

Abstract
Changes in mitochondrial DNA (mtDNA) integrity have been reported in many cancers, however, the contribution of mtDNA integrity to tumorigenesis is not well understood. We used a transgenic mouse model that is haploinsufficient for the apurinic/apyrimidinic endonuclease 1 (Apex1+/-) gene, which encodes the base excision repair (BER) enzyme APE1, to determine its role in protecting mtDNA from the effects of azoxymethane (AOM), a carcinogen used to induce colorectal cancer (CRC). Repair kinetics of AOM-induced mtDNA damage was evaluated using quantitative PCR after a single AOM dose and a significant induction in mtDNA lesions in colonic crypts from both wild type (WT) and Apex1+/-animals were observed. However, Apex1+/- mice had slower repair kinetics in addition to decreased mtDNA abundance. Tumors were also induced using multiple AOM doses and both WT and Apex1+/-animals exhibited significant loss in mtDNA abundance. Surprisingly, no major differences in mtDNA lesions were observed in tumors from WT and Apex1+/-animals, whereas a significant increase in nuclear DNA lesions was detected in tumors from Apex1+/- mice. Finally, tumors from Apex1+/-mice displayed an increased proliferative index and histological abnormalities. Taken together, these results demonstrate that APE1 is important for preventing changes in mtDNA integrity during AOM-induced CRC.
Implications: Azoxymethane, a colorectal cancer carcinogen, generates damage to the mitochondrial genome and the BER enzyme APE1 is required to maintain its integrity.
Keywords: DNA Repair, Base Excision Repair, Colorectal Cancer
Introduction
Colorectal cancer (CRC) is a global health problem causing significant morbidity and mortality. The Surveillance, Epidemiology, and End Results Program (seer.cancer.gov) estimates that in 2016 there will be in the U.S. 134,490 new CRC cases and 49,190 CRC related deaths. Thus, a great deal of effort has been invested in animal models of CRC to help identify molecular targets for pharmacological interventions.
Studies using mouse models of CRC have helped elucidate molecular mechanisms underlying tumor initiation and promotion (1). A commonly used mouse model is based in exposure to the carcinogen azoxymethane (AOM) (2). AOM induces mutagenic lesions within target cells by alkylating DNA primarily at positions N7 of guanine (N7MeG) and N3 of adenine (N3MeG) and to a lesser extent, at position O6 of guanine (O6MeG) (3). O6MeG is primarily repaired by the suicide enzyme O6-methylguanine-DNA methyltransferase (MGMT) (4), whereas the above mentioned N-alkylated bases are repaired by the base excision repair (BER) pathway. BER is a multistep process that repairs damage induced by reactive oxygen species and alkylating agents, which involves the coordinate actions of glycosylases, endonucleases and other end-trimming enzymes, DNA polymerases and DNA ligases (5).
Within BER, the apurinic/apyrimidinic endonuclease 1 (APE1) is the major AP endonuclease. APE1 is a multifunctional enzyme involved in DNA repair, transcriptional regulation, and redox signaling. APE1 is normally localized in the cytoplasm and upon NADPH-dependent ROS generation it mobilizes to the cell nucleus (6). In addition, a N-terminal truncation of the full-length APE1 translocates to the mitochondria in response to oxidative stress (7).
Mice carrying a homozygous null mutation in the Apex1 gene (which encodes APE1) are embryonically lethal, indicating that APE1 is necessary for normal embryonic development (8). In contrast, Apex1 heterozygous mice (Apex1+/-) are viable and have a normal lifespan, although these mice present increase markers of oxidative stress. In addition, Apex1+/- mice show enhanced mutagenesis in liver, spleen, and germ cells (9,10). Also, Apex1+/- mice exhibit an age-dependent accumulation of mtDNA lesions in germ cells as compared to WT mice suggesting that APE1 may play a role in preventing mtDNA damage during aging (10).
MtDNA mutations, deletions, and copy number alterations are associated with cancer (11). Among CRC, loss of mtDNA has been identified in the form of missense mutations, chain terminations, and frame shift mutations (12). In addition, changes in mtDNA copy number have been reported in CRC (13,14). However, how these mtDNA changes contribute to carcinogenesis is not totally understood. Our study seeks to determine the contribution of APE1 in preventing nuclear and mtDNA alterations that could drive the tumorigenic process. We hypothesize that the effects of AOM treatment are more pronounced in the Apex1+/-mice than in WT mice due to decreased nuclear and mtDNA repair capacity. To test this hypothesis, we measured changes in mtDNA abundance, and mtDNA and nDNA damage in colonic crypts after AOM treatment. We also determined the characteristics of AOM-induced tumors. By focusing in APE1, we show that BER is important for the repair of AOM-induced mtDNA and nDNA damage and for the subsequent development of specific tumor characteristics.
Materials and Methods
Animals
Six (6)-month-old WT and Apex1+/- in the C57BL/6J background were used in this study (8). The original breading pair in our mouse colony was kindly provided by Dr. Christi Walter (University of Texas Health Science Center at San Antonio). Mice were housed 2 per cage with a 12-hour light-dark cycle and ad libitum access to food and water. Genotyping analysis was performed collecting 5mm of tissue from the tail from each mouse at 21 days of birth. DNA was extracted from tail samples following the protocol from Manual Archive Pure DNA purification (ArchivePure™ Trademark of 5 PRIME). PCR was performed using a set of primers that recognize a Neomycin resistance transgene present only in the Apex1 knockout mice, as previously described (8,9). The University of Puerto Rico Medical Sciences Campus Institutional Animal Care and Use Committee approved the studies.
AOM Treatment
WT and Apex1+/- mice were randomized into two groups; AOM (Sigma-Aldrich Chemical, St. Louis, MO) treated or saline treated. To measure AOM-induced DNA damage and repair kinetics, mice were injected intraperitoneally with a single dose of AOM (10 mg/kg of body weight) and sacrificed 24, 48 and 72 hours after treatment. Colonic crypts were isolated followed by the isolation of genomic DNA as described below.
To study AOM-induced tumor formation, mice were treated with 10 mg/kg AOM, once a week during four weeks as previously reported (15). Six months after the first AOM injection, colon tissues were removed, cut longitudinally and examined for tumor incidence (percent of animals with tumors), multiplicity (average number of tumors per mouse) and tumor volume. Tumor volume (expressed in mm3) was determined by measuring the length, width and height of each tumor with a caliper and then obtaining the product of these dimensions.
Isolation of colonic crypts
After euthanization of mice by cervical dislocation, the abdomen was opened followed by colon removal. The colon was cut along its length and rinsed with ice-cold phosphate buffer saline (PBS). The colon was then incubated in a 15 ml tube containing 1× (PBS), 1.5 mM ethylenediaminetetraacetic acid (EDTA) and 0.5 mM of dithiothreitol (DTT) for 90 minutes at room temperature. The incubation buffer was replaced with 1 ml 1× PBS and the sample was vortex for 30 seconds to obtain the crypts. The crypt suspension was transferred to a fresh tube, washed two times with 1 ml 1× PBS and pelleted by centrifugation at 1,000 rpm for 5 min. The supernatant was removed and the crypt containing pellets were stored at -80°C until further processing.
DNA isolation and quantification of genomic DNA from colonic crypts
DNA isolation was performed using a high molecular weight genomic DNA purification kit as described by the manufacturer (Qiagen). DNA was quantitated using the Picogreen® dsDNA quantitation assay (Molecular Probes/Life Technologies) using a microplate reader (Wallac 1420 VICTOR™ F) with a 535 nm excitation filter and a 485 nm emission filter. A standard curve with a Lambda DNA was constructed in order to determine the correct DNA concentration of the samples. Before performing the DNA damage analysis, DNA samples (100 ng) were visualized by 1% agarose gel electrophoresis, stained with ethidium bromide to ensure that no degradation occurred during the isolation procedure.
