was gently vacuum-distilled into an ice-chilled flask while the vertical condenser was slowly warmed. The distillate was dried over CaCO3 to yield 1-bromobutane,11 3A, as a clear liquid (1.13 g, 8.25 mmol, 61%). 1H NMR (400 MHz, CDCl3) δ 3.42 (t, J = 6.8 Hz, 2H), 1.84 (p, J = 6.9 Hz, 2H), 1.48 (hept, J = 7.5 Hz, 2H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 34.83, 33.72, 21.37, 13.23.
1-Bromo-[2H9]-butane, 3B
A solution of 48% HBr (2.50 g) was added to a flame-dried 25 mL round bottomed flask. Dropwise addition of H2SO4 (0.40 mL) was followed by stirring until the flask was cool to the touch. Dropwise addition of commercially available [2H9]-butan-1-ol (1.00 g, 12.0 mmol), was followed by addition of H2SO4 (0.35 mL). A reflux condenser was attached and the solution was heated at reflux for 3 h. A vacuum distillation apparatus was set up through the top of the original reflux condenser. The product was vacuum distilled into an ice-chilled flask while the vertical condenser was slowly warmed. The distillate was dried over CaCO3 to yield 1-bromo-[2H9]-butane,12 3B, as a clear liquid (0.99 g, 6.78 mmol, 56.4%).
tert-Butyldimethyl(pentadecyloxy)silane, 4A
Magnesium turnings (33 mg, 1.37 mmol) were added to a 25 mL round bottomed flask that was fitted with a stirbar and the apparatus was flame-dried under a nitrogen atmosphere. After cooling, the Mg turnings were suspended in dry THF (5 mL) and 1-bromobutane (3A, 0.13 mL, 1.20 mmol) was rapidly added with vigorous stirring. The reaction was stirred until the flask cooled to ambient temperature and the flask was subsequently chilled to 0 °C. The Grignard reagent was added to a mixture of ((11-bromoundecyl)oxy)(tert-butyl)dimethylsilane (2, 0.250 g,0.684 mmol), LiCl (0.88 mg, 0.021 mmol), copper (II) chloride (2.8 mg, 0.021 mmol), and N-methyl-2-pyrrolidone (NMP, 0.30 mL, 3.11 mmol) dissolved in THF (5 mL) and held at 0 °C. The mixture was slowly warmed to ambient temperature and stirred for 1 h. The reaction was quenched on ice with 1M HCl (10 mL) before being transferred to a separatory funnel with EtOAc (15 mL) and H2O (10 mL). The layers were separated and the aqueous layer was extracted once with EtOAc (15 mL). The organic layers were combined and then washed with 1M HCl (10 mL), H2O (2 X 10 mL), and brine (10 mL). The organic phase was dried with MgSO4, gravity filtered, and solvents removed in vacuo. The crude product was purified by column chromatography on silica gel (hexane) to yield tert-butyldimethyl(pentadecyloxy)silane, 4A, as a clear liquid (0.218 g, 0.636 mmol, 93.0%). 1H NMR (400 MHz, CDCl3) δ 3.60 (t, J = 6.6 Hz, 2H), 1.52 (d, J = 6.8 Hz, 2H), 1.26 (s, 24H), 0.96–0.79 (m, 12H), 0.05 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 63.41, 32.97, 31.99, 29.75, 29.72, 29.51, 29.42, 26.05, 25.87, 22.75, 14.18, -5.19. HRMS (AccuTOF): [MH]+ Calc’d for C21H46OSi m/z 343.3396. Found: m/z 343.3373.
tert-Butyldimethyl([12,12,13,13,14,14,15,15,15-2H9]-pentadecyloxy)silane, 4B
Magnesium turnings (133 mg, 5.48 mmol) were added to a 50 mL round bottom flask fitted with a stirbar and the apparatus was flame-dried under a nitrogen atmosphere. After cooling, the Mg turnings were suspended in dry THF (10 mL) and 1-bromo-[2H9]-butane (3B, 0.52 mL, 4.80 mmol) was rapidly added with vigorous stirring. The reaction was stirred until the flask cooled to ambient temperature and the temperature was lowered to 0 °C. The Grignard reagent was added to a mixture of ((11-bromoundecyl)oxy)(tert-butyl)dimethylsilane (1.00 g, 2.74 mmol), LiCl (5.8 mg, 0.14 mmol), copper (II) chloride (9.2 mg, 0.069 mmol), and NMP (1.98 mL, 20.6 mmol) dissolved in THF (10 mL) and the reaction mixture was maintained at 0 °C. The mixture was slowly warmed to ambient temperature and stirred for 1 h. The reaction was quenched on ice with 1M HCl (25 mL) before being transferred to a separatory funnel with EtOAc (50 mL) and H2O (50 mL). The layers were separated and the aqueous layer was extracted once with EtOAc (50 mL). The organic layers were combined and washed with 1M HCl (25 mL), H2O (2 X 25 mL), and brine (25 mL). The organic phase was dried with MgSO4, gravity filtered, and solvents removed in vacuo. The crude product was purified by column chromatography on silica gel (hexane) to yield tert-butyldimethyl([12,12,13,13.14,14,15,15,15-2H9]-pentadecyloxy)silane, 4B, as a clear liquid (0.949 g, 2.69 mmol, 98.3%). 1H NMR (400 MHz, CDCl3) δ 3.60 (t, J = 6.6 Hz, 2H), 1.51 (t, J = 6.8 Hz, 2H), 1.25 (s, 18H), 0.89 (s, 9H), 0.05 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 63.42, 32.96, 29.76, 29.72, 29.68, 29.52, 29.47, 26.05, 25.87, 18.44, -5.19. HRMS (AccuTOF): [MH]+ Calc’d for C21H37D9OSi m/z 352.3961. Found: m/z 352.3949.
Pentadecan-1-ol, 5A
tert-Butyldimethyl(pentadecyloxy)silane (4A, 678 mg, 1.98 mmol) was dissolved in 25 mL of dry THF under nitrogen. The solution was cooled to 0 °C and was treated with 1 M TBAF (4.95 mL, 4.95 mmol). The reaction was slowly warmed to ambient temperature and stirred for 6 h. The solvent was removed under reduced pressure, and the resulting oil was purified by column chromatography (15% EtOAc:Hexanes) to yield ethyl 3-hydroxyheptadecanoate,13 5A, as a white solid (395 mg, 1.73 mmol, 87.4 %), MP 44-46 °C. 1H NMR (400 MHz, CDCl3) δ 3.64 (t, J = 6.6 Hz, 2H), 1.57 (t, J = 7.2, 6.7 Hz, 2H), 1.35–1.23 (m, 25H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 63.17, 32.88, 31.98, 29.74, 29.72, 29.66, 29.49, 29.41, 25.80, 22.75, 14.17. HRMS (AccuTOF): [M2H]+ Calc’d for C15H32O m/z 457.4985. Found: m/z 457.4998.
