

Abstract
Genetic diseases of blood cells are prime candidates for treatment through ex vivo gene editing of CD34+ hematopoietic stem/progenitor cells (HSPCs), and a variety of technologies have been proposed to treat these disorders. Sickle Cell Disease (SCD) is a recessive genetic disorder caused by a single nucleotide polymorphism (SNP) in the β-globin gene (HBB). Sickle hemoglobin damages erythrocytes, causing vasoocclusion, severe pain, progressive organ damage, and premature death. Here we optimize design and delivery parameters of a ribonucleoprotein (RNP) complex comprising Cas9 protein and unmodified sgRNA together with a single-stranded DNA oligonucleotide donor (ssODN) to enable efficient replacement of the SCD mutation in human HSPCs. Corrected HSPCs from SCD patients produce less sickle hemoglobin RNA and protein and correspondingly increased wild-type hemoglobin when differentiated into erythroblasts. When engrafted in immunocompromised mice, ex vivo treated human HSPCs maintain SCD gene edits throughout sixteen weeks at a level likely to have clinical benefit. These results demonstrate that an accessible approach combining Cas9 RNP with an ssODN can mediate efficient HSPC genome editing, enables investigator-led exploration of gene editing reagents in primary hematopoietic stem cells, and suggests a path towards the development of new gene editing treatments for SCD and other hematopoietic diseases.
Introduction
Sickle Cell Disease (SCD) is a recessive genetic disorder that affects at least 90,000 predominantly African-American individuals in the US and hundreds of thousands worldwide (1, 2). The genetics and molecular basis of SCD have been understood for nearly 70 years, but curative treatments have lagged (3, 4). SCD is caused by a single nucleotide polymorphism (SNP) in the seventh codon of the gene for β-globin (HBB), one of two globins that make up the major adult form of hemoglobin. The resulting glutamate-to-valine substitution renders hemoglobin prone to polymerization under hypoxic conditions, producing characteristic “sickle” shaped red blood cells (RBCs). Sickle RBCs have a markedly reduced lifespan in the bloodstream, damage the vasculature, and cause vasoocclusion. Major clinical manifestations of SCD are chronic anemia, severe pain episodes, and progressive damage to vital organs such as the brain, lung, and kidney. In the United States, the disease causes a 30-year decrement in lifespan and a greatly diminished quality of life (2, 5–7).
RBCs are produced from re-populating hematopoietic stem cells (HSCs) in the bone marrow, and allogeneic hematopoietic cell transplantation (HCT) from an unaffected HLA-matched donor is currently the only lasting cure for SCD (8). However, HCT has been used sparingly because of the difficulty in identifying donors, risks associated with the toxicity of the transplant regimen (requiring preparation with chemotherapy and immune suppression), and potentially fatal graft-versus-host disease (9, 10). Recent transplant advances have reduced these risks in children (11) and have extended treatment to selected adults (12) and individuals for whom only a haploidentical HLA donor is available (13). Still, the vast majority of individuals with SCD do not pursue allogeneic HCT because of an unfavorable risk-reward profile, especially during early childhood. A curative treatment for SCD that can be safely applied to more people remains an urgent need.
Gene editing has recently emerged as a promising avenue to treat genetic diseases affecting hematopoietic cells (14–16). Ex vivo editing of autologous HSPCs would be followed by re-implantation of edited cells, bypassing donor requirements and eliminating the risk of graft-versus-host disease and post-grafting immunosuppression. Because sickle RBCs have a markedly shorter lifespan in circulation compared to wild type RBCs, even low levels of genotypic correction are predicted to generate a clinical benefit (17). Indeed, observations in patients after allogeneic HCT suggest that clinical improvement may occur when as few as 2 – 5 of long-term engrafted cells carry a normal HBB allele (18–20). An ideal gene editing treatment would exceed this modest target, but to date even this level of gene editing has not been achieved (14, 21).
During gene editing, a targeted nuclease creates a double strand break (DSB) that can be repaired by one of two mechanisms: error-prone non-homologous end joining (NHEJ) that results in genomic insertions and deletions (indels), or templated homology-directed repair (HDR) to precisely insert, delete, or replace a genomic sequence (22). The recent development of CRISPR-Cas9, a programmable RNA-targeted DNA endonuclease, has ignited an explosion of interest in gene editing to cure many genetic disorders, including SCD (23, 24) Guided by a single guide RNA (sgRNA), the Cas9 nuclease can be programmed to cut a target locus within the genome, allowing rapid iteration and optimization not possible with other gene editing approaches (23, 25).
Optimized methods for efficient ex vivo gene editing of human HSPCs are required to enable a CRISPR/Cas9-based treatment for blood disorders such as SCD. Recent work has demonstrated that Cas9 can be used for in vitro reversion of the SCD mutation in laboratory cell lines (26, 27) and induced pluripotent stem cells (iPSCs) (28), as well as efficient knockout of an erythroid enhancer in an immortalized cell line (29). Cas9-mediated in vitro knockout at the HBB locus in hematopoietic stem/progenitor cells (HSPCs) has so far been reported only using Cas9 mRNA and chemically protected sgRNAs (27). Correction of the SCD mutation in iPSCs has been reported using TALENs (30, 31), and most recently zinc finger nucleases (ZFNs) have been used to correct the SCD mutation in HSPCs, albeit at levels of less than 1% in the long-term re-populating stem cell population (21). To date, gene editing has yet to achieve long-term correction of the SCD mutation in these cells at levels greater than 1%, based on engraftment in immunocompromised mice (21). Previous approaches to HSPC gene editing have required specialized technologies that are not broadly available, including highly engineered and proprietary ZFNs, chemically protected Cas9 guide RNAs, and viral HDR donors (14, 15, 21, 27). Although these factors are not inherently a barrier to clinical translation, they limit the discovery of efficient gene editing reagents for disease mutations, including SCD.
Electroporation of a pre-assembled ribonucleoprotein complex (RNP), composed of recombinant Cas9 protein and unmodified in vitro transcribed sgRNA, can be used for gene knockout in a variety of cell types. We have recently extended this approach to enable extremely efficient HDR sequence replacement via rationally designed ssDNA donors in laboratory cell lines (32–35). Given the efficiency and speed of Cas9 RNP-based editing, we reasoned that a Cas9 RNP-based approach to gene editing could form the basis for an accessible protocol to correct mutations in human HSPCs. We paired the Cas9 RNP with single stranded oligonucleotide HDR donors (ssODNs), which are generally available, easily designed, and able to mediate efficient sequence replacement in immortalized cell lines (35). Our goal is not only to develop an approach to edit HSPCs as a potential treatment for SCD, but also to use reagents that are inexpensive and accessible to a wide variety of researchers, enabling investigator-led studies and rapid optimization to address a multitude of genetic hematopoietic diseases, including polygenic diseases.