Detection of DNA damage and mtDNA abundance using the quantitative polymerase chain reaction (QPCR) assay
Levels of DNA lesions (nDNA and mtDNA) and mtDNA abundance in AOM-treated and saline-treated WT and Apex1+/- mice were measured by QPCR as described previously (16). The rationale of using the QPCR assay for the quantification of lesions is based on the ability of various DNA lesions to act as blocks to the action of the PCR polymerase, resulting in decreased amplification of the template. The QPCR assay detects alkylation lesions as well as oxidative DNA damage such as AP sites, strand breaks, and thymine glycol, all of which block the movement of the thermostable polymerase along the DNA template. The PCR amplification was performed using the Master Amp XL Polymerase (Epicentre). Prior to QPCR analysis we performed a cycle test to determine the optimal number of amplification cycles and a template test to determine the optimal initial DNA concentration (data not shown).
The mouse mitochondrial (10 kb) and nuclear (6.9 kb) fragments were amplified using an initial denaturation for 45 seconds at 94°C, followed by 22 cycles (mtDNA amplicon) and 28 cycles (nDNA amplicon) of denaturation for 15 seconds at 94°C, and annealing/extension at 64°C and 68°C for 12 minutes, respectively. A final extension at 72°C was performed for 10 minutes at the completion of the profile. The primer nucleotide sequences used for the amplification of the 10 kb mtDNA fragment were the following: 5′-CCA GTC CAT GCA GGA GCA TC-3′ (5733 sense; relative to sequence NC_005089) and 5′CGA GAA GAG GGG CAT TGG TG-3′ (15733 antisense). The primer nucleotide sequences used for the amplification of a 6.9 kb mouse hypoxanthine phosphoribosyltransferase (HPRT) (17) gene fragment were the following: 5′-CCA CCA GGC GTC ACC CTT GA-3′ (9349 sense) and 5′-TGG GAG GCA GGG ATC TGA AGC-3′ (16246 antisense). We normalized the amplification of the 10 kb mtDNA fragment with the levels of mtDNA abundance (see below).
Steady state levels of mtDNA abundance were measured by QPCR amplification of a small, 116 bp mtDNA fragment. Because the probability of finding a lesion in a small mtDNA fragment is nearly null, changes in the amplification of the small mtDNA fragment provides an accurate measure of the abundance of mtDNA molecules. The PCR amplification profile for the 116 bp mouse mtDNA fragment was performed with an initial denaturation for 45 seconds at 94°C, followed by 22 cycles of denaturation for 15 seconds at 94°C, and annealing/extension at 61°C for 45 seconds and 45 seconds at 72°C. A final extension at 72°C was performed for 10 minutes at the completion of the profile. The primer nucleotide sequences were the following: 5′CCC AGC TAC TAC CAT CAT TCA AGT-3′ (sense) and 5′GAT GGT TTG GGA GAT TGG TTG ATG T-3′ (antisense).
The initial DNA template concentration for the amplification of the 116 bp mtDNA fragment, the 10 kb mtDNA fragment, and the 6.9 kb nDNA fragment were 15, 10, and 7.5 ng, respectively. We amplified a DNA containing 50% of the initial template concentration as a control for amplification, therefore, under optimal amplification conditions, a 50% reduction in the amount of template amplification is expected. We considered QPCR reactions to be within the linear range of DNA amplification if the resulting amplicon was within 40-60% amplification. The 10 kb and 6.9 kb PCR products were resolved on 1% agarose gels, while the 116 bp PCR products were resolved on 6% polyacrylamide gels. DNA was visualized under UV-light using ethidium bromide-stained gels. Quantification of the PCR products was performed using the Picogreen® dsDNA quantitation assay.
DNA damage was calculated using the Poisson equation, where a decrease in the amplification of the damaged template is converted to lesion frequency (16). According to the Poisson's law if there is a random distribution of measurable independent events (i.e. DNA lesions), we can predict or estimate the number of lesions or how likely is that certain number of lesions are present in the damaged template (16). DNA damage was expressed as lesion frequency per 10 kb per strand.
Histopathology analyses
Histochemical staining using hematoxylin and eosin (H&E; Sigma) was performed using standard procedures (18). Briefly, colon and tumor tissues were fixed in 10% formaldehyde and embedded in paraffin. The paraffin blocks were then sectioned in 5 mm thickness. The histopathology analyses were performed blinded and the mean number of nuclear/cytoplasmic ratio, epithelial stratification, nuclear dispolarity, goblet depletion and structural abnormality of WT and Apex1+- tumors was analyzed as previously described (19). The grade of histological abnormality was scored using the following five parameters: a) nuclear/cytoplasmic ratio (<25%: 0, 25-50%: 1, >50%: 2); b) epithelial stratification (none: 0, mild: 1, severe: 2); c) nuclear dispolarity (none: 0, mild: 1, severe: 2); d) goblet depletion (null to mild: 0, moderate: 1, severe: 2); e) structural abnormality (none: 0, mild: 1, severe: 2) (19). Using all mice from each experimental group, four different and independent microscopic fields for each parameter were selected from each slide/mice and analyzed at 400× magnification with a phase contrast microscope and the mean number was established for each parameter. The histopathology analyses and quantification of samples were performed blinded by two pathologists.
For immunohistochemistry (IHC) staining, tissue sections were deparaffinized with xylene and rehydrated through a series of descending graded ethanol solutions following standard IHC methods. Tumor antigens were retrieved using Antigen Unmasking Solution (1:100 dilution) (Vector Laboratories, Burlingame, Ca, USA) followed by quenching of endogenous peroxidase with 3% v/v H2O2. Primary antibodies diluted in 10% fetal bovine serum (FBS) were incubated overnight. Sections were stained for monoclonal mouse anti β-catenin (1:50; BD Bioscience) monoclonal anti Ki-67 (1:1000; Vector Laboratories), monoclonal anti cytochrome oxidase subunit I (COX1) (1:500; Invitrogen) and polyclonal anti Nox1 (1:200; Bioss). A biotinylated secondary antibody and an avidin-biotin complex with horseradish peroxidase was used followed by the addition of the chromogen 3,3′-diaminobenxidine (DAB). Finally, slides were counterstained with hematoxylin and observed under a light microscope.
We used quantitative criteria to evaluate the Ki-67 positive staining by visualizing the slides by brown nuclear staining and assessed as the percent of Ki-67 positive cells per tissue sections. The percent of positive cells were calculated as follows: % positive cells = positive cells staining/ total cells × 100. The strength staining intensity scores for Ki-67 were semi-qualitatively calculated based on a scale of three (3) for high intensity, two (2) for medium intensity and one (1) for low intensity of Ki-67 positive cell staining. Four different random tissue fields per section were selected from each mouse and counted for Ki-67 positive cells staining and intensity.
The criteria used to evaluate β-catenin, COX1, and NOX1 staining in the colon and tumor sections were made following a semi-quantitative scoring system as previously described (20). For β-catenin analysis, we estimated the proportion of positive cells in the nucleus. For COX1 analysis we estimated membrane staining. For NOX1 analysis we estimated the cytoplasmic staining. The semi-quantitative score was made following parameters in a 1-4 scale of positive staining: 1 = <25% staining, 2 = 25-50% staining, 3 = 50-75% staining, 4 = >75% staining. Four different random tissue fields per section were selected from each mouse and counted for each marker positive cells staining and intensity. All the immunohistochemical scoring was performed as blinded studies.
RNA isolation and real-time reverse transcriptase-polymerase chain reaction (RT-PCR)
RNA was isolated from mouse colonic crypts using the RNeasy Mini Kit (Qiagen Inc., Valencia CA) according to the manufacturer's instructions. The RNA was spectrophotometrically qualified and quantified at 260 nm and 280 nm on a NanoDrop 2000 Spectrophotometer (Thermo Scientific, Wilmington, DE). RNA (75ng) was reversed-transcribed using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) following the manufacturer's instructions. Thermal cycling was performed using a Step One Plus Real-time PCR System (Applied Bio-systems, Carlsbad; CA, USA). The real-time PCR was performed by using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) in a total volume of 10μL, at 95°C for 15 seconds and 60°C for 1 minute. PCR efficiency was examined by serially diluting the template cDNA and the melting curve data were collected to check the PCR specificity. An internal loading control of GAPDH expression was used for Apex1 1 gene. Results were quantified using the ΔΔCT method. No PCR product was detected in control samples in which reverse transcriptase was omitted. The PCR primers (IDT, Coralville, Iowa) were: glyceraldehyde phosphodehydrogenase (GAPDH) antisense, 5′-AGTGGGAGTTGCTGTTGAAGTC-3′, and sense, 5′-CGTGCCGCCTGGAGAAAC-3′; Apex1 sense, 5′-TTGGTCTCTGGCTCCGA-3′ and antisense, 5′-GGTCTCTGGCTTCGTTGG -3”.