[12,12,13,13.14,14,15,15,15-2H9]-Pentadecan-1-ol, 5B
tert-Butyldimethyl([12,12,13,13.14,14,15,15,15-2H9]-pentadecyloxy)silane (4B, 961 mg, 2.73 mmol) was dissolved in 25 mL of dry THF under nitrogen. The solution was cooled to 0 °C and was treated with 1 M TBAF (6.83 mL, 6.83 mmol). The reaction was slowly warmed to ambient temperature and stirred for 6 h. The solvent was removed under reduced pressure, and the resulting oil was purified by column chromatography (15% EtOAc:Hexanes) to yield [12,12,13,13.14,14,15,15,15-2H9]-pentadecan-1-ol, 5B, as a white solid (643 mg, 2.71 mmol, 99.1 %), MP 42-45 °C. 1H NMR (400 MHz, CDCl3) δ 3.64 (t, J = 6.6 Hz, 2H), 1.57 (p, J = 6.9 Hz, 2H), 1.39–1.22 (m, 19H). 13C NMR (101 MHz, CDCl3) δ 63.18, 32.89, 29.72, 29.67, 29.49, 25.80. HRMS (AccuTOF): [M-OH]+ Calc’d for C15H23D9O m/z 220.2991. Found: m/z 220.2975.
Pentadecanal, 6A
4-(Acetyamino)-2,2,6,6-tetramethyl-1-oxo-piperidinium tetrafluoroborate5 (Bobbit’s salt, 3.552 g, 11.84 mmol) was added to pentadecan-1-ol (5A, 2.351 g, 10.29 mmol) dissolved in 200 mL of dry DCM. An equal mass of silica was slowly added to the stirring mixture. The bright yellow solution stirred at ambient temperature until the color faded, giving a solution with a pale yellow tint. The solution was filtered to remove the silica, and the filtrate was concentrated in vacuo. The crude product was purified by column chromatography (5% EtOAc:hexanes) to yield pentadecanal,14 6A, as a clear solid, (2.116 g, 9.346 mmol, 90.8%), MP ~25 °C. 1H NMR (400 MHz, CDCl3) δ 9.76 (t, J = 1.9 Hz, 1H), 2.41 (td, J = 7.3, 1.9 Hz, 2H), 1.63 (p, J = 7.3 Hz, 2H), 1.35–1.22 (m, 22H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 203.01, 43.98, 31.98, 29.70, 29.63, 29.48, 29.41, 29.23, 22.74, 22.16, 14.17. HRMS (AccuTOF): [M-H]+ Calc’d for C15H30O m/z 225.2218. Found: m/z 225.2216.
[12,12,13,13.14,14,15,15,15-2H9]-Pentadecanal, 6B
4-(Acetyamino)-2,2,6,6-tetramethyl-1-oxo-piperidinium tetrafluoroborate5 (Bobbit’s salt, 829 mg, 2.76 mmol) was added to [12,12,13,13.14,14,15,15,15-2H9]-pentadecan-1-ol (5B, 625 mg, 2.63 mmol) dissolved in 100 mL of dry DCM. An equal mass of silica was slowly added to the stirring mixture. The bright yellow solution stirred at ambient temperature until the color faded, giving a solution with a pale yellow tint. The solution was filtered to remove the silica, and the filtrate was concentrated in vacuo. The crude product was purified by column chromatography (5% EtOAc:hexanes) to yield [12,12,13,13.14,14,15,15,15-2H9]-pentadecanal, 6B, as a clear solid, (468 mg, 1.99 mmol, 75.6%), MP, ~25 °C. 1H NMR (400 MHz, CDCl3) δ 9.76 (t, J = 2.0 Hz, 1H), 2.41 (td, J = 7.4, 1.9 Hz, 2H), 1.64 (p, J = 7.2 Hz, 2H), 1.32–1.20 (m, 16H). 13C NMR (101 MHz, CDCl3) δ 203.01, 43.98, 29.73, 29.69, 29.64, 29.48, 29.46, 29.41, 29.23, 22.16. HRMS (AccuTOF): [MH]+ Calc’d for C15H21D9O m/z 234.2783. Found: m/z 234.2808.
Ethyl 3-oxoheptadecanoate, 7A
Tin (II) chloride dihydrate (52 mg, 0.231 mmol) was suspended in 20 mL of dry DCM. Ethyl diazoacetate (319 mg, 2.43 mmol) was added, dropwise via syringe, and the reaction was stirred for 15 min. Pentadecanal (6A, 524 mg, 2.31 mmol) was dissolved in 10 mL of DCM and added to the reaction, dropwise, via cannula. The mixture was stirred at ambient temperature for about 6 h, until nitrogen evolution ceased. The reaction mixture was diluted with 100 mL of DCM and washed with a saturated brine solution (2 X 50 mL). The aqueous layers were combined and extracted with DCM (2 X 25 mL). All organic layers were combined, dried with MgSO4, filtered, and solvents evaporated at reduced pressure. The resulting crude product was purified via column chromatography (5% EtOAc:hexanes) to yield ethyl 3-oxoheptadecanoate,15 7A, as a clear solid (642 mg, 2.07 mmol, 89.6%), MP 37-39 °C. 1H NMR (400 MHz, CDCl3) δ 4.20 (q, J = 7.1 Hz, 2H), 3.42 (s, 2H), 2.53 (t, J = 7.4 Hz, 2H), 1.59 (p, J = 7.1 Hz, 2H), 1.34–1.22 (m, 25H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 203.03, 167.32, 61.38, 49.38, 43.12, 31.98, 29.71, 29.65, 29.50, 29.41, 29.09, 23.54, 22.75, 14.17. HRMS (AccuTOF): [MH]+ Calc’d for C19H36O3 m/z 313.2743. Found: m/z 313.2724.
