

Abstract
AIM
To investigate the therapeutic potential of tesevatinib (TSV), a unique multi-kinase inhibitor currently in Phase II clinical trials for autosomal dominant polycystic kidney disease (ADPKD), in well-defined rodent models of autosomal recessive polycystic kidney disease (ARPKD).
METHODS
We administered TSV in daily doses of 7.5 and 15 mg/kg per day by I.P. to the well characterized bpk model of polycystic kidney disease starting at postnatal day (PN) 4 through PN21 to assess efficacy and toxicity in neonatal mice during postnatal development and still undergoing renal maturation. We administered TSV by oral gavage in the same doses to the orthologous PCK model (from PN30 to PN90) to assess efficacy and toxicity in animals where developmental processes are complete. The following parameters were assessed: Body weight, total kidney weight; kidney weight to body weight ratios; and morphometric determination of a cystic index and a measure of hepatic disease. Renal function was assessed by: Serum BUN; creatinine; and a 12 h urinary concentrating ability. Validation of reported targets including the level of angiogenesis and inhibition of angiogenesis (active VEGFR2/KDR) was assessed by Western analysis.
RESULTS
This study demonstrates that: (1) in vivo pharmacological inhibition of multiple kinase cascades with TSV reduced phosphorylation of key mediators of cystogenesis: EGFR, ErbB2, c-Src and KDR; and (2) this reduction of kinase activity resulted in significant reduction of renal and biliary disease in both bpk and PCK models of ARPKD. The amelioration of disease by TSV was not associated with any apparent toxicity.
CONCLUSION
The data supports the hypothesis that this multi-kinase inhibitor TSV may provide an effective clinical therapy for human ARPKD.
Keywords: Autosomal recessive, Autosomal dominant, Polycystic kidney disease, Therapy, Kinase inhibition, Multi-kinase inhibitor, Phosphorylation, Renal cysts, Biliary, G-protein coupled receptor
Core tip: This study examined the effect of a multi-kinase inhibitor, tesevatinib (TSV) on cyst development and growth in rodent models of autosomal recessive polycystic kidney disease (ARPKD). Tesevatinib which targets epidermal growth factor receptors, Src and KDR is currently in clinical trials for autosomal dominant polycystic kidney disease (ADPKD) and given the molecular and cellular interactions of the ADPKD and ARPKD genes and proteins we sought to determine if TSV would ameliorate ARPKD TSV was tested in two well described models of ARPKD, the BPK a phenocopy, and an orthologous rat model of ARPKD, the PCK. Of particular interest was the effect of TSV’s inhibition of VEGFR2 or KDR during early post-natal development and renal maturation.
INTRODUCTION
Renal cystic diseases encompass a broad group of disorders with variable phenotypic expression. The term polycystic kidney disease (PKD) explicitly refers to two genetically defined renal cystic diseases: Autosomal recessive polycystic kidney disease (ARPKD) and autosomal dominant polycystic kidney disease (ADPKD).
ARPKD (OMIM 263200) belongs to a group of congenital hepatorenal fibrocystic diseases characterized by dual renal and hepatic involvement[1-5]. ARPKD has an incidence estimated at 1/20000 to 1/40000 with a wide spectrum of phenotypic expression[2,4-6]. ARPKD occurs as the result of a mutations in a single gene, polycystic kidney and hepatic disease 1 (PKHD1)[7,8]; encodes a protein called fibrocystin or polyductin (FPC)[9,10]; and is commonly diagnosed in utero through early childhood[2,3,5].
ADPKD is generally an adult-onset multisystem disorder, characterized by bilateral renal cysts, vascular abnormalities including intracranial aneurysms, and cysts in other organs including the liver. ADPKD is increasingly diagnosed in childhood and adolescents[2,11]. Substantial variability in the severity of renal disease and other extrarenal manifestations occurs even within the same family. ADPKD is more common than ARPKD with an incidence of estimated at 1 in 600 and 1 in 1000 live births[12]. ADPKD is a heterogenetic disorder caused by mutations in either the PKD1 gene (85%) (OMIM173900) encoding the protein polycystin 1 (PC1), or in the PKD2 gene (15%) (OMIM173910), encoding polycystin 2 (PC2). The recent report of a third ADPKD gene has been reported but it represents a very small number of ADPKD patients that did not map to PKD1 or PKD2 locus[13]. ADPKD is characterized by the progressive development and expansion of fluid-filled cysts derived from renal tubule epithelia and typically leads to ESRD by late middle age[14,15].
Clinically, a significant overlap of symptoms between ARPKD and ADPKD has been observed in children. Recent investigations at the cellular and molecular level provide compelling rationale for this clinical observation as ARPKD and ADPKD share many common pathophysiological features[2,5,16]. Such studies reveal: (1) PKHD1, PKD1 and PKD2 proteins are expressed in multimeric “cystoprotein” complexes at specific locations within epithelial cells[5,17-19]; (2) complex interactions between all three genes and their protein products occur at a molecular and cellular level[15,20-23]; (3) cyst formation not only requires a PKD mutation but also requires simultaneous cell proliferation; (4) cyst development in both ARPKD and ADPKD begins in utero[24-26]; and (5) clinical manifestations of both disorders are seen in newborns, children, and adolescents[3,5,27-30]. These findings suggest that therapeutic interventions at an early stage of disease may provide the best opportunity for beneficial long-term clinical outcomes for patients with PKD[3,5,31].
Cyst initiation and expansion are complex phenomena comprised of multiple interacting processes. The precise mechanisms leading to enlarged cystic kidneys and liver abnormalities are unclear. However, various cellular defects have been identified that are common to cystic renal epithelia in both ARPKD and ADPKD. These include: (1) abnormalities of expression, function and polarity of one or more members of the epidermal growth factor (EGF) family of receptors and ligands [the (EGFR)-axis]; (2) abnormal activity of c-Src (pp60c-Src); decreased intracellular calcium leading to aberrant intracellular cAMP signaling; (3) abnormal structure and/or function of the primary cilia; (4) alterations in cell-cell, and cell-matrix interactions; and (5) activation of interstitial macrophages leading to progressive fibrosis.
Tesevatinib (TSV, previously been known as XL7647, XL647, PRIM-001 and KD-019) is a unique multi-kinase inhibitor that is being utilized in a phase II clinical trial for treatment of ADPKD (https//ClinicalTrials.gov/ADPKD: NCT01559363)[32,33]. The basis of this therapeutic approach is that the use of a multi-kinase inhibitor provides “combination therapy” targeting multiple abnormal signal transduction events in PKD. TSV is a particularly appealing candidate for therapy due to its inhibition profile of c-Src (pp60c-Src) which decreases activity of both the EGFR axis and cAMP pathways[34] as well as its inhibition of KDR that will target abnormal angiogenesis necessary for cyst growth.
