

Abstract
DNA single strand breaks (SSB) are the most common form of DNA damage, requiring repair processes that to initiate must overcome chromatin barriers. The FACT complex comprised of the SSRP1 and SPT16 proteins are important for maintaining chromatin integrity, with SSRP1 acting as an histone H2A/H2B chaperone in chromatin disassembly during DNA transcription, replication and repair. In this study, we show that SSRP1 but not SPT16 is critical for cell survival after ionizing radiation or methyl methanesulfonate-induced single-strand DNA damage. SSRP1 is recruited to SSB in PARP-dependent manner and retained at DNA damage sites by N-terminal interactions with the DNA repair protein XRCC1. Mutational analyses showed how SSRP1 function is essential for chromatin decondensation and histone H2B exchange at sites of DNA strand breaks, which are both critical to prime chromatin for efficient SSB repair and cell survival. By establishing how SSRP1 facilitates SSB repair, our findings provide a mechanistic rationale to target SSRP1 as a general approach to selectively attack cancer cells.
Keywords: FACT, SSRP1, single strand break repair, PARP, chromatin, histone exchange
Introduction
DNA single strand breaks (SSBs) arise tens of thousands of times per cell per day, resulting from environmental or endogenous agents such as reactive oxygen species (ROS). Unrepaired SSBs in proliferating cells will most likely collapse DNA replication forks and form DNA double strand breaks (DSBs), leading to genome instability (1). DSBs, the most lethal type of DNA damage, are repaired by homologous recombination (HR) or non-homologous end joining (NHEJ) in human cells. In non-proliferating cells, such as post-mitotic neurons with high transcriptional demand, SSBs lead to stalled transcription and trigger cell death (2). Most SSBs can be removed by global SSB repair (SSBR), which includes four major steps: SSB recognition/detection, DNA end processing, gap filling, and ligation (2). PARP1 recognizes SSBs and synthesizes poly-ADP-ribose (PAR), modifying itself as well as other target proteins. PAR may help trigger the relaxation of chromatin around SSBs, and promote the accumulation of X-ray repair cross-complementing protein 1 (XRCC1) and other SSBR proteins at the opened chromatin site (3,4). Targeting PARP activation has been shown as an effective cancer therapeutic approach, meanwhile rational combinations exploiting alternate mechanisms of defective DNA repair, imploring additional functions of PARP, and identifying the optimal combination partner will present novel opportunities for further treatment options for patients (5). XRCC1, a scaffold protein in SSBR, also physically interacts with PARPs (PARP1 and PARP2), APEI, and other repair factors, thereby organizing SSBR in cells in a temporal and spatial manner (6).
The DNA damage response in cells is referred to as the “Access/Prime-Repair-Restore” process for successful repair (7). Access and restore require the disassembly and reassembly of the chromatin structure around damage sites, which involves chromatin remodeling factors and histone chaperones (8,9). The high-mobility group (HMG)-1-like structure-specific recognition protein-1 (SSRP1) is one of the critical components of the Facilitate Chromatin Transcription (FACT) complex together with suppressor of Ty homolog-16 (SPT16) (10), which also has been implicated in cancers (11,12). FACT is a histone H2A/H2B chaperone that regulates chromatin dynamics during transcription (13), replication (14,15) and DNA damage repair. FACT is known to mediate the turnover of histone H2A/H2A.X, which is regulated by both the phosphorylation of H2A.X and the ADP-ribosylation of SPT16 in vitro (16). FACT promotes transcription restart by enhancing histone H2A/H2B exchange at UV damage sites, driven by the larger component SPT16 but not SSRP1 (17). SPT16 also remodels chromatin through interaction with RNF20 upon DNA damage to promote HR (18). Although SSRP1 is not involved in histone exchange upon UVC-induced damage (17), SSRP1 interacts with cisplatin-damaged DNA (19). In addition, SSRP1 depletion is associated with increased Rad51 foci, which indicates that SSRP1 is necessary for efficient HR (20). It is not known whether SSRP1 also plays a direct role in repairing the most frequent type of DNA damage, SSBs. In this study, we elucidated the molecular pathways of if and how SSRP1 is involved in single strand break repair and promotion of chromatin priming at damage sites so as to facilitate efficient SSBR. We show that SSRP1 accumulates at SSBs in a PAR-dependent manner. How SSRP1 remains at damage sites by interacting with XRCC1 is shown based on a ALFFSRI RIR motif mediated interaction. Finally, the role of SSRP1 as a histone chaperone, priming the chromatin around damage sites for successful SSBR thus will be a potential cancer treatment target is discussed.
Experimental procedures
Plasmids, transfections, and chemicals
SSRP1, Histone H2B cDNAs, and the deletions were amplified using XhoI/SalI and NotI, then subcloned into pEGFP-C1 (Clonetech). RFP-XRCC1, Flag-XRCC1, and the XRCC1 deletions were cloned previously (21). Cherry-H2B was purchased from Addgene. Plasmids were transfected into cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Olaparib (10 nM, Catalog No. A4154, APExBIO), ABT88 (10 nM, APExBio), and PJ34 (20 nM, Sigma) were used in imaging and immunoprecipitation (IP) as well as for survival assays. MMS (129925-5G, Sigma) was used with the indicated dose in survival assays. Bleocin (CALBIOCHEM, 5 μg/μl 1:1000) was used to induce damage in HeLa cells.
Site-directed Mutagenesis
The N-terminal of XRCC1 was subcloned from pEGFP-C1-XRCC1 into the plasmid of pBlueScript KS (+) via Sal I and Kpn I digestion. Site-directed mutagenesis was performed by polymerase chain reactions (PCR) with mutated pairs of primers and KOD hot start DNA polymerase (71086-3, Novagen, MA, USA), using the subcloned pBS-SK-XRCC1 as the template. After digestion with Dpn I (R0176S, NEB, USA), the mutated plasmids were transformed into TOP10 competent cells and screened on plates with ampicillin. Subsequently, the isolated plasmid DNA was sent to the Genomic Core Facilities of the University of Pittsburgh for sequencing to further confirm the mutations. Finally, all three mutated genes were transferred back onto the vector of pEGFP-C1 via Sal and KpnI. The mutation primers used are as follows, pBS-XRCC1-F67A-For: 5′-GGAATGATGGCTCAGCTGCCGTGGAGGTGCTGGCGGG-3′; pBS-XRCC1-F67A-Rev: 5′-CCCGCCAGCACCTCCACGGCAGCTGAGCCATCATTCC-3′ pBS-XRCC1-FF191/192AA-For:5′-CAACTCTCTGAGGCCGGGGGCTCTCgcCgcCAGCCGGATCAACAAGACATCCCCAG-3′; and pBS-XRCC1-FF191/192AA-Rev: 5′-CTGGGGATGTCTTGTTGATCCGGCTGgcGgcGAGAGCCCCCGGCCTCAGAGAGTTG-3′
Cell lines and siRNA/shRNAs
U2OS, HeLa, and FLP-in-293 cells were purchased from ATCC in 2012. XPA-C2 and XPA-UVDE Cells was originally derived in Dr. Akira Yasui’s lab in 2010, in which a foreign UV damage endonuclease (UVDE) or a control vector was stably introduced into human xeroderma pigmentosum group A (XPA)-deficient cells to make XPA-UVDE or XPA-C2 cells. In the described experiments, we cultured the cells stocked in Nitrogen liquid tank for 3–4 weeks. Number of passages are varying from 3–10. The cells lines were tested by mycoplasma testing kit (AccuSEQ Thermo Fisher Scientific) to exclude the possibility of mycoplasma contamination. All cell lines were cultured in Dulbecco’s modified e’s medium (DMEM, Lonza) with 10% fetal bovine serum (Atlanta Biologicals) at 37°C and 5% CO2. The siRNAs were transfected into HeLa cells using DharmaFECT1 (Thermo) in the colony-forming assay. The SSRP1 siRNA (h) (SC-37877) (A: Sense: GCAAGACCUUUGACUACAAtt, Antisense: UUGUAGUCAAAGGUCUUGCtt; B: Sense: CGUUGACUCUGAACAUGAAtt, Antisense: UUCAUGUUCAGAGUCAACGtt; C: Sense: GGAUCCAAAUCCUCAUCUUtt, Antisense: AAGAUGAGGAUUUGGAUCCtt) and SPT16 siRNA (h) (SC-37875) (A: Sense: GAAGAGCACAUCAGAAAGAtt, Antisense: UCUUUCUGAUGUGCUCUUCtt; B: Sense: GUCAUUGGGUAGUGAAGAAtt, Antisense: UUCUUCACUACCCAAUGACtt; C: Sense: GAUGGCUUCUGACAUCUUAtt, Antisense: UAAGAUGUCAGAAGCCAUCtt) were purchased from Santa Cruz Biotechnology. shRNAs of SSRP1 (human) (SHCLNG-NM_003146) were purchased from Sigma Aldrich, including: shSSRP1-1, TRCN0000019271; shSSRP1-2, TRCN0000343894. The shCtrl stably expressed the vector Plko.1 (Sigma mission).