Ethyl 3-oxo-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoate, 7B
Tin (II) chloride dihydrate (46 mg, 0.206 mmol) was suspended in 15 mL of dry DCM. Ethyl diazoacetate (0.275 mL, 2.27 mmol) was added, dropwise via syringe, and the reaction was stirred for 15 min. The [12,12,13,13.14,14,15,15,15-2H9]-pentadecanal (6B, 486 mg, 2.06 mmol) was dissolved in 10 mL of DCM and added to the reaction, dropwise, via cannula. The mixture was stirred at ambient temperature for about 6 h, until nitrogen evolution ceased. The reaction mixture was diluted with 100 mL of DCM and washed with a saturated brine solution (2 X 50 mL). The aqueous layers were combined and extracted with DCM (2 X 25 mL). All organic layers were combined, dried with MgSO4, filtered, and solvents evaporated at reduced pressure. The resulting crude product was purified via column chromatography (5% EtOAc:hexanes) to yield ethyl 3-oxo-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoate,15 7B, as a clear solid (534 mg, 1.66 mmol, 80.6%), MP 36-38 °C. 1H NMR (400 MHz, CDCl3) δ 4.19 (q, J = 7.1 Hz, 2H), 3.42 (s, 2H), 2.52 (t, J = 7.4 Hz, 2H), 1.59 (t, J = 7.2 Hz, 2H), 1.29–1.24 (m, 19H). 13C NMR (101 MHz, CDCl3) δ 203.05, 167.32, 61.38, 49.38, 43.12, 29.73, 29.70, 29.65, 29.50, 29.46, 29.41, 29.09, 23.54, 14.16. HRMS (AccuTOF): [MH]+ Calc’d for C19H27D9O3 m/z 322.3308. Found: m/z 322.3324.
Ethyl 3-hydroxy-heptadecanoate, 8A
A solution of ethyl 3-oxoheptadecanoate (7A, 620 mg, 1.98 mmol) in 2 mL of ethanol and 18 mL of THF was cooled to 0 °C on an ice bath. Sodium borohydride (60 mg, 1.59 mmol) was added and the resulting slurry was stirred vigorously for 1 h. The reaction was quenched by the slow addition of 10 mL of a 10 % aq. citric acid solution. Subsequent neutralization with satd. K2CO3 solution was followed by extraction with EtOAc (3 X 20 mL). The organic layers were washed with 10 mL of satd. brine solution, dried with MgSO4, filtered, and the solvents were removed at reduced pressure. The crude product was purified by column chromatography (10% EtOAc:Hexanes) to yield ethyl 3-hydroxy-heptadecanoate, 8A, as a white solid (455 mg, 1.45 mmol, 77.6 % brsm) MP 41-43 °C. 1H NMR (400 MHz, CDCl3) δ 4.17 (q, J = 7.1 Hz, 2H), 3.99 (tt, J = 7.9, 3.9 Hz, 1H), 2.90 (d, J = 4.0 Hz, 1H), 2.50 (dd, J = 16.4, 3.1 Hz, 1H), 2.39 (dd, J = 16.4, 9.0 Hz, 1H), 1.43 (dd, J = 8.5, 4.3 Hz, 2H), 1.38–1.19 (m, 27H), 0.88 (t, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 173.19, 68.11, 60.71, 41.35, 36.59, 31.99, 29.74, 29.71, 29.64, 29.59, 29.42, 25.54, 22.75, 14.25, 14.18. HRMS (AccuTOF): [MH]+ Calc’d for C19H38O3 m/z 315.2899. Found: m/z 315.2899.
Ethyl 3-hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoate, 8B
A solution of ethyl 3-oxo-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoate (7B, 515 mg, 1.60 mmol) in 2 mL of ethanol and 18 mL of THF was cooled to 0 °C on an ice bath. Sodium borohydride (48 mg, 1.28 mmol) was added and the resulting slurry was stirred vigorously for 1 h. The reaction was quenched by the slow addition of 10 mL of a 10 % aq. citric acid solution. Subsequent neutralization with satd. K2CO3 solution was followed by extraction with EtOAc (3 X 20 mL). The organic layers were washed with 10 mL of satd. brine solution, dried with MgSO4, filtered, and the solvents were removed at reduced pressure. The crude product was purified by column chromatography (10% EtOAc:Hexanes) to yield ethyl 3-hydroxy- [14,14,15,15,16,16,17,17,17-2H9]-heptadecanoate, 8B, as a white solid (392 mg, 1.21 mmol, 79.7 % brsm) MP 38-40 °C. 1H NMR (400 MHz, CDCl3) δ 4.17 (q, J = 7.2 Hz, 2H), 4.00 (ddt, J = 12.0, 7.3, 4.0 Hz, 1H), 2.90 (d, J = 4.0 Hz, 1H), 2.50 (dd, J = 16.4, 3.2 Hz, 1H), 2.39 (dd, J = 16.4, 9.0 Hz, 1H), 1.47–1.39 (m, 2H), 1.39–1.12 (m, 21H). 13C NMR (101 MHz, CDCl3) δ 173.19, 68.11, 60.72, 41.35, 36.59, 29.75, 29.71, 29.64, 29.59, 29.47, 25.54, 14.25. HRMS (AccuTOF): [MH]+ Calc’d for C19H29D9O3 m/z 324.3464. Found: m/z 324.2442.
3-Hydroxyheptadecanoic acid, 9A
Hydroxy ester 8A (440 mg, 1.40 mmol) was dissolved in 2 mL of MeOH and 4 mL of THF and 1 M aqueous LiOH (1.68 mL, 1.61 mmol) were added. The mixture was stirred overnight at ambient temperature. The reaction was then acidified with 1 M HCl and extracted with EtOAc (3 X 15 mL). The organic layers were washed with 10 mL of brine, dried with MgSO4, filtered, and the solvents were removed at reduced pressure. The crude solid was recrystallized from hexanes to yield 3-hydroxy-heptadecanoic acid, 9A, as a white solid (335 mg, 1.17 mmol, 83.6%), MP 86-88 °C. 1H NMR (400 MHz, CDCl3) δ 4.04 (tt, J = 8.1, 3.8 Hz, 1H), 2.59 (dd, J = 16.6, 3.2 Hz, 1H), 2.48 (dd, J = 16.6, 8.9 Hz, 1H), 1.55–1.43 (m, 3H), 1.40–1.15 (m, 24H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 175.72, 68.07, 40.73, 36.62, 31.98, 29.75, 29.71, 29.63, 29.60, 29.52, 29.42, 25.48, 22.75, 14.18. HRMS (AccuTOF): [MH]+ Calc’d for C17H34O3 m/z 287.2586. Found: m/z 287.2605.
3-Hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoic acid, 9B
Ethyl 3-hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoate, 8B (390 mg, 1.21 mmol) was dissolved in 2 mL of MeOH and 4 mL of THF and 1 M aqueous LiOH (1.45 mL, 1.39 mmol) were added. The mixture was stirred overnight at ambient temperature. The reaction was then acidified with 1 M HCl and extracted with EtOAc (3 X 15 mL). The organic layers were washed with 10 mL of brine, dried with MgSO4, filtered, and the solvents were removed at reduced pressure. The crude solid was recrystallized from hexanes to yield 3- hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoic acid, 9B, as a white solid (318 mg, 1.08 mmol, 89.3%), MP 79-81 °C. 1H NMR (400 MHz, CDCl3) δ 4.03 (s, 1H), 2.58 (dd, J = 16.6, 3.2 Hz, 1H), 2.48 (dd, J = 16.6, 8.9 Hz, 1H), 1.58–1.40 (m, 3H), 1.25 (s, 18H). 13C NMR (101 MHz, CDCl3) δ 175.79, 68.06, 40.76, 36.63, 29.71, 29.63, 29.60, 29.53, 29.47, 25.49. HRMS (-TOF MS): [M-H]- Calc’d for C17H25D9O3 m/z 294.2995. Found: m/z 294.2982.
