

Abstract
Basal cell carcinoma (BCC) is the most common skin malignancy. Deregulated hedgehog signaling plays a central role in BCC development; therefore hedgehog inhibitors have been approved to treat locally advanced or metastatic BCC. However, the development of resistance to hedgehog inhibitors is the major challenge in effective treatment of this disease. Herein, we evaluated the efficacy of a natural agent silibinin to overcome resistance with hedgehog inhibitors (Sant-1 and GDC-0449) in BCC cells. Silibinin (25–100 μM) treatment for 48 hrs strongly inhibited growth and induced death in ASZ001, Sant-1 resistant (ASZ001-Sant-1) and GDC-0449 resistant (ASZ001-GDC-0449) BCC cells. Furthermore, colony forming ability of ASZ001, ASZ001-Sant-1 and ASZ001-GDC-0449 cells was completely inhibited by silibinin treatment. Molecular analysis showed that silibinin treatment decreased the level of phosphorylated-EGFR (Tyrosine-1173) and total EGFR in ASZ001-Sant-1 cells; key signaling molecules responsible for BCC resistance towards hedgehog inhibitors. Further, silibinin treatment decreased the phosphorylated-Akt (Serine-473), phosphorylated-ERK1/2 (Threonine 202/Tyrosine 204), cyclin D1 and Gli-1 level but increased the SUFU expression in ASZ001-Sant-1 resistant cells. Silibinin treatment of ASZ001-Sant-1 resistant cells also decreased bcl-2 but increased cleaved caspases 3 and PARP cleavage, suggesting induction of apoptosis. Together, these results support silibinin use to target hedgehog inhibitors resistant BCC cells.
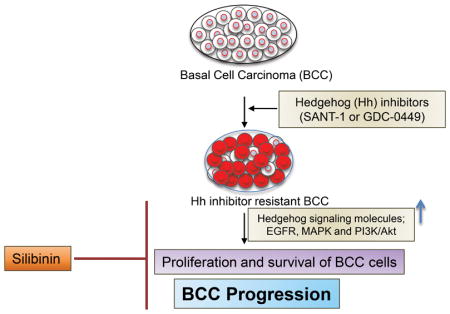
Graphical Abstract
In the present study, we evaluated the efficacy of a natural agent silibinin to overcome resistance against hedgehog inhibitors (Sant-1 or GDC-0449) in basal cell carcinoma (BCC) cells. The silibinin treatment strongly inhibited the cell growth and induced death in ASZ001, ASZ001-Sant-1 resistant and ASZ001-GDC-0449 resistant BCC cells. Colony forming ability of the hedgehog inhibitor resistant BCC cells was completely inhibited with silibinin treatment. Mechanistic studies revealed that silibinin inhibits EGFR-MAPK-Akt and hedgehog signaling in resistant BCC cells. Silibinin treatment also targeted the key cell death associated molecules in the resistant BCC cells.
INTRODUCTION
Basal cell carcinoma (BCC) is the most common form of skin malignancy with approximately 2.8 million new cases diagnosed each year in the United States (1). BCC arises in the skin’s basal cells that line the deepest layer of the epidermis. The disease is mostly slow growing except in patients with basal cell nevus syndrome (BCNS). These patients are genetically pre-disposed to the early development of BCC. Untreated BCC lesions can become aggressive and even metastasize to other organs. The incidence of BCC is more common in men than in women and tend to appear in patients after the age of 50 years (2). The higher prevalence of BCC is mainly attributed to exposure to ultraviolet (UV) radiation, aging population and exposure to environmental factors such as drinking arsenic contaminated water (3, 4). These exposures could cause genetic and epigenetic changes as well as aberrant activation of various mitogenic and pro-survival cellular signaling pathways leading to BCC development (5, 6).
Hedgehog (Hh) signaling plays an important role in embryonic development as well as adult tissue maintenance. Hedgehog signal transduction is initiated by binding of hedgehog ligand (Sonic, Indian or Desert) to its receptor Patched (PTCH1 or PTCH2). In the absence of hedgehog ligand, the PTCH receptor inhibits the activation of SMO and subsequent activation of downstream effector target molecules such as Gli-1 (glioma-associated oncogene homolog 1), a zinc finger transcription factor regulating the expression of genes associated with proliferation, angiogenesis, stemness and metastasis (7–9). The tumor suppressor SUFU (suppressor of fused) negatively regulates the hedgehog signaling through binding to c-terminal region of Gli (10). Deregulated activation of hedgehog signaling pathway through the loss of function mutation (PTCH1) and gain of function mutation (SMO) is the key mechanism of BCC development (11–15).
Considering the key role of hedgehog signaling in BCC development, several inhibitors of hedgehog signaling such as cyclopamine, GDC-0449 (Vismodegib), and Sant-1 which target SMO have been developed and tested for their therapeutic efficacy against BCC (16–18). However, BCC cells ultimately develop resistance against hedgehog inhibitors via activation of various signaling pathways such as EGFR, MAPK and Akt (19–22). Toxicity associated with these agents (loss of body weight, taste and hairs; muscle spasm, nausea, and fatigue) is generally mild to moderate etc., but can be chronic and persistent and are the main reason for therapy discontinuation (23, 24). For example, in the BCNS prevention study, 54% of patients on Vismodegib discontinued therapy because of adverse effects (25). Therefore, non-toxic agents are needed that could be useful to reduce the toxicity of hedgehog inhibitors and/or could target resistant BCC cells.
Silibinin is a non-toxic flavonoid (MW 482.84) isolated mainly from the seeds of Milk thistle plant (Silybum marianum) (26, 27). Silibinin has shown strong efficacy against several cancers including skin cancer (28). Recently, we reported that silibinin treatment inhibits growth and induces death in BCC cells both in vitro and in vivo (3). Additionally silibinin has been extensively studied for its protective efficacy against hepatotoxicity, nephrotoxicity, and cardiotoxicity caused by chemotherapeutic drugs and radiation damage (29–33). Results show that silibinin could effectively inhibit the growth of hedgehog inhibitor resistant BCC cells via targeting several proliferation and survival pathways.