Microscope and laser micro-irradiation
The Olympus FV/1000 confocal microscopy system (Olympus) and FV/1000 software were used for image acquisition and analysis and described in a previous study (22). Damage was induced with a 405 nm laser. The laser passed through a PLAON 60X oil lens. The output power was 5 mW/scan. Cells transfected with GFP-tagged proteins were incubated at 37°C on a thermos plate in normal media during observation. For the evaluation of accumulation and kinetics, the mean intensity of each accumulated point or line was obtained after subtraction and quantified by ImageJ.
XPA-UVDE system induced DNA SSBs after local UVC irradiation
XPA-UVDE and XPA-C2 cells were cultured in 35-mm glass-bottomed culture dishes (poly-d-lysine coated; MatTek, Ashland, OR). Twenty-four hours after transfection, cell monolayers were covered with a polycarbonate isopore membrane filter with 3 μm diameter pores (Millipore) and UV irradiated with a germicidal lamp (GL-10; Toshiba; predominantly 254-nm UV) at a dose of 100 J/m2. Cells were imaged before and after local UV light exposure using Olympus confocal microscopy.
Colony formation assay
Approximately 350 HeLa cells were replated on 6-cm dishes 48 h after siRNA transfection. For treatment with methyl methanesulfonate (MMS) (Sigma), cells were incubated in DMEM (10% FBS) containing MMS for 9 days. For treatment with UVC light and ionizing radiation (IR), cells were washed and irradiated with the indicated dose 8 h after passaging. Colonies were stained with 0.3% crystal violet/methanol and counted 9 days after treatments. Each experiment was performed at least 3 times and the standard errors were calculated and indicated in the graphs. Results were normalized for plating efficiencies.
Immunoprecipitation, immunofluorescence and Western blots
Two days after transfection, HEK-293T cells were washed once with pre-cooled PBS and lysed on ice for 30 min in lysis buffer (10 mM Hepes, pH 7.6, 250 mM NaCl, 0.1% Nonidet P-40, 5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1:1000 protease inhibitor). For immunoprecipitation, 2.4 μg anti-GFP monoclonal antibody (Roche) and 30 μl of protein G-Sepharose beads (GE Healthcare Bio-Sciences) or 30 μl anti-FLAG M2 agarose from mouse (Sigma A2220) were added to each lysate. Mixtures were incubated overnight at 4°C with rotation; the supernatant was removed, and protein beads were washed three times using 0.5 m lysis buffer. Cells were fixed for immunofluorescence staining in PBS 3.7% formaldehyde for 15 min and permeabilized with 0.2% Triton X-100 for 5 min at room temperature. After being washed with PBS 3 times, cells were incubated in DMEM+0.02% Azide with the appropriate primary antibodies, then incubated with Alexa-405/488/594 conjugated second antibodies (Invitrogen). For Western blot analysis, samples were subjected to electrophoresis in 8% or 10% SDS-polyacrylamide gels and immunoblotted using the polyclonal antibody specific to anti-FLAG antibody, anti-GFP antibody, anti-cherry antibody, anti-SSRP1 antibody (PA 5-22186, rabbit, Thermo), or anti-SPT16 antibody (H-300, sc-28734, Santa Cruz). Anti-mouse IgG VeriBlot for IP secondary antibody (HRP) (ab131368, Abcam) was used.
Histone exchange assay with FRAP
The FRAP assay was modified according to the previous method (17). SSRP1 KD cell lines were transfected with GFP-tagged H2B. Twenty-four hours later, cells were imaged using the Olympus microscope. After bleaching half of the cell nucleus with 10 scans of a 488 nm laser, a 405 nm laser was used to induce point damage in the bleached half. Then, the recovery of GFP fluorescence was traced immediately after damage was induced for 15 min. Data was analyzed using Matlab. The damage sites were fitted with the elliptical Gaussian function (due to the movement distortion). The background was defined as the average background intensity per pixel. A is the peak of each Gaussian function in each frame. A is linear to the highest intensity at damage sites after removal of the background signal. Amax is the biggest A among 30 frames. Three independent experiments were performed. N>10 cells.
Quantification of chromatin compaction via fluorescence lifetime (FL) imaging microscopy (FLIM)
For fluorescence lifetime imaging microscopy (FLIM) experiments, a Leica TCS SP5 inverted laser scanning confocal microscope and a 100x (1.4 NA) oil immersion objective was used. Samples were excited with a Ti:sapphire mode-locked, pulsed infrared laser (Chameleon, Coherent) system for the multiphoton excitation source (1 W, average) tuned to 825 nm with pulse-widths of <140 fs delivered at 90 MHz. For emission, a FLIM-specific photomultiplier tube (PMT) was used and the spectrum from 404–536 nm was collected. Fluorescence lifetime data was acquired and analyzed using previously published methods (23) with a suite of software from Becker & Hickl SPC-830 for time-correlated single photon counting (TCSPC) with 10 ps resolution along with 220 time channels and a 10.8 ns measurement window. The decay rate of the fluorescence lifetime can be modeled as an exponential decay (Equation 1), where t is time, τ is the lifetime and I0 is the number of photons at t = 0, respectively. I(t) is the number of photons detected per unit time, t. . The heat maps of the fluorescence lifetimes were created in Becker & Hickl SPCImage software along with data analysis. For comparison of treatment conditions, we segmented the nuclei in each field of view to isolate only nuclear pixel signals for data analysis using MATLAB. We analyzed the fluorescence lifetime fits using a χ2 test, with DAPI best modeled by a single exponential decay. The average fluorescence lifetime was calculated from all nuclear pixels. Magnitudes of the average fluorescence lifetimes for segmented nuclei in each sample condition were compared using Student’s t-test. The fluorescence lifetime at distinct DNA damage sites was determined by obtaining the fluorescence lifetime values at corresponding DNA damage sites labeled for 53BP1 manually in the Becker & Hickl SPCImage software. The average fluorescence lifetime of DNA damage sites in wild type (WT) and SSRP1 repressed cells was calculated and compared using Student’s t-test.
Sample preparation for STORM imaging
For immunofluorescence staining, cells were fixed with 3.7% (v/v) formaldehyde for 12 min at room temperature and permeabilized with 0.1% Triton X-100 for 10 min. After washed with PBS, cells were incubated with anti-Cherry rabbit primary antibodies at a concentration of 1:400 overnight at 4°C. The cells were washed three times with PBS and then Alexa 647-conjugated donkey anti-rabbit secondary antibodies (Jackson ImmunoResearch, 121165) at a concentration of 1:200 were added to the sample and incubated for 2 hours, protected from light. Cells were washed with PBS again and stored in PBS before imaging. Prior to STORM imaging, the 100 nm fluorescent nanospheres (F8803, Thermo Fisher Scientific) were added to the petri-dish at a desired concentration to ensure 5–8 fiduciary markers in the field of view used for drift correction. Immediately before STORM imaging, the PBS buffer is switched to the STORM imaging buffer containing 50 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.14M β-mercaptoethanol, 10% w/v glucose, 0.56 mg/mL glucose oxidase and 0.17 mg/mL catalase.
STORM imaging
The STORM imaging is performed on our home-built fluorescence microscopy on an Olympus IX71 inverted microscope, equipment with 4 lasers including 405 nm (DL405-050, CrystaLaser), 488 nm (DL488-150, CrystaLaser), 561 nm (VFL-P-200-560-OEM1, MPB Communications) and 642 nm laser (VFL-P-1000-642-OEM3, MPB Communications). The laser beam is expanded by a 10X beam expander (T81-10X, Newport), focused onto the back focal plane of a 100X, NA = 1.4 oil immersion objective (UPLSAPO 100XO, Olympus) by an achromatic lens, and illuminates the sample. The emitted fluorescence is collected by the objective, passing through the dichroic mirror (FF660-Di02, Semrock), a band-pass emission filter (ET700/75m, Chroma), and finally focused by the tube lens and a 0.5X C-mount adapter onto a sCMOS camera (pco.edge 4.2, PCO-Tech), corresponding to a pixel size of 130 nm on the sample plane. A closed-loop piezo nanopositioner (Nano-F100S, Mad City Labs) was used for drift correction by real-time adjustment of the axial position of the objective. Data acquisition, laser intensity control and drift correction were all integrated in our custom-designed software written in LabVIEW (National Instruments) and MATLAB (MathWorks). During STORM imaging of biological cells, the 642 nm excitation laser was used at a power density of 3kW/cm2, and 30,000 frames were acquired at the exposure time of 20 millisecond for each frame. The drift correction was performed using 488 nm excitation laser at a power density of 75W/cm2 for every 200 imaging frames. The image was reconstructed using our custom-written software written in C that implements single-molecule Gaussian function model (24).