Statistical Analysis
To determine the statistical significance of the effect of AOM treatment in the number of nDNA and mtDNA lesions and mtDNA abundance and for histological analyses we made comparisons using one-way ANOVA and Tukey's honestly significant difference (HSD) test for post hoc analyses. The independent variables were the treatment groups and the dependent variables were the lesion number. For tumor incidence, size and multiplicity, we performed the Person's Chi-squared with Yates' continuity correction test. All statistical testing was performed at a pre-set alpha of 0.05.
Results
Effect of AOM on the levels of mtDNA damage and mtDNA abundance in colonic crypts from WT and Apex1+/- mice
AOM is an alkylating agent widely used for the induction of CRC in rodents (2,21). It is well known that this agent induces mutations in nuclear genes, however, little is known about the effects of AOM on mtDNA and how this may lead to CRC. To define the role of the BER enzyme APE1 on the repair of mtDNA and nDNA damage in WT and Apex1+/- mice, we studied the formation and repair of mtDNA and nDNA lesions induced by AOM using quantitative PCR (QPCR). Mice were administered intraperitoneally with a single dose of AOM (10 mg/kg of body weight) and DNA was isolated from colonic crypts 24, 48, and 72 hours after treatment. In WT mice we observe a significant 3.3-fold increase in mtDNA lesions 48 hours after AOM treatment, however, the number of lesions was reduced to control levels 72 hours after treatment (Figure 1A). Similarly, in Apex1+/- mice there was a 2.4-fold increase in mtDNA lesions 48 hours after treatment but, contrary to WT mice, mtDNA lesions persisted after 72 hours of treatment (Figure 1B).
Figure 1. Detection of DNA damage and APE1 mRNA levels induced by AOM.
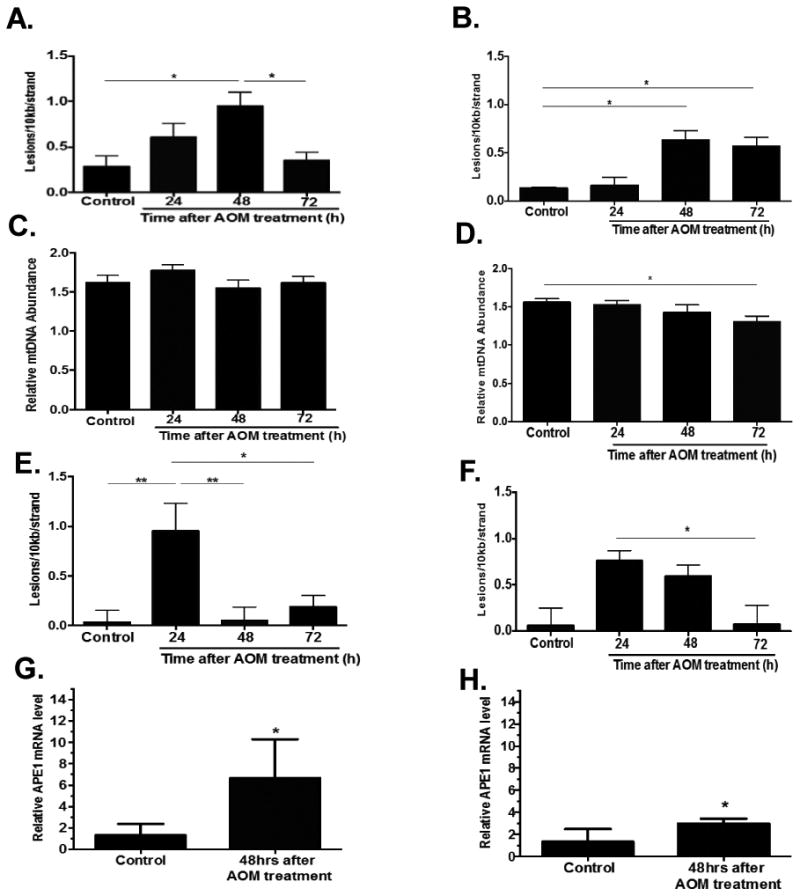
Total DNA from colonic crypts was isolated 24, 48, and 72 hrs after AOM treatment from WT and Apex1+/- mice and analyzed for mtDNA damage, mtDNA abundance, and nDNA damage by QPCR. (A) Frequency of mtDNA lesions in WT mice. (B) Frequency of mtDNA lesions in Apex1+/- mice. C) Relative mtDNA abundance in WT. (D) Relative mtDNA abundance in Apex1+/- mice. (E) Frequency of nDNA lesions in WT mice. (F) Frequency of nDNA lesions in Apex1+/- mice. Results are expressed as mean ± SEM values for 3 QPCRs performed in triplicate. (G) Relative APE1 mRNA levels in WT mice 48 hours after AOM treatment. (H) Relative APE1 mRNA levels in Apex1+/- mice 48 hours after AOM treatment. WT and Apex1+/- mice: N=8 for Saline treated mice; N=9 mice 24 hr AOM treatment; N=9 mice 48 hr AOM treatment; N=8 mice, 72 hr AOM treatment. Asterisks (*) denote statistical significance (One-way ANOVA, *P<0.05, **P<0.01).
Besides qualitative changes in mtDNA (i.e. DNA lesions and mutations) quantitative changes such as mtDNA copy number have been proposed as key determinant factors in mitochondrial-related pathogenesis (22). We evaluated whether AOM treatment could exert changes in mtDNA abundance in the colonic crypts using QPCR. We observed that WT mice show no significant changes in mtDNA abundance 24, 48 and 72 hours after AOM treatment (Figure 1C). However, we found a statistically significant 16% decrease in the abundance of mtDNA molecules in Apex1+/- mice 72 hours after AOM treatment (Figure 1D).
Repair kinetics of AOM-induced nuclear DNA lesions in Apex1+/- mice colonic crypts
We sought to analyze the levels of AOM-induced damage to the nuclear genome in colonic crypts from WT and Apex1+/- mice. We found that WT mice treated with AOM exhibited a significant 27-fold increase in the number of lesions 24 hours after treatment. The lesion number was significantly reduced to levels similar to WT control 48 hours after treatment (Figure 1E). Similarly, in Apex1+/- mice, lesion number peaked 24 hours after treatment (a 12.8-fold increase over saline treated mice). In contrast to WT animals, nDNA lesions remained elevated in Apex1+/- mice 48 hours after treatment and returned to control levels 72 hours after treatment (Figure 1F).
APE1 mRNA levels after AOM treatment
To determine whether AOM treatment results in changes in APE1 mRNA levels, we isolated RNA from colonic crypts and performed RT-PCR analysis. We chose to analyze samples 48 hours after AOM treatment since at this time point, lesions in both nDNA and mtDNA were still present (except in nDNA from WT mice). We observe that in WT animals, there is a statistically significant 5.1-fold increase in APE1 mRNA levels 48 hours after AOM treatment. In contrast, in colonic crypts from Apex1+/- mice there was only a 2.2-fold increase.
Effect of AOM treatment in tumor burden
To analyze the contribution of mtDNA damage in the induction of CRC we exposed mice to four 10 mg/kg AOM doses (once per week) as described previously (15). Six months after the first AOM treatment the number of mice bearing tumors (tumor incidence), tumor number and tumor size was determined. We observed that 65% of Apex1+/- mice exhibited tumors, all in the distal part of the colon compared to 55% of WT mice (Table 1). Although the percent of Apex1+/- mice exhibiting tumors is higher than in WT mice, this difference was not statistically significant. Similarly, tumor multiplicity and size were not statistically significant between WT and Apex1+/- mice.
Table 1.