We developed a pipeline to enable efficient, RNP/ssODN-based correction of the SCD mutation without introducing a selective marker (Fig. 1A). We first used an erythroleukemia cell line to explore a panel of Cas9 RNPs that cut near the SCD mutation. We then used the most effective RNPs to develop ex vivo editing methods in human HSPCs, achieving up to 33% sequence replacement. We demonstrate efficient correction of the sickle mutation in SCD HSPCs, with corresponding production of wild type adult hemoglobin (HbA) RNA and protein in edited, differentiated erythroblasts. After the edited human HSPCs are engrafted in immunocompromised mice, sequence replacement at the SCD locus is retained four months after engraftment at levels likely to have clinical benefit.
Figure 1. Editing the SCD SNP in K562 cells.
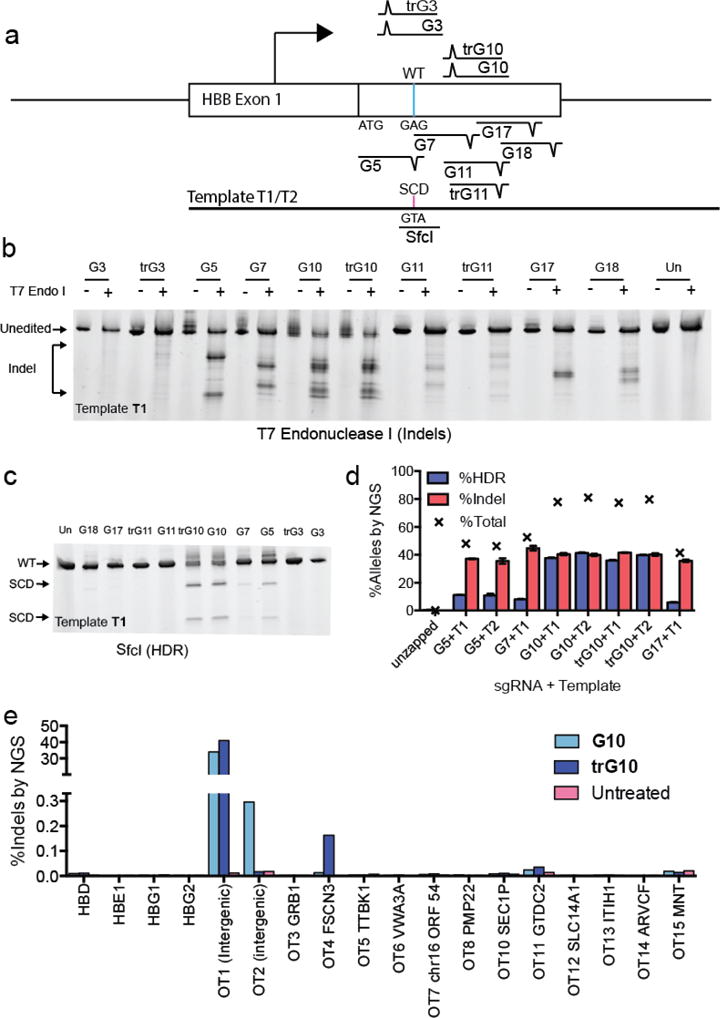
A) Schematic depicting the experimental approach to editing in K562 cells. A panel of 10 sgRNAs that cut within 100 bp of the SCD SNP was selected. A WT-to-SCD edit was programmed by an ssDNA template (T1) bearing silent PAM mutations for the sgRNAs (except G7), which also introduces a SfcI restriction site. B) T7 endonuclease assay showing indel formation in pools of cells edited by candidate RNPs. C) HDR editing of candidate sgRNAs detected by SfcI digestion. G5, G10 and a truncated variant, trG10, efficiently yield HDR products in K562 cells. D) Gene modification of select sgRNAs and templates at the SCD SNP, assessed by NGS. See Fig. S1 for definitions of donors T1 and T2. (average of 3 biological replicates, error bars indicate standard deviation). E) Analysis of off-target cutting by the G10 RNP at sites predicted by the online CRISPR-Design tool (reference), in K562 cells, determined by NGS.
Results
Prioritizing SCD editing reagents in a model cell line
Pairing Cas9 with various sgRNAs can lead to different activities on the same gene target (36). For HDR-mediated editing, each sgRNA must in turn be paired with an HDR donor template that encodes the desired nucleotide changes. Coupled with subtleties of donor template design and reagent delivery, the complexity of this problem expands rapidly. We used inexpensive, commercially-available single-stranded DNA oligonucleotide donors (ssODNs), and co-delivered these with the Cas9 RNP by electroporation. Using these components, we were able to quickly iterate combinations of sgRNA, HDR donor, and editing condition. The rules governing the choice of sgRNA and ssODN are still unclear, though we have developed guidelines based on the mechanism of Cas9’s cleavage activity (35).
We searched for maximally active sgRNAs and ssODNs in K562 cells, a human erythroleukemia line that resembles early committed hematopoietic progenitors. Two key restrictions when designing sgRNAs and ssODNs for HDR experiments are the distance between the sgRNA recognition site and the mutation, and the ability to silently ablate the sgRNA protospacer adjacent motif (PAM). This latter constraint ensures that Cas9 cannot re-cut corrected alleles, thus preventing the introduction of indels into the corrected allele.
To find maximally active sgRNAs compatible with SCD editing, we identified targets in the first exon of the HBB gene and chose to test six of them for which the PAM could be silently mutated (G3, G5, G10, G11, G17, G18) and one for which it could not (G7) (Fig. 1A). Some of these sgRNAs have been tested previously (26, 27), but to our knowledge they have not been used in a Cas9 RNP format. We also tested truncated versions of three guides (trG3, trG10, trG11; table S1), a modification that is reported to reduce off-target cleavage without compromising on-target efficiency (37). We designed an initial ssODN (T1) that contained silent mutations in the PAMs of all sgRNAs and a WT-to-SCD edit (K562 cells are WT at HBB, fig. S1A). The combination of the WT-to-SCD SNP mutation and removal of the G5 PAM generates a silent SfcI restriction site, allowing easy tracking of HDR-mediated editing (fig. S1A).