Results
SSRP1 is recruited to SSBs in a PAR-dependent manner
To investigate the role of FACT, we suppressed SSRP1 or SPT16 knock down cell lines with stable expression of shRNA for SSRP1 or SPT16 in HeLa cells. ShSSRP1 largely affected cell shape and led to severe cell proliferation defects (Fig. 1A) and shSPT16 cells were not viable. We then examined whether SSRP1 is recruited to sites of DNA strand breaks in U2OS cells in real time. We used laser micro-irradiation, which at low dosages (less than 100 ms) mainly induces DNA SSBs (25). Both GFP-SSRP1 and GFP-XRCC1 accumulated at sites of SSBs induced by 405 nm laser micro irradiation for 100 ms (Fig. 1B). Given that the accumulation of XRCC1 at laser-induced damage is triggered by an amplified PAR signal, we tested if SSRP1’s recruitment is dependent on PARP activation. The PARP inhibitors ABT888 and olaparib eliminated the recruitment of SSRP1 as well as that of XRCC1 (Fig. 1C). However, the accumulation of SSRP1 was not affected after the suppression of XRCC1 by siRNA (Fig. 1D). These results suggest that accumulation of SSRP1 at laser-induced damage is largely dependent on PARP activation. It is known that FACT’s nucleosome binding activity is regulated by poly-ADP-ribosylation of SPT16 through the interaction with PARP1 in vitro (16), and we inferred that PARylation of SPT16 by PARP1 is critical for the recruitment of SPT16. In addition, siSPT16 did not inhibit the recruitment of SSRP1 (Fig. 1D), indicating that it is dependent on PAR but not on SPT16 or PARylation of SPT16. Given that CK2 phosphorylation of SSRP1 is commonly found when SSRP1 is involved in DNA damage and repair (26–29), we further tested whether the recruitment of SSRP1 is also affected by CK2 phosphorylation. Compared to the DMSO control and PARPi, the CK2 inhibitor (TBB) did not show any effect on the recruitment of SSRP1 (Fig. 1D). We concluded that PARP activation but not CK2 phosphorylation is critical for FACT’s accumulation at laser-induced damage.
Figure 1. SSRP1 maintains cell survival after ionizing radiation (IR) and methyl methanesulfonate (MMS) treatments.
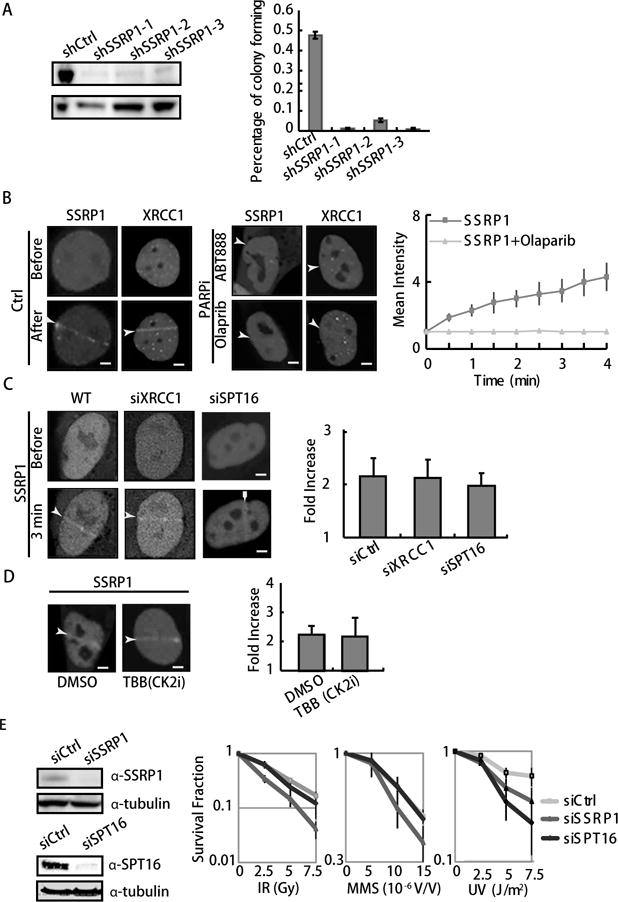
A. Suppression of SSRP1 by shRNA 1–3 is shown by WB (left panel). The colony-forming ability of the SSRP1 suppressed cells is shown by the percentage of colonies formed in 350 cells. Error bars are derived from SEMs in three repeated experiments (right panel). B. Accumulation of GFP-tagged XRCC1 and SSRP1 at sites of damage before and after 100 ms laser micro-irradiation in U2OS cells (left and middle panels). Cells were pretreated with ABT888 or olaparib for 30 min, then imaged (lower panels). PARP inhibitors suppress the accumulation of GFP-tagged SSRP1 at sites of damage. Kinetics of GFP-SSRP1 after laser micro-irradiation with and without PARPi in U2OS cells. The relative intensity vs. background is shown as a fold intensity, which was obtained from the mean damage site/background intensity. Error bars were derived from SDs in three independent experiments (right panel). C–D. Repression of SPT16, XRCC1 or CK2 phosphorylation does not inhibit the recruitment of SSRP1 at laser induced damage sites. Accumulation of GFP-tagged SSRP is shown at sites of damage 3 min after 100 ms laser micro-irradiation in U2OS cells. The cells used were either knocked down w/o siXRCC1 or siSPT16 (C), or pretreated with a CK2 inhibitor, TBB (D). Fold increase of intensity is shown. E. Clonogenic survival assay in SSRP1 or SPT16 suppressed HeLa cells by siRNA. Cells were pretreated with IR, MMS or UVC, and further incubated for 9 days before staining with 0.3% of crystal violet/methanol for the assay. For Western blot (WB), HeLa cells 48 h after treatment with siRNA for SSRP1 and SPT16 (left panel). For IR and UVC, cells were treated with the indicated dose 8 h after passaging. Suppression of SSRP1 but not SRP16 significantly sensitizes cells to IR and MMS treatment. Error bars are derived from SDs in three independent experiments (right Panel).
SSRP1 maintains cell survival after ionizing radiation (IR) and methyl methanesulfonate (MMS) treatments
A previous study indicated that depletion of SPT16 enhanced the sensitivity of MCF7 cells to IR (18) and sensitized HeLa cells to UVC damage (17). In contrast, SSRP1 depletion did not sensitize cells to UVC-induced damage as much as SPT16 depletion (17). To evaluate the importance of FACT in oxidative DNA damage and its derived DNA strand breaks, we further determined how each subunit of FACT is required for repair of MMS and IR induced damage compared to UVC damage (Fig. 1A). Both IR and MMS treatments induce large amount of DNA SSBs, that can directly or indirectly transformed to the lethal type of DNA damage double strand breaks (30–35). Because the long-term suppression of SSRP1 consequent to stable expression of shRNA for SSRP1 in HeLa cells largely affected cell shape and led to severe cell proliferation defects (Fig. 1A), we used siRNA rather than the stable knockdown cell lines to conduct the colony formation assay. SSRP1 suppression in HeLa cells led to substantially greater sensitivity to IR and MMS treatments compared to siSPT16 treatment (Fig. 1E right panel, left and middle). In contrast, siSPT16 suppression led to increased sensitivity to UVC treatment compared to siSSRP1 (Fig. 1E right panel, right). These results indicate the important role of SSRP1 in cell survival in the face of MMS and IR induced damage. Conversely, the suppression of SPT16 significantly sensitized HeLa cells to UVC damage, which is consistent with a previous study (17). These data together suggest a novel function of SSRP1 in maintaining cell survival in response to MMS and IR induced damage.