Incidence, multiplicity and size of AOM-induced tumors in WT and Apex1+/- mice.
| Genotype | Incidence (%) | Multiplicity | Size (mm3) |
|---|---|---|---|
| Wild Type | 55 | 1.36 ± 0.28 | 24.98 ± 11.03 |
| Apex1+/- | 65 | 1.54 ± 0.21 | 21.99 ± 5.32 |
No statistically significant differences were observed between genotypes as determined by Person's Chi-squared with Yates' continuity correction. N=18 for WT mice and n=17 for Apex1+/- mice.
Immunohistochemical characteristics of AOM-induced tumors
While normal colon tissue from Apex1+/- mice show no significant histopathological changes, tumors from both WT and Apex1+/- AOM-treated animals show statistically higher values in all the parameters examined (mean number of nuclear/cytoplasmic ratio, epithelial stratification, nuclear dispolarity, goblet depletion and structural abnormalities) when compared to normal colon (Table 2). Interestingly, a statistically significant increase in epithelial stratification was observed in tumors from Apex1+/- animals compared to tumors from WT mice. This increase in epithelial stratification describes the occurring changes in the thickness of the abnormal epithelium characteristics of dysplasia.
Table 2.
Histopathological analysis of AOM-induced tumors in WT and Apex1+/- mice.
| Parameters | |||||
|---|---|---|---|---|---|
| Groups | Nuclear/cytoplasmic ratio | Epithelial stratification | Nuclear dispolarity | Goblet depletion | Structural abnormality |
| WT control tissue | 0±0.10 | 0±0.13 | 0±0.16 | 0.17±0.10 | 0±0.10 |
| WT tumor | 1.8±0.10a | 1.6±0.13a,b | 1.6±0.16a | 2.0±0.10a | 1.8±0.10a |
| Apex1+/- control tissue | 0±0 | 0±0 | 0±0 | 0±0 | 0±0 |
| Apex1+/- tumor | 2.0±0.09a | 2.0±0.12 a | 1.8±0.16a | 2.0±0.09a | 2.0±0.09a |
The grade of histological abnormality was scored using the following five parameters: a) nuclear/cytoplasmic ratio(<25%:0, 25-50%:1, >50%:2; b) epithelial stratification (none: 0,mild: 1,severe: 2); c) nuclear dispolarity (none: 0, mild: 1, severe: 2); d) goblet depletion (null to mild: 0, moderate: 1, severe: 2); e) structural abnormality (none: 0, mild: 1,severe: 2). Values represented mean ± SEM. One-way ANOVA,
P<0.05 compared to WT control tissue group;
P<0.05 compared to Apex1+/- tumor group.
We examined the expression of the proliferation marker Ki-67 in AOM-induced tumors from WT and Apex1+/- mice. Colon from saline treated WT mice shows 11.7% of Ki67 positive cells, whereas tumors from WT mice show 25.3% (a 2.2 fold increase) (Figures 2A, B and C). Colon from saline treated Apex1+/- mice show 14.5% of Ki67 positive cells whereas tumors from Apex1+/- mice show 34.3% (a 2.4-fold increase). A comparison in the number of Ki67 positive cells between tumors from WT and Apex1+/- mice reveals a statistically significant 1.3-fold increase in the Apex1+/- animals compared to WT. Moreover, determination of the intensity of Ki-67 staining showed that tumors from Apex1+/- mice exhibit a statistically significant 1.6 fold increase compared to WT tumor (Figure 2A, B and D).
Figure 2. Immunohistochemical detection of Ki-67 and β-catenin in AOM-induced tumors from WT and Apex1+/- mice.
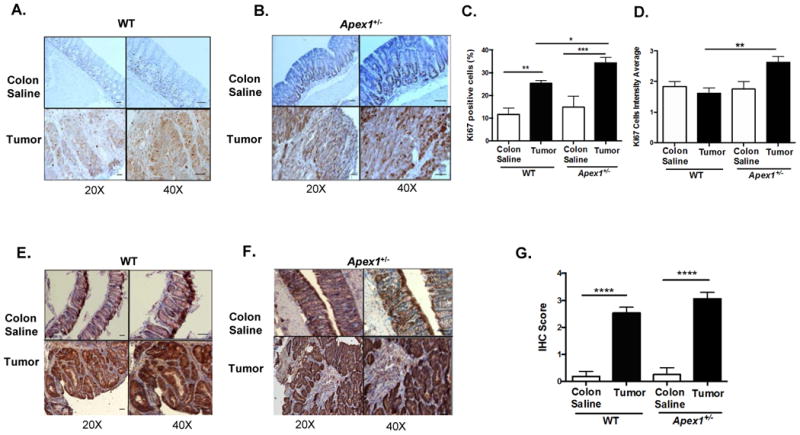
(A) and (B) Representative pictures of Ki-67 staining in colonic and tumor tissues from WT and Apex1+/- mice, respectively. Brown staining represents positive cells. (C) and (D) Quantification of Ki-67 positive cells and Ki-67 staining intensity, respectively. (E) and (F) Representative pictures of β-catenin staining in tumor and colonic tissues from WT and Apex1+/- mice, respectively. Brown nuclear staining represent positive cells. (G) Quantification of β-catenin staining in colonic and tumor tissues. Results are expressed as mean ± SEM values. The parameters for IHC score were as follows: 1 = <25% staining; 2 = 25-50% staining; 3 = 50-75% staining; 4 = >75% staining. N=4 WT colon saline and N=2 Apex1+/- colon saline, N=4 AOM-induced tumors from both WT and Apex1+/- mice. Scale bars represent 20 μm and 50 μm at 20 × and 40 × magnification, respectively. Asterisks (*) denote statistical significance (One-way ANOVA, *P<0.05; **P<0.01; ***p<0.001; ****P<0.0001).
We analyzed the expression of β-catenin in AOM-induced tumors and observe a 13.1 fold increase in the nuclear staining of this marker in tumors obtained from WT mice compared to colon from saline treated animals (Figure 2E and G). Similarly, AOM-induced tumors obtained from Apex1+/- mice exhibit a 12.4 fold increase in β-catenin nuclear staining compared to colon from saline treated Apex1+/- mice (Figures 2F and G). No significant differences were observed between β-catenin expression in AOM-induced tumors from WT and Apex1+/- mice.
Reduced expression in colonic crypts of the mitochondrial encoded COX1 have been associated to increased CRC risk (23). AOM-induced tumors from WT show a 39% reduction in COX1 staining when compared to colon from saline treated WT mice (Figure 3A and C). Similarly, there is 29% reduction in COX1 staining in Apex1+/- mice tumors when compared to colon from saline treated Apex1+/- mice (Figure 3B and C). Interestingly, a comparison between AOM-induced tumors from WT and Apex1+/- mice reveal that tumors from the Apex1+/- mice exhibit a 17% decrease in COX1 staining. Colon from saline treated Apex1+/- mice shows a 28% reduction in COX1 staining when compared to colon from saline treated control WT animals.
Figure 3. Immunohistochemical detection of cytochrome C subunit I in AOM-induced tumors from WT and Apex1+/- mice.