MATERIALS AND METHODS
Cell culture and reagents
ASZ001 cells were received as a generous gift from Dr. Ervin Epstein (Children’s Hospital & Research Center Oakland) and cultured in M154F (Gibco, USA) media supplemented with 2% chelexed heat-inactivated FBS, 1% penicillin - streptomycin, and 0.05 M calcium chloride. Silibinin, dimethyl sulfoxide (DMSO) and trypan blue dye were purchased from Sigma-Aldrich (St. Louis, MO). Sant-1 was obtained from Tocris Biosciences (Bristol, UK); GDC-0449 was purchased from LC laboratories (Woburn, MA). The antibodies for phosphorylated-EGFR (Tyrosine-1173), EGFR, phosphorylated-Akt (Serine-473), phosphorylated-ERK1/2 (Threonine 202/Tyrosine 204), ERK1/2, cyclin D1 and HRP-conjugated secondary antibodies were purchased from Cell Signaling technology (Beverly, CA). Antibodies for Gli-1, SMO and SUFU were purchased from Abcam (Cambridge, UK). HRP-conjugated β-actin was purchased from Santa Cruz Biotechnology (CA, USA).
Generation of ASZ001 Sant-1 and GDC-0449 resistant cells
ASZ001 cells were initially treated either with hedgehog inhibitors Sant-1 (10 μM) or GDC-0449 (5 μM), and then regularly treated with increasing concentrations of Sant-1 or GDC-0449. Treatment was continued for 8–10 weeks until inhibitors had no or minimal effect on cell growth and death. The resistance of cells against Sant-1 and GDC-0449 was achieved at a concentration of 60 μM and 40 μM, respectively. Hereafter, Sant-1 resistant ASZ001 cells have been abbreviated as ASZ001-Sant-1; and GDC-0449 resistant ASZ001 cells have been abbreviated as ASZ001-GDC-0449.
Cell growth assay
1x105 Cells (ASZ001, ASZ001-Sant-1 and ASZ001-GDC-0449) were seeded per well in six-well plate under regular growth conditions. Next day, cells were treated with vehicle (DMSO), silibinin (25–100 μM) and/or Sant-1 (60 μM) and/or GDC-0449 (40 μM) for 48 hrs. At the end, cells were collected by brief trypsinization; and live and dead cells were counted using a hemocytometer by trypan blue dye exclusion method.
Clonogenic assay
ASZ001, ASZ001-Sant-1 and ASZ001-GDC-0449 cells were seeded in 6-well plates (0.5 x 103 cells/well) in regular growth medium. After 24 hrs of seeding, cells were treated with DMSO or silibinin (25–100 μM) and/or Sant-1 (30 and 60 μM) and/or GDC-0449 (20 and 40 μM). At the end of the 7th day, cells were washed with 1x PBS and fixed with 4% formalin for 20 min and stained with 0.1% crystal violet for 20 min. The plates were washed with distilled water and the numbers of colonies with greater than 50 cells were counted.
Immunoblotting
ASZ001 Sant-1 resistant cells were grown to 60% confluency under regular growth media. Cells were treated with DMSO or silibinin (100 μM) and/or Sant-1 (60 μM) and/or GDC-0449 (40 μM) for 48 hrs. At the end, cell lysates were prepared as previously described (3, 34), and protein concentration was determined by Bio-Rad DC protein assay kit (Manufacturer’s protocol). Equal protein per sample was resolved on Tris-glycine gels, and transferred onto nitrocellulose membranes. Cell lysates were probed with specific primary antibodies followed by appropriate peroxidase-conjugated secondary antibody and visualized by enhanced chemiluminescence detection system (GE healthcare). To ensure equal protein loading, membranes were stripped and re-probed with appropriate loading control. The bands were scanned with Adobe Photoshop 6.0 (Adobe Systems, San Jose, CA). The band intensity was analysed by using Image J (NCI, USA) tool and presented as fold change to that of their respective controls. The expression in each group was also normalized to their respective loading control.
Statistical analysis
SigmaStat software version 3.5 (Systat Software, Inc., Richmond, CA) was used for all statistical analyses. Quantitative data are presented as mean ± SEM. Statistical significance of difference between control and treatment groups was determined through one-way analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons and p<0.05 was considered significant.
RESULTS
Silibinin inhibits the growth of ASZ001, ASZ001-Sant-1 and ASZ001-GDC-0449 BCC cells
The efficacy of silibinin against the cell growth was evaluated in ASZ001, ASZ001-Sant-1 resistant and ASZ001-GDC-0449 resistant BCC cells with or without the treatment of Sant-1 or GDC-0449. In ASZ001 cells, total cell number was decreased by 13–71% with 50–100 μM doses of silibinin (Figure 1A, left panel). Treatment of ASZ001 cells with Sant-1 (60 μM) and GDC-0449 (40 μM) decreased the total cell number by 47% and 52%, respectively (Figure 1A, left panel). The combination of silibinin (25–100 μM) with Sant-1(60 μM) or GDC-0449 (40 μM) strongly decreased the total cell number, greater than any single agent treatment alone (Figure 1A, left panel). The inhibition of cell proliferation by silibinin in combination with Sant-1 or GDC-0449 was 41–66% and 29–67% respectively, compared with individual treatment. Similarly, silibinin (25–100 μM) treatment induced cell death in ASZ001 cells; but its effect was variable in combination (Figure 1A, right panel). There was significant increase in cell death in combination at 100 μM silibinin dose but a decrease or no effect at lower doses (25–75 μM) compared to Sant-1 or GDC-0449 alone (Figure 1A, right panel).
Figure 1.