We assembled Cas9 RNPs from each candidate sgRNA and individually delivered them to K562 cells together with the ssODN by electroporation. We used a dose of 100 pmol of each RNP and template per 150,000 K562 cells, based on previous work (32). A T7 endonuclease I digest of PCR amplicons from edited cell pools, which in this case detects mismatches arising from both NHEJ-and HDR-mediated repair of Cas9 DSBs, revealed sequence modifications with all but two sgRNAs (G3 and trG3) (Fig. 1B). SfcI digest showed appreciable HDR-mediated editing of the SCD SNP with three sgRNAs, G5, G10, and trG10 (G10 with an 18-nt guide sequence) (Fig. 1C).
We quantified the frequency of HDR and indel formation at the SCD SNP by next-generation sequencing (NGS) of PCR amplicons derived from genomic DNA extracted from pools of edited cells (Fig. 1D), and by droplet digital PCR (ddPCR, fig. S1B). When the trG10 Cas9 RNP was provided without an ssODN, 90% of NGS reads contained indels, indicating excellent delivery of the RNP and efficacious gene knockout (fig. S1C). We performed experiments using both the T1 ssODN and a derivative of T1 bearing only the WT-to-SCD edit and the G5 and G10 PAM mutations (T2) (fig. S1A). Most candidate sgRNAs induced substantial quantities of indels (35–40% of reads) (Figs. 1B and 1D), and G5, G10, and trG10 also yielded high levels of WT-to-SCD HDR (11% of reads for G5, 41% of reads for G10) from multiple donor designs (Fig. 1 and S1). A phosphorothioate-protected ssODN did not improve HDR frequencies (fig. S1D). The G5 guide targets a sequence very similar to one found in the closely related hemoglobin-delta (HBD) gene, and we experimentally verified that this guide induces indels in HBD (fig. S1E). Hence, we selected the truncated trG10 guide for further testing.
To optimize an ssODN for HDR, we drew guidance from recent work in our laboratory showing that upon binding its target, Cas9 releases the PAM-distal non-target strand, and that asymmetric homology arms taking advantage of this property can increase HDR efficiency (35). For sgRNAs that target the sense strand such as G10, the best template to employ thus matches the sense strand (annealing to the antisense strand) and has a long 5′ homology arm and a shorter 3′ annealing arm (fig. S1F). Based on these principles, we will henceforth name templates by the lengths of their 5′ and 3′ homology arms relative to the G10 cut site. For example, the initial template T2 is termed T88-107. Unless noted, all templates bear only the G5 and G10 PAM mutations. We tested the effects of asymmetric templates containing the SCD-to-WT edit on HDR-mediated editing in K562 cells (fig. S1C). We found that a template with a 111 nt 5′ arm and a 57 bp 3′ arm (T111-57) yielded an HDR frequency modestly higher than the original template T88-107 (33% vs. 28.5% in the same experiment). We also found low but measureable rates of conversion (0.5–1%) between the HBB coding sequence and the homologous region in HBD, but this was only observed in samples treated with G7, G10, and trG10 (fig. S1G).
The possibility that Cas9 may cut at off-target sites is a concern for the development of Cas9-based therapies (37, 38). This tendency may be reduced by the use of Cas9 RNP delivery, truncated sgRNAs, and other emerging techniques (32, 39, 40). We selected off-target candidates by sequence similarity using a popular off-target prediction tool (38), and used NGS of PCR amplicons to analyze the off-target activity G10 and trG10 Cas9 RNPs in K562 cells (Fig. 1E). The two top-scoring off-targets (which are both intergenic), the top 13 exonic off-target, and predicted off-target sites within the four globin genes (HBD, HBE1, HBG1, and HBG2, fig. S2) (41) were assessed.
We observed very little exonic off-target activity, with most off-target sites showing no Cas9-dependent indel formation at rates within the limit of detection (~0.001%) (Fig. 1E). Substantial off-target activity was observed for both G10 and trG10 at a top-scoring intergenic site (OT1, chr9:104,595,865, 55% indel formation), which lies ~3 kb from the nearest annotated genic sequence. High off-target activity by G10 at this site has been observed previously (26). Off-target activity was detected with G10 at the other intergenic site (OT2, chr17:66,624,238, 0.30%), but this was absent with the truncated guide trG10. Low but detectable off-target activity was observed with both the G10 and trG10 RNPs at two exonic off-targets, FSCN3 and GTDC2. Indel formation was lower for the full-length G10 RNP. (FSCN3: 0.013% and 0.16% indel for G10 and trG10, respectively; GTDC2: 0.025% and 0.035% indel for G10 and trG10, respectively). Very few indels were observed at HBD (0.010% and 0.011% indel for G10 and trG10 respectively), and indels at other β-globin genes (HBE1, HBG1, HBG2) were not detected over background (Fig. 1E).
Optimizing candidate Cas9 RNPs and ssODNs to edit CD34+ HSPCs
The results of RNP delivery to K562 cells encouraged us to investigate using this approach with HSPCs, recognizing that editing outcomes can differ between cell types. We used the trG10 sgRNA, which showed robust HDR and few off-target effects in K562, and re-optimized HDR in adult mobilized peripheral blood HSPCs, which are the cells most relevant for therapeutic gene editing to address SCD. Because these cells were obtained from healthy wild-type (WT) donors, our initial experiments used ssODNs bearing a WT-to-SCD mutation.
We first explored optimization of electroporation conditions, template design, and RNP/ssODN dose in HSPCs (figs. S3A–S3B). To selectively interrogate editing in viable cells, we used NGS to assay HSPCs cultured in erythroid expansion conditions for 7 days after editing (erythroid-expanded), along with edited HSPCs cultured for only 2 days (un-expanded). During initial treatments, we found that treatment with even 100 pmol of RNP led to decline in viability (fig. S3C), and so we used 75 pmol RNP as a lower dose in subsequent in vitro experiments. In addition, we tested the effects of the small molecule SCR7 on editing in HSPCs. This NHEJ inhibitor has been reported to increase HDR-mediated editing in some cell types, but we did not observe an improvement in HDR in HSPCs (fig. S3A) (42, 43).