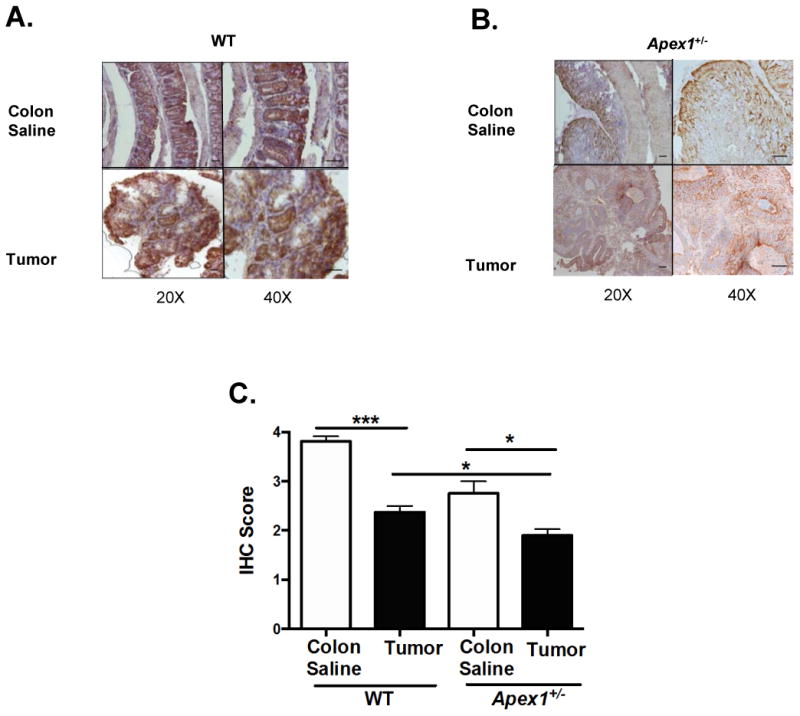
(A) and (B) Representative pictures of cytochrome C subunit I staining in tumor and colonic tissues from WT and Apex1+/- mice. (C) Quantification of cytochrome C subunit I staining in tumor tissues. Results are expressed as mean ± SEM values. The parameters for IHC score were as follows: 1 = <25% staining; 2 = 25-50% staining; 3 = 50-75% staining; 4 = >75% staining. N=4 WT colon saline and N=2 Apex1+/- colon saline. N=6 for AOM-induced tumors from both WT and Apex1+/- mice. Scale bars represent 20 μm and 50 μm at 20 × and 40 × magnification, respectively. Asterisks (*) denote statistical significance (One-way ANOVA, * p<0.05), *** p<0.001).
Damage to mtDNA, nDNA and mtDNA abundance in tumors from AOM-treated mice
We examined tumor tissues to determine relative abundance of mtDNA molecules, mtDNA damage, and nDNA damage. We observed that tumors from WT and Apex1+/- show a 56% and 44% reduction in mtDNA abundance respectively, as compared with colon from their respective saline treated controls (Figure 4A). No statistically significant differences were observed in mtDNA abundance between AOM-induced tumors from WT and Apex1+/- mice.
Figure 4. Analysis of changes in mtDNA and nDNA in AOM-induced tumors using QPCR.
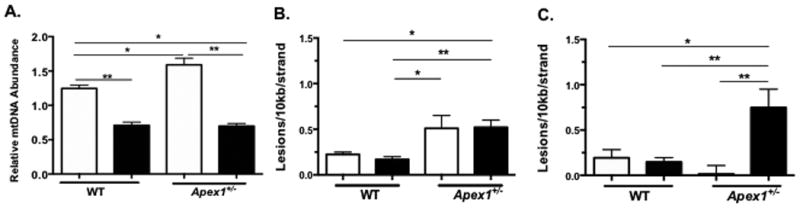
(A) AOM-induced changes in mtDNA abundance in tumors from WT and Apex1+/- mice. (B) Mitochondrial DNA lesions in AOM-induced tumors from WT and Apex1+/- mice. (C) Nuclear DNA lesions in AOM-induced tumors from WT and Apex1+/- mice. Open histograms represent normal tissue and closed histograms represent tumor tissue. Results are expressed as mean ± SEM values for 3 QPCRs performed in triplicate. N=6 for saline treated mice; N=8 mice for AOM treated mice. Asterisks (*) denote statistical significance (One-way ANOVA, * P<0.05; ** P<0.01; *** P<0.001).
When we measured the frequency of mtDNA lesions, we found no significant changes in the levels of mtDNA lesions in tumors from WT mice as compared to colonic tissue from saline treated WT mice (Figure 4B). Similarly, no increase in the frequency of mtDNA lesions is observed in tumors from Apex1+/- mice, compared to colonic tissue from saline treated Apex1+/- mice (Figure 4B). However, tumors from Apex1+/- mice show a statistically significant 2.1-fold increase in mtDNA damage compared to tumors from WT mice.
We also measured nDNA damage and found no differences in AOM-induced tumors from WT mice as compared to colonic tissue from saline treated WT animals. In contrast, we observe a significant 47.5-fold increase in the number of nDNA lesions in tumors from Apex1+/- mice as compared to colonic tissue from saline treated Apex1+/- mice (Figure 4C). Taken together these results indicate that AOM-induced tumors show mtDNA depletion and that tumors from Apex1+/- mice exhibit increased burden of nDNA damage.
Expression of NADPH oxidase 1 (NOX1) in tumors from WT and Apex1+/- mice
To examine if damage to the nDNA (Figure 4C) might be caused by other endogenous sources of ROS besides mitochondria, we turned our attention to NADPH oxidases. NADPH oxidases have been proposed as an important contributors of ROS in cancer (24). To test the hypothesis that nDNA damage was due to the action of NADPH oxidases, we performed IHC analysis to detect the expression of NOX1, which has a perinuclear location and has been suggested to play a role in ROS production in colon cancer (25). We observe abundant expression of NOX1 in normal colonic tissue from both WT and Apex1+/- mice. Moreover, the expression of NOX1 remains elevated in AOM-induced tumors in both WT and Apex1+/- mice (Figure 5) with approximately 50-75% of positive cell staining. However, no statistically significant differences are observed in NOX1 expression between tumors from WT and Apex1+/- mice.
Figure 5. Immunohistochemical detection of NADPH oxidase subunit I in AOM-induced tumors from WT and Apex1+/- mice.
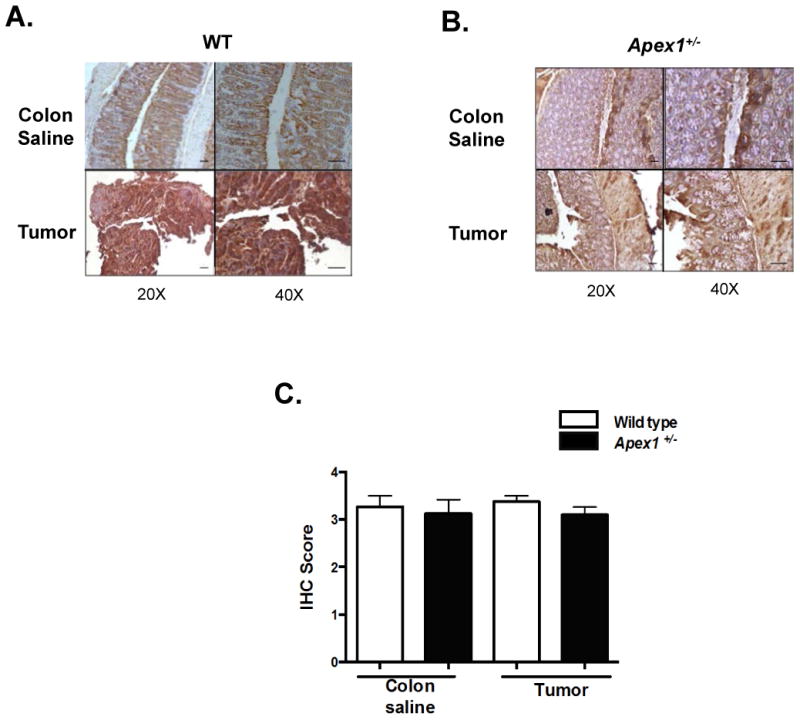
(A) and (B) Representative pictures of NADPH oxidase subunit I staining in tumor and saline colonic tissues from WT and Apex1+/- mice, respectively. Brown perinuclear staining represent positive cells. (C) Quantification of NADPH oxidase subunit I staining in tumor and saline colonic tissues. Results are expressed as mean ± SEM values. The parameters for IHC score were as follows: 1 = <25% staining; 2 = 25-50% staining; 3 = 50-75% staining; 4 = >75% staining. N=4 WT colon saline and N=4 Apex1+/- colon saline. N=4 for AOM-induced tumors from both WT and Apex1+/- mice. Scale bars represent 20 μm and 50 μm at 20 × and 40 × magnification, respectively.