Effect of silibinin treatment on cell growth in ASZ001, ASZ001 Sant-1 and ASZ001-GDC-0449 BCC cells with or without Sant-1 or GDC-0449 treatment. (A) Effect of silibinin (25–100 μM) treatment for 48 hrs, with or without Sant-1 (60 μM) or GDC-0449 (40 μM), on total cell number (left panel) and cell death (right panel) in ASZ001 cells. (B) Effect of silibinin (25–100 μM) treatment for 48 hrs, with or without Sant-1 (60 μM) or GDC-0449 (40 μM), on total cell number (left panel) and cell death (right panel) in ASZ001-Sant-1 cells. (C) Effect of silibinin (25–100 μM) treatment for 48 hrs, with or without Sant-1 (60 μM) or GDC-0449 (40 μM), on total cell number (left panel) and cell death (right panel) in ASZ001-GDC-0449 cells. Quantitative data are represented as Mean±SEM of three samples. # p≤0.05; $ p≤0.005; * p≤0.001 versus respective control.
In ASZ001-Sant-1 cells, silibinin treatment reduced total cell number in a dose-dependent manner and significantly increased the growth inhibitory effect of Sant-1 or GDC-0449, when used in combination (Figure 1B, left panel). The combination of silibinin (25–100 μM) with Sant-1 and GDC-0449 inhibited the total cell number by 23–65% and 27–57%, respectively, compared with individual inhibitor treatment group (Figure 1B, left panel). Sant-1 (60 μM) alone treatment caused only a slight decrease in total cell number confirming the resistance of these cells towards this hedgehog inhibitor. Importantly, ASZ001-Sant-1 cells were also resistant to GDC-0449 treatment, suggesting the development of cross-resistance. Silibinin treatment alone induced modest cell death at 100 μM in ASZ001-Sant-1 cells (26% compared to 12% in DMSO-treated control cells) but significantly increased the cell death when combined with Sant-1 or GDC-0449 (Figure 1B, right panel). The cell death was increased to 42% and 34% in combination treatment group (silibinin plus Sant-1 or silibinin plus GDC-0449) compared to 17% and 23% in Sant-1 or GDC-0449 alone treated group, respectively (Figure 1B, right panel).
Similarly, in ASZ001-GDC-0449 cells, silibinin treatment reduced the total cell number in a dose-dependent manner; and significantly increased the growth inhibitory effect of Sant-1 or GDC-0449 when used in combination (Figure 1C, left panel). The combination of silibinin (25–100 μM) with Sant-1 and GDC-0449 strongly inhibited the total cell number by 19–33% and 18–55% respectively, as compared with the individual treatment group (Figure 1C, left panel). In this assay, Sant-1 (60 μM) alone treatment reduced total cells by 35% and GDC-0449 (40 μM) alone treatment showed an increase in cell number suggesting that these cells are resistant to GDC-0449 but not to Sant-1. Further, silibinin alone treatment induced only modest cell death at 100 μM dose in ASZ001-GDC-0449 cells (21% compared to 10% in DMSO-treated control cells) but significantly increased the cell death when combined with Sant-1 or GDC-0449 (Figure 1C, right panel). Cell death was increased to 40% and 42% in combination treatment groups (silibinin plus Sant-1 or GDC-0449) compared to 13% and 19% of Sant-1 and GDC-0449 alone treated groups, respectively (Figure 1C, right panel). In these cells, even lower doses of silibinin (50 and 75 μM) were effective in increasing the cell death when combined with GDC-0449 (Figure 1C, right panel).
Overall, these results show silibinin inhibits cell growth in hedgehog inhibitor resistant ASZ001 cells similar to hedgehog inhibitor sensitive ASZ001 cells. Furthermore, silibinin combinations improved the efficacy of hedgehog inhibitors against both sensitive and resistant BCC cells, in terms of cell growth inhibition and induction of cell death.
Silibinin inhibits the clonogenicity of ASZ001, ASZ001-Sant-1 and ASZ001-GDC-0449 BCC cells
Next, the effect of silibinin on colony forming potential of ASZ001, ASZ001-Sant-1 and ASZ001-GDC-0449 cells was assessed. Silibinin (25–100 μM) treatment strongly inhibited (28–100%) the formation of colonies by ASZ001 cells. The combination of silibinin (25–100 μM) with Sant-1 30 μM and 60 μM also inhibited the clonogenic potential by 33–100% and 51–100%, respectively, as compared to 33% and 42% for Sant-1 30 μM and 60 μM alone in ASZ001 cells. Similarly, silibinin (25–100 μM) in combination with GDC-0449 20 μM and 40 μM inhibited the colony formation by 72–100% and 95–100%, respectively as compared to 63% and 65% for GDC-0449 20 μM and 40 μM alone in ASZ001 cells (Figure 2A).
Figure 2.

Effect of silibinin treatment on the clonogenicity of ASZ001, ASZ001-Sant-1 and ASZ001-GDC-0449 BCC cells with or without Sant-1 or GDC-0449 treatment. In each case, 500 cells per well (six-well plate) were seeded and cultured for 24 hrs in complete medium. Next day, cells were treated with indicated doses of silibinin and/or Sant-1 and/or GDC-0449 for a week. After which plates were washed with cold 1X PBS, fixed, stained and scored for colonies as discussed in ‘materials and methods’. Average number of colonies is present for (A) ASZ001 cells, (B) ASZ001 Sant-1 resistant cells and (C) ASZ001 GDC-0449 resistant cells. Quantitative data are represented as Mean±SEM of three samples. # p≤0.05; $ p≤0.005; * p≤0.001 versus respective control.
In ASZ001-Sant-1 cells, silibinin treatment inhibited the clonogenicity in a dose-dependent manner with 10% to near complete inhibition (Figure 2B). Further, silibinin (25–100 μM) treatment in combination with Sant-1 or GDC-0449 sensitized the Sant-1 resistant cells to almost complete inhibition of formation of colonies. Similarly, silibinin treatment alone or in combination with Sant-1 or GDC-0449 reduced the colony forming ability of ASZ001-GDC-0449 cells. Importantly, the strong clonogenicity inhibition in combination studies was mainly driven by silibinin treatment in both ASZ001-Sant-1 and ASZ001-GDC-0449 cells.