We obtained appreciable initial levels of total editing at the sickle SNP in HSPCs, with HDR rates between 6–11% (Figs. 2A and 2B). After 5 days of erythroid expansion, HDR rates increased, with up to 33% editing at both high and low doses of RNP (150 and 75 pmol, respectively). Total editing in expanded HSPCs was between 66 and 72%, indicating good delivery of the trG10 RNP to HSPCs. In general, higher editing was accompanied by some reduction in viability as measured by fewer viable cells remaining after treatment, particularly at a 150 pmol dose of RNP (fig. S3C). The asymmetric ssODN that was most effective in K562 cells (T111-57) drove HDR more efficiently at a lower Cas9 dose of 75 pmol RNP per 150,000 HSPCs, whereas a shorter template (T111-27) was more efficient at a higher dose of Cas9 (150 pmol RNP per 150,000 HSPCs) (Fig. 2A). In a separate experiment, high rates of HDR were confirmed by ddPCR (fig. S3D). These experiments demonstrate efficient in vitro editing of CD34+ HSPCs using the Cas9 RNP, including HDR-mediated sequence replacement using ssODNs without a selection marker. As was the case with K562 cells, in HSPCs we saw low but measureable conversion of HBB coding sequence to HBD (0.2 to 2%), and rates of conversion increased after expansion of edited cells (fig. S3E).
Figure 2. Editing of wild-type human CD34+ HSPCs by the Cas9 RNP.
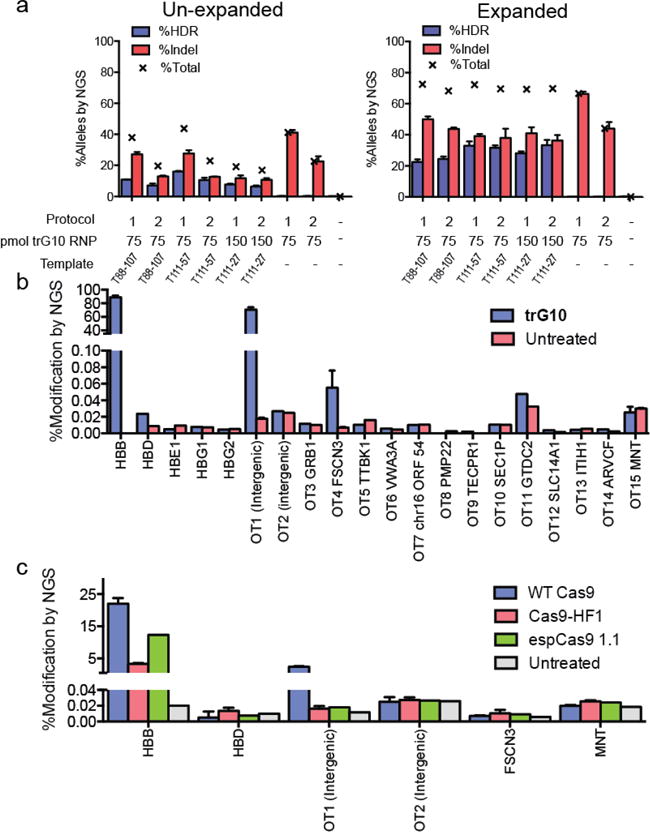
A) Analysis of editing in un-expanded HSPCs (left) and erythroid-expanded HSPCs (right), using trG10 RNP and conditions as indicated. Templates, which are asymmetric about the G10 cut site, were designed as described in the text. All samples are 3 biological replicates, error bars are ± standard deviation. B) Modification (HDR+Indel) at off-target sites in HSPCs edited with the trG10 RNP and template T88-107, compared to untreated cells. Samples for HBB, OT1, FSCN3, and MNT are from 3 biological replicates, with error bars ± standard deviation. Targets were selected using the online CRISPR-design tool (reference). C) Indel formation at on-and off-target sites by Cas9 mutants with increased specificity (HF1 and espCas9 1.1), compared to WT Cas9, all complexed to the G10 sgRNA and no ssODN. All samples are n=3 biological replicates, with error bars ± standard deviation.
To analyze how allele frequencies in the HSPC population translate to the editing of alleles in individual HSPCs, we repeated our best editing condition in CD34+ HSPCs (75 pmol trG10 RNP and 100 pmol T111-57), recovered the cells in HSPC expansion medium for two days, and plated single edited HSPCs by limiting dilution in erythroid expansion medium. After 14 days of growth, 96 edited clones were individually genotyped by multiplexed next generation sequencing (Table 1). In this experiment, we observed 21% HDR alleles in the cell pool. However, these alleles were spread across 32% of cells. This increased prevalence of edited cells relative to edited alleles is predicted by the independent assortment of alleles within a population (although we observed that homozygous genotypes were still overrepresented relative to prediction) (44).
Table 1. Zygosity of clonal colonies of CD34+ HSPCs edited with the trG10 RNP.
HSPCs were edited with 75 pmol of the trG10 RNP (similar to Figure 2), and cloned by limiting dilution. 96 of the resulting clones were then genotyped by NGS. The fraction of all three alleles, the frequency of clones with at least one copy of each of the three alleles, and the frequency of all six genotypes are indicated.
| N = 96 colonies | ||||||
|---|---|---|---|---|---|---|
|
|
||||||
| %WT | %Indel | %HDR | ||||
|
|
||||||
| All Alleles | 46 | 33 | 21 | |||
| Clones with one allele | %WT/_ | %Indel/_ | %HDR/_ | |||
|
|
||||||
| Actual | 60 | 48 | 32 | |||
| Predicted | 71 | 55 | 37 | |||
| Clones by genotype | % WT/WT | % WT/Indel | %WT/HDR | %Indel/Indel | %Indel/HDR | %HDR/HDR |
|
| ||||||
| Actual | 32 | 18 | 10 | 18 | 13 | 9.4 |
| Predicted | 21 | 30 | 19 | 11 | 14 | 4.3 |
We analyzed off-target activity of the trG10 RNP in HSPCs using the target selection criteria described above for K562 cells (Fig. 2B). Most predicted genic off-targets showed no detectable indel formation, although cutting at the previously observed intergenic site remained high (OT1, 44% indel). The sites of off-target cleavage observed in HSPCs generally corresponded to observations in K562 cells, though the rates were often reduced (for example, FSCN3 ~0.05% in HSPCs vs. up to 0.16% in K562s).
As an additional test of the effects of off-target activity of the trG10 RNP, we used over-amplification PCR to detect the presence or absence of translocations between the on-target site at HBB and selected off-target sites (OT1, HBD, FSCN3, and MNT), in both K562 cells and HPSCs edited with trG10 (fig. S4). Translocations between OT1 and HBD with HBB were observed in K562 cells. One translocation may have occurred between OT1 and HBB in HSPCs as well.