MATERIALS AND METHODS
Animal models
All animal experiments were conducted in accordance with policies of the NIH Guide for the Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee of the Medical College of Wisconsin.
The BALB/c-(Bicc1/Bicc1) model
The BALB/c-(Bicc1/Bicc1) (BPK) model, a murine phenocopy of ARPKD, has been extensively characterized[35-37]. Affected animals die at postnatal day (PN) 23 (average) with a range of PN-21 to PN-25. Extrarenal manifestations include biliary ductal ectasia (BDE). BPK animals were identified utilizing a PCR based genotyping method that has been described in detail[38]. BALB/c wildtype (WT) controls were utilized in all experiments.
Both WT and cystic (bpk) male mice were administered the HCL salt of TSV, by intraperitoneal (IP) injections at 7.5 and 15 mg/kg per day starting at PN4. Kidney and liver (left lobe) tissues were harvested at PN-21, 2 h following the last dose, and assessed for the following: Body weight; total kidney weight (TKW); kidney weight to body weight (KW/BW) ratios; morphometric determination of a cystic index (CI); target validation was confirmed by western analysis; and renal function was assessed by determining serum BUN; creatinine (CR); and urinary concentrating ability (UCA).
A colony of bpk animals have been inbred in the Avner laboratory since 1991 without significant phenotypic drift.
PCK rat model
The PCK rat is an orthologous model of human ARPKD characterized by slowly progressing renal cyst formation, impairment of renal function and development of congenital hepatic fibrosis. The PCK arose from a spontaneous mutation in the rat homolog of human PKHD1 in a Sprague Dawley colony in Japan at the Education and Research Center of Animal Models for Human Diseases of Fujita Health University and subsequently transferred to Charles River United States[10,39].
Similar to human disease, previous reports in PCK rats have demonstrated an increased renal expression of active ErbB2 (pY1221/1222)[34], active c-Src (pY418)[34,40] and hepatic sensitivity to EGFR inhibition[41]. Therefore, the PCK is a valuable orthologous model for testing therapeutic intervention in ARPKD.
Male PCK and Sprague Dawley controls were administered TSV daily by oral gavage, from PN30 to PN90 (61 doses). Similar to the bpk, PCK and SD kidney and liver (left lobe) tissues were harvested two hours following the final dose and assessed for the following parameters: Body weight; total kidney weight (TKW); KW/BW ratios; morphometric determination of a CI; target validation by western analysis; and renal function was assessed by determining serum BUN; CR; and UCA.
A colony of PCK and SD animals has been maintained at the Medical College of Wisconsin 2004 without significant phenotypic drift.
TSV
TSV (formerly named XL7647, XL647, PRIM-001 and KD-019) is an orally bioavailable compound with a MW of 663.59 g/mole. TSV is a 4-anilinoquinazoline bearing an octahydro-cyclopentapyrrol substituted methoxy group at the C-7 position. It is a mono-p-toluenesulfonic acid salt with a MW of 663.59 g/m. Data from pharmacodynamics in vivo experiments show that inhibition spectrum of TSV includes key tyrosine kinases (EGFR, HER2/ErbB2, c-Src) that promote epithelial cell proliferation and VEGFR2/KDR which promotes angiogenesis and/or neovascularization[32,33].
TSV preparation and administration
Compound solutions EtOH:PEG 400:distilled water (5:45:50) were prepared under sterile conditions at concentrations of 5 mg/mL (for 7.5 mg/kg per day dose) or 10 mg/mL (for 15 mg/kg per day dose) to keep injection volumes equivalent despite the different dosing levels. Solutions were prepared in vehicle and stored at 4 °C for 3 d. Solutions were made fresh every 4 d.
BPK (cystic) and BALB/c (wild-type control) littermates received TSV at 7.5 or 15 mg/kg per day by intraperitoneal (i.p.) injections. This dosage was based on company supplied pharmacologic and pharmacokinetic data of TSV treatment in mice and preliminary dose-response studies (data not shown). Animals were treated from PN4 to PN21 (18 doses). Kidney and liver were routinely harvested at P21 (2 h post last injection) and heart, spleen, pancreas, stomach, and thymus were periodically harvested at PN21 to evaluate possible toxicity of vehicle or TSV. Both BALB/c and bpk animals were divided into four groups each for testing and analysis. These groups included: (1) untreated or sham wildtype control; (2) wildtype treated with vehicle only; (3) wildtype treated with TSV at 7.5 mg/kg per day; (4) wildtype treated with TSV at 15 mg/kg per day; (5) untreated or sham cystic (bpk); (6) vehicle treated bpk; (7) TSV treated bpk at 7.5 mg/kg per day beginning at PN4 to PN21; and (8) bpk treated with TSV at 15 mg/kg per day beginning at PN4 to PN21.
PCK treatment
SD and PCK males were administered TSV by oral gavage from PN30 to PN90. SD sham, vehicle treated and TSV treated at 7.5 and 15 mg/kg per day along with the same groups of PCK animals were examined. Two hours following the last dose, renal and the left lobe of the liver were routinely harvested at P90 (2 h post last substitute dose for injection). Heart, spleen, pancreas, stomach, and thymus were periodically harvested at PN90 to evaluate possible toxicity of vehicle or TSV. The same parameters evaluated in bpk were assessed in the PCK. First urine samples were collected in metabolic cages after 88 d to assess UCA after 12 h without water. TSV treatment of PCK animals resulted in significant improvement of all assessed parameters compared to untreated PCK without evidence of toxicity or detrimental effects of KDR inhibition.
Western analysis
Control and cystic kidneys were first chopped in RIPA buffer supplemented with protease and phosphatase inhibitors (RIPA+), to minimize the contribution of cyst fluid protein to the total cellular protein. Tissue lysates were then extracted in fresh RIPA+ as previously described[34] and were adjusted to an equal amount of protein (1.5 mg) (determined by BCA assay, Pierce, Rockford, IL, United States) per milliliter based on the least concentrated sample.
For Western analysis, 30 μg of the original tissue lysate (after volume adjustment), were denatured with 5 × sample buffer; and subjected to SDS-PAGE on 4% to 20% gradient gels, and were transferred onto membrane by Western blotting. The levels of total protein and active (phosphorylated) proteins were determined using specific primary antibodies, followed by peroxidase-conjugated appropriate secondary antibody and visualized by ECL (GE Healthcare Bio-Sciences Corp, Piscataway, NJ, United States). Transfer efficiency and molecular weight determinations were performed with Rainbow® MW markers (GE Healthcare Bio-Sciences Corp, Piscataway, NJ, United States). Bands were semi-quantitatively compared using NIH Image 1.62. The densitometry value of wildtype untreated control was arbitrarily set at 1 and the densitometry of the other bands was compared to this value. Both Westerns and respective densitometry data shown are representative of three independent experiments with reproducible findings.