These results suggested that silibinin treatment alone is quite potent in inhibiting the clonogenicity of ASZ001, ASZ001-Sant-1 and ASZ001-GDC-0449 BCC cells.
Silibinin inhibits multiple signaling pathways in ASZ001-Sant-1 BCC cells
Recent studies have identified the role of several signaling pathways in the development of resistance in BCC cells towards hedgehog inhibitors (19, 20, 35, 36); therefore, next we characterized the effect of silibinin on several such important molecules in ASZ001-Sant-1 cells. As shown in figure 3A, silibinin (100 μM) treatment either alone or in combination with Sant-1 (60 μM) or GDC-0449 (40 μM) reduced the level of activating phosphorylation of EGFR (Tyrosine-1173). Further, silibinin treatment alone as well as in combination with both inhibitors strongly decreased the total EGFR. Similarly, silibinin treatment strongly reduced the phosphorylated Akt (Ser-473) either alone or in combination; but neither had an effect or only modest effect on phosphorylated and total ERK1/2 (Figure 3A). Further, silibinin treatment also decreased the level of proliferation marker cyclin D1 in combination with Sant-1 (Figure 3A).
Figure 3.

Effect of silibinin treatment on various signaling molecules in ASZ001-Sant-1 cells. (A–B) ASZ001-Sant-1 resistant cells were grown up to 60% confluency and treated with indicated doses of silibinin and/or Sant-1 and/or GDC-0449 for 48 hrs. Total cell lysate was prepared and analyzed by immunoblotting for indicated proteins. β-actin is presented as loading control. Densitometry value of each band is also presented.
Next, we examined whether silibinin could target resistant BCC cells as well as improve the efficacy of hedgehog inhibitors (Sant-1 and GDC-0449) via inhibiting the hedgehog downstream signaling molecules. As shown in figure 3B, silibinin treatment decreased the Gli-1 expression both alone and in combination with Sant-1or GDC-0449 in ASZ001-Sant-1 cells. Silibinin treatment showed only a slight decrease in SMO expression that was induced by Sant-1or GDC-0449 in ASZ001-Sant-1 cells. Importantly, silibinin treatment increased the expression of SUFU in combination with GDC-0449, suggesting an inhibitory effect of silibinin on hedgehog signaling.
Silibinin treatment activates apoptosis-related pathway in ASZ001-Sant-1 BCC cells
Since most of the drug-resistant cells become resistant to induction of cell death, we next characterized the effect of silibinin treatment on apoptosis pathway related molecules in ASZ001-Sant-1 cells. As shown in figure 4A, silibinin treatment strongly decreased the level of anti-apoptotic molecule bcl-2 and slightly affected the pro-apoptotic molecules Bax. It is known that a higher ratio of bax/bcl-2, as caused by silibinin treatment, induces apoptotic death in cells (37). Further, silibinin treatment either alone or in combination (Sant-1 or GDC-0449) strongly increased the level of cleaved caspase 3 which is involved in apoptotic cell death. Silibinin treatment, alone or in combination (Sant-1 or GDC-0449), caused PARP cleavage, an established biomarker for apoptosis. Together, these results suggested that silibinin treatment could activate apoptotic-death machinery in ASZ001-Sant-1 cells.
Figure 4.

Effect of silibinin treatment on apoptosis-related signaling molecules in ASZ001-Sant-1 cells. (A) ASZ001-Sant-1 resistant cells were grown up to 60% confluency and treated with indicated doses of silibinin and/or Sant-1 and/or GDC-0449 for 48 hrs. Total cell lysates were prepared and analyzed by immunoblotting for indicated proteins. β-actin is presented as loading control. Densitometry value of each band is also presented. (B) Proposed Model: Silibinin treatment targets multiple signaling pathways responsible for resistance towards hedgehog inhibitors; thereby, inhibits proliferation and survival and induces cell death in BCC cells.
DISCUSSION
BCC is a major health problem around the globe with increasing incidence due to prolonged exposure to sun, ultraviolet radiation exposure, aging population, arsenic exposure, immune suppression and lack of effective preventive strategies (3, 38). The enormity of this disease could be gauged from the fact that annual BCC incidences alone are higher than all other cancer incidences combined. Ultraviolet radiation (UVR) is the major etiological risk factor in the development of skin cancer including BCC, and accounts for about 50–90% of total reported cases of skin malignancy (39). The UV radiation which reaches to the surface of earth mainly comprise of UVA (320–400 nm) and UVB (280–320 nm), and UVB predominantly causes chronic skin damage and is considered as complete carcinogen inducing initiation, promotion and progression (40, 41). UVB is absorbed in skin keratinocytes resulting in DNA lesions including cyclobutane pyrimidine dimers (CPD) and 6-4 photoproducts which are major contributors to skin carcinogenesis (42). UVB also generates reactive oxygen species (ROS) in skin and induces oxidative DNA damage such as 8-hydroxy -2′-deoxyguanosine (43, 44). The absorption of the UV radiation by nucleotides causes the formation of pyrimidine dimers which leads to mutations in the TP53 and PTCH1 genes (45, 46). In 40% cases of BCC, alterations in PTCH and SMOH genes are corresponded to UV signature mutations (47).