We also tested whether trG10 has off-target activity at cancer-associated genes. Though these sites bear little similarity to the trG10 protospacer, even low levels of activity in such locations would be a concern. We used a capture library (Illumina TruSight Cancer) to sequence 94 genes and 290 cancer-associated SNPs to ~8,000 fold coverage each. Relative to unedited cells, we found a small number of indel mutations enriched in K562 cells edited with the trG10 RNP, typically at less than 1% of alleles (table S2). Almost all of these mutations were present in unedited cells. In contrast, no indels were detected in similarly edited HSPCs. These results highlight the importance of performing rare off-target event detection in the target primary cell type and suggest that the trG10 RNP may not have substantial genotoxic liabilities, although this has not been tested at clinical scale.
Two mutant variants of the Cas9 protein have been reported to reduce off-target effects in cell lines, even at sites such as OT1 for the G10 RNP’s OT1 off-target site (45, 46). We expressed and purified the eSpCas9-1.1 and HF1 variants of Cas9, paired them with the G10 sgRNA to form RNPs, and used them to edit wild type CD34+ HSPCs in the absence of an ssODN. We used NGS to determine the on-and off-target editing frequencies (Fig. 2C). As expected, for all enzymes the on-target indel formation was reduced in the absence of an ssODN (Fig. 2B vs 2C) (47). Compared to wild type Cas9, both HF1 and eSpCas9-1.1 showed even further decreased on-target indel formation at the HBB locus, including an almost five-fold decrease in indels for HF1. However, both modified enzymes also completely eliminated all previously observed off-target events, including the prevalent OT1 intergenic off-target (Fig. 2C).
Correction of the SCD mutation in HSPCs results in production of wild type hemoglobin
Our success in WT-to-SCD editing in HSPCs implies that the same method could be used to edit SCD to WT in HSPCs derived from SCD patients. Because human erythropoiesis does not occur when human HSPCs are xenografted into mice, and the availability of SCD HSPCs is limited, we evaluated the effects of correcting the SCD mutation in HSPCs in vitro, by carrying out erythroid differentiation of edited HSPCs. We obtained CD34+ HSPCs from whole blood discarded after exchange transfusion of SCD patients. Because the HF1 and eSpCas9-1.1 proteins yielded reduced levels of on-target editing and the predominant off-target from wild type Cas9 lies in an intergenic region with no known function, we focused on experiments using more efficacious wild-type Cas9. We corrected the SCD mutation using the trG10 RNP and ssODNs carrying an SCD-to-WT edit. These SCD-to-WT templates, denoted by the suffix “S”, encode the same number of mutations as the WT-to-SCD templates, with the base identity different only at the SCD SNP. Measuring editing by both NGS and ddPCR, we found that the SCD HSPCs were edited at levels similar to those observed in WT HSPCs from mobilized blood, with up to 25% of alleles corrected to WT at high RNP dose and 18% corrected at low RNP dose (Figs. 3A and S5).
Figure 3. Correction of the SCD mutation in SCD HSPCs.
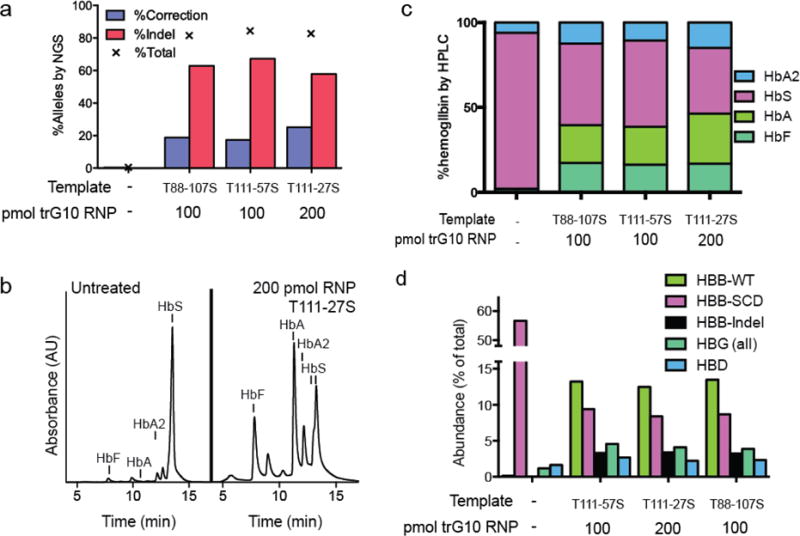
A) Editing the SCD mutation in un-expanded CD34+ HSPCs from the whole blood of SCD patients, assessed by NGS. B) HPLC trace depicting hemoglobin production in SCD HSPCs edited with 200 pmol of the trG10 RNP and the T111-27S donor, compared to untreated HSPCs, after differentiation into erythroblasts. Significant increases in HbA, HbF, and HbA2 are apparent. C) Stacked bars showing HPLC results, with HSPCs edited as indicated after differentiation into erythroblasts. D) Globin gene levels in SCD HSPCs edited as in Fig. 3C, determined by RNA-seq.
To analyze the hemoglobin production potential of corrected HSPCs, we differentiated pools of treated HSPCs into enucleated erythrocytes and late-stage erythroblasts; we measured hemoglobin by HPLC (21, 28, 48). We found that corrected HSPC pools produce substantial amounts of wild-type adult hemoglobin (HbA), with a concomitant decrease in sickle hemoglobin (HbS) (22.2%–22.4% HbA, 48.0%–50.6% HbS at low observed a substantial increase in fetal hemoglobin (HbF) in edited cell pools (16.3%–17.4% HbF in edited cells vs 2.0% HbF in unedited cells).
We used RNA-seq to measure globin transcript abundance in pools of edited SCD HSPCs differentiated to erythrocytes (49). Globin transcript levels showed a trend similar to protein levels after editing, with sickle HBB transcripts decreasing from 56.7% of all transcripts to ~9% and wild-type HBB transcripts increasing from 0.1% to 13% across all three editing conditions (Fig. 3D). Consistent with the increase in HbF protein, we observed a ~3-fold increase in the expression of γ-globin (HBG1 and HBG2) mRNA. Thus, corrected pools of cells produce decreased sickle β-globin, increased adult wild-type β-globin, and increased fetal β-like (γ) globin.
Edited HSPCs repopulate in vivo
The trG10 Cas9 RNP and an ssODN donor template can efficiently edit the SCD mutation in CD34+ HSPCs, and erythrocytes derived from these cells have altered hemoglobin levels consistent with substantial gene correction. In order for gene correction to manifest in vivo, edited re-populating stem cells must engraft and repopulate within a recipient (50). A powerful method of assaying re-populating stem cells is through long-term xenograft in an immunodeficient mouse model, such as the NOD/SCID/IL-2rγnull (NSG) mouse (51). In this model, edited human stem cells must engraft in the mouse and persist over several months. After sixteen weeks, progenitor cells should be lost from the system and human cells in the bone marrow should be derived from long-term re-populating stem cells within the initial HSPC population (21, 51). Because SCD HSPCs are difficult to obtain at the necessary scale, we edited wild-type HSPCs to SCD and implanted them in NSG mice.