Methyl-O-(tert-butyldiphenylsilyl)-N-((3-hydroxy-heptadecanoyl)glycyl)-l-serinate, 11A
N-Hydroxy succinimide (185 mg, 1.61 mmol) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (308 mg, 1.61 mmol) were added to a stirring solution of 3-hydroxyheptadecanoic acid (9A, 401 mg, 1.40 mmol) in 25 mL of dry DCM. The solution was stirred for 5 h at ambient temperature, filtered, and the filtrate was concentrated in vacuo. The resulting white solid was dissolved in 25 mL of methanol and this solution was added via cannula to a solution of methyl N-(((benzyloxy)carbonyl)glycyl)-O-(tert-butyldiphenylsilyl)-l-serinate (10, 960 mg, 1.75 mmol) and Pd/C (298 mg) in 50 mL of methanol. The reaction vessel was flushed with H2 and the mixture was stirred overnight, filtered through Celite, and concentrated under reduced pressure. The crude product was purified via column chromatography (2% MeOH:DCM) to yield methyl O-(tert-butyldiphenylsilyl)-N-((3-hydroxy-heptadecanoyl)glycyl)-l-serinate, 11A, as a clear oil (641 mg, 0.938 mmol, 67.0%). 1H NMR (400 MHz, CDCl3) δ 7.58 (t, J = 7.0 Hz, 4H), 7.50–7.33 (m, 6H), 6.88 (dd, J = 28.7, 8.2 Hz, 1H), 6.39 (dt, J = 42.3, 5.5 Hz, 1H), 4.67 (dt, J = 7.7, 2.5 Hz, 1H), 4.19–3.76 (m, 5H), 3.75 (s, 3H), 3.43 (dd, J = 38.5, 3.6 Hz, 1H), 2.41 (dt, J = 14.6, 2.2 Hz, 1H), 2.27 (dd, J = 13.9, 9.4 Hz, 1H), 1.52–1.35 (m, 3H), 1.33–1.22 (m, 24H), 1.04 (s, 9H), 0.88 (t, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.80, 172.75, 170.73, 168.71, 168.71, 168.53, 135.56, 135.52, 132.98, 132.93, 132.69, 132.64, 130.08, 127.97, 127.94, 127.90, 127.87, 68.93, 64.31, 64.27, 54.34, 52.63, 43.06, 43.02, 42.93, 42.81, 37.19, 37.15, 31.98, 29.75, 29.72, 29.64, 29.57, 29.42, 26.80, 25.49, 22.75, 19.33, 14.17. HRMS (AccuTOF): [MH]+ Calc’d for C39H62N2O6Si m/z 683.4455. Found: m/z 683.4432.
Methyl O-(tert-butyldiphenylsilyl)-N-((3-hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoyl)glycyl)-l-serinate, 11B
N-Hydroxy succinimide (130 mg, 1.13 mmol) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (216 mg, 1.13 mmol) were added to a stirring solution of 3- hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoic acid (9B, 303 mg, 1.03 mmol) in 10 mL of dry THF. The solution was stirred for 5 h at ambient temperature, filtered, and the filtrate was concentrated in vacuo. The resulting white solid was dissolved in 25 mL of methanol and this solution was added via cannula to a solution of methyl N-(((benzyloxy)carbonyl)glycyl)-O-(tert-butyldiphenylsilyl)-l-serinate (10, 563 mg, 1.03 mmol) and Pd/C (273 mg) in 50 mL of methanol. The reaction vessel was flushed with H2 and the mixture was stirred overnight, filtered through Celite, and concentrated under reduced pressure. The crude product was purified via column chromatography (2% MeOH:DCM) to yield methyl O-(tert-butyldiphenylsilyl)-N-((3-hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoyl)glycyl)-l-serinate, 11B, as a clear oil (555 mg, 0.802 mmol, 78.2%). 1H NMR (400 MHz, CDCl3) δ 7.59 (tt, J = 8.1, 1.6 Hz, 4H), 7.49–7.33 (m, 6H), 6.86 (dd, J = 26.2, 8.2 Hz, 1H), 6.34 (dt, J = 41.6, 5.4 Hz, 1H), 4.67 (dtd, J = 7.6, 2.9, 1.2 Hz, 1H), 4.19–3.78 (m, 5H), 3.75 (s, 3H), 3.41 (dd, J = 35.9, 3.6 Hz, 1H), 2.40 (dt, J = 14.6, 2.5 Hz, 1H), 2.26 (ddd, J = 14.6, 9.5, 2.1 Hz, 1H), 1.53–1.34 (m, 3H), 1.32–1.22 (m, 17H), 1.04 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 172.78, 172.73, 170.70, 168.66, 168.49, 135.57, 135.52, 132.98, 132.93, 132.70, 132.64, 130.09, 127.96, 127.93, 127.90, 127.87, 68.92, 64.31, 64.26, 54.34, 52.61, 43.05, 43.02, 42.94, 42.82, 37.19, 37.14, 29.72, 29.65, 29.57, 29.47, 26.80, 25.49, 19.33. HRMS (AccuTOF): [MH]+ Calc’d for C39H53D9N2O6 m/z 692.5020. Found: m/z 692.5007.
Methyl (3-hydroxyheptadecanoyl)glycyl-l-serinate, 12A
Methyl O-(tert-butyldiphenylsilyl)-N-((3-hydroxy-heptadecanoyl)glycyl)-l-serinate (11A, 124 mg, 0.182 mmol) was dissolved in 10 mL of dry THF under nitrogen. The solution was cooled to 0°C and was treated with 1 M tetrabutylammonium fluoride (TBAF, 0.20 mL, 0.200 mmol). The reaction was slowly warmed to ambient temperature and stirred for 6 h. The solvent was removed under reduced pressure, and the resulting oil was purified by column chromatography (2.5 % MeOH:DCM) to yield methyl (3-hydroxyheptadecanoyl)glycyl-l-serinate 12A, as a white solid, (65 mg, 0.146 mmol, 80.5 %), MP, 104-111 °C. 1H NMR (400 MHz, CDCl3) δ 7.36 (d, J = 7.8 Hz, 1H), 6.71 (d, J = 5.5 Hz, 1H), 4.66 (dt, J = 7.3, 3.4 Hz, 1H), 4.22–3.80 (m, 6H), 3.79 (s, 3H), 3.55 (d, J = 23.0 Hz, 1H), 2.46 (dt, J = 13.9, 2.8 Hz, 1H), 2.36–2.24 (m, 1H), 1.52–1.36 (m, 3H), 1.32–1.24 (m, 24H), 0.88 (t, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 173.30, 173.10, 171.07, 170.89, 169.39, 169.18, 69.59, 69.31, 62.92, 54.87, 54.83, 52.89, 43.69, 43.50, 43.46, 43.32, 37.48, 37.27, 31.98, 29.75, 29.72, 29.65, 29.55, 29.42, 25.61, 25.54, 22.75, 14.17. HRMS (AccuTOF): [MH]+ Calc’d for C23H44N2O6 m/z 445.3278. Found: m/z 444.3278.