The majority of BCC, if not all, shows abnormal activation of hedgehog signaling pathway which drives the progression of the disease (48, 49). The aberrant activation of hedgehog signaling could be attributed to loss of function mutation in hedgehog receptor patched (PTCH1 and PTCH2) and gain of function mutation in smoothened (SMO). Hedgehog ligands especially sonic hedgehog (shh) is sufficient to induce BCC in mice (13, 50). Several inhibitors of hedgehog pathway, in particular SMO inhibitors, have been extensively studied, and a few of them [GDC-0449 (Vismodegib) and LDE225 (Sonidegib)] have received FDA approval to treat BCC, while several other hedgehog inhibitors are currently under clinical trials. Around 48% cases of advanced and metastatic BCCs respond to Vismodegib (17, 51) but 20% of patients developed resistance during the first year of treatment itself (52). The development of resistance to these inhibitors, which primarily occurs through SMO mutation and/or amplification of downstream signaling molecules, has become a major problem when treating BCC (53, 54). Herein, we developed hedgehog inhibitor (GDC-0449 and Sant-1) resistant ASZ001 BCC cell lines, and used them to evaluate the efficacy of a natural non-toxic agent, silibinin, against these cells. Silibinin has been extensively studied for its chemopreventive efficacy against several cancers including BCC (3). The adverse photodamage and photocarcinogenic effects of UVB are significantly reduced by the silibinin treatment in cell culture and animal models. For example, silibinin treatment strongly inhibited the UVB-induced CPD formation in SKH-1 hairless mice and JB6 epidermal cells (55, 56). Further, silibinin treatment also increased the p53-positive, TUNEL and cleaved caspase-3- positive cells in SKH-1 hairless mouse skin epidermis either before or after exposure of UVR or its dietary feeding (57). The in vitro efficacy of silibinin against A431 cells was through inhibiting the activation of ERK1/2, JNK and p38 kinase when it was added post irradiation (58). Overall, silibinin offers a chemopreventive option to manage and reduce skin cancer burden. As silibinin has already shown strong efficacy against BCC, the use of silibinin against hedgehog inhibitor resistant BCC cells seemed logical; and outcomes of the present study clearly support that silibinin possesses strong efficacy against hedgehog inhibitor resistant BCC cells.
Results show that silibinin inhibits cell growth and clonal expansion of hedgehog inhibitor resistant ASZ001 cells. The observed reduced cell growth in resistant cells could be through silibinin-mediated inhibition of EGFR-MAPK-Akt and hedgehog signaling which are essential components of mitogenic and survival signaling pathways as well as play important role in the development of resistant BCC cells (19, 36). Previous studies have shown the co-operation between EGFR and Hh/GLI signaling pathways in BCC and pancreatic cancer. It was reported that a synergism between these two pathways is required for oncogenic transformation and inhibition of EGFR and GLI reduced the BCC growth (59, 60). We observed that silibinin treatment decreased the level of phosphorylated-EGFR in hedgehog inhibitor resistant BCC cells. Interestingly, silibinin treatment also reduced the total EGFR level in hedgehog inhibitor resistant BCC cells as well as in hedgehog inhibitor sensitive ASZ001 cells (3). Further studies are required to understand the mechanism underlying this strong reduction of total EGFR level by silibinin in resistant cells.
Besides EGFR, silibinin treatment also inhibited the Akt and ERK1/2 activation; both known to regulate several biological processes including cell proliferation, survival and drug-resistance (61–64). Furthermore, silibinin strongly inhibited the hedgehog signaling in hedgehog inhibitor resistant BCC cells as we observed a strong decrease in Gli1 level by silibinin treatment. This was also reflected in silibinin-mediated inhibition of cyclin D1 expression, which is one of the target genes regulated by Gli1. The crosstalk between EGFR/Hh signaling induces GLI1 expression in medulloblastoma cells through non-canonical HH/GLI profile (65). Also, the nuclear localization and transcriptional activity of GLI1 is regulated by endogenous RAS-MEK and Akt signaling in melanoma (66). It will be interesting to see whether silibinin-mediated inhibition of EGFR/MAPK/Akt signaling is related to decrease in Gli1 expression. Interestingly, we also observed an increase in SUFU level in hedgehog inhibitor resistant cells following silibinin treatment; and SUFU is known to negatively regulate hedgehog signaling by binding with Gli1. This could be an additional mechanism for silibinin-mediated inhibition of hedgehog signaling in resistant BCC cells.
To summarize, results from present study showed that silibinin has strong efficacy against hedgehog inhibitor resistant BCC cells, both as a single agent and in combination with hedgehog inhibitors, and strongly inhibits their proliferation, clonogenicity and induces cell death. Moreover, silibinin targeted a multitude of signaling processes in hedgehog inhibitor resistant BCC cells which makes it as an important agent to use either alone or in combination with hedgehog inhibitors to better manage and treat advanced BCC.
Acknowledgments
We acknowledge the grant support from NCI (R01 CA140368 to RA) and Skaggs School of Pharmacy ADR seed grant (to GD). University grant commission, India is acknowledged for senior research fellowship to AD.
Footnotes
This article is part of the Special Issue honoring Dr. Hasan Mukhtar’s 70th Birthday and his outstanding contributions to various aspects of photobiology research, including photocarcinogenesis and chemoprevention.