We injected NSG mice with pools of WT CD34+ HSPCs edited with the trG10 RNP and the T88-107 template (7 mice over three treatments, 1×106 cells per mouse), along with two uninjected NSG mice (51). Engraftment was monitored by FACS analysis of blood draws at five and eight weeks after injection. Final engraftment was assessed at sixteen weeks after injection, when mice were sacrificed and bone marrow (BM) cells were harvested and subjected to FACS-based lineage analysis (6 mice, Figs. 4A and S6. Substantial numbers of hematopoietic (CD45+) cells were detectable in BM of all injected mice at 16 weeks (37% ± 21% human CD45, mean ± s.d.), demonstrating that edited cells maintain long-term repopulating potential. Within BM, engrafted human CD45+ cells were primarily B cells (CD19+, 57% ± 18%, mean ± s.d.), along with a minority of myeloid cells (CD33+, 25% ± 18%, mean ± s.d.) (fig. S6B).
Figure 4. Engraftment of edited HSPCs into NSG mice.
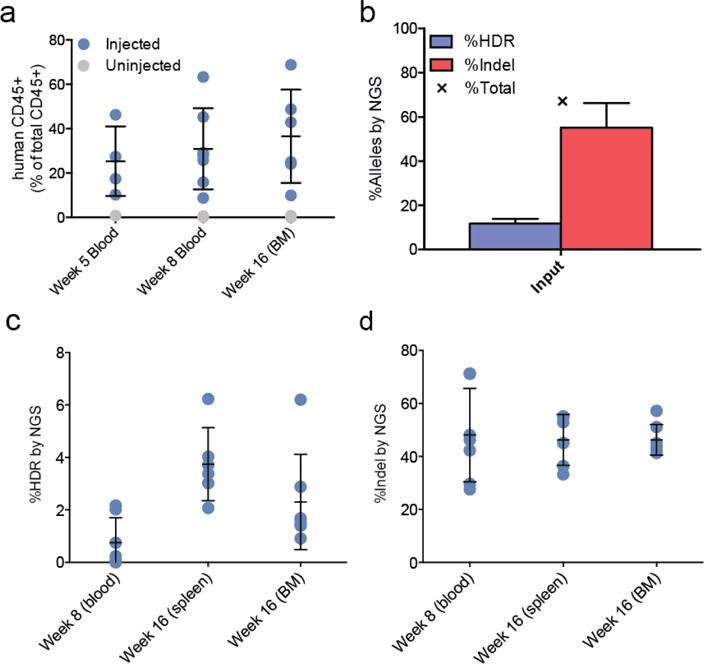
A) Engraftment of human CD45+ cells in NSG mice injected with edited HSPCs, compared to 2 un-injected mice. B) Analysis by NGS of editing at the SCD SNP in cells prior to engraftment. Error bars indicate mean ± standard deviation of three separate experiments with cells from two healthy donors. C) HDR-mediated editing (left) and indel formation (right) at the SCD SNP in human cells engrafted in mouse blood, spleen, and bone marrow (BM), at 5 and 16 weeks post-injection. Error bars indicate mean ± standard deviation over either 6 mice (spleen) or 7 mice (BM and blood).
Genotyping of edited HSPCs was assessed by NGS immediately after editing and before injection (Fig. 4B), and from mice at weeks 8 (blood) and 16 (BM and spleen, Figures 4C–4D). The input populations had 55% ± 19% indel alleles and 11.8% ± 3.7% HDR alleles (mean ± s.d., Fig. 4B). NGS analysis revealed consistently high levels of indel alleles (46 ± 6% in BM, mean ± s.d.), along with maintenance of HDR-mediated editing at the SCD SNP throughout the lifetime of the xenograft in both BM and spleen (2.3 ± 1.8% in bone marrow, 3.7 ± 1.4% in spleen, mean ± s.d.). Human cells engrafted in one mouse maintained HDR-mediated editing in BM at a remarkably high level (6.2%), although the reasons for this difference are unclear. To ensure that editing was present in the progenitor (CD34+) cells and not over-represented in the B lymphocyte (CD19+) populations, marrow from two mice was sorted for these markers and genotyped by NGS. Editing was maintained within both populations, indicating durable editing of the long-term stem cell (HSC) population (fig. S6). Despite the decreased HDR rate, this level of sequence replacement using Cas9 RNP and ssODNs is more than 9-fold greater than previously reported editing of the SCD mutation in HSCs using ZFN mRNA electroporation and ssODNs (21).
Discussion
The treatment of genetic diseases by gene editing to replace an endogenous sequence is a long-standing goal of regenerative medicine. Unlike gene therapy using integrating viral vectors, where regulation of the introduced gene may be compromised and endogenous genes may be disrupted, gene editing corrects the disease mutation at the endogenous locus. In the case of most genetic blood disorders, lasting correction requires HDR-mediated editing of endogenous genes in repopulating hematopoietic stem cells. Ideally, these edits should be efficient enough to operate without the introduction of a non-native selection marker. This is technically challenging, particularly since the edits do not confer a selective advantage in the target cell compartment, as is the case for correction of the HBB sickle mutation in bone marrow stem cells (14, 21).
Here, we used the Cas9 RNP and ssODNs to develop a rapid and extensible gene editing pipeline to introduce SNPs into human adult HSPCs, focusing on the SCD mutation. We used K562 cells to iterate combinations of Cas9 RNPs and ssODNs that edit the HBB gene and then applied these reagents to human HSPCs. These reagents efficiently induce HDR-mediated editing of the SCD mutation in HSPCs with minimal genic off-target activity. Previous reports of gene knockout in HSPCs by Cas9 mRNA delivery required chemical protection of the sgRNA (27), but we found that RNP delivery yields efficient knockout and HDR even with unmodified sgRNAs and ssODNs. Although we used a truncated guide (trG10) here, we found no apparent decrease in off-target activity compared to the full-length guide. RNP delivery has been associated with reduced off-target effects (39), suggesting that this modality could allow efficient editing of disease mutations in HSPCs using full-length unmodified sgRNAs such as G10.