Antibodies
Rabbit polyclonal antibody to pan Src (#2108), p-Src (#2101), EGFR (#4267), p-EGFR (pY1068) (#3777), ERK1/2 (#4695), p-ERK1/2 (#4376), KDR (#9698, #2472), p-KDR (#3770), and β-actin (#3700) were purchased from Cell Signaling Technology (Beverly, MA, United States). Mouse anti-rat (#550300) and rat anti-mouse (#553708) CD-31 (PECAM1) (#550300) was purchased from BD Biosciences (San Jose CA, United States) and anti-mouse CD-31 (ab28364), ErbB2 (ab16901), and p-ErbB2 (ab131102) was purchased from Abcam Inc. (Cambridge, MA, United States) and used to probe Western blots. A rabbit polyclonal to β-actin (ab8227) was used to monitor protein loading of Westerns.
Histology, immunohistology, and determination of segmental nephron cyst localization
All kidney and liver tissues were fixed in 4.0% paraformaldehyde in phosphate buffer (pH = 7.4) for 30 min at 4 °C, dehydrated and embedded in paraffin. Segmental nephron cyst localization and collecting tubule (CT) CI were quantitated in each experimental group by combining morphometric analysis with light microscopy and immunohistology as previously described[34,35,37,42].
Kidney weight/body weight ratio and renal CI
At P21, or PN90 control and cystic, treated and untreated animals were given the last dose weight, as were both excised kidneys. Total kidney weights to body weight ratios were calculated. The degree of CT cyst formation was quantitatively assessed by segment specific morphometric analysis as previously described[34,37,42] and expressed as the CI (see below). Cyst localization was determined by segment-specific lectin binding using Dolichos biflorus agglutinin (DBA) as a marker for CTs and Lotus tetragonolobus agglutinin (LTA) as a marker for proximal tubules[34,37,42]. For each treatment group, a CT CI was determined on 5 to 6 affected pups.
The CI is a model specific means to assess the degree of disease burden or progression based on the natural progression of the renal disease and hepatic disease (BDE in bpk) and (LW/BW in PCK) In the bpk, the CI and BDE index are on a scale of 1 to 5 based on the disease burden normally present at PN0, 5, 10, 15, and 20. The renal index in bpk measures the number and size of PT and CT cysts and the BDE (also a 1-5 scale) is based on the number of abnormal portal triads in the left lobe of the liver that is seen during the natural progression in the bpk.
The PCK CI is on a scale of 1 to 10 based on the number and size of cystic lesions seen at 15 d intervals beginning at PN15 and ending at PN150. Since all cysts occur in CT segments the number and size of these lesions are evaluated. The hepatic lesions in the PCK are biliary cysts and all are affected to varying degrees so we use a simple LW/BW ratio to assess hepatic disease progression.
The kidney and left lobe of the liver in bpk and kidney in PCK is embedded medulla side down and a minimum of 3, 4 μmol/L thick sections at depths 100 µMs apart (25 sections) are evaluated as previously described[34,37,42].
Analysis of renal function and UCA
Animals were deprived of water for 12 h prior to collection of urine samples for determination of UCA. Urinary concentrating was determined by freeze point using a freezing point osmometer (Multi-Osmette; Model 2430, Precision Instruments Inc.
Blood samples were obtained by cardiac puncture for serum BUN and CR measurements. Serum BUN and CR was determined on an automated clinical chemistry analyzer as previously described[37].
Statistical analysis
The significance of differences between experimental groups was determined by a one-way analysis of variance. Results are expressed as means ± SD.
RESULTS
BPK
The in vivo activity of TSV was evaluated in the bpk mouse, a phenocopy of ARPKD based on the dual organ abnormalities seen in this model. It is a rapidly progressing model that results in ESRD and death by PN 24. The rapid progression simulates the clinical course of severe ARPKD presenting in newborns. The bpk model provides a relatively rapid means of assessing the in vivo efficacy of a potential therapeutic compound. Given the reported activity of TSV against the vascular endothelial growth factor receptor 2 (VEGFR2 or KDR), the bpk model is also a good model to evaluate the inhibition of angiogenesis inhibition during postnatal cystic kidney development.
Both wildtype (WT) (BALB/c(Bicc+/Bicc+)) and cystic (bpk) (bpk(Bicc-/Bicc-)) mice were administered the HCL salt of TSV, by intraperitoneal (IP) injections at 7.5 and 15 mg/kg per day starting at PN4. At PN21 the animals were weighted for the last time and kidney and liver tissues were harvested 2 h following the last injection of TSV. The following parameters were assessed: body weight, total kidney weight (TKW); KW/BW ratios; and morphometric determination of a CI. Renal function was assessed by: Serum BUN; CR; and a 12 h UCA. Validation of reported targets including the level of angiogenesis and inhibition of angiogenesis (active VEGFR2/KDR) was assessed by Western analysis. TSV treatment of bpk mice resulted in significant improvement of all assessed parameters compared to untreated or vehicle treated cystic animals without significant changes in body weight, evidence of toxicity or detrimental effects of the inhibition of KDR (Table 1).
Table 1.
BPK summary data
| WT PN-21 Sham n = 22 | WT PN-21 VEH n = 10 | WT PN-21 TSV 7.5 n = 10 | WT PN-21 TSV 15 n = 10 | BPK PN-21 Sham n = 17 | BPK PN-21 VEH n = 12 | BPK PN-21 TSV 7.5 n = 10 | BPK PN-21 TSV 15 n = 14 | |
| BW (g) | 10.31 ± 0.55 | 10.76 ± 0.72 | 10.57 ± 0.80 | 10.10 ± 0.29 | 10.46 ± 0.60 | 10.29 ± 0.45 | 10.50 ± 0.96 | 10.11 ± 0.50 |
| TKW (g) | 0.15 ± 0.01 | 0.16 ± 0.02 | 0.141 ± 0.02 | 0.13 ± 0.01 | 2.03 ± 0.28 | 2.01 ± 0.22 | 1.59 ± 0.19e | 1.21 ± 0.09f,e |
| KW/BW (%) | 1.41 ± 0.10 | 1.50 ± 0.12 | 1.33 ± 0.08 | 1.25 ± 0.09 | 19.32 ± 1.71 | 19.57 ± 1.62 | 15.12 ± 1.1e | 11.96 ± 1.22f,e |
| CI | NA | NA | NA | NA | 4.7 ± 0.50 | 4.8 ± 0.42 | 3.6 ± 0.52e | 2.50 ± 0.35f,e |
| BUN (mg/dL) | 19.25 ± 1.14 | 21.20 ± 1.99 | 18.60 ± 2.07 | 19.40 ± 1.5 | 108.3 ± 17.8 | 112 ± 23.3 | 63.6 ± 20.3e | 40.9 ± 13.7d,e |
| Cr (mg/dL) | 0.24 ± 0.05 | 0.29 ± 0.06 | 0.24 ± 0.05 | 0.28 ± 0.06 | 0.55 ± 0.14 | 0.58 ± 0.10 | 0.41 ± 0.1b | 0.28 ± 0.07c,e |
| 12 h UCA | 1043 ± 35 | 1065 ± 52 | 1026 ± 39 | 1024 ± 27 | 465 ± 83 | 460 ± 92 | 581 ± 67a | 786 ± 185.2d,e |
| BDE | NA | NA | NA | NA | 4.8 ± 0.4 | 4.5 ± 0.5 | 3.70 ± 0.4b | 2.8 ± 0.4d,e |
P < 0.05,
P < 0.01,
P < 0.001 vs untreated (Sham) BPK;
P < 0.05,
P < 0.01,
P < 0.001 vs BPK TSV treatment at 7.5. CI: Cystic index; UCA: Urinary concentration ability at 12 h; BDE: Biliary ductal ectasia; KW/BW: Kidney weight to body weight; NA: Not applicable; TKW: Total kidney weight.