References
- 1.Wu S, Han J, Li WQ, Li T, Qureshi AA. Basal-cell carcinoma incidence and associated risk factors in U.S. women and men. American journal of epidemiology. 2013;178:890–897. doi: 10.1093/aje/kwt073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doan HQ, Silapunt S, Migden MR. Sonidegib, a novel smoothened inhibitor for the treatment of advanced basal cell carcinoma. OncoTargets and therapy. 2016;9:5671–5678. doi: 10.2147/OTT.S108171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tilley C, Deep G, Agarwal C, Wempe MF, Biedermann D, Valentová K, Kren V, Agarwal R. Silibinin and its 2,3-dehydro-derivative inhibit basal cell carcinoma growth via suppression of mitogenic signaling and transcription factors activation. Molecular Carcinogenesis. 2016;55:3–14. doi: 10.1002/mc.22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zak-Prelich M, Narbutt J, Sysa-Jedrzejowska A. Environmental Risk Factors Predisposing to the Development of Basal Cell Carcinoma. Dermatologic Surgery. 2004;30:248–252. doi: 10.1111/j.1524-4725.2004.30089.x. [DOI] [PubMed] [Google Scholar]
- 5.Katiyar SK, Singh T, Prasad R, Sun Q, Vaid M. Epigenetic Alterations in Ultraviolet Radiation-Induced Skin Carcinogenesis: Interaction of Bioactive Dietary Components on Epigenetic Targets() Photochemistry and photobiology. 2012;88:1066–1074. doi: 10.1111/j.1751-1097.2011.01020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spurgers KB, Chari NS, Bohnenstiehl NL, McDonnell TJ. Molecular mediators of cell death in multistep carcinogenesis: a path to targeted therapy. Cell Death Differ. 2006;13:1360–1370. doi: 10.1038/sj.cdd.4401986. [DOI] [PubMed] [Google Scholar]
- 7.Michaud EJ, Yoder BK. The primary cilium in cell signaling and cancer. Cancer research. 2006;66:6463–6467. doi: 10.1158/0008-5472.CAN-06-0462. [DOI] [PubMed] [Google Scholar]
- 8.Nilsson M, Unden AB, Krause D, Malmqwist U, Raza K, Zaphiropoulos PG, Toftgard R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:3438–3443. doi: 10.1073/pnas.050467397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properies and tumor mircroenvironment. Mol Cancer. 2016;15:24. doi: 10.1186/s12943-016-0509-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han Y, Shi Q, Jiang J. Multisite interaction with Sufu regulates Ci/Gli activity through distinct mechanisms in Hh signal transduction. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:6383–6388. doi: 10.1073/pnas.1421628112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Otsuka A, Levesque MP, Dummer R, Kabashima K. Hedgehog signaling in basal cell carcinoma. J Dermatol Sci. 2015;78:95–100. doi: 10.1016/j.jdermsci.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Sekulic A, Mangold AR, Northfelt DW, LoRusso PM. Advanced basal cell carcinoma of the skin: targeting the hedgehog pathway. Curr Opin Oncol. 2013;25:218–223. doi: 10.1097/CCO.0b013e32835ff438. [DOI] [PubMed] [Google Scholar]
- 13.Oro AE, Higgins KM, Hu Z, Bonifas JM, Epstein EH, Jr, Scott MP. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science. 1997;276:817–821. doi: 10.1126/science.276.5313.817. [DOI] [PubMed] [Google Scholar]
- 14.Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, Rosenthal A, Epstein EH, Jr, de Sauvage FJ. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 15.Tang JY, Aszterbaum M, Athar M, Barsanti F, Cappola C, Estevez N, Hebert J, Hwang J, Khaimskiy Y, Kim A, Lu Y, So PL, Tang X, Kohn MA, McCulloch CE, Kopelovich L, Bickers DR, Epstein EH., Jr Basal cell carcinoma chemoprevention with nonsteroidal anti-inflammatory drugs in genetically predisposed PTCH1+/− humans and mice. Cancer Prev Res (Phila) 2010;3:25–34. doi: 10.1158/1940-6207.CAPR-09-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Athar M, Li C, Tang X, Chi S, Zhang X, Kim AL, Tyring SK, Kopelovich L, Hebert J, Epstein EH, Jr, Bickers DR, Xie J. Inhibition of smoothened signaling prevents ultraviolet B-induced basal cell carcinomas through regulation of Fas expression and apoptosis. Cancer research. 2004;64:7545–7552. doi: 10.1158/0008-5472.CAN-04-1393. [DOI] [PubMed] [Google Scholar]
- 17.Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, Solomon JA, Yoo S, Arron ST, Friedlander PA, Marmur E, Rudin CM, Chang ALS, Low JA, Mackey HM, Yauch RL, Graham RA, Reddy JC, Hauschild A. Efficacy and Safety of Vismodegib in Advanced Basal-Cell Carcinoma. New England Journal of Medicine. 2012;366:2171–2179. doi: 10.1056/NEJMoa1113713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atwood SX, Li M, Lee A, Tang JY, Oro AE. Gli activation by aPKC iota/lambda regulates basal cell carcinoma growth. Nature. 2013;494:484–488. doi: 10.1038/nature11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brechbiel J, Miller-Moslin K, Adjei AA. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat Rev. 2014;40:750–759. doi: 10.1016/j.ctrv.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C, Regl G, Kroismayr R, Moriggl R, Sibilia M, Aberger F. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer research. 2009;69:1284–1292. doi: 10.1158/0008-5472.CAN-08-2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kasper M, Schnidar H, Neill GW, Hanneder M, Klingler S, Blaas L, Schmid C, Hauser-Kronberger C, Regl G, Philpott MP, Aberger F. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Molecular and cellular biology. 2006;26:6283–6298. doi: 10.1128/MCB.02317-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, Hsiao K, Yuan J, Green J, Ospina B, Yu Q, Ostrom L, Fordjour P, Anderson DL, Monahan JE, Kelleher JF, Peukert S, Pan S, Wu X, Maira SM, Garcia-Echeverria C, Briggs KJ, Watkins DN, Yao YM, Lengauer C, Warmuth M, Sellers WR, Dorsch M. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Science translational medicine. 2010;2:51ra70. doi: 10.1126/scitranslmed.3001599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, Solomon JA, Yoo S, Arron ST, Friedlander PA, Marmur E, Rudin CM, Chang AL, Low JA, Mackey HM, Yauch RL, Graham RA, Reddy JC, Hauschild A. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. The New England journal of medicine. 2012;366:2171–2179. doi: 10.1056/NEJMoa1113713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basset-Seguin N, Sharpe HJ, de Sauvage FJ. Efficacy of Hedgehog pathway inhibitors in Basal cell carcinoma. Molecular cancer therapeutics. 2015;14:633–641. doi: 10.1158/1535-7163.MCT-14-0703. [DOI] [PubMed] [Google Scholar]