SSRP1 retains at sites of damage dependently on XRCC1
The rapid and PARP activation-dependent damage response of SSRP1 at laser-induced damage sites strongly suggested its role in the repair of DNA SSBs. To further examine the specificity of the damage response of SSRP1 to SSBs, we applied the XPA-UVDE system to produce pure SSBs in cells. In this system, a foreign UV damage endonuclease (UVDE) or a control vector was stably introduced into human xeroderma pigmentosum group A (XPA)-deficient cells to make XPA-UVDE or XPA-C2 cells (36). XPA plays a role to recruit endonuclease for removing UVC damage in the nucleotide excision repair (NER) pathway. After UVC irradiation of XPA cells through a micro pore filter, the UVDE enzyme then introduces nicks 5′ to unrepaired UVC damage (Fig. 2A). In summary, only SSBs will be induced after local UVC (LUVC) irradiation in XPA-UVDE cells. XPA-UVDE cells showed co-localization of GFP-SSRP1 with RFP-XRCC1 at local UVC induced damage, but there is no colocalization in the control XPA-C2 cells, indicating that SSRP1 is recruited to pure DNA SSBs (Fig. 2B). The PAR signals are quickly degraded by PARG after triggering the recruitment of repair factors (21,37,38). However, we observed that SSRP1, together with XRCC1, remained at damage sites after the degradation of PAR as long as 4 h after damage production (Fig. 2C). The dissociation kinetics of SSRP1 from XPA-UVDE after local UVC induced SSBs seemed to be consistent with those of XRCC1; they were both dissociated by 6 h after damage induction (Fig. 2), and could not form foci any more than cells pretreated by the PAR inhibitor, olaparib (Fig. 2D). These results indicate that although the initial recruitment of SSRP1 and XRCC1 is dependent on PAR, SSRP1 is retained independently of PAR at SSBs. Thus, SSRP1 might function together with XRCC1 after PAR degradation.
Figure 2. FACT is enriched at SSBs together with XRCC1 at damage sites after PAR degradation.
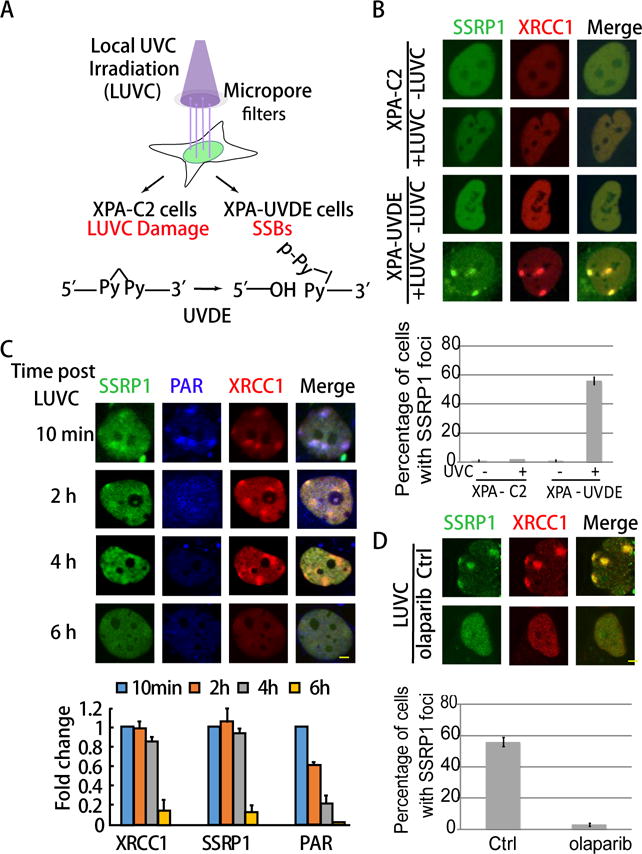
A. Scheme of the assay used to induce specific SSBs in XPA-UVDE cells after local UVC irradiation. XPA-UVDE/C2 cells were exposed to 254 nm UVC light through a polycarbonate isopore member filter. After local UVC (LUVC) irradiation, UV damage was produced and unrepaired in both XPA-C2 and XPA-UVDE cells. In XPA-UVDE cells, SSBs were introduced by UV damage endonuclease (UVDE) at the unrepaired UV damage sites. B. Recruitment of GFP-SSRP1 at SSBs produced in XPA-UVDE cells after 100 J/m2 local UVC irradiation. No enrichment of SSRP1 was observed in XPA-C2 cells. Quantification of percentage of foci positive cells of SSRP1 is shown. Cells showing > 3 foci were defined as positive cells. The error bar represents three independent experiments with 50 cells in each. C. Staining of pAR in GFP-tagged FACT and RFP-XRCC1 expressing XPA-UVDE cells at 10 min, 2 h, 4 h, and 6 h after 100 J/m2 LUVC irradiation. Percentage of foci positive cells of PAR, FACT and XRCC1 in XPA-UVDE cells at 10 min is normalized as 100%, fold decrease of foci positive cells at 2 h, 4 h, and 6 h after 100 J/m2 LUVC irradiation relative to 10 min foci formation is shown. The PAR signal disappeared while FACT and XRCC1 were retained at SSBs 2 h after damage production. D. Co-localization of GFP-tagged SSRP1 and RFP-tagged XRCC1 w/o PARPi. Quantification of percentage of foci positive cells of SSRP1 is shown. Cells showing > 3 foci were defined as positive cells. The error bar represents three independent experiments with 50 cells in each.
The N-terminal of SSRP1 interacts with the RIR motif of XRCC1 in cells
To test if SSRP1 forms a complex with XRCC1 after PAR degradation, we performed immunoprecipitation for XRCC1 and SSRP1 and found XRCC1 pulled down endogenous SSRP1 (Fig. 3A). Cells were further treated with MMS or IR, while the interaction was not significantly increased after damage (Fig. 3A). Given that cells are consistently exposed to endogenous oxidative DNA damage, the interaction itself between SSRP1 and XRCC1 may not need extra stimulations. Next we mapped the domains of XRCC1 interacting with SSRP1 using IP in 293 cells. As shown in Fig. 3B, C, SSRP1 interacts with the N-terminus of XRCC1 (1–537 a.a. and 1–300 a.a.). The minimum 1–300 a.a. N-terminal of XRCC1 necessary for interaction does not overlap with the BRCT1 domain of XRCC1, which is responsible for interaction with PARPs. Since CK2 phosphorylation of XRCC1 is important for its dissociation from SSBs (21), we examined the interaction of SSRP1 with wild type (WT) versus a CK2 phosphorylation sites-mutated XRCC1 (CKM-XRCC1) mutated at seven sites: Ser408Ala, Ser409Gly, Ser410Ala, Thr453Ala, Thr488Ala, Ser519Ala, and Thr523Ala. The CKM-XRCC1 could no longer be phosphorylated by CK2 (39). Here we found that the CKM-XRCC1 co-immunoprecipitated myc-SSRP1 as well as WT XRCC1 (Fig. 3D). Taken together, these data show that SSRP1 interacts with XRCC1 via its N-terminal 1–300 a.a., independent of both the interaction of XRCC1 with PARP1, and of the phosphorylation of XRCC1. There are a few critical amino acids in XRCC1 for protein-protein interactions. F67A is a mutation selected on the N terminal of XRCC1 that is involved in the interaction between XRCC1 and polymerase β (Pol β) (40). FF191/192AA is another mutation on XRCC1, located on an RIR (Rev1 Interacting Region) motif, which is not only consensus through Pol κ, Pol η and Pol ι, but also highly conserved among different species (40). Importantly, mutation of FF191/192AA on the RIR motif aborted the binding of Rev1 with Pol κ, Pol ι, and Pol η (41). We created these mutants and found that the F67A and FF191/192AA XRCC1 formed foci at sites of SSBs in XPA-UVDE cells, indicating that the mutations did not destroy the protein structure and remain functional upon foci formation (Fig 4A). We next examined their interactions with XRCC1 (Fig. 4B). FF191/192AA showed a significantly decreased level of interaction (Fig. 4B) with XRCC1, indicating that the RIR motif is responsible for XRCC1-SSRP1 interaction. Importantly, FF191/192AA XRCC1 expression led to dissociation of SSRP1 at sites of SSBs 4h after UVC irradiation (Fig. 4C), indicating that the retention of SSRP1 is dependent on the XRCC1-SSRP1 interaction.
Figure 3. SSRP1 interacts with the N-terminus of XRCC1 independent of XRCC1 phosphorylation or interaction with PARP1.