Figure 1 reveals that TSV administration to bpk animals from PN4 to PN21 (18 doses) results in a dose dependent reduction in whole kidney size when compared to untreated bpk cystic kidneys. The change in whole kidney size is also reflected in the dose dependent significant decrease in TKW from 2.03 ± 0.28 g in untreated bpk (column E, Table 1) to 1.59 ± 0.19 (P < 0.001) and 1.21 ± 0.10 g (P < 0.001) with TSV at 7.5 mg/kg per day (column G, Table 1) and 15 mg/kg per day (column H, Table 1) respectively.
Figure 1.
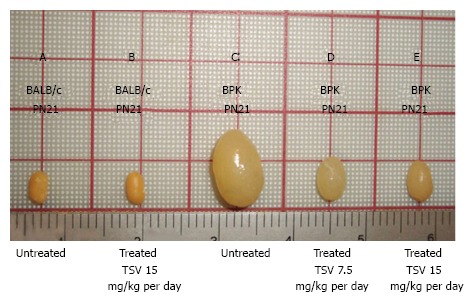
Tesevatinib treatment in the BPK model of autosomal recessive polycystic kidney disease. Compared to wild-type BALB/c control kidneys (A) at PN21, BPK cystic kidneys (C) are extremely enlarged by PN21. TSV treatment resulted in a dose-dependent reduction in the overall kidney size at doses of 7.5 mg/kg per day (C) and 15 mg/kg per day (D) when compared to PN21 BPK untreated cystic kidneys (C). Treatment of wildtype BALB/c with 15 mg/kg per day (B) did not result in significant reduction of overall kidney size when compared to untreated BALB/c kidneys (A). This correlates directly with the total kidney weight (TKW) of each group listed in Table 1. Further, the morphology of wildtype TVS treated kidneys seen in Figure 2A shows no obvious signs of toxicity (Treatment interval: PN4-PN21). TSV: Tesevatinib.
The reduction in whole kidney size and TKW for bpk kidneys is associated with markedly improved renal morphology as shown in Figure 2. Accordingly, the CI was significantly reduced by treatment as seen in Table 1.
Figure 2.
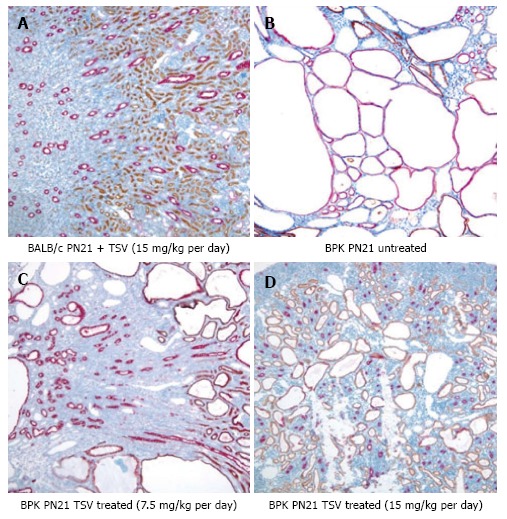
Renal morphology of tesevatinib-treated BPK kidneys. Immuno-histological analysis of BPK kidneys reveals a marked decrease in the size and number of collecting tubule cysts (stained red) following treatment with TSV at 7.5 mg/kg per day (C) and 15 mg/kg per day (D) when compared to the untreated cystic kidneys (B). Treatment of control BALB/c kidneys with TSV at 15 mg/kg per day (A) demonstrated no obvious histological abnormalities (Nephron segment-specific lectin binding of lotus tetragonolabus agglutin shows proximal tubules in brown, and binding of dolichus biflorus agglutin shows collecting tubules in red). TSV: Tesevatinib.
The CI index was reduced by 25% in bpk mice from 4.80 ± 0.4 to 3.60 ± 0.52 (P < 0.001) treated with TSV at 7.5 mg/kg per day and by 48% (2.5 ± 0.5, P < 0.001) in BPK mice treated with TSV at 15 mg/kg per day compared to untreated bpk kidneys (Table 1).
The morphology of bpk kidney and liver shown in Figures 2 and 3 respectively, demonstrates a dose dependent reduction in the progression of disease in both kidney and liver respectively. The red labelled CTs cysts in PN21 untreated bpk animals (Figure 2B) are very large with little normal parenchyma in between. TSV treatment reduces the number and size of the CT cysts in kidneys treated with TSV at 7.5 mg/kg per day (Figure 2C) and 15 mg/kg per day (Figure 2D). Kidneys shown in Figure 2C and D reveal LTA labelled proximal tubule cysts (brown) that are indicative of a “younger” PKD disease course as previously described[34,35,43]. The improved morphology with TSV treatment correlates with the reduced whole kidney size (Figure 1) as well as with the reduced TKW, CI and BDE index in Table 1.
Figure 3.
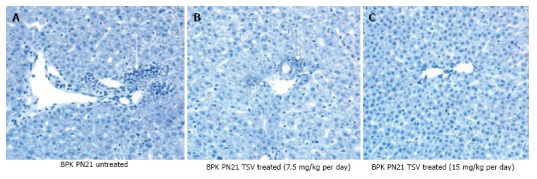
Liver morphology of tesevatinib-treated BPK mice. Histological analysis of hematoxylin stained BPK livers reveals a marked decrease in the biliary ductal ectasia (arrow) following treatment with TSV at 7.5 mg/kg per day (B) (arrow) and 15 mg/kg per day (C) when compared to untreated BPK livers (A) (Treatment interval: PN4-PN21).