- 25.Rudin CM. Vismodegib. Clin Cancer Res. 2012;18:3218–3222. doi: 10.1158/1078-0432.CCR-12-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dhanalakshmi S, Mallikarjuna GU, Singh RP, Agarwal R. Silibinin prevents ultraviolet radiation-caused skin damages in SKH-1 hairless mice via a decrease in thymine dimer positive cells and an up-regulation of p53-p21/Cip1 in epidermis. Carcinogenesis. 2004;25:1459–1465. doi: 10.1093/carcin/bgh152. [DOI] [PubMed] [Google Scholar]
- 27.Mallikarjuna G, Dhanalakshmi S, Singh RP, Agarwal C, Agarwal R. Silibinin Protects against Photocarcinogenesis via Modulation of Cell Cycle Regulators, Mitogen-Activated Protein Kinases, and Akt Signaling. Cancer research. 2004;64:6349–6356. doi: 10.1158/0008-5472.CAN-04-1632. [DOI] [PubMed] [Google Scholar]
- 28.Singh RP, Agarwal R. Mechanisms and preclinical efficacy of silibinin in preventing skin cancer. European journal of cancer (Oxford, England : 1990) 2005;41:1969–1979. doi: 10.1016/j.ejca.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 29.Agarwal R, Agarwal C, Ichikawa H, Singh RP, Aggarwal BB. Anticancer potential of silymarin: from bench to bed side. Anticancer Research. 2006;26:4457–4498. [PubMed] [Google Scholar]
- 30.Deep G, Agarwal R. Chemopreventive efficacy of silymarin in skin and prostate cancer. Integr Cancer Ther. 2007;6:130–145. doi: 10.1177/1534735407301441. [DOI] [PubMed] [Google Scholar]
- 31.Raina K, Agarwal R. Combinatorial strategies for cancer eradication by silibinin and cytotoxic agents: efficacy and mechanisms. Acta Pharmacol Sin. 2007;28:1466–1475. doi: 10.1111/j.1745-7254.2007.00691.x. [DOI] [PubMed] [Google Scholar]
- 32.Prabu SM, Muthumani M. Silibinin ameliorates arsenic induced nephrotoxicity by abrogation of oxidative stress, inflammation and apoptosis in rats. Mol Biol Rep. 2012;39:11201–11216. doi: 10.1007/s11033-012-2029-6. [DOI] [PubMed] [Google Scholar]
- 33.Ezhilarasan D, Karthikeyan S, Vivekanandan P. Ameliorative effect of silibinin against N-nitrosodimethylamine-induced hepatic fibrosis in rats. Environ Toxicol Pharmacol. 2012;34:1004–1013. doi: 10.1016/j.etap.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 34.Deep G, IS, Agarwal R. Methods to analyze chemopreventive effect of silibinin on prostate cancer biomarkers protein expression in Cancer Prevention: Dietary Factors and Pharmacology. Humana Press; 2013. pp. 85–105. [Google Scholar]
- 35.Metcalfe C, Alicke B, Crow A, Lamoureux M, Dijkgraaf GJ, Peale F, Gould SE, de Sauvage FJ. PTEN loss mitigates the response of medulloblastoma to Hedgehog pathway inhibition. Cancer research. 2013;73:7034–7042. doi: 10.1158/0008-5472.CAN-13-1222. [DOI] [PubMed] [Google Scholar]
- 36.Metcalfe C, de Sauvage FJ. Hedgehog fights back: mechanisms of acquired resistance against Smoothened antagonists. Cancer research. 2011;71:5057–5061. doi: 10.1158/0008-5472.CAN-11-0923. [DOI] [PubMed] [Google Scholar]
- 37.Kauntz H, Bousserouel S, Gosse F, Marescaux J, Raul F. Silibinin, a natural flavonoid, modulates the early expression of chemoprevention biomarkers in a preclinical model of colon carcinogenesis. International Journal of Oncology. 2012;41:849–854. doi: 10.3892/ijo.2012.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubin AI, Chen EH, Ratner D. Basal-cell carcinoma. The New England journal of medicine. 2005;353:2262–2269. doi: 10.1056/NEJMra044151. [DOI] [PubMed] [Google Scholar]
- 39.Kumar R, Deep G, Agarwal R. An Overview of Ultraviolet B Radiation-Induced Skin Cancer Chemoprevention by Silibinin. Current pharmacology reports. 2015;1:206–215. doi: 10.1007/s40495-015-0027-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dhanalakshmi S, Mallikarjuna GU, Singh RP, Agarwal R. Dual efficacy of silibinin in protecting or enhancing ultraviolet B radiation-caused apoptosis in HaCaT human immortalized keratinocytes. Carcinogenesis. 2004;25:99–106. doi: 10.1093/carcin/bgg188. [DOI] [PubMed] [Google Scholar]
- 41.Kraemer KH. Sunlight and skin cancer: Another link-revealed. Proceedings of the National Academy of Sciences. 1997;94:11–14. doi: 10.1073/pnas.94.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Gruijl FR, Rebel H. Early Events in UV Carcinogenesis—DNA Damage, Target Cells and Mutant p53 Foci†. Photochemistry and Photobiology. 2008;84:382–387. doi: 10.1111/j.1751-1097.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 43.Bickers DR, Athar M. Oxidative stress in the pathogenesis of skin disease. The Journal of investigative dermatology. 2006;126:2565–2575. doi: 10.1038/sj.jid.5700340. [DOI] [PubMed] [Google Scholar]
- 44.Cadet J, Douki T, Pouget JP, Ravanat JL, Sauvaigo S. Effects of UV and visible radiations on cellular DNA. Current problems in dermatology. 2001;29:62–73. doi: 10.1159/000060654. [DOI] [PubMed] [Google Scholar]