Methyl (3-hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoyl)glycyl-l-serinate, 12B
Methyl O-(tert-butyldiphenylsilyl)-N-((3-hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoyl)glycyl)-l-serinate, (11B, 192 mg, 0.277 mmol) was dissolved in 10 mL of dry THF under nitrogen. The solution was cooled to 0°C and was treated with 1 M tetrabutylammonium fluoride (TBAF, 0.30 mL, 0.304 mmol). The reaction was slowly warmed to ambient temperature and stirred for 6 h. The solvent was removed under reduced pressure, and the resulting oil was purified by column chromatography (2.5 % MeOH:DCM) to yield methyl (3-hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoyl)glycyl-l-serinate, 12B, as a white solid, (107 mg, 0.236 mmol, 85.1 %), MP, 95-102 °C. 1H NMR (400 MHz, CDCl3) δ 7.47 (dd, J = 30.8, 7.7 Hz, 1H), 6.93 (d, J = 6.2 Hz, 1H), 4.65 (dt, J = 7.4, 3.5 Hz, 1H), 4.23–3.78 (m, 6H), 3.78 (s, 3H), 2.45 (dt, J = 14.0, 2.4 Hz, 1H), 2.36–2.25 (m, 1H), 1.54–1.37 (m, 3H), 1.33–1.23 (m, 17H). 13C NMR (101 MHz, CDCl3) δ 173.36, 173.21, 171.17, 170.97, 169.54, 169.39, 69.62, 69.30, 62.76, 54.92, 54.87, 52.86, 43.72, 43.57, 43.35, 43.26, 37.49, 37.28, 29.76, 29.73, 29.67, 29.58, 29.47, 25.65, 25.59. HRMS (AccuTOF): [MH]+ Calc’d for C23H35D9N2O6 m/z 454.3843. Found: m/z 454.3814.
(3-Hydroxyheptadecanoyl)glycyl-l-serine, 13A
A solution of 1 M LiOH (0.12 mL, 0.12 mmol) was added to a solution of methyl (3-hydroxyheptadecanoyl)glycyl-l-serinate (12A, 47 mg, 0.11 mmol) in 2 mL of methanol at 0 °C, and 4 mL of THF was subsequently added. The resulting solution was warmed to ambient temperature and stirred for 2 h. The solution was then neutralized with 10% HCl and diluted with 25 mL of H2O. The solution was then extracted with EtOAc (3 X 25 mL) and the organic layers were concentrated in vacuo. The resulting crude material was recrystallized from hot EtOAc to yield (3-hydroxyheptadecanoyl)glycyl-l-serine, 13A, as a crystalline solid (38 mg, 0.088 mmol, 84 %), MP, 119-122 °C. 1H NMR (400 MHz, CDCl3) δ 4.64–4.40 (m, 1H), 4.14–3.69 (m, 5H), 2.43–2.33 (m, 1H), 2.30–2.20 (m, 1H), 1.52–1.20 (m, 26H), 0.83 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 171.95, 171.25, 168.89, 67.48, 61.38, 54.54, 51.82, 43.63, 41.83, 36.86, 31.27, 29.08, 29.03, 28.98, 28.68, 25.10, 22.07, 13.93. HRMS (-TOF MS): [M-H]- Calc’d for C22H42N2O6 m/z 429.2965. Found: m/z 429.2989.
(3-Hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoyl)glycyl-l-serine, 13B
A solution of 1 M LiOH (0.28 mL, 0.28 mmol) was added to a solution of methyl (3- hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoyl)glycyl-l-serinate (12B, 117 mg, 0.258 mmol) in 2 mL of methanol at 0 °C, and 4 mL of THF was subsequently added. The resulting solution was warmed to ambient temperature and stirred for 2 h. The solution was then neutralized with 10% HCl and diluted with 25 mL of H2O. The solution was then extracted with EtOAc (3 X 25 mL) and the organic layers were concentrated in vacuo. The resulting crude material was recrystallized from hot EtOAc to yield ((3-hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoyl)glycyl-l-serine, 13B, as a crystalline solid (83 mg, 0.19 mmol, 73 %), MP, 118-122 °C. 1H NMR (400 MHz, CDCl3) δ 4.50 (dt, J = 11.0, 3.4 Hz, 1H), 4.17–3.65 (m, 5H), 2.37 (ddd, J = 13.6, 7.9, 2.8 Hz, 1H), 2.29–2.19 (m, 1H), 1.49–1.16 (m, 20H). 13C NMR (101 MHz, DMSO) δ 171.78, 171.22, 168.96, 67.47, 61.31, 54.51, 51.83, 43.60, 41.75, 36.84, 29.08, 29.04, 28.99, 28.74, 25.10. HRMS (AccuTOF): [MH]+ Calc’d for C22H33D9N2O6 m/z 440.3686. Found: m/z 440.3689.