All morphometric measures of renal disease and liver abnormalities associated with ARPKD, as well as all renal functional parameters improved in a dose dependent manner in bpk with TSV administration. In the bpk, at PN21 this included an improvement in: BUN of 41% (P < 0.001) and 62% (P < 0.001); serum CR improved 25.5% (P < 0.01) and 49% (P < 0.001); and the UCA improved by 25% (P < 0.05) and 69.4% (P < 0.001) with 7.5 and 15 mg/kg per day respectively as seen in Table 1.
There was also a marked change in biliary ductal ectasia (BDE) with TSV treatment as assessed by histological evaluation of hepatic morphology in the bpk (Figure 3 and Table 1). BDE at PN21 of TSV treated cystic mice (Table 1) was significantly reduced by 25% from 4.80 ± 0.50 to 3.70 ± 0.40 (P < 0.01) with 7.5 mg/kg per day and was furthered reduced by 42% to 2.80 ± 0.40 (P < 0.001) with TSV treatment at 15 mg/kg per day.
Treatment of BALB/c wildtype control animals showed no statistical difference in any assessed parameters including renal function at 7.5 or 15 mg/kg per day dose when compared to untreated wildtype animals. The morphology of both the kidney (Figure 2A) and liver (Figure 3A) at the 15 mg/kg per day dose and the lack of change in renal function (Table 1) at either dose indicated no obvious signs of toxicity with TSV administration.
Kinase target validation was assessed in the bpk kidneys treated with TSV. As seen in the Western blots of Figure 4, TSV targeted the kinases previously shown to play a role in proliferation and cystic disease in renal epithelia. The use of TSV resulted in a significantly reduced phosphorylation level of EGFR (Figure 4A3, and 4A4), c-Src (Figure 4C3 and 4C4) and ERK1/2 (Figure 4E3 and 4E4). For these key kinases, TSV treatment of 15 mg/kg per day resulted in phosphorylation levels similar to untreated control kidneys (Figure 4, lanes A4, C4 and E4 compared to wildtype control levels as seen in Figure 4 lane 1 of panel A, C and E).
Figure 4.

Target validation in BPK. Western analysis of the MAPK pathway activity of EGFR (p-EGFR), cSrc (pSrc), and ERK1/2 (p-ERK ½) reveals that BPK cystic kidneys have significantly increased levels of active or phosphorylated p-EGFR (A2), pSrc (C2) and ERK1/2 (E2) compared to wild-type BALB/c control levels in lanes (A1, C1 and E1) respectively. TSV treatment at 7.5 or 15 mg/kg per day resulted in a dose dependent reduction in the phosphorylation or activity of p-EGFR (A3 and A4), and p-Src (C3 and C4) that correlated directly to a reduction in the activity of ERK1/2 (p-ERK1/2: E3 and E4).The reduced level of phosphorylated proteins occurred without changes in the level of total EGFR (B2, B3 and B4), total Src (D2, D3 and D4) or total ERK1/2 (F2, F3 and F4). The numbers below the band in each lane represents the average ± SD (n = 3) of the relative density of each band when normalized to the wildtype BALB/c control value arbitrarily set at 1.
An obvious consideration with the potential use of TSV in pediatric patients is deleterious side effects due to disruption of developmentally critical proteins. A particular concern is the effect of TSV on angiogenesis. The inhibition of KDR or VEGFR2 ideally would not cause a reduction in activity of the receptor much below wildtype levels where inhibition of angiogenesis would likely have significant detrimental effects on normal kidney development. The rapidly progressive bpk was used to evaluate the inhibitory effect of KDR during postnatal development.
To investigate the level of angiogenesis inhibition we measured the change in VEGFR2 (KDR) activity in bpk (Figure 5). As Figure 5 shows, the amount of active p-KDR in TSV-treated kidneys at 15 mg/kg per day is not significantly different from the level of untreated wild type controls. The expression data of CD-31 or PECAM-1, an endothelial cell protein reveal a decreased expression in TSV treated bpk cystic kidneys. These data demonstrate that inhibition of angiogenesis as assessed by p-KDR translated into a reduced expression of CD-31. As seen in Figure 6, the level of CD-31, is increased around cystic CT (6B) when compared to age-matched BALB/c animals (6A). It appears that TSV treatment eliminates abnormal peritubular CD-31 expression but still permits the robust expression of CD-31 in glomeruli (6C) of TSV treated bpk kidneys.
Figure 5.
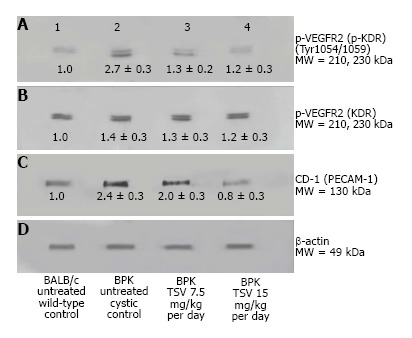
VEGFR2 (KDR) and CD-31 expression in BPK. Western analysis of the renal expression of VEGF2 (KDR) in BPK (B), and active (p-KDR) in BPK kidneys (A) demonstrate an increased phosphorylation of KDR in untreated cystic kidneys (2) compared to BALB/c untreated wildtype controls (A1). TSV treatment of BPK at 7.5 (A3) and 15 mg/kg per day (A4) respectively, reduced the level of p-KDR in a dose dependent manner to near normal (wildtype) levels. This reduced p-KDR correlated directly with expression levels of CD-31 shown in (C) which was also reduced in a dose dependent manner with TSV treatment. The numbers below the band in each lane represents the average ± SD of the relative density of each band when normalized to the wildtype BALB/c control value arbitrarily set at 1. D is the loading control. Each average represents a minimum of n = 3 individual westerns. These data are confirmed by the morphology of CD-1 stained BPK kidneys shown in Figure 6.
Figure 6.
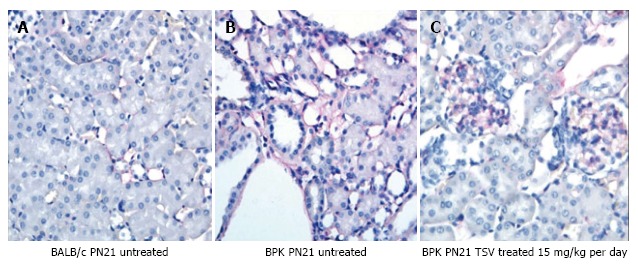
Immuno-histological analysis of CD-31 expression in tesevatinib-treated BPK kidneys. Expression of CD-31 (PECAM1) (red), a marker of endothelium, in PN21 wild-type BALB/c kidneys (A) reveals fine thin lines of endothelial elements that run along the nephrons. Untreated BPK kidneys (B) demonstrate prominent expression of CD-31 along the tubules and pronounced expression surrounding cystic lesions. TSV-treated cystic kidneys at 15 mg/kg per day (C) reveal a marked reduction in CD31 expression along the tubules to near normal levels but maintain robust glomerular staining of CD-31.