Discussion
Our results show that treatment with a single dose of AOM leads to increased mtDNA damage in both WT and Apex1+/- mice, demonstrating that AOM also targets mitochondria (Figure 1A and 1B). WT mice exhibit efficient repair of AOM-induced mtDNA lesions, whereas Apex1+/- mice show delayed repair kinetics. These results indicate that mice haploinsufficient for APE1 (and therefore, their BER capacity is low), have reduced repair kinetics of alkylation damage (or an intermediate BER lesion such as abasic sites) in the mitochondrial genome. In addition to deficient mtDNA damage, we observe that colonic crypts from Apex1+/- mice show decrease mtDNA abundance 72 hours after AOM treatment (Figure 1D). No changes in mtDNA abundance were observed in colonic crypts from WT animals (Figure 1C), which is in agreement with previous results showing that treatment of BER competent mouse embryonic fibroblasts with the alkylating agent methyl methanesulfonate did not lead to mtDNA depletion (26). These results indicate that deficient mtDNA repair may lead to decreased mtDNA abundance. Persistent mtDNA damage and/or decreased mtDNA abundance may result in deficient mitochondrial bioenergetics and energy metabolism, thus, contributing to carcinogenesis (27). This idea is supported by experiments performed in animals carrying simultaneous heterozygous mutations in Tfam (Tfam+/-) and Apc (ApcMin/+), which showed increased tumorigenesis indicating that the mtDNA depletion phenotype observed in Tfam+/- mice modifies the tumorigenic phenotype of ApcMin/+ mice (28).
In our study, both WT and Apex1+/- mice exhibit faster kinetics of nDNA damage compared to mtDNA since the peak of nDNA lesions occurred 24 hours after AOM treatment (Figure 1E and 1F) vs 48 hours in the mtDNA (Figure 1A vs 1B). This faster DNA repair kinetics in the nucleus is consistent with studies measuring O6-MeG, an alkylating lesion which is completely repaired 48 hours after AOM treatment but it is not detected by the QPCR assay employed in our study (29). We speculate that this slower mtDNA damage kinetics could be due to a slower AOM distribution inside the mitochondria as compared to the nucleus. In addition to the fast kinetics of AOM-induced nDNA damage there was fast and efficient repair kinetics in colonic crypts from WT mice (Figure 1E). This could be due to AOM-induced up regulation of the Apex1 gene in the distal colon as part of a DNA damage response (30). Indeed, our RT-PCR analyses of APE1 mRNA expression show that there is a strong AOM-dependent induction in APE1 mRNA levels in colonic crypts from WT mice (Figure 1G). This DNA damage response response to AOM is blunt in colonic crypts from Apex1+/- mice (Figure 1H). In addition, efficient DNA repair in the nucleus can result from the overlapping DNA repair pathways (BER, nucleotide excision repair and recombination) that can act on the repair of abasic sites, an intermediate during the repair of AOM-induced lesions (31-33). Nevertheless, APE1 haploinsuffiency results in delayed repair of both AOM-induced nDNA and mtDNA lesions. The partial repair activity observed in the Apex1+/- mice could be attributed to the functional Apex1 allele in these animals. Altogether these results suggest that the APE1 haploinsufficiency sensitizes both mtDNA and nDNA to alkylating agents.
To analyze how deficient BER may impact the development of CRC, we induced CRC with multiple AOM doses as previously reported (15). No major differences between AOM-induced tumors from WT and Apex1+/- mice were observed. Although tumor incidence was higher in the Apex1+/- mice (65% vs 55%), this difference was not statistically significant (Table 1). An important consideration is the genetic background of the animals that were used in this study. We conducted our studies in the C57BL/6 genetic background, which is not as sensitive as the A/J or SWR/J backgrounds (15). Thus it is possible that a more permissive genetic background is needed in order to detect statistically significant differences in tumor formation between WT and Apex1+/- mice.
When we analyzed the histological characteristics of colon and tumor tissues in WT and Apex1+/- mice we observe that tumors of Apex1+/- mice present a higher structural abnormality score and that these tumors were less differentiated than WT tumor tissues (Table 2). Moreover, we observe that AOM-induced tumors from Apex1+/- mice present increased number and staining intensity of Ki-67 positive cells as compared to tumors from WT mice (Figure 2A-C). This observation indicates that AOM-induced tumors in Apex1+/- mice are poorly differentiated and have a higher risk of becoming metastatic, particularly to liver tissue (34–36). To answer this question further studies are needed in which tumors are allowed to progress into the metastatic stage.
The role of Wnt/β-catenin signaling pathway in CRC is well documented (37). Our studies show that AOM-induced tumors from both WT and Apex1+/- mice exhibit increased levels of β-catenin staining confirming that the nuclear DNA paradigm for CRC is prominent in the AOM-induced CRC model. However, it is interesting to note that even though the β-catenin levels are similar between tumors from WT and Apex1+/- mice, tumors from Apex1+/- mice exhibit increased expression of the cell proliferation marker, Ki-67. These results indicate that, besides the well-established effects mediated by the Wnt/β-catenin, other factors could be influencing the expression of cell proliferation markers such as Ki-67.
Studies have shown that mitochondrial dysfunction is present in cancer cells and contribute to the process of carcinogenesis (38). Mitochondrial dysfunction could result from mutations in genes encoding proteins that participate in the electron transport chain. For example, mutations in the mitochondria encoded COX1 results in reduced cytochrome c oxidase activity, decreased mitochondrial respiration and increased apoptotic resistance, processes that contribute to CRC (23,39,40). We analyzed the immunohistochemical expression of COX1 in AOM-induced tumors from WT and Apex1+/- mice and observe that there is significant loss of COX1 expression when compared to their respective control colon tissue (Figure 3). Furthermore, the loss of the COX1 expression in AOM-induced tumors from Apex1+/- mice is more pronounced than in WT mice. Even control saline treated tissue from Apex1+/- mice shows a significant reduction in COX1 when compared to control saline tissue from WT animals. These results indicate that APE1 is required for the prevention of mtDNA mutations in COX1 and that reduced APE1 levels leads to increase susceptibility to AOM-induced mutations in the mtDNA. The significance of these observations is that COX1 mutations have been shown to result in increased ROS generation and increased tumor growth potential in a prostate cancer model (41). This supports the hypothesis that mtDNA mutations contribute to CRC by generating ROS that could generate a positive cell growth signal and/or that can inflict further damage to the mitochondrial and the nuclear genome, thus favoring tumorigenesis (42). In support of this notion, ApcMin/+ mice carrying a transgene which targets catalase to mitochondria results in decrease intestinal polyposis (28). Although the results presented in this study point to a positive correlation between mtDNA damage/repair and tumorigenesis, further studies are needed to determine if mtDNA mutations cause CRC.
AOM-induced tumors had both expected and unexpected characteristics in terms of mtDNA abundance, mtDNA damage, and nDNA damage. We expected changes in mtDNA abundance in AOM-induced tumors from WT and Apex1+/- mice and we indeed observed significant mtDNA depletion in both cases (Figure 4A). However, there were no differences between the two genotypes, that is, AOM-induced tumors from WT and Apex1+/- mice display the same levels of mtDNA depletion. This finding is contrary to what we observed when we examined the acute effects of AOM treatment, in which we detect a small but statistically significant decrease in mtDNA abundance 72 hr after AOM treatment only in the Apex1+/- mice (Figure 1D). A plausible explanation for these results is that there is a threshold of mtDNA depletion that a cell (within the context of a tumor) can sustain, and tumors from both WT and Apex1+/- mice reached that threshold. A MGMT-dependent threshold in alkylation-induced CRC has been proposed but whether this is related to mtDNA repair is unknown (43). However, thresholds in mtDNA damage-induced apoptosis have been reported previously (44). We propose that a threshold in mtDNA abundance also exits in the cells that constitute a tumor. Variations in mtDNA copy number during cancer have been reported in numerous studies, however there are controversies in whether low or high mtDNA copy number is associated with increased or decrease risk of cancer (45). In our study we observe that mtDNA copy number decreases significantly in AOM-induced tumors from both WT and Apex1+/- mice. Decreased mtDNA abundance can be due to mutations in the D-loop region of the mitochondrial genome, which controls mtDNA replication; mutations in nuclear encoded genes involved in mtDNA replication, such as Polγ, and; deficiency in p53 signaling, which can lead to disturbed mitochondrial ROS homeostasis (45).