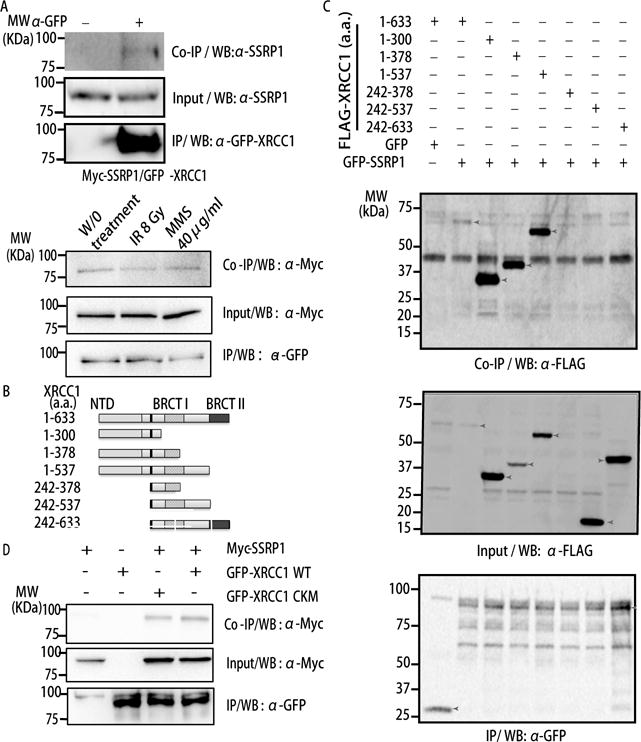
A. Schematic representation of XRCC1 domains used in this study. B. 293 cells expressing both Flag-tagged XRCC1 deletions and a pEGFP-SSRP1/pEGFP-C1 vector were immunoprecipitated by GFP antibody. Western blot (WB) with anti-Flag and anti-GFP is shown. C. 293 cells expressing GFP-SSRP1 and Flag-tagged XRCC1 deletions were immunoprecipitated using GFP antibody. WB with anti-Flag and anti-GFP is shown. D. 293 cells expressing GFP-tagged WT XRCC1 (left); CKM-XRCC1 and myc-SSRP1 (right) were immunoprecipitated by GFP antibody. WB with anti-SSRP1, anti-Flag and anti-GFP is shown.
Figure 4. The RIR motif of XRCC1 is responsible for SSRP1 interaction and retention at SSB foci.
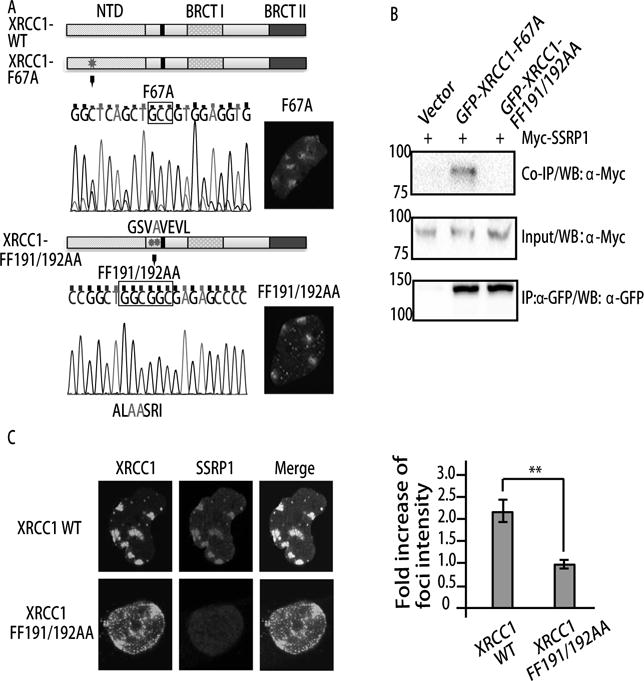
A. Scheme of two mutants at the N-terminus of XRCC1. Mutations of F67A (Polbeta interacting site) and FF191/192AA (Rev1 interacting site in RIR motif) were confirmed by Sanger sequencing. Both mutants form foci after LUVC treatment in XPA-UVDE cells. B. GFP-tagged XRCC1 WT, F67A, or FF191/192AA was co-transfected with Myc-tagged SSRP1 in 293 cells. The cell lysates were immunoprecipitated by anti-GFP mouse serum. Western blot (WB) with anti-GFP and anti-Myc is shown. C. Retention of SSRP1 at SSBs 4 h after 100 J/m2 local UVC irradiation. GFP-tagged XRCC1 WT or FF191/192AA was co-transfected with Myc-tagged SSRP1 in XPA-UVDE cells 24h before UVC irradiation. The relative intensity of SSRP1 at damage sites vs. background is shown as a fold intensity. Error bars were derived from SDs of multiple foci (n=7).
We inversely determined which domain of SSRP1 is necessary for interacting with XRCC1 using the SSRP1 deletions described above. SSRP1 contains an HMG domain, which is necessary for the recognition of damaged DNA (19,42,43). We divided SSRP1 into the evolutionarily conserved N-terminus (1–546 a.a) and HMG included C-terminus (547–709 a.a.) (Fig. 5A). As shown in Fig. 5B, the N-terminus of SSRP1 strongly interacted with XRCC1. Furthermore, the same N-terminus SSRP1 accumulated at sites of SSBs induced by UVDE in XPA-UVDE cells (Fig. 5C). Therefore, the N-terminus of SSRP1 but not the HMG domain primarily interacts with XRCC1 and responds to SSBs.
Figure 5. SSRP1’s N-terminus primarily interacts with XRCC1 and responds to SSBs.
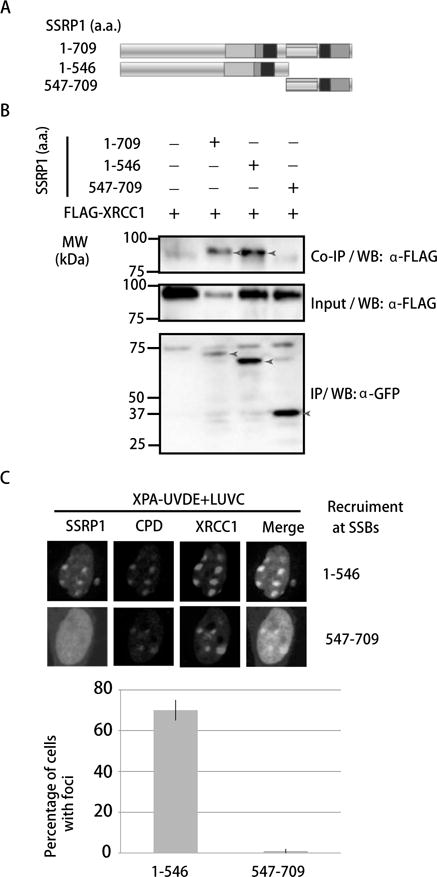
A. Schematic representation of SSRP1 domains. B. 293 cells expressing both Flag-tagged XRCC1 and GFP-tagged SSRP1 deletions were immunoprecipitated by anti-Flag antibody. WB with anti-Flag and GFP is shown. C. The recruitment of GFP-tagged SSRP1 deletions at SSBs induced by UVDE in XPA-UVDE cells. The N-terminus is highly enriched while the C-terminus is not. Quantification of percentage of foci positive cells of SSRP1 is shown. Cells showing > 3 foci were defined as positive cells. The error bar represents three independent experiments with 50 cells in each.
SSRP1 facilitates chromatin decondensation after damage
Given the role of SSRP1 in affecting chromatin structure, we next analyzed the chromatin states. The fluorescence lifetime imaging assay is a quantitative and spatially resolved measurement of chromatin condensation states for cell nuclei in situ (23). The fluorescent-tagged chromatin of the interphase cell nucleus is associated with a spatially heterogeneous distribution of fluorescence lifetimes, indicating differential fluorophore environments corresponding to varying levels of chromatin condensation. Decondensed chromatin is associated with increased fluorescence lifetimes (FL). Using fluorescence lifetime imaging microscopy (FLIM) of DAPI-labeled DNA, we examined the role of SSRP1 in chromatin condensation after damage. Increased FL represents the relaxed chromatin structure. As expected, we observed chromatin decondensation (FL from 2.35 to 2.44) in shCtrl cells. In two SSRP1-suppressed cell lines, chromatin was much more condensed, as shown by the reduced FL observed relative to control cell nuclei (FL=2.35 in shCtrl versus FL=2.06 in shSSRP1) (Fig. 6A). This result indicates that SSRP1 is necessary for global chromatin decondensation in cells after damage.
Figure 6. SSRP1 promotes cell survival and histone H2B exchange at damage sites.