PCK
The orthologue of human ARPKD, the PCK, and Sprague Dawley wildtype controls were administered TSV by oral gavage from PN30 to PN90 and the same parameters assessed following TSV administration as in the bpk.
Figure 7 reveals that TSV administration to PCK animals from PN30 to PN90 (61 doses) results in a dose dependent reduction in whole kidney size when compared to untreated PCK. The change in whole kidney size is also reflected in the dose dependent significant decrease in TKW from 8.02 ± 0.32 g in untreated PCK to 6.21 ± 0.44 (P < 0.001) and 5.06 ± 0.43 g (P < 0.001) with TSV at 7.5 and 15 mg/kg per day respectively as shown in Table 2. The reduction in whole kidney size and TKW is associated with markedly improved renal morphology shown in Figure 8. Accordingly, the CI was significantly reduced by treatment as seen in Table 2. The improved renal morphology was mirrored by a dose dependant improvement in hepatic morphology as seen in Figure 9 which was reflected in a reduced LW/BW ratio listed in Table 2.
Figure 7.
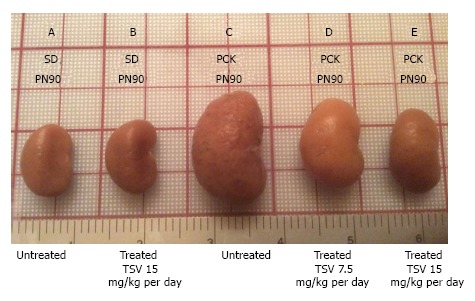
Tesevatinib treatment in the PCK model of autosomal recessive polycystic kidney disease. Compared to wild-type SD control kidneys (A) at PN90, PCK cystic kidneys (C) are significantly enlarged by PN90. TSV treatment results in a dose-dependent reduction in the overall kidney size at doses of 7.5 mg/kg per day (D) and 15 mg/kg per day (E) when compared to untreated PCK cystic kidneys at PN90 (C). Treatment of wildtype SD with 15 mg/kg per day did not result in significant reduction of overall kidney size (B) compared to untreated wildtype SD animals. This correlates directly with the total kidney weight (TKW) of each group listed in Table 2 (Treatment interval: PN30-PN90). TSV: Tesevatinib.
Table 2.
PCK summary data
| SD PN-90 Sham n = 12 | SD PN-90 VEH n = 10 | SD PN-90 7.5 mg/kg per day n = 8 | SD PN-90 15 mg/kg per day n = 8 | PCK PN-90 Sham n = 10 | PCK PN-90 VEH n = 12 | PCK PN-90 7.5 mg/kg per day n = 10 | PCK PN-90 15 mg/kg per day n = 12 | |
| BW (g) | 375.9 ± 12.1 | 380 ± 19.46 | 375 ± 23.23 | 377 ± 13.31 | 434 ± 16.6 | 417.8 ± 21.0 | 413.4 ± 18.7 | 406.0 ± 18.5 |
| TKW (g) | 3.63 ± 0.46 | 3.70 ± 0.22 | 3.71 ± 0.16 | 3.85 ± 0.32 | 8.02 ± 0.32 | 7.54 ± 0.52 | 6.21 ± 0.44e | 5.06 ± 0.43e,f |
| KW/BW (%) | 0.96 ± 0.10 | 0.97 ± 0.04 | 0.99.± 0.6 | 0.91 ± 0.33 | 1.84 ± 0.09 | 1.81 ± 0.05 | 1.50 ± 0.11b | 1.18 ± 0.1c,e |
| CI | NA | NA | NA | NA | 7.44 ± 0.8 | 7.0 ± 0.6 | 5.3 ± 1.0e | 3.8 ± 0.9d,e |
| BUN (mg/dL) | 21.17 ± 1.2 | 22.40 ± 1.6 | 23.50 ± 1.7 | 23.6 ± 1.9 | 49.2 ± 12.7 | 48.88 ± 15.3 | 36.40 ± 4.4b | 24.17 ± 8.7c,e |
| Cr (mg/d) | 0.33 ± 0.08 | 0.34 ± 0.12 | 0.36 ± 0.05 | 0.34 ± 0.08 | 0.67 ± 0.1 | 0.66 ± 0.12 | 0.51 ± 0.08b | 0.38 ± 0.07c,e |
| UCA 12 h | 2778 ± 159 | 2983 ± 421 | 2749 ± 63.1 | 2712 ± 113 | 1390 ± 221.9 | 1448 ± 236 | 2196 ± 169e | 2630 ± 214d,e |
| LW/BW (%) | 4.3 ± 0.47 | 4.2 ± 0.7 | 4.13 ± 0.57 | 4.00 ± 0.65 | 6.02 ± 0.35 | 6.08 ± 0.42 | 5.40 ± 0.21b | 4.92 ± 0.27c,e |
aP < 0.05,
P < 0.01,
P < 0.001 vs untreated (Sham) PCK;
P < 0.05,
P < 0.01;
P < 0.001 vs TSV treatment at 7.5. CI: Cystic index; UCA: Urinary concentration ability at 12 h; LW/BW: Measure of cyst burden; KW/BW: Kidney weight to body weight; TKW: Total kidney weight.
Figure 8.
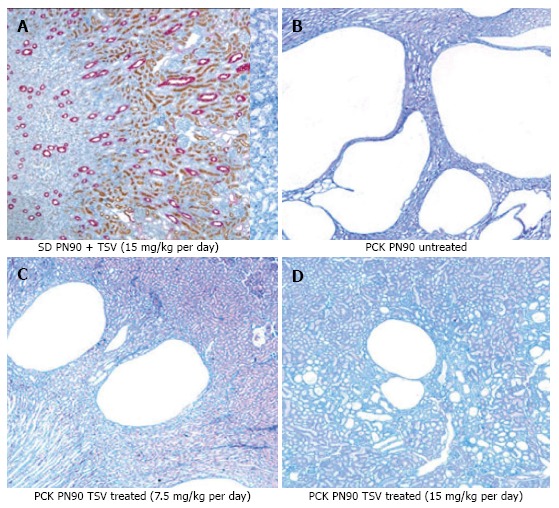
Renal morphology of tesevatinib-treated PCK kidneys. Histological analysis of HE stained PCK kidneys reveals a marked decrease in the size and number of collecting tubule cysts following treatment with TSV at 7.5 mg/kg per day (C) and 15 mg/kg per day (D) when compared to untreated cystic kidneys (B). Treatment of control Sprague Dawley kidneys at 15 mg/kg per day (A) revealed no histological abnormalities (Treatment Interval: PN30-PN90). TSV: Tesevatinib.