(6S)-6-(Methoxycarbonyl)-2,2-dimethyl-8,11-dioxo-3,3-diphenyl-4-oxa-7,10-diaza-3-silaheptacosan-13-yl 13-methyltetradecanoate, 14A
13-Methyltetradecanoic acid (82 mg, 0.338 mmol) was dissolved in 15 mL of dry DCM and stirred at 0 °C. To the solution was then treated with DCC (87 mg, 0.423 mmol) and a catalytic amount of DMAP. After stirring for 15 min, methyl O-(tert-butyldiphenylsilyl)-N-((3-hydroxy-heptadecanoyl)glycyl)-l-serinate, (11A, 231 mg, 0.338 mmol) dissolved in 5 mL of dry DCM was added to the reaction via cannula. The resulting solution was warmed to ambient temperature and stirred overnight. The solvent was removed under reduced pressure and the resulting oil was redissolved in 20 mL of diethyl ether. The resulting precipitate was filtered, and the filtrate washed with satd NaHCO3 (3 X 10 mL). The organic layer was dried, filtered, and solvents evaporated under reduced pressure. Purification via column chromatography (1% MeOH:DCM) yielded (6S)-6-(methoxycarbonyl)-2,2-dimethyl-8,11-dioxo-3,3-diphenyl-4-oxa-7,10-diaza-3-silaheptacosan-13-yl 13-methyltetradecanoate, 14A, as a clear wax (123 mg, 0.136 mmol, 63.4% brsm). 1H NMR (400 MHz, CDCl3) δ 7.58 (t, J = 7.1 Hz, 4H), 7.46–7.37 (m, 6H), 6.54 (t, J = 6.7 Hz, 1H), 6.37 (d, J = 4.9 Hz, 1H), 5.16 (p, J = 6.3 Hz, 1H), 4.66 (d, J = 7.8 Hz, 1H), 4.14–3.82 (m, 5H), 3.74 (s, 3H), 2.55–2.42 (m, 2H), 2.29 (t, J = 7.5 Hz, 2H), 1.50 (hept, J = 6.6 Hz, 1H), 1.31–1.24 (m, 42H), 1.15–1.10 (m, 4H), 1.03 (s, 9H), 0.89–0.84 (m, 9H). 13C NMR (101 MHz, CDCl3) δ 173.45, 170.47, 169.98, 168.26, 135.56, 135.52, 132.82, 132.64, 130.11, 130.06, 127.97, 127.88, 71.12, 64.15, 54.28, 52.58, 49.26, 42.88, 41.43, 39.12, 34.56, 34.18, 34.00, 31.98, 30.01, 29.75, 29.62, 29.57, 29.42, 29.36, 29.21, 28.03, 27.48, 26.78, 25.67, 25.32, 25.05, 24.99, 22.75, 22.72, 14.17. HRMS (AccuTOF): [MH]+ Calc’d for C54H90N2O7Si m/z 907.6596. Found: m/z 907.6570.
(6S)-6-(Methoxycarbonyl)-2,2-dimethyl-8,11-dioxo-3,3-diphenyl-4-oxa-7,10-diaza-3-sila- [24,24,25,25,26,26,27,27,27-2H9]-heptacosan-13-yl 13-methyltetradecanoate, 14B
13-Methyltetradecanoic acid (130 mg, 0.535 mmol) was dissolved in 15 mL of dry DCM and stirred at 0 °C. To the solution was then treated with DCC (110 mg, 0.535 mmol) and a catalytic amount of DMAP. After stirring for 15 min, methyl O-(tert-butyldiphenylsilyl)-N-((3- hydroxy-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoyl)glycyl)-l-serinate (11B, 322 mg, 0.465 mmol) dissolved in 5 mL of dry DCM was added to the reaction via cannula. The resulting solution was warmed to ambient temperature and stirred overnight. The solvent was removed under reduced pressure and the resulting oil was redissolved in 20 mL of diethyl ether. The resulting precipitate was filtered, and the filtrate washed with satd NaHCO3 (3 X 10 mL). The organic layer was dried, filtered, and solvents evaporated under reduced pressure. Purification via column chromatography (1% MeOH:DCM) yielded (6S)-6-(methoxycarbonyl)- 2,2-dimethyl-8,11-dioxo-3,3-diphenyl-4-oxa-7,10-diaza-3-sila-[24,24,25,25,26,26,27,27,27-2H9]-heptacosan-13-yl 13-methyltetradecanoate, 14B, as a clear wax (216 mg, 0.236 mmol, 79.6% brsm). 1H NMR (400 MHz, CDCl3) δ 7.63–7.55 (m, 4H), 7.48–7.36 (m, 6H), 6.52 (d, J = 6.8 Hz, 1H), 6.33 (d, J = 5.0 Hz, 1H), 5.20–5.13 (m, 1H), 4.66 (dd, J = 7.6, 3.4 Hz, 1H), 4.13 (dd, J = 10.3, 2.9 Hz, 1H), 4.01–3.81 (m, 3H), 3.75 (s, 3H), 2.54–2.44 (m, 2H), 2.29 (t, J = 7.5 Hz, 2H), 1.51 (hept, J = 13.2, 6.6 Hz, 1H), 1.39–1.16 (m, 38H), 1.15 (q, J = 6.7 Hz, 2H), 1.03 (s, 9H), 0.86 (d, J = 6.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 173.42, 170.44, 170.40, 169.95, 168.21, 135.56, 135.52, 132.83, 132.65, 130.11, 130.06, 127.97, 127.88, 71.10, 64.15, 54.28, 54.26, 52.57, 42.88, 41.43, 39.12, 34.56, 34.18, 30.01, 29.72, 29.62, 29.57, 29.47, 29.42, 29.36, 29.21, 28.03, 27.48, 26.78, 25.32, 25.06, 22.72, 19.31. HRMS (AccuTOF): [MH]+ Calc’d for C54H81D9N2O7Si m/z 916.7160. Found: m/z 916.7154.
1-((2-(((S)-3-Hydroxy-1-methoxy-1-oxopropan-2-yl)amino)-2-oxoethyl)amino)-1- oxoheptadecan-3-yl 13-methyltetradecanoate, 15A
(6S)-6-(Methoxycarbonyl)-2,2-dimethyl-8,11-dioxo-3,3-diphenyl-4-oxa-7,10-diaza-3-silaheptacosan-13-yl 13-methyltetradecanoate (14A, 123 mg, 0.136 mmol) was dissolved in 5 mL of dry THF under nitrogen. The solution was cooled to 0 °C and was treated with 1 M TBAF (0.15 mL, 0.15 mmol). The reaction was slowly warmed to ambient temperature and stirred for 1 h. The solvent was removed under reduced pressure, and the resulting oil was purified by column chromatography (5% MeOH:DCM) to yield 1-((2-(((S)-3-hydroxy-1-methoxy-1-oxopropan-2-yl)amino)-2-oxoethyl)amino)-1-oxoheptadecan-3-yl 13- methyltetradecanoate, 15A, as a white solid (69 mg, 0.103 mmol, 76.1 % brsm) MP 84-92 °C. 1H NMR (400 MHz, CDCl3) δ 7.06 (dd, J = 46.7, 7.5 Hz, 1H), 6.44 (s, 1H), 5.24–5.12 (m, 1H), 4.66 (t, J = 3.4 Hz, 1H), 4.11–3.89 (m, 4H), 3.79 (s, 3H), 3.20 (d, J = 68.5 Hz, 1H), 2.49 (dd, J = 6.0, 3.2 Hz, 2H), 2.31 (td, J = 7.6, 2.1 Hz, 2H), 1.51 (hept, J = 6.6 Hz, 1H), 1.38–1.20 (m, 40H), 1.15 (q, J = 6.8 Hz, 2H), 0.93–0.79 (m, 9H). 13C NMR (101 MHz, CDCl3) δ 174.55, 174.39, 170.78, 170.71, 170.66, 170.60, 168.97, 168.93, 71.53, 62.96, 55.02, 52.76, 43.43, 42.04, 41.95, 39.12, 34.62, 34.55, 31.99, 30.01, 29.76, 29.71, 29.62, 29.57, 29.42, 29.38, 29.33, 29.21, 28.03, 27.48, 25.32, 25.06, 25.00, 22.76, 22.72, 14.17. HRMS (-TOF MS): [M-H]- Calc’d for C38H72N2O7 m/z 667.5261. Found: m/z 667.5243.