When we analyzed the levels of mtDNA damage in AOM-induced tumors we observe that mtDNA damage was not different when comparing normal tissue versus tumor tissue (Figure 4B). In contrast, we observe high nDNA damage in tumors from Apex1+/- mice versus tissue from saline treated mice (Figure 4C). These results indicate that reduced APE1 levels makes nDNA more sensitive to damage than mtDNA. The source of DNA damage in these tumors cannot be AOM itself since tumors were harvested five months after the last AOM dose. Thus, these DNA lesions must be the result of ROS generated within the tumor microenvironment. Mitochondria have been postulated to be the major source of ROS in the cell (27). Based on this observation one should expect increased DNA damage in both the mitochondrial and nuclear compartments in Apex1+/- mice. However, what we observe is a differential accumulation of DNA damage in nDNA. These results indicate that in AOM-induced tumors, in addition to mitochondrial generated ROS, non-mitochondrial sources of ROS that primarily inflict nDNA damage. Since APE1 also acts as a transcriptional coactivator of several transcription factors through a redox function, reduced APE1 levels in Apex1+/- mice may render nDNA more susceptible to the effects of ROS generated in the tumor microenvironment. Further studies are necessary to define the role of mitochondrial and non-mitochondrial ROS flux in AOM-induced tumors.
In recent years the family of NADPH oxidases have been postulated as a prominent endogenous source of ROS in cancerous tissues (24,46). Of particular interest is NOX1, which is expressed in colonic tissue and has functions in host defense by ROS-mediated bacterial killing and whose overexpression has been reported during CRC (47,48). In our study, AOM-induced tumors from WT and Apex1+/- mice are equally positive for NOX1 staining (Figure 5). These results indicate that this NAPDH oxidase isoform could be a non-mitochondrial source of ROS responsible for inflicting DNA damage in AOM-induced tumors. Since we observed extensive nDNA damage only in tumors from Apex1+/- mice, these results suggest that reduced levels of APE1, which results in deficient BER, sensitizes nDNA to the effects of ROS within the tumor microenvironment. Studies performed in an inflammation-induced CRC model show that another BER enzyme, alkyandenine glycosylase (AAG), prevents DNA damage and tumorigenesis (49). Two other DNA glycosylases with overlapping DNA damage specificity with AAG, ALKBH2 and ALKBH3, also protect against inflammation-induced CRC (50). Thus, our studies in APE1 haploinsufficient mice support the notion that CRC involves an inflammatory microenvironment that promotes DNA damage.
In summary, we show that diminished DNA repair capacity due to APE1 haploinsufficiency in Apex1+/- mice leads to increase nDNA and mtDNA lesions in colonic crypts after acute AOM treatment. In addition, we demonstrate that AOM-treated Apex1+/- mice show a time dependent decrease in levels of mtDNA abundance. Moreover, these changes in mtDNA abundance become more profound in AOM-induced tumors in both WT and Apex1+/- mice. AOM induced tumors from Apex1+/- mice display increased markers of dysplasia and cell proliferation such as epithelial stratification and Ki-67 staining compared to tumors from WT animals. Finally, we observe that levels of nDNA damage in AOM-induced tumors from Apex1+/- mice are higher compared to tumors from WT mice. A possible source of nDNA damage in these tumors could be the NADPH oxidase isoform, NOX1. Thus, Ape1 haploinsufficiency leads to increased sensitivity of nDNA to the deleterious effects of ROS produced within the tumor microenvironment. We conclude that AOM-induced CRC involves both nDNA and mtDNA damage and highlight the importance of BER in maintaining mtDNA integrity during carcinogenesis.
Acknowledgments
We are grateful of Jose G. Conde, MD MPH for advise on the statistical analyses of tumor burden data.
Financial Support: This research project was supported in part by grants from the National Institutes of Health R25GM061838, 2G12RR003051, U54CA096297 and 5SC3GM084759.
Footnotes
Conflict of Interest Statement: None declared.
References
- 1.Taketo MM. Mouse models of gastrointestinal tumors. Cancer Sci. 2006;97:355–61. doi: 10.1111/j.1349-7006.2006.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg DW, Giardina C, Tanaka T. Mouse models for the study of colon carcinogenesis. Carcinogenesis. 2009;30:183–96. doi: 10.1093/carcin/bgn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beranek TD. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res. 1990;231:11–30. doi: 10.1016/0027-5107(90)90173-2. [DOI] [PubMed] [Google Scholar]
- 4.Fahrer J, Kaina B. O6-methylguanine-DNA methyltransferase in the defense against N-nitroso compounds and colorectal cancer. Carcinogenesis. 2013;34:2435–42. doi: 10.1093/carcin/bgt275. [DOI] [PubMed] [Google Scholar]
- 5.Bauer NC, Corbett AH, Doetsch PW. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015;43:10083–101. doi: 10.1093/nar/gkv1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pines A, Perrone L, Bivi N, Romanello M, Damante G, Gulisano M, et al. Activation of APE1/Ref-1 is dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. Nucleic Acids Res. 2005;33:4379–94. doi: 10.1093/nar/gki751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chattopadhyay R, Wiederhold L, Szczesny B, Boldogh I, Hazra TK, Izumi T, et al. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006;34:2067–76. doi: 10.1093/nar/gkl177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ludwig DL, MacInnes MA, Takiguchi Y, Purtymun PE, Henrie M, Flannery M, et al. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat Res - DNA Repair. 1998;409:17–29. doi: 10.1016/s0921-8777(98)00039-1. [DOI] [PubMed] [Google Scholar]
- 9.Huamani J, McMahan CA, Herbert DC, Reddick R, McCarrey JR, MacInnes MI, et al. Spontaneous mutagenesis is enhanced in Apex heterozygous mice. Mol Cell Biol. 2004;24:8145–53. doi: 10.1128/MCB.24.18.8145-8153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vogel KS, Perez M, Momand JR, Acevedo-Torres K, Hildreth K, Garcia RA, et al. Age-related instability in spermatogenic cell nuclear and mitochondrial DNA obtained from Apex1 heterozygous mice. Mol Reprod Dev. 2011;78:906–19. doi: 10.1002/mrd.21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu M. Somatic mitochondrial DNA mutations in human cancers. Adv Clin Chem. 2012;57:99–138. doi: 10.1016/b978-0-12-394384-2.00004-8. [DOI] [PubMed] [Google Scholar]
- 12.Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, et al. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet. 1998;20:291–3. doi: 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- 13.Feng SHI, Xiong L, Ji Z, Cheng WEI, Yang H. Correlation between increased copy number of mitochondrial DNA and clinicopathological stage in colorectal cancer. Oncol Lett. 2011;2:899–903. doi: 10.3892/ol.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osch FHMV, AM V, LJ S, RW G, CC S, Engeland V, et al. Mitochondrial DNA copy number in colorectal cancer: between tissue comparisons, clinicopathological characteristics and survival. Carcinogenesis. 2015;36:1502–1510. doi: 10.1093/carcin/bgv151. [DOI] [PubMed] [Google Scholar]
- 15.Bissahoyo A, Pearsall RS, Hanlon K, Amann V, Hicks D, Godfrey VL, et al. Azoxymethane is a genetic background-dependent colorectal tumor initiator and promoter in mice: effects of dose, route, and diet. Toxicol Sci. 2005;88:340–5. doi: 10.1093/toxsci/kfi313. [DOI] [PubMed] [Google Scholar]
- 16.Ayala-Torres S, Chen Y, Svoboda T, Rosenblatt J, Van Houten B. Analysis of gene-specific DNA damage and repair using quantitative polymerase chain reaction. Methods. 2000;22:135–47. doi: 10.1006/meth.2000.1054. [DOI] [PubMed] [Google Scholar]
- 17.Acevedo-Torres K, Berríos L, Rosario N, Dufault V, Skatchkov S, Eaton MJ, et al. Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington's disease. DNA Repair. 2009;8:126–36. doi: 10.1016/j.dnarep.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Avwioro G. Histochemical uses of Haematoxylin - A review. Jpcs. 2011;1:24–34. [Google Scholar]