Figure 9.
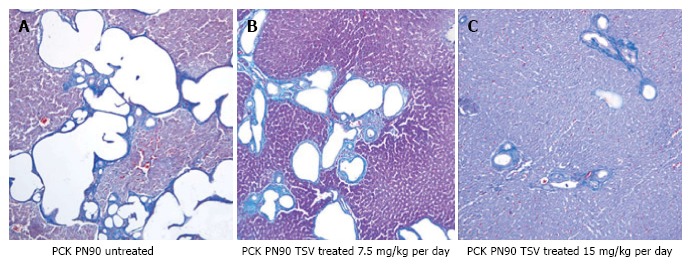
Liver morphology of tesevatinib-treated PCK mice. Histological analysis of PCK livers reveals a marked decrease in the size and number of biliary ductal cysts following treatment with tesevatinib at 7.5 mg/kg per day (B) and 15 mg/kg per day (C) when compared to untreated PCK livers (A) (Treatment interval: PN30-PN90).
Results summarized in Table 2, demonstrate that compared to untreated PCK animals (n = 10), cystic animals treated with TSV showed an 18%, (P < 0.01) and 36% (P < 0.001) reduction in KW/BW ratio, reductions in CI of 29%, (P < 0.001) and 49%, (P < 0.001) and a 10% (P < 0.05) and 18% (P < 0.001) reduction in LW/BW ratios. There was a dose dependent improvement in KW/BW of 26% and CI of 28% with increased dosage from 7.5 to 15 mg/kg per day. Compared to untreated PCK animals, renal function parameters also demonstrated significant dose dependent improvement with TSV treatment. BUN levels decreased by 26% (P < 0.01), and 49% (P < 0.001), CR levels decreased by 24% (P < 0.01) and 43% (P < 0.001) while UCA improved by 36% (P < 0.001) and 47% (P < 0.001) following TSV treatment with 7.5 and 15 mg/kg per day respectively. Treatment of SD controls shows no significant differences in assessed parameters at either TSV dose.
The improvement in morphology and renal function correlates with target verification shown in Figure 10. The activity (phosphorylation) of ErbB2 (Figure 10A3 and A4) Src (Figure 10C3 and 10C4) ERK1/2 (Figure 10E3 and 10E4) are reduced in a dose dependent manner with treatment with TSV at 7.5 and 15 mg/kg per day respectively. Figure 11 demonstrates that TSV reduces pKDR levels which correlates with a reduced CD-31 level with TSV therapy.
Figure 10.
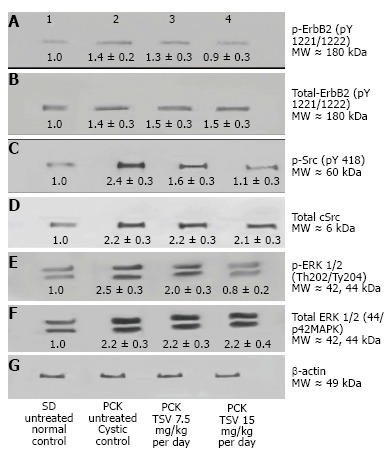
Target validation in BPK. Western analysis of the MAPK pathway activity of EGFR (p-EGFR), cSrc (pSrc), and ERK1/2 (p-ERK ½) reveals that BPK cystic kidneys have significantly increased levels of active or phosphorylated p-EGFR (A2), pSrc (C2) and ERK1/2 (E2) compared to wild-type BALB/c control levels in lanes (A1, C1 and E1) respectively. TSV treatment at 7.5 or 15 mg/kg per day resulted in a dose dependent reduction in the phosphorylation or activity of p-EGFR (A3 and A4), and p-Src (C3 and C4) that correlated directly to a reduction in the activity of ERK1/2 (p-ERK1/2: E3 and E4).The reduced level of phosphorylated proteins occurred without changes in the level of total EGFR (B2, B3 and B4), total Src (D2, D3 and D4) or total ERK1/2 (F2, F3 and F4). The numbers below the band in each lane represents the average ± SD (n = 3) of the relative density of each band when normalized to the wildtype BALB/c control value arbitrarily set at 1.
Figure 11.
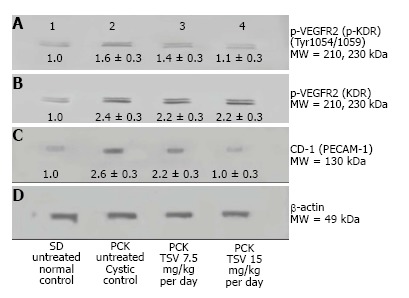
VEGFR2 (KDR) and CD-31 expression in PCK. Western analysis of the renal expression of VEGF2 (KDR) in PCK (B), and active (p-KDR) in PCK kidneys (A) demonstrate an increased phosphorylation of KDR in untreated cystic PCK kidneys (A2) compared to SD untreated wildtype controls (A1). TSV treatment of PCK at 7.5 (A3) and 15 mg/kg per day (A4) respectively, reduced the level of p-KDR in a dose dependent manner to near normal (wildtype) levels. This reduced p-KDR correlated directly with expression levels of CD-31 shown in C, which was also reduced in a dose dependent manner with TSV treatment. The numbers below the band in each lane represents the average ± SD of the relative density of each band when normalized to the wildtype SD control value arbitrarily set at 1. D is the loading control. Each average represents a minimum of n = 3 individual westerns.
Toxicology
Treatment of bpk mice and PCK rats with TSV demonstrated no apparent morphological evidence of renal or hepatic toxicity, no compound-related deaths, and no microscopic changes in heart, spleen, stomach, pancreas or thymus with 7.5 mg/kg per day or 15 mg/kg per day dosing.
In sum, the use of TSV at 7.5 or 15 mg/kg per day results in a significant improvement in all assessed parameters compared to untreated or vehicle treated cystic animals in both bpk and PCK animals without obvious evidence of toxicity or detrimental effect of KDR inhibition.
DISCUSSION
A long-recognized clinical observation is the overlap of phenotypic features that at times leads to difficulty delineating ARPKD from ADPKD in pediatric patients[3,5,16]. Data now reveal that the pathophysiology of the two diseases have significant overlap. Recent evidence revealed a complex set of interactions between PKHD1, PKD1 and PKD2 that occur both at the molecular and cellular level[5,15,20-23]. Despite the identification of the causative genes responsible for ARPKD and ADPKD, the precise function of these genes and their protein products is still unclear.