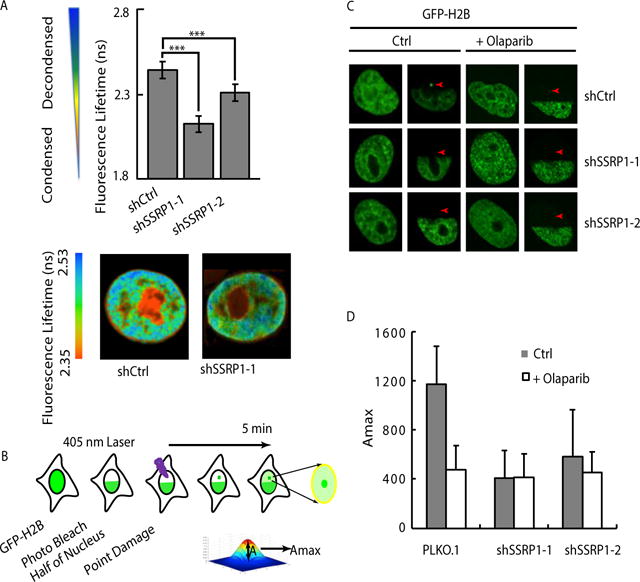
A. Decondensation of chromatin within a nucleus after bleocin treatment using the fluorescence lifetime imaging assay. The representative heat map of chromatin with or without SSRP1 suppression and the quantification of the lifetime of the whole nucleus are shown. B. FRAP assay for measuring histone H2B exchange at sites of damage induced by a 405 nm laser. Half of the nuclei of HeLa cells transfected with GFP-H2B were photo bleached with a 488 nm laser, then irradiated with a 405 nm laser at the indicated point within the bleached region. C–D. The fluorescence recovery of histone H2B at the 405 nm laser-induced damage sites was determined within 5 min after damage. The representative image shows the GFP-H2B recovery in shCtrl and shSSRP1 cells with or without pretreatment by olaparib. The maximum fold increase of mean intensity of H2B at damage sites vs. bleached region without damage is quantified. The graph shows histone H2B exchange in shCtrl and shSSRP1 cells. Error bars are derived from SEMs in more than 20 cells. Three independent experiments were conducted.
SSRP1 promotes histone H2B exchange at damage sites
To further explore the function of SSRP1 in repair of strand breaks, we next focused on its role as a histone H2A/H2B chaperone. Since the damage repair process needs to first “open” the chromatin at damage sites to allow access of the repair machinery, we tested whether SSRP1 plays a more critical role to facilitate histone exchange at sites of strand breaks. We modified the fluorescence recovery after photo bleaching (FRAP) assay based on a previous study (17), which was designed to compare histone H2A/H2B exchange at UVC-induced DNA damage sites. In this study, instead of using a short-wave length laser to induce CPDs, we used a 405 nm laser to induce DNA strand breaks in living cells after photo bleaching. By monitoring the fluorescence recovery of GFP-H2B at the damage sites compared to the photo-bleached half of the nucleus in a single cell nucleus, we could calculate the rate of histone exchange at damage sites relative to the undamaged region. We monitored the dynamics of GFP-H2B and observed a faster recovery of H2B incorporation at sites of damage compared to that in the bleached area. The higher histone exchange rate was coupled with an increased local fluorescence. We further recorded the highest fluorescence at each damage site, which was indicated by the linear index Amax (Fig. 6B). After bleaching, the fluorescence at damage sites in the shCtrl cells recovered fairly rapidly and to a higher intensity, which indicated that more GFP-H2B was accumulated at the sites of damage. In contrast, in the PARP inhibitor treated and shSSRP1 KD cells, the recovered GFP-H2B fluorescence at damage sites was much less enriched compared to the bleached background (Fig. 6C, D). Therefore, shSSRP1 knockdown significantly reduced the dynamics of GFP-H2B at damage sites. In addition, the PAPRi treatment did not further decrease the H2B exchange at damage sites in shSSRP1 cells, indicating that the function of SSRP1 to promote H2B exchange is dependent on PARP activation. To test the effect of XRCC-FF191/192AA mutant, N-terminal part of SSRP1 (1–546) and the C-terminus of SSRP1 on chromatin states, we measured the decondensation of chromatin using H2B in the cells with or without expression above deletions or mutants. We use stochastic optical reconstruction microscopy (STORM) to visualize the nanoscale organization of chromatin influenced by the treatment. First, our STORM images reveal that the nucleosomes were assembled in heterogeneous clusters with varying sizes, in agreement with the previous report (44). As shown in Fig. 7A, significantly less dense nucleosome clusters with smaller size can be seen in cells after the SSRP1 knockdown, which is in consistence with the observation shown in Fig. 6 using FLIM. When we express N-terminus SSRP1 fragments in shSSRP1 cells, the nucleosome cluster density was partially recovered (Fig. 7A, B). The H2B level was rescued by the expression of WT SSRP1, N-terminus of SSRP1 but not the C-terminus of SSRP1 (Fig. 7B). In addition, XRCC1 FF191/192AA mutant overexpression in WT cells led to the a similar diffused H2B distribution and reduced H2B intensity, indicating that overexpression of loss of interaction mutant did affect chromatin decondensation as well as the KD of SSRP1 did (Fig. 7A, B). Thus, N-termins of SSRP1 (1–546) which is recruited to the damage sites via binding with XRCC1 seems to be more important than C-terminus SSRP1 for chromatin priming. Collectively, these results show that SSRP1 facilitates histone H2B exchange via its N-terminus in both PARP activation- and XRCC1 interaction- dependent manner at strand breaks in live cells after damage.
Figure 7. A model of how FACT functions in repair of DNA strand breaks.
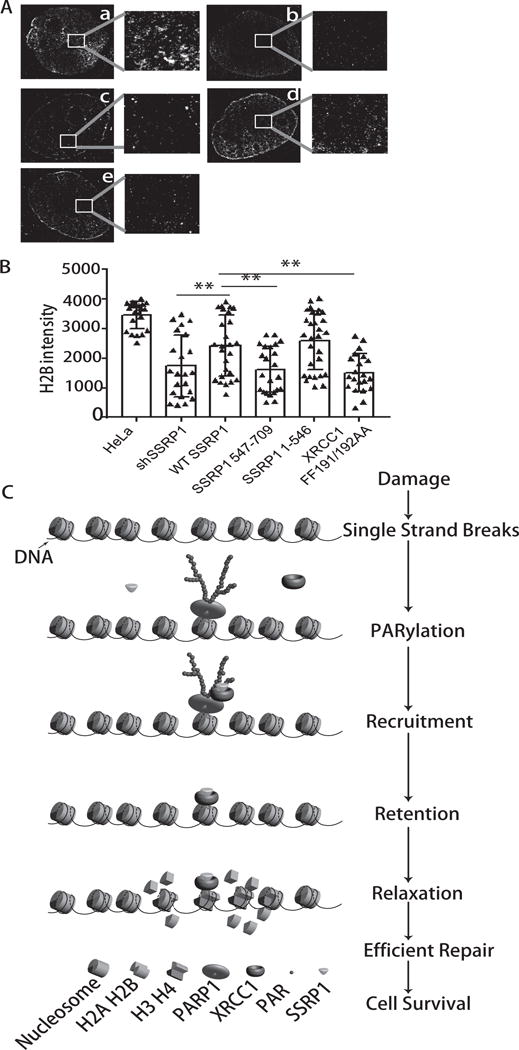
A and B. Images and intensity of H2B in HeLa cells or shSSRP1 HeLa cells were co-expressed with cherry-H2B with genes indicated. A) STORM images of cherry-H2B in below cells: a. HeLa; b. shSSRP1 HeLa; c. shSSRP1 expressing 547-709 a.a. SSRP1; d. shSSRP1 expressing 1–546 a.a. SSRP1; e. HeLa expressing FF191/192AA XRCC1. B) The representative H2B intensity was measured in images taken by confocal microscopy. Error bars are derived from SEMs in 20 cells. C. PARP activation facilitates the recruitment of FACT to sites of SSBs. Though PAR is quickly degraded, FACT remains at damage sites together with XRCC1 through a physical interaction in SSBR. The function of FACT at strand breaks is to facilitate histone exchange at damage sites and to decondense chromatin, thereby facilitating efficient DNA SSB repair.
Discussion
Here we have shown that the involvement of SSRP1 in the repair of DNA strand breaks, especially SSBs, is dependent on PARP activation and is in an association with XRCC1 through a physical interaction (Fig. 1–5). Importantly, although SSRP1 is not necessary for the repair of UVC-induced damage (17), in this study we showed that it is essential for cell survival and chromatin remodeling in the case of DNA strand break repair (Fig. 1). We further demonstrated that SSRP1 is important for histone exchange and chromatin decondensation at sites of DNA strand breaks. We have proposed a model to show how FACT is recruited and retained at SSBs and its possible function in SSBR (Fig. 7C). At the initiating step of repair, PARP activation facilitates the recruitment of FACT to sites of SSBs. Although PAR is quickly degraded by PARG (45), FACT remains at damage sites together with XRCC1 through a physical interaction. This interaction is mediated by the N-terminus of XRCC1, which does not overlap with its BRCTI domain (which mediates the interaction between XRCC1 and PARP1). Therefore, the interaction of SSRP1 and XRCC1 is not mediated by PARP1, which is consistent with the observation of PAR-independent retention of FACT after PAR degradation for 2–4 h after damage production. Therefore, our study reveals a novel mechanism of SSRP1 in SSBR and maintaining genome stability.