- 19.Alizadeh AM, Khaniki M, Azizian S, Mohaghgheghi MA, Sadeghizadeh M, Najafi F. Chemoprevention of azoxymethane-initiated colon cancer in rat by using a novel polymeric nanocarrier-curcumin. Eur J Pharmacol. 2012;689:226–32. doi: 10.1016/j.ejphar.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 20.Tan BL, Esa NM, Rahman HS, Hamzah H, Karim R. Brewers' rice induces apoptosis in azoxymethane-induced colon carcinogenesis in rats via suppression of cell proliferation and the Wnt signaling pathway. BMC Complement Altern Med. 2014;14:304. doi: 10.1186/1472-6882-14-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nandan MO, Yang VW. Genetic and Chemical Models of Colorectal Cancer in Mice. Curr Colorectal Cancer Rep. 2010;6:51–9. doi: 10.1007/s11888-010-0046-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tyynismaa H, Suomalainen A. Mouse models of mitochondrial DNA defects and their relevance for human disease. EMBO Rep. 2009;10:137–43. doi: 10.1038/embor.2008.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Payne CM, Holubec H, Bernstein C. Crypt-restricted loss and decreased protein expression of cytochrome c oxidase subunit I as potential hypothesis-driven biomarkers of colon cancer risk. Cancer Epidemiol Biomarkers Prev. 2005;14:2066–75. doi: 10.1158/1055-9965.EPI-05-0180. [DOI] [PubMed] [Google Scholar]
- 24.Fukuyama M, Rokutan K, Sano T, Miyake H, Shimada M, Tashiro S. Overexpression of a novel superoxide-producing enzyme, NADPH oxidase 1, in adenoma and well differentiated adenocarcinoma of the human colon. Cancer Lett. 2005;221:97–104. doi: 10.1016/j.canlet.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 25.O'Leary DP, Bhatt L, Woolley JF, Gough DR, Wang JH, Cotter TG, et al. TLR-4 Signalling Accelerates Colon Cancer Cell Adhesion via NF-kB Mediated Transcriptional Up-Regulation of Nox-1. PLoS One. 2012;7 doi: 10.1371/journal.pone.0044176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furda AM, Marrangoni AM, Lokshin A, Houten B, Van Van, Houten B. Oxidants and not alkylating agents induce rapid mtDNA loss and mitochondrial dysfunction. DNA Repair. 2012;11:1–9. doi: 10.1016/j.dnarep.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Moura MB, dos Santos LS, Van Houten B. Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environ Mol Mutagen. 2010;51:391–405. doi: 10.1002/em.20575. [DOI] [PubMed] [Google Scholar]
- 28.Woo DK, Green PD, Santos JH, D'Souza AD, Walther Z, Martin WD, et al. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC Min/+ mice. Am J Pathol Elsevier Inc. 2012;180:24–31. doi: 10.1016/j.ajpath.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nyskohus LS, Watson AJ, Margison GP, Le Leu RK, Kim SW, Lockett TJ, et al. Repair and removal of azoxymethane-induced O6-methylguanine in rat colon by O6-methylguanine DNA methyltransferase and apoptosis. Mutat Res - Genet Toxicol Environ Mutagen BV. 2013;758:80–6. doi: 10.1016/j.mrgentox.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 30.Kerr CA, Hines BM, Shaw JM, Dunne R, Bragg LM, Clarke J, et al. Genomic homeostasis is dysregulated in favour of apoptosis in the colonic epithelium of the azoxymethane treated rat. BMC Physiol BMC Physiology. 2013;13:2. doi: 10.1186/1472-6793-13-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swanson RL, Morey NJ, Doetsch PW, Jinks-Robertson S. Overlapping specificities of base excision repair, nucleotide excision repair, recombination, and translesion synthesis pathways for DNA base damage in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:2929–35. doi: 10.1128/mcb.19.4.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gellon L, Barbey R, Van der Kemp PA, Thomas D, Boiteux S. Synergism between base excision repair, mediated by the DNA glycosylases Ntg1 and Ntg2, and nucleotide excision repair in the removal of oxidatively damaged DNA bases in Saccharomyces cerevisiae. Mol Genet Genomics. 2001;265:1087–96. doi: 10.1007/s004380100507. [DOI] [PubMed] [Google Scholar]
- 33.Torres-Ramos C, Johnson RE, Prakash L, Prakash S. Evidence for the involvement of nucleotide excision repair in the removal of abasic sites in yeast. Mol Cell Biol. 2000;20:3522–8. doi: 10.1128/mcb.20.10.3522-3528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawakami M, Yamaguchi T, Takahashi K, Matsumoto H, Yasutome M, Horiguchi S, et al. Assessment of SMAD4, p53, and Ki-67 alterations as a predictor of liver metastasis in human colorectal cancer. Surg Today. 2010;40:245–50. doi: 10.1007/s00595-009-4028-3. [DOI] [PubMed] [Google Scholar]
- 35.Kitabatake T, Kojima K, Fukasawa M, Beppu T, Futagawa S. Correlation of thymidine phosphorylase staining and the Ki-67 labeling index to clinicopathologic factors and hepatic metastasis in patients with colorectal cancer. Surg Today. 2002;32:322–8. doi: 10.1007/s005950200046. [DOI] [PubMed] [Google Scholar]
- 36.Kuniyasu H, Oue N, Shigeishi H, Ito R, Kato Y, Yokozaki H, et al. Prospective study of Ki-67 labeling index in the mucosa adjacent to cancer as a marker for colorectal cancer metastasis. J Exp Clin Cancer Res. 2001;20:543–8. [PubMed] [Google Scholar]
- 37.Novellasdemunt L, Antas P, Li VSW. Targeting Wnt signaling in colorectal cancer. Am J Physiol - Cell Physiol. 2015;309:511–21. doi: 10.1152/ajpcell.00117.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wallace DC. Mitochondria and cancer. Nat Rev Nature Publishing Group. 2012;12:685–98. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gallardo Esther M, Loshuertos-Moreno R, Lopez C, Casqueiro M. m.6267G4A: A recurrent mutation in the human mitochondrial DNA that reduces cytochrome C oxidase activity and is associated with tumors. Hum Mutat. 2006;27:575–82. doi: 10.1002/humu.20338. [DOI] [PubMed] [Google Scholar]
- 40.Greaves LC, Preston SL, Tadrous PJ, Taylor RW, Barron MJ, Oukrif D, et al. Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. Proc Natl Acad Sci U S A. 2006;103:714–9. doi: 10.1073/pnas.0505903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A. 2005;102:719–24. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wallace DC, Fan W. The pathophysiology of mitochondrial disease as modeled in the mouse. Genes Dev. 2009:1714–36. doi: 10.1101/gad.1784909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fahrer J, Frisch J, Nagel G, Kraus A, Dörsam B, Thomas AD, et al. DNA repair by MGMT, but not AAG, causes a threshold in alkylation-induced colorectal carcinogenesis. Carcinogenesis. 2015;36:1235–44. doi: 10.1093/carcin/bgv114. [DOI] [PubMed] [Google Scholar]
- 44.Santos JH, Hunakova L, Chen Y, Bortner C, Van Houten B. Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. J Biol Chem. 2003;278:1728–34. doi: 10.1074/jbc.M208752200. [DOI] [PubMed] [Google Scholar]
- 45.Yu M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 2011;89:65–71. doi: 10.1016/j.lfs.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 46.Szanto I, Rubbia-Brandt L, Kiss P, Steger K, Banfi B, Kovari E, et al. Expression of NOXI, a superoxide-generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J Pathol. 2005;207:164–76. doi: 10.1002/path.1824. [DOI] [PubMed] [Google Scholar]
- 47.Krause KH. Tissue distribution and putative physiological function of NOX family NADPH oxidases. Jpn J Infect Dis. 2004;57:155–6. [PubMed] [Google Scholar]
- 48.Laurent E, McCoy IIIJW, Macina RA, Liu W, Cheng G, Robine S, et al. Nox1 is over-expressed in human colon cancers and correlates with activating mutations in K-Ras. Int J cancer. 2008;123:100–7. doi: 10.1002/ijc.23423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meira L, Bugni J, Green S. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516–25. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Calvo JA, Meira LB, Lee C-YI, Moroski-Erkul CA, Abolhassani N, Taghizadeh K, et al. DNA repair is indispensable for survival after acute inflammation. J Clin Invest. 2012;122:2680–9. doi: 10.1172/JCI63338. [DOI] [PMC free article] [PubMed] [Google Scholar]