1-((2-(((S)-3-Hydroxy-1-methoxy-1-oxopropan-2-yl)amino)-2-oxoethyl)amino)-1-oxo- [14,14,15,15,16,16,17,17,17-2H9]-heptadecan-3-yl- 13-methyltetradecanoate, 15B
(6S)-6-(Methoxycarbonyl)-2,2-dimethyl-8,11-dioxo-3,3-diphenyl-4-oxa-7,10-diaza-3- sila-[24,24,25,25,26,26,27,27,27-2H9]-heptacosan-13-yl 13-methyltetradecanoate (14B, 192 mg, 0.277 mmol) was dissolved in 5 mL of dry THF under nitrogen. The solution was cooled to 0 °C and was treated with 1 M TBAF (0.30 mL, 0.15 mmol). The reaction was slowly warmed to ambient temperature and stirred for 1 h. The solvent was removed under reduced pressure, and the resulting oil was purified by column chromatography (5% MeOH:DCM) to yield 1-((2-(((S)-3-hydroxy-1-methoxy-1-oxopropan-2-yl)amino)-2-oxoethyl)amino)-1-oxo-[14,14,15,15,16,16,17,17,17-2H9]-heptadecan-3-yl 13-methyltetradecanoate, 15B, as a white solid (107 mg, 0.236 mmol, 85.1 % brsm) MP 82-88 °C. 1H NMR (400 MHz, CDCl3) δ 7.10 (dd, J = 43.2, 7.8 Hz, 1H), 6.48 (d, J = 4.2 Hz, 1H), 5.18 (dt, J = 19.0, 6.4 Hz, 1H), 4.66 (t, J = 3.4 Hz, 1H), 4.14–3.89 (m, 4H), 3.79 (s, 3H), 3.27 (d, J = 65.9 Hz, 1H), 2.49 (dd, J = 6.1, 3.1 Hz, 2H), 2.31 (td, J = 7.6, 2.3 Hz, 2H), 1.50 (t, J = 7.0 Hz, 1H), 1.41–1.18 (m, 34H), 1.15 (q, J = 6.4 Hz, 2H), 0.86 (d, J = 6.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 174.56, 174.39, 170.79, 170.73, 170.67, 170.61, 168.98, 168.93, 71.53, 62.96, 62.92, 55.02, 52.76, 43.44, 43.40, 42.05, 41.94, 39.12, 34.62, 34.55, 30.01, 29.78, 29.73, 29.62, 29.57, 29.48, 29.38, 29.33, 29.22, 29.19, 28.03, 27.48, 25.32, 25.06, 25.00, 22.71. HRMS (AccuTOF): [MH]+ Calc’d for C38H63D9N2O7 m/z 678.5983. Found: m/z 678.5992.
(3-((13-Methyltetradecanoyl)oxy)heptadecanoyl)glycyl-l-serine 2A
A stirring solution of 1-((2-(((S)-3-hydroxy-1-methoxy-1-oxopropan-2-yl)amino)-2-oxoethyl)amino)-1-oxoheptadecan-3-yl 13-methyltetradecanoate (15A, 48 mg, 0.078 mmol), in 3 mL of dry DMF, was cooled to 0 °C. Potassium trimethylsilanoate (11 mg, 0.083 mmol) was added, and the reaction was stirred on the ice bath for 1.5 h before being warmed to ambient temperature and stirred for an additional 1.5 h. The solution was diluted with 10 mL of 10 mL of H2O and acidified with 10% HCl. The resulting cloudy precipitate was extracted with ethyl acetate (3 X 15mL). The organic layer was dried, filtered, and solvents evaporated under reduced pressure. The resulting solid was purified by column chromatography (10% MeOH:DCM) to yield (3-((13-methyltetradecanoyl)oxy)heptadecanoyl)glycyl-l-serine, 2A, as a white sticky solid (33 mg, 0.050 mmol, 65 % brsm). 1H NMR (500 MHz, CDCl3) δ 7.07 (d, J = 8.4 Hz,0.5H), 6.77 (d, J = 8.2 Hz,0.5H), 5.17–5.07 (m, 1H), 4.26 (s, 1H), 4.11–3.60 (m, 4H), 3.36–3.30 (m, 1H), 2.47 (s, 2H), 2.23 (s, 2H), 1.67–1.41 (m, 6H), 1.21 (s, 41H), 0.82 (t, J = 7.0 Hz, 9H). 13C NMR spectrum could not be obtained due to liposome formation. HRMS (-TOF MS): [M-H]- Calc’d for C37H70N2O7 m/z 653.5105. Found: m/z 653.5077.
(3-((13-Methyltetradecanoyl)oxy)-[14,14,15,15,16,16,17,17,17-2H9]-heptadecanoyl)glycyl-l-serine, 2B
A stirring solution of 1-((2-(((S)-3-hydroxy-1-methoxy-1-oxopropan-2-yl)amino)-2- oxoethyl)amino)-1-oxo-[14,14,15,15,16,16,17,17,17-2H9]heptadecan-3-yl 13-methyltetradecanoate (15A, 98 mg, 0.144 mmol), in 3 mL of dry DMF, was cooled to 0 °C. Potassium trimethylsilonoate (18 mg, 0.144 mmol) was added, and the reaction was stirred on the ice bath for 1.5 h before being warmed to ambient temperature and stirred for an additional 1.5 h. The solution was diluted with 10 mL of 10 mL of H2O and acidified with 10% HCl. The resulting cloudy precipitate was extracted with ethyl acetate (3 X 15mL). The organic layer was dried, filtered, and solvents evaporated under reduced pressure. The resulting solid was purified by column chromatography (10% MeOH:DCM) to yield (3-((13-methyltetradecanoyl)oxy)- [14,14,15,15,16,16,17,17,17-2H9]-heptadecanoyl)glycyl-l-serine, 2B, as a white sticky solid (55 mg, 0.083 mmol, 58% brsm). 1H NMR (400 MHz, CDCl3) δ 7.09 (d, J = 8.1 Hz,0.5H), 6.78 (d, J = 8.4 Hz,0.5H), 5.24–5.08 (m, 1H), 4.30 (s, 1H), 4.14–3.55 (m, 4H), 3.43–3.28 (m, 1H), 2.52 (s, 2H), 2.25 (t, J = 7.4 Hz, 2H), 1.57 (d, J = 13.1 Hz, 4H), 1.51–1.44 (m, 1H), 1.42–1.15 (m, 30H), 1.12 (q, J = 6.8 Hz, 2H), 0.83 (d, J = 6.6 Hz, 6H). 13C NMR spectrum could not be obtained due to liposome formation. HRMS (-TOF MS): [M-H]- Calc’d for C37H61D9N2O7 m/z 662.5670. Found: m/z 662.5640.