We subjected the trG10 RNP to extensive off-target analysis in HSPCs. We found few genic off-target events, and one major intergenic event that had been described previously (26, 27). We did not find clear evidence of frequent translocation events between targeted loci in HSPCs. Similarly, we found low frequency mutations at a handful of cancer-associated loci in edited K562 cells, typically at sites with measurable mutations in unedited cells, but did not observe these mutations in HSPCs. We conclude that off-target analysis should be performed in the target primary cell type and not cancer cell lines. To reduce off-target activity, we tested several recently-characterized high fidelity Cas9 variants as RNPs, but their low on-target activity was concerning (45, 46).
Recent gene editing efforts in hematopoietic cells have focused on the use of viral HDR donors that are often delivered at very high multiplicity of infection (15, 16, 21). Advances in viral delivery technology have greatly improved safety, and viral donors are a valid option for gene correction. But genomic insertion of even non-integrating vector sequences has been observed during genome editing, and high levels of off-target integration have been observed in HSPCs (52, 53). Furthermore, the intensive engineering associated with viral donor design restricts optimization. By contrast, the co-delivered RNP/ssODN approach described here is non-viral, modular, and readily optimized.
We have achieved levels of sequence replacement in long-term stem cells that may be clinically relevant (21). However, HDR was still 5-fold diminished relative to the input CD34+ populations. This has been observed previously, and remains a major impediment to bringing HDR-based therapies to the clinic (14, 15, 21). It may be that HDR-edited HSCs do not engraft as well as unedited cells, that the HDR donors are themselves toxic, or that HSCs are intrinsically more difficult to edit than other CD34+ cells. We found that relatively high doses of ssDNA donor do not affect indel formation, implying that donor delivery is not in itself toxic, but that the stem cells may instead be intrinsically refractory to HDR-based editing.
Corrected SCD HSPCs produced greatly reduced sickle, and increased wild-type, hemoglobin and globin mRNA. Edited HSPCs also produced more γ-globin mRNA and fetal hemoglobin. Although the cause of this increase is unclear, it is possible that indels within HBB cause selective expansion of fetal hemoglobin-expressing cells during differentiation or that indels within HBB stimulate a cell-intrinsic up-regulation of fetal hemoglobin. The molecular basis by which alterations at β-globin cause changes in fetal hemoglobin abundance warrants further investigation.
Observations of transient mixed chimerism (2–5%) after allogeneic HCT for SCD suggest that correcting the sickle allele in relatively few bone marrow stem cells could translate into a clinically meaningful increase in the numbers of non-sickling circulating RBCs (18–20) (see Supplemental Discussion). The abrogation of ineffective erythropoiesis intrinsic to sickle cell anemia, and the greatly increased lifespan in circulation of wild type RBCs carrying a WT β-globin allele imply that even a small minority of corrected HSCs can increase the hemoglobin level and lower the HbS fraction in the blood. Furthermore, as expected, we found that alleles assort largely independently within the edited population, such that a given level of correction on the allele level corresponds to correction of nearly twice as many cells. This should be taken into account when comparing the allelic correction data presented here with observations of mixed chimerism (for example, correction of 1–3% of alleles may correct 2–6% of cells, table S3).
An approach to SCD treatment that integrates this method will have several limitations. First, it is possible that the allelic correction frequency we observed in the HSPC population will ameliorate but not eliminate the clinical expression of SCD. Second, although autologous HSCT should be safer than allogeneic HSCT, it is still an intensive procedure, requiring myeloablative conditioning, which can have serious side effects. Finally, clinical translation of this therapy in developing countries, where SCD is most prevalent, will be a major barrier to wider application of the therapy.
The approach described here allows researchers to edit SNPs at endogenous loci in human adult HSPCs using readily available reagents that are conducive to rapid iteration and optimization. Given the low barrier to entry, we anticipate that these democratizing methods will enable investigator-led gene editing studies in a wide variety of disease areas. We predict that the methods outlined can be improved and scaled up for a gene editing treatment for SCD.
Supplementary Material
Supplementary Discussion
Materials and Methods
Supplementary Figures S1–S6
Supplementary Tables S1–S11
Acknowledgments
We thank Chris Jeans of the QB3 Macro Lab at UC Berkeley for expression and purification of the Cas9 protein variants used in this study. We thank Shana McDevitt and the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, which is supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303. We thank Holly Turner and Mee J. Kim (CHORI) for advice and assistance with mouse work, and Lorelle Parker (CHORI) for advice and assistance with flow cytometry. We thank Jennifer Doudna and Michael Botchan (Berkeley) for their contributions to the initiation of this project.
Funding: J.E.C., M.A.D., D.C., N.L.B., T.M., T.W., are supported by the Li Ka Shing Foundation. M.A.D. was a CIRM post-doctoral fellow under training program TG2-01164. D.C. is supported by NIH grant R01 GM078571, and the Siebel Scholars Fund at Berkeley. D.I.M., D.B.O., and S.H. were supported by NIH grants 5R21AA022753 and 1R56ES022377-01; D.I.K.M., W.M., M.C.W., and D.P.M. were supported by the Jordan Family Fund at CHORI. D.B.K. and F.U. were supported by a research grant from the Doris Duke Charitable Foundation to D.B.K. (Innovations in Clinical Research Award ##2013158).
Footnotes
Author Contributions. J.E.C., D.C., M.C.W. and D.K.M. conceived the project. J.E.C, D.I.M., M.A.D., D.C., D.P.M., D.B., W.M., D.K., and J.R.B. designed experiments. W.M., M.A.D., D.C., T.W., S.H., and F.U. performed experiments. N.L.B., T.M., S.H., M.A.D., W.M. and D.C. analyzed data. M.A.D., J.E.C., D.I.M., M.C.W., and D.C. wrote the manuscript.
Competing Interests. J.R.B. is an employee of Bio-Rad, Inc. All other authors declare that they have no competing interests.
Materials and Data Availability: All next-generation sequencing data have been deposited at the NCBI Sequence Read Archive under accession number XXXXXX.