Mechanistically, we uncovered the molecular mechanism involved in the interaction of SSRP1 and XRCC1. The HMG domain of SSRP1 has been implicated in the DNA damage response based on its DNA binding affinity (19). In addition to the HMG domain, we also found that the functionally unknown but highly conserved N-terminus of SSRP1 is important for the DNA damage response and for protein-protein interactions, e.g., with the SSBR scaffold protein XRCC1. The RIR motif in XRCC1 is necessary for SSRP1 binding. The newly identified RIR motif of XRCC1 is in its N-terminus; this motif has been shown to be responsible for interacting with post translational error-prone polymerases including pol κ, pol η and pol ι (40). It was interesting that the XRCC1 F67A mutant, which is necessary for interaction between XRCC1 and Pol β(40), did not affect the interaction with XRCC1 and SSRP1. It is possible that the SSRP1 binding at the RIR motif of XRCC1 also contributes to error-free repair by competing with the binding of error-prone polymerase mediated translesion synthesis.
Damage-induced chromatin relaxation at a global level throughout the cell nucleus due to histone modifications and chromatin remodeling factors has been shown in previous studies (46). We predict that the complex of SSRP1 and XRCC1 at damage sites might cooperate to facilitate repair processing by promoting chromatin relaxation. Our results, using multiple approaches, have provided evidence that: 1) SSRP1 facilitates decondensation of chromatin, and 2) SSRP1 mediates histone H2B exchange at damage sites (Fig. 6). It has been shown that SSRP1 is necessary for both spontaneous and replication-associated DNA damage responses, since SSRP1 depletion leads to increased Rad51 and γH2AX foci. The role of SSRP1 in facilitating SSBR indicates that the unrepaired SSB could be transformed to double strand breaks at collapsed replication forks without SSRP1, thus leading to increased damage foci formation. Meanwhile, It is known that SSRP1 interacts with a key HR repair protein, Rad54, both in vitro and in vivo. Branch migration studies demonstrated that SSRP1 inhibits Rad54-promoted branch migration of Holliday junctions in vitro (20). Therefore, over expression of SSRP1 decreases the foci formation of Rad51 and γH2AX spontaneously upon hydroxyurea (HU) treatment (20). How SSRP1 contributes to SSBs and double strand breaks differently need further investigation to understand our evidence of SSRP1 in maintaining cell survival upon IR and MMS.
FACT is highly expressed in undifferentiated cells, and its deficiency leads to zebrafish developmental defects (47) and mouse embryo lethality (48). Interestingly, FACT is frequently upregulated in undifferentiated aggressive tumors, indicating a potential role as an anti-cancer target (49–51). A well-studied drug, Curaxin, is toxic to cancer cells by simultaneously suppressing NF-kB and activating p53, resulting in the “chromatin trapping” of FACT (50). The mechanism of FACT’s role in SSBR sheds light on its possibility for targeted cancer therapy. Upregulated FACT may protect tumor cells, especially less differentiated tumor stem cells, from DNA damage-induced cell death by enhancing efficient repair. Here we show that PARP1 inhibitors could inhibit the recruitment of FACT at strand breaks; therefore FACT suppression should have a similar effect as PARP inhibitors in cancer cells. Recently it also has been shown that Rev7 is responsible for the resistance of BRCA1-deficient tumors to PARP inhibitors (52). Given the role of SSRP1 in promoting repair, over-expression of SSRP1 might lead to increased resistance to PARP inhibitors. Accordingly, it is known that FACT is highly expressed in breast and ovarian tumors (12,53). FACT inhibition or combination therapy with Curaxin and PARP inhibitors could be effective in treating FACT-upregulated aggressive breast and ovarian tumors.
Acknowledgments
We thank Dr. Akira Yasui for providing the cDNA of SSRP1.
Funding: This work was supported in part by grants from the National Institutes of Health GM118833 and AG045545-01 to L. Lan. Funding for the open access charge: National Institutes of Health/GM118833. This project used the University of Pittsburgh Cancer Institute Cytometry Facility, supported in part by National Institutes of Health P30CA047904.
Footnotes
Conflict of interest: Authors declare no conflict of interest.
References
- 1.Kuzminov A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci U S A. 2001;98(15):8241–6. doi: 10.1073/pnas.131009198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9(8):619–31. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 3.Poirier GG, de Murcia G, Jongstra-Bilen J, Niedergang C, Mandel P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc Natl Acad Sci U S A. 1982;79(11):3423–7. doi: 10.1073/pnas.79.11.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fang Q, Inanc B, Schamus S, Wang XH, Wei L, Brown AR, et al. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase beta. Nat Commun. 2014;5:5513. doi: 10.1038/ncomms6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abdel-Magid AF. Potential Use of Inhibitors of Tankyrases and PARP-1 as Treatment for Cancer and Other Diseases. ACS Med Chem Lett. 2016;7(3):209–10. doi: 10.1021/acsmedchemlett.6b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caldecott KW. XRCC1 and DNA strand break repair. DNA Repair (Amst) 2003;2(9):955–69. doi: 10.1016/s1568-7864(03)00118-6. [DOI] [PubMed] [Google Scholar]
- 7.Soria G, Polo SE, Almouzni G. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell. 2012;46(6):722–34. doi: 10.1016/j.molcel.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Avvakumov N, Nourani A, Cote J. Histone chaperones: modulators of chromatin marks. Mol Cell. 2011;41(5):502–14. doi: 10.1016/j.molcel.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 9.Smerdon MJ. DNA repair and the role of chromatin structure. Curr Opin Cell Biol. 1991;3(3):422–8. doi: 10.1016/0955-0674(91)90069-b. [DOI] [PubMed] [Google Scholar]
- 10.Orphanides G, Wu WH, Lane WS, Hampsey M, Reinberg D. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature. 1999;400(6741):284–8. doi: 10.1038/22350. [DOI] [PubMed] [Google Scholar]
- 11.Dermawan JK, Hitomi M, Silver DJ, Wu Q, Sandlesh P, Sloan AE, et al. Pharmacological Targeting of the Histone Chaperone Complex FACT Preferentially Eliminates Glioblastoma Stem Cells and Prolongs Survival in Preclinical Models. Cancer Res. 2016;76(8):2432–42. doi: 10.1158/0008-5472.CAN-15-2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koman IE, Commane M, Paszkiewicz G, Hoonjan B, Pal S, Safina A, et al. Targeting FACT complex suppresses mammary tumorigenesis in Her2/neu transgenic mice. Cancer Prev Res (Phila) 2012;5(8):1025–35. doi: 10.1158/1940-6207.CAPR-11-0529. [DOI] [PubMed] [Google Scholar]
- 13.Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;301(5636):1090–3. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- 14.Abe T, Sugimura K, Hosono Y, Takami Y, Akita M, Yoshimura A, et al. The histone chaperone facilitates chromatin transcription (FACT) protein maintains normal replication fork rates. J Biol Chem. 2011;286(35):30504–12. doi: 10.1074/jbc.M111.264721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan BC, Chien CT, Hirose S, Lee SC. Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J. 2006;25(17):3975–85. doi: 10.1038/sj.emboj.7601271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heo K, Kim H, Choi SH, Choi J, Kim K, Gu J, et al. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol Cell. 2008;30(1):86–97. doi: 10.1016/j.molcel.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 17.Dinant C, Ampatziadis-Michailidis G, Lans H, Tresini M, Lagarou A, Grosbart M, et al. Enhanced chromatin dynamics by FACT promotes transcriptional restart after UV-induced DNA damage. Mol Cell. 2013;51(4):469–79. doi: 10.1016/j.molcel.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 18.Oliveira DV, Kato A, Nakamura K, Ikura T, Okada M, Kobayashi J, et al. Histone chaperone FACT regulates homologous recombination by chromatin remodeling through interaction with RNF20. J Cell Sci. 2014;127(Pt 4):763–72. doi: 10.1242/jcs.135855. [DOI] [PubMed] [Google Scholar]