Scheme 1.

Synthesis of non-deuterated 2A
Reagents: (a) TBDMSCl, imidazole, 61%; (b) (i) 1-bromobutane (3A), Mg0, THF (ii) CuCl2, LiCl, NMP, 93%; (c) TBAF, 87.4%; (d) Bobbitt’s salt, SiO2, 90.8%; (e) SnCl2, ethyl diazoacetate, 89.6%; (f) NaBH4, 77.6%; (g) LiOH, 83.6%; (h) (i) NHS, EDC, DMAP (ii) Pd/C, H2, 67%; (i) TBAF, 80.5% (j) LiOH, 84%; (k) 13-methylpentadecanoic acid, EDC, DMAP, 63.4%; (l) TBAF, 76.1% (m) KOSi(CH)3, 65%.
Scheme 2.

Synthesis of Deuterated 2B
Reagents: (a) TBDMSCl, imidazole, 56.4%; (b) (i) 1-bromo-[2H9]-butane (3B), Mg0, THF (ii) CuCl2, LiCl, NMP, 98.3%; (c) TBAF, 99.1%; (d) Bobbitt’s salt, SiO2, 75.6%; (e) SnCl2, ethyl diazoacetate, 80.6%; (f) NaBH4, 79.7%; (g) LiOH, 89.3% (h) (i) 10, NHS, EDC, DMAP (ii) Pd/C, H2, 78.2%; (i) TBAF, 85.1% (j) LiOH, 73%; (k) 13-methylpentadecanoic acid, EDC, DMAP, 79.6%; (l) TBAF, 85.1% (m) KOSi(CH)3, 58%.
Acknowledgments
The work was supported, in part, by NIH Grant DE 021055. We thank the UCONN Mass Spectra facility, under the direction of Dr. You-Jun Fu, for their efforts.
References
- 1.Dietz C, Hart TK, Nemati R, Yao X, Nichols FC, Smith MB. Tetrahedron. 2016;72:7557–7569. Also see Nichols FC, Riep B, Mun J, Morton MD, Kawai T, Dewhirst FE, Smith MB. J Lipid Res. 2006;47:844–853. doi: 10.1194/jlr.M500542-JLR200.; Nichols FC, Riep B, Mun J, Morton MD, Bojarski MT, Dewhirst FE, Smith MB. J Lipid Res. 2004;45:2317–2330. doi: 10.1194/jlr.M400278-JLR200.; Clark RB, Cervantes JL, Maciejewski MW, Farrokhi V, Nemati R, Yao X, Anstadt E, Fujiwara M, Wright KT, Riddle C, La Vake CJ, Salazar JC, Finegold S, Nichols FC. Infect Immun. 2013;81:3479–3489. doi: 10.1128/IAI.00803-13..
- 2.Holt SC, Kesavalu L, Walker S, Genco CA. Periodontol 2000. 1999;20:168–238. doi: 10.1111/j.1600-0757.1999.tb00162.x. [DOI] [PubMed] [Google Scholar]
- 3.Nichols FC, Yao X, Bajrami B, Downes J, Finegold SM, Knee E, Gallagher JJ, Housley WJ, Clark RB. PLoS One. 2011;6:e16771. doi: 10.1371/journal.pone.0016771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang YH, Nemati R, Anstadt E, Liu Y, Son Y, Zhu Q, Yao X, Clark RB, Rowe DW, Nichols FC. Bone. 2015;81:654–661. doi: 10.1016/j.bone.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith MB. March’s Advanced Organic Chemistry. 7. Wiley; NJ: 2013. p. 1449.; Bobbitt JM, Flores MCL. Heterocycles. 1988;27:509–533.; Ma Z, Bobbitt JM. J Org Chem. 1991;56:6110–6114..
- 6.Holmquist CR, Roskamp EJ. J Org Chem. 1989;54:3258–260.; Brockwell JC, Holmquist CR. J Chem Ed. 1992;69:68.; Mun J, Onorato A, Nichols FC, Morton MD, Saleh AI, Welzel M, Smith MB. Org Biomol Chem. 2007;5:3826–3833. doi: 10.1039/b712707c..
- 7.Bergbreiter DE, Whitesides GM. J Org Chem. 1975;40:779–782. doi: 10.1021/jo00894a605.; Cahiez G, Chaboche C, Jezequel M. Tetrahedron. 2000;56:2733–2737..
- 8.Shute RE, Rich DH. J Chem Soc. 1987;15:1155–1156. [Google Scholar]
- 9.Lovric M, Cepanec I, Litvic M, Bartolincic A, Vinkovic V. Croatica Chemica Acta CCACAA. 2007;80:109–115. [Google Scholar]
- 10.Bowler J, Lilley TJ, Pittam JD, Wakeling AL. Steroids. 1989;54:71–99. doi: 10.1016/0039-128x(89)90076-7. [DOI] [PubMed] [Google Scholar]
- 11.Savile CK, Reed DW, Meesapyodsuk D, Covello PS, Buist PH. J Chem Soc Perkin Trans. 2001;1:1116–1121. [Google Scholar]
- 12.van der Veken BJ, Odeurs RO, Brown A, Mckean DC, Morrisson AR. J Mol Str. 1986;147:57–66. [Google Scholar]
- 13.Jorapur YR, Chi DY. J Org Chem. 2005;70:10774–10777. doi: 10.1021/jo051722h.; Chandrasekhar S, Shyamsunder T, Chandrashekar G, Narsihmulu C. Synlett. 2004;3:522–524..
- 14.Paul B, Das D, Ellington B, Marsh ENG. J Am Chem Soc. 2013;135:5234–5237. doi: 10.1021/ja3115949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mai A, Rotili D, Tarantino D, Ornaghi P, Tosi F, Vicidomini C, Sbardella G, Nebbioso A, Miceli M, Altucci L, Filetici P. J Med Chem. 2006;49:6897–6907. doi: 10.1021/jm060601m. [DOI] [PubMed] [Google Scholar]