- 45.Situm M, Buljan M, Bulat V, Lugovic Mihic L, Bolanca Z, Simic D. The role of UV radiation in the development of basal cell carcinoma. Collegium antropologicum. 2008;32(Suppl 2):167–170. [PubMed] [Google Scholar]
- 46.Heitzer E, Lassacher A, Quehenberger F, Kerl H, Wolf P. UV fingerprints predominate in the PTCH mutation spectra of basal cell carcinomas independent of clinical phenotype. The Journal of investigative dermatology. 2007;127:2872–2881. doi: 10.1038/sj.jid.5700923. [DOI] [PubMed] [Google Scholar]
- 47.Reifenberger J, Wolter M, Knobbe CB, Kohler B, Schonicke A, Scharwachter C, Kumar K, Blaschke B, Ruzicka T, Reifenberger G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. The British journal of dermatology. 2005;152:43–51. doi: 10.1111/j.1365-2133.2005.06353.x. [DOI] [PubMed] [Google Scholar]
- 48.Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nature reviews Cancer. 2008;8:743–754. doi: 10.1038/nrc2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gailani MR, Stahle-Backdahl M, Leffell DJ, Glynn M, Zaphiropoulos PG, Pressman C, Unden AB, Dean M, Brash DE, Bale AE, Toftgard R. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nature genetics. 1996;14:78–81. doi: 10.1038/ng0996-78. [DOI] [PubMed] [Google Scholar]
- 50.Fan H, Oro AE, Scott MP, Khavari PA. Induction of basal cell carcinoma features in transgenic human skin expressing Sonic Hedgehog. Nature medicine. 1997;3:788–792. doi: 10.1038/nm0797-788. [DOI] [PubMed] [Google Scholar]
- 51.Axelson M, Liu K, Jiang X, He K, Wang J, Zhao H, Kufrin D, Palmby T, Dong Z, Russell AM, Miksinski S, Keegan P, Pazdur R. U.S. Food and Drug Administration approval: vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:2289–2293. doi: 10.1158/1078-0432.CCR-12-1956. [DOI] [PubMed] [Google Scholar]
- 52.Chang AS, Oro AE. INitial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Archives of Dermatology. 2012;148:1324–1325. doi: 10.1001/archdermatol.2012.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, Tsui V, Durham AB, Dlugosz AA, Haverty PM, Bourgon R, Tang JY, Sarin KY, Dirix L, Fisher DC, Rudin CM, Sofen H, Migden MR, Yauch RL, de Sauvage FJ. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer cell. 2015;27:327–341. doi: 10.1016/j.ccell.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kasper M, Jaks V, Hohl D, Toftgard R. Basal cell carcinoma — molecular biology and potential new therapies. The Journal of Clinical Investigation. 122:455–463. doi: 10.1172/JCI58779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gu M, Dhanalakshmi S, Singh RP, Agarwal R. Dietary feeding of silibinin prevents early biomarkers of UVB radiation-induced carcinogenesis in SKH-1 hairless mouse epidermis. Cancer Epidemiol Biomarkers Prev. 2005;14:1344–1349. doi: 10.1158/1055-9965.EPI-04-0664. [DOI] [PubMed] [Google Scholar]
- 56.Roy S, Deep G, Agarwal C, Agarwal R. Silibinin prevents ultraviolet B radiation-induced epidermal damages in JB6 cells and mouse skin in a p53-GADD45alpha-dependent manner. Carcinogenesis. 2012;33:629–636. doi: 10.1093/carcin/bgr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mallikarjuna G, Dhanalakshmi S, Singh RP, Agarwal C, Agarwal R. Silibinin protects against photocarcinogenesis via modulation of cell cycle regulators, mitogen-activated protein kinases, and Akt signaling. Cancer research. 2004;64:6349–6356. doi: 10.1158/0008-5472.CAN-04-1632. [DOI] [PubMed] [Google Scholar]
- 58.Mohan S, Dhanalakshmi S, Mallikarjuna GU, Singh RP, Agarwal R. Silibinin modulates UVB-induced apoptosis via mitochondrial proteins, caspases activation, and mitogen-activated protein kinase signaling in human epidermoid carcinoma A431 cells. Biochem Biophys Res Commun. 2004;320:183–189. doi: 10.1016/j.bbrc.2004.05.153. [DOI] [PubMed] [Google Scholar]
- 59.Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C, Regl G, Kroismayr R, Moriggl R, Sibilia M, Aberger F. Epidermal Growth Factor Receptor Signaling Synergizes with Hedgehog/GLI in Oncogenic Transformation via Activation of the MEK/ERK/JUN Pathway. Cancer research. 2009;69:1284–1292. doi: 10.1158/0008-5472.CAN-08-2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eberl M, Klingler S, Mangelberger D, Loipetzberger A, Damhofer H, Zoidl K, Schnidar H, Hache H, Bauer HC, Solca F, Hauser-Kronberger C, Ermilov AN, Verhaegen ME, Bichakjian CK, Dlugosz AA, Nietfeld W, Sibilia M, Lehrach H, Wierling C, Aberger F. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO molecular medicine. 2012;4:218–233. doi: 10.1002/emmm.201100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 0000;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 62.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA, D’Assoro AB, Salisbury JL, Mazzarino MC, Stivala F, Libra M. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Advances in enzyme regulation. 2006;46:249–279. doi: 10.1016/j.advenzreg.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 64.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochimica et biophysica acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Götschel F, Berg D, Gruber W, Bender C, Eberl M, Friedel M, Sonntag J, Rüngeler E, Hache H, Wierling C, Nietfeld W, Lehrach H, Frischauf A, Schwartz-Albiez R, Aberger F, Korf U. Synergism between Hedgehog-GLI and EGFR Signaling in Hedgehog-Responsive Human Medulloblastoma Cells Induces Downregulation of Canonical Hedgehog-Target Genes and Stabilized Expression of GLI1. PLoS ONE. 2013;8:e65403. doi: 10.1371/journal.pone.0065403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, Beermann F, Ruiz IAA. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5895–5900. doi: 10.1073/pnas.0700776104. [DOI] [PMC free article] [PubMed] [Google Scholar]