References and notes
- 1.Panepinto JA, Bonner M. Health-related quality of life in sickle cell disease: past, present, and future. Pediatr Blood Cancer. 2012;59:377–85. doi: 10.1002/pbc.24176. [DOI] [PubMed] [Google Scholar]
- 2.Adewoyin AS. Management of sickle cell disease: a review for physician education in Nigeria (sub-saharan Africa) Anemia. 2015;2015:791498. doi: 10.1155/2015/791498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pauling L, Itano HA, Singer SJ, Wells IC. Sickle Cell Anemia, a Molecular Disease. Science (80-) 1949;110:543–548. doi: 10.1126/science.110.2865.543. [DOI] [PubMed] [Google Scholar]
- 4.Neel JV. The Inheritance of Sickle Cell Anemia. Science. 1949;110:64–6. doi: 10.1126/science.110.2846.64. [DOI] [PubMed] [Google Scholar]
- 5.McClish DK, et al. Health related quality of life in sickle cell patients: the PiSCES project. Health Qual Life Outcomes. 2005;3:50. doi: 10.1186/1477-7525-3-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Platt OS, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–44. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 7.Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore) 2005;84:363–76. doi: 10.1097/01.md.0000189089.45003.52. [DOI] [PubMed] [Google Scholar]
- 8.Walters MC, et al. Indications and Results of HLA-Identical Sibling Hematopoietic Cell Transplantation for Sickle Cell Disease. Biol Blood Marrow Transplant. 2016;22:207–211. doi: 10.1016/j.bbmt.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yawn BP, et al. Management of Sickle Cell Disease. JAMA. 2014;312:1033. doi: 10.1001/jama.2014.10517. [DOI] [PubMed] [Google Scholar]
- 10.Appelbaum FR, Forman SJ, Negrin RS, Blume KG, editors. Thomas’ Hematopoietic Cell Transplantation. Wiley-Blackwell; Oxford, UK: 2009. http://doi.wiley.com/10.1002/9781444303537. [Google Scholar]
- 11.Bernaudin F, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007;110:2749–56. doi: 10.1182/blood-2007-03-079665. [DOI] [PubMed] [Google Scholar]
- 12.Hsieh MM, et al. Nonmyeloablative HLA-Matched Sibling Allogeneic Hematopoietic Stem Cell Transplantation for Severe Sickle Cell Phenotype. JAMA. 2014;312:48. doi: 10.1001/jama.2014.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolaños-Meade J, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012;120:4285–91. doi: 10.1182/blood-2012-07-438408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Genovese P, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–40. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol. 2015 doi: 10.1038/nbt.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sather BD, et al. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci Transl Med. 2015;7:307ra156–307ra156. doi: 10.1126/scitranslmed.aac5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu CJ, et al. Evidence for ineffective erythropoiesis in severe sickle cell disease. Blood. 2005;106:3639–45. doi: 10.1182/blood-2005-04-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walters M, et al. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant. 2001;7:665–673. doi: 10.1053/bbmt.2001.v7.pm11787529. [DOI] [PubMed] [Google Scholar]
- 19.Wu CJ, et al. Mixed haematopoietic chimerism for sickle cell disease prevents intravascular haemolysis. Br J Haematol. 2007;139:504–7. doi: 10.1111/j.1365-2141.2007.06803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iannone R, et al. Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and β-thalassemia. Biol Blood Marrow Transplant. 2003;9:519–528. doi: 10.1016/s1083-8791(03)00192-7. [DOI] [PubMed] [Google Scholar]
- 21.Hoban MD, et al. Correction of the sickle-cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015 doi: 10.1182/blood-2014-12-615948. blood-2014-12-615948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15:321–34. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- 23.Jinek M, et al. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science (80-) 2014;346:1258096–1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 25.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41:9584–92. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hendel A, et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang X, et al. Production of gene-corrected adult beta globin protein in human erythrocytes differentiated from patient iPSCs after genome editing of the sickle point mutation. Stem Cells. 2015 doi: 10.1002/stem.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Canver MC, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature advance on. 2015 doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun N, Zhao H. Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnol Bioeng. 2014;111:1048–53. doi: 10.1002/bit.25018. [DOI] [PubMed] [Google Scholar]
- 31.Ramalingam S, Annaluru N, Kandavelou K, Chandrasegaran S. TALEN-mediated generation and genetic correction of disease-specific human induced pluripotent stem cells. Curr Gene Ther. 2014;14:461–72. doi: 10.2174/1566523214666140918101725. [DOI] [PubMed] [Google Scholar]
- 32.Lin S, Staahl B, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 2014;3:e04766. doi: 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schumann K, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci. 2015;112:201512503. doi: 10.1073/pnas.1512503112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang X, et al. Rapid and Highly Efficient Mammalian Cell Engineering via Cas9 Protein Transfection. J Biotechnol. 2015;208:44–53. doi: 10.1016/j.jbiotec.2015.04.024. [DOI] [PubMed] [Google Scholar]
- 35.Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. Cas9’s asymmetric interaction with target DNA can be exploited to promote high efficiency homology-directed genome editing. Nat Biotechnol. 2016 doi: 10.1038/nbt.3481. In Press. [DOI] [PubMed] [Google Scholar]
- 36.Doench JG, et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu Y, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–6. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–32. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–9. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–84. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kent WJ, et al. The Human Genome Browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chu VT, et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- 43.Maruyama T, et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hardy GH. MENDELIAN PROPORTIONS IN A MIXED POPULATION. Science. 1908;28:49–50. doi: 10.1126/science.28.706.49. [DOI] [PubMed] [Google Scholar]
- 45.Slaymaker IM, et al. Rationally engineered Cas9 nucleases with improved specificity. Science (80-) 2015;aad5227 doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kleinstiver BP, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.C J, Richardson CD, Ray GJ, Bray NL. Non-homologous DNA increases gene disruption efficiency by altering DNA repair outcomes. Nat Commun. 2016 doi: 10.1038/ncomms12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giarratana MC, et al. Ex vivo generation of fully mature human red blood cells from hematopoietic stem cells. Nat Biotechnol. 2005;23:69–74. doi: 10.1038/nbt1047. [DOI] [PubMed] [Google Scholar]
- 49.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol advance on. 2016 doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 50.Notta F, et al. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. 2011;333:218–21. doi: 10.1126/science.1201219. [DOI] [PubMed] [Google Scholar]
- 51.Drake AC, Chen Q, Chen J. Engineering humanized mice for improved hematopoietic reconstitution. Cell Mol Immunol. 2012;9:215–24. doi: 10.1038/cmi.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corrigan-Curay J, et al. Genome editing technologies: defining a path to clinic. Mol Ther. 2015;23:796–806. doi: 10.1038/mt.2015.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–21. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iannone R. Effects of mixed hematopoietic chimerism in a mouse model of bone marrow transplantation for sickle cell anemia. Blood. 2001;97:3960–3965. doi: 10.1182/blood.v97.12.3960. [DOI] [PubMed] [Google Scholar]
- 55.Franco RS, et al. The effect of fetal hemoglobin on the survival characteristics of sickle cells. Blood. 2006;108:1073–6. doi: 10.1182/blood-2005-09-008318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Discussion
Materials and Methods
Supplementary Figures S1–S6
Supplementary Tables S1–S11