- 19.Yarnell AT, Oh S, Reinberg D, Lippard SJ. Interaction of FACT, SSRP1, and the high mobility group (HMG) domain of SSRP1 with DNA damaged by the anticancer drug cisplatin. J Biol Chem. 2001;276(28):25736–41. doi: 10.1074/jbc.M101208200. [DOI] [PubMed] [Google Scholar]
- 20.Kumari A, Mazina OM, Shinde U, Mazin AV, Lu H. A role for SSRP1 in recombination-mediated DNA damage response. J Cell Biochem. 2009;108(2):508–18. doi: 10.1002/jcb.22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei L, Nakajima S, Hsieh CL, Kanno S, Masutani M, Levine AS, et al. Damage response of XRCC1 at sites of DNA single strand breaks is regulated by phosphorylation and ubiquitylation after degradation of poly(ADP-ribose) J Cell Sci. 2013;126(Pt 19):4414–23. doi: 10.1242/jcs.128272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lan L, Nakajima S, Wei L, Sun L, Hsieh CL, Sobol RW, et al. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Res. 2014;42(4):2330–45. doi: 10.1093/nar/gkt1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spagnol ST, Dahl KN. Active cytoskeletal force and chromatin condensation independently modulate intranuclear network fluctuations. Integr Biol (Camb) 2014;6(5):523–31. doi: 10.1039/c3ib40226f. [DOI] [PubMed] [Google Scholar]
- 24.Ma H, Xu J, Jin J, Gao Y, Lan L, Liu Y. Fast and Precise 3D Fluorophore Localization based on Gradient Fitting. Sci Rep. 2015;5:14335. doi: 10.1038/srep14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, et al. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc Natl Acad Sci U S A. 2004;101(38):13738–43. doi: 10.1073/pnas.0406048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keller DM, Lu H. p53 serine 392 phosphorylation increases after UV through induction of the assembly of the CK2.hSPT16.SSRP1 complex. J Biol Chem. 2002;277(51):50206–13. doi: 10.1074/jbc.M209820200. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Keller DM, Scott JD, Lu H. CK2 phosphorylates SSRP1 and inhibits its DNA-binding activity. J Biol Chem. 2005;280(12):11869–75. doi: 10.1074/jbc.M413944200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krohn NM, Stemmer C, Fojan P, Grimm R, Grasser KD. Protein kinase CK2 phosphorylates the high mobility group domain protein SSRP1, inducing the recognition of UV-damaged DNA. J Biol Chem. 2003;278(15):12710–5. doi: 10.1074/jbc.M300250200. [DOI] [PubMed] [Google Scholar]
- 29.Keller DM, Zeng X, Wang Y, Zhang QH, Kapoor M, Shu H, et al. A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol Cell. 2001;7(2):283–92. doi: 10.1016/s1097-2765(01)00176-9. [DOI] [PubMed] [Google Scholar]
- 30.Sage E, Shikazono N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic Biol Med. 2016 doi: 10.1016/j.freeradbiomed.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 31.Shiraishi I, Shikazono N, Suzuki M, Fujii K, Yokoya A. Efficiency of radiation-induced base lesion excision and the order of enzymatic treatment. Int J Radiat Biol. 2016:1–8. doi: 10.1080/09553002.2017.1239849. [DOI] [PubMed] [Google Scholar]
- 32.Chomicz L, Furmanchuk A, Leszczynski J, Rak J. Electron induced single strand break and cyclization: a DFT study on the radiosensitization mechanism of the nucleotide of 8-bromoguanine. Phys Chem Chem Phys. 2014;16(14):6568–74. doi: 10.1039/c3cp55411b. [DOI] [PubMed] [Google Scholar]
- 33.Nikitaki Z, Mavragani IV, Laskaratou DA, Gika V, Moskvin VP, Theofilatos K, et al. Systemic mechanisms and effects of ionizing radiation: A new ‘old’ paradigm of how the bystanders and distant can become the players. Semin Cancer Biol. 2016:37–38. 77–95. doi: 10.1016/j.semcancer.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 34.Kashkina E, Qi T, Weinfeld M, Young D. Polynucleotide kinase/phosphatase, Pnk1, is involved in base excision repair in Schizosaccharomyces pombe. DNA Repair (Amst) 2012;11(8):676–83. doi: 10.1016/j.dnarep.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 35.Praveen Kumar MK, Shyama SK, Sonaye BS, Naik UR, Kadam SB, Bipin PD, et al. Evaluation of gamma-radiation-induced DNA damage in two species of bivalves and their relative sensitivity using comet assay. Aquat Toxicol. 2014;150:1–8. doi: 10.1016/j.aquatox.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 36.Okano S, Lan L, Caldecott KW, Mori T, Yasui A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol Cell Biol. 2003;23(11):3974–81. doi: 10.1128/MCB.23.11.3974-3981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fisher AE, Hochegger H, Takeda S, Caldecott KW. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol Cell Biol. 2007;27(15):5597–605. doi: 10.1128/MCB.02248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zahradka P, Ebisuzaki K. A shuttle mechanism for DNA-protein interactions. The regulation of poly(ADP-ribose) polymerase. Eur J Biochem. 1982;127(3):579–85. [PubMed] [Google Scholar]
- 39.Loizou JI, El-Khamisy SF, Zlatanou A, Moore DJ, Chan DW, Qin J, et al. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004;117(1):17–28. doi: 10.1016/s0092-8674(04)00206-5. [DOI] [PubMed] [Google Scholar]
- 40.Marintchev A, Gryk MR, Mullen GP. Site-directed mutagenesis analysis of the structural interaction of the single-strand-break repair protein, X-ray cross-complementing group 1, with DNA polymerase beta. Nucleic Acids Res. 2003;31(2):580–8. doi: 10.1093/nar/gkg159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohashi E, Hanafusa T, Kamei K, Song I, Tomida J, Hashimoto H, et al. Identification of a novel REV1-interacting motif necessary for DNA polymerase kappa function. Genes Cells. 2009;14(2):101–11. doi: 10.1111/j.1365-2443.2008.01255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bruhn SL, Housman DE, Lippard SJ. Isolation and characterization of cDNA clones encoding the Drosophila homolog of the HMG-box SSRP family that recognizes specific DNA structures. Nucleic Acids Res. 1993;21(7):1643–6. doi: 10.1093/nar/21.7.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kasai N, Tsunaka Y, Ohki I, Hirose S, Morikawa K, Tate S. Solution structure of the HMG-box domain in the SSRP1 subunit of FACT. J Biomol NMR. 2005;32(1):83–8. doi: 10.1007/s10858-005-3662-3. [DOI] [PubMed] [Google Scholar]
- 44.Ricci MA, Manzo C, Garcia-Parajo MF, Lakadamyali M, Cosma MP. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell. 2015;160(6):1145–58. doi: 10.1016/j.cell.2015.01.054. [DOI] [PubMed] [Google Scholar]
- 45.Davidovic L, Vodenicharov M, Affar EB, Poirier GG. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268(1):7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- 46.Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell. 2013;152(6):1344–54. doi: 10.1016/j.cell.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koltowska K, Apitz H, Stamataki D, Hirst EM, Verkade H, Salecker I, et al. Ssrp1a controls organogenesis by promoting cell cycle progression and RNA synthesis. Development. 2013;140(9):1912–8. doi: 10.1242/dev.093583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao S, Bendall H, Hicks GG, Nashabi A, Sakano H, Shinkai Y, et al. The high-mobility-group box protein SSRP1/T160 is essential for cell viability in day 3.5 mouse embryos. Mol Cell Biol. 2003;23(15):5301–7. doi: 10.1128/MCB.23.15.5301-5307.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia H, Fleyshman D, Kolesnikova K, Safina A, Commane M, Paszkiewicz G, et al. Expression of FACT in mammalian tissues suggests its role in maintaining of undifferentiated state of cells. Oncotarget. 2011;2(10):783–96. doi: 10.18632/oncotarget.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gasparian AV, Burkhart CA, Purmal AA, Brodsky L, Pal M, Saranadasa M, et al. Curaxins: anticancer compounds that simultaneously suppress NF-kappaB and activate p53 by targeting FACT. Sci Transl Med. 2011;3(95):95ra74. doi: 10.1126/scitranslmed.3002530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsieh FK, Kulaeva OI, Orlovsky IV, Studitsky VM. FACT in Cell Differentiation and Carcinogenesis. Oncotarget. 2011;2(11):830–2. doi: 10.18632/oncotarget.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521(7553):541–4. doi: 10.1038/nature14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hudson ME, Pozdnyakova I, Haines K, Mor G, Snyder M. Identification of differentially expressed proteins in ovarian cancer using high-density protein microarrays. Proc Natl Acad Sci U S A. 2007;104(44):17494–9. doi: 10.1073/pnas.0708572104. [DOI] [PMC free article] [PubMed] [Google Scholar]