This complexity, in part, is due in part to many novel attributes of the genes including: (1) the complexity of their protein structures; (2) the large size of PKHD1 and PKD1; (3) the multiple transcripts produced by these genes; (4) the interaction of the PKD proteins; (5) the multiple intracellular sites of PKD protein localization; and (6) the participation of these proteins in a number of multimeric protein complexes[5,17-19,44]. The lack of functional PC1, PC2 or fibrocystin is embryonically lethal, indicating and PKD begins in utero[25]. ARPKD and ADPKD share common phenotypic abnormalities and intersecting signaling pathways whose disruption leads to cyst formation[3,5,16,23,34]. Despite this rudimentary understanding, emerging molecular and cellular insights into the pathophysiology of PKD is beginning to translate into unique disease-specific, targeted therapies.
Previous work in our laboratory demonstrated that the combination of therapies that inhibit both the epidermal growth factor receptor (EGFR) autophosphorylation and EGFR ligand availability provide a novel example of effective combination therapy in murine PKD[37]. Extending this concept, we reasoned that identification of signaling intermediates which: (1) regulate multiple steps in a single pathophysiological pathway; in addition to acting as; and (2) an integration point for multiple signaling cascades may provide even greater benefit than EGFR combination therapy noted above. Key integration points for targeting to prevent disease progression include the activation of the EGFR axis, c-Src, and VEGFR2 by specific kinases[3,34,40,45-50] and cAMP signaling[51-53]. This led us to examine the potential of a novel multi-kinase inhibitor, TSV, in preventing progression of ARPKD.
Given the differences in route of administration, the speed of disease development and the fact that the measure of the disease burden in the kidney and liver are model specific make direct comparison of the results in bpk and PCK unreliable. It is also difficult to compare these results to other interventional therapies we have attempted in these models because we tested two doses only on the basis of recommendations from Kadmon. Never the less the current study demonstrates that TSV ameliorates the progression of renal cystic disease in two well characterized rodent models of ARPKD. Further, TSV’s inhibition of the phosphorylation of c-Src, EGFR and ErbB2 led to significant reduction of MAPK activity (ERK 1/2 phosphorylation), and resultant cellular proliferation. In addition to amelioration of renal cystic disease, TSV also led to significant reduction in the biliary ductal abnormalities characteristic of ARPKD in BPK and in the LW/BW ratios of PCK. The results of the current study also indicate that the use of a multi-kinase inhibitor that targets angiogenesis driven by KDR or VEGFR2 can be an effective and safe therapy during postnatal renal maturation and development.
Preliminary data from the ongoing phase II clinical trial of TSV (KD-019) for treatment of ADPKD (https//ClinicalTrials.gov/ADPKD: NCT01559363) presented at the American Society of Nephrology 2015 annual meeting reported: (1) QTc prolongation to ≥ 485 ms was seen in 2/8 pts treated with 100 mg/d; (2) The half-life in adults allowed for intermittent dosing which improved tolerability; (3) The most common AEs were those associated with EGFR kinase inhibition: diarrhea; nausea; and acneiform rash; and (4) 50 mg/d appeared to be safe and well tolerated and additional patients were being enrolled at this dose to confirm the safety profile[54].
On balance, these data suggest that TSV may be a safe and effective therapy for childhood ARPKD.
COMMENTS
Background
The study detailed here was designed to ascertain if a multi-kinase inhibitor “tesevatinib” would effectively slow the growth of renal cysts and reduce the loss of renal function in two well characterized models of autosomal recessive polycystic kidney disease (ARPKD). This compound targets the EGFR axis, cSrc and it inhibits angiogenesis by preventing phosphorylation of VEGFR2 (also called KDR). The bpk mouse was chosen to examine the effect of KDR inhibition on post-natal development and the orthologous PCK was chosen to examine the effect of oral dosing on inhibition of the target signaling pathways. Tesevatinib inhibit renal cyst growth in both models without overt signs of toxicity.
Research frontiers
This study demonstrated that there is indeed considerable overlap in the aberrant signaling cascades in ARPKD and autosomal dominant polycystic kidney disease (ADPKD). This raises the hope that the lessons learned from therapeutic interventions in ADPKD can be adapted for ARPKD patients.
Innovations and breakthroughs
The most exciting result of this study is that inhibition of angiogenesis, an important component of cyst growth, can now be added to arsenal to slow cyst growth when activity (phosphorylation) is cautiously reduced to normal or wildtype levels. This inhibition with tesevatinib contributed to the amelioration of cyst growth without overt signs of toxicity even in early post-natal development.
Applications
The value of these results is that effective therapies developed and tested for ADPKD patients can be for use in ARPKD patients. A multi-kinase drug with multiple targets theoretically would reduce the amount of compound necessary to achieve equivalent results compared to amounts needed with single target drugs. This effectively reduces the exposure to the compounds and would result in reduced toxicity and potentially allow long term use of the therapeutic compound. This should accelerate the use of therapeutic compounds in clinical trials of ARPKD patients.
Terminology
The activity of a kinase refers to the phosphorylation state of the molecule. It generally means the target molecule becomes phosphorylated and this results in increased activity until it is de-phosphorylated by another molecule. A multi-kinase inhibitor is a compound that targets multiple signaling components simultaneously and can prevent phosphate from binding to and activating the signaling components thus rendering the molecule useless. This occurs simultaneously and provides a means hitting two or more targets with a single compound.
Peer-review
This paper was well-written.
Footnotes
Supported by The PKD research program is provided by the Children’s Research Institute, the Lillian Goldman Charitable Trust, Ellsworth Family and Children’s Foundation of Children’s Hospital and Health System of Wisconsin.
Institutional animal care and use committee statement: All animal experiments were conducted in accordance with policies of the NIH Guide for the Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee (IACUC) of the Medical College of Wisconsin. The IACUC at the Medical College of Wisconsin is properly appointed according to PHS policy IV.A.3.a and is qualified through the experience and expertise of its members to oversee the Institution’s animal care and use program. The Animal Welfare Assurance for the Medical College of Wisconsin is A3102-01. Specific protocols used in this study were approved by the Medical College of Wisconsin IACUC (approved protocols are AUA 4278 and AUA 4179).
Conflict-of-interest statement: The authors have no conflict of interest to declare. Conflict of Interest in Research statements are on file with the institution as per Medical College of Wisconsin policy #RS.GN.020.
Data sharing statement: Data sets and statistical methods are available upon request from the corresponding author.
Manuscript source: Invited manuscript
Specialty type: Urology and nephrology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): A, A, A
Grade B (Very good): 0
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: February 15, 2017
First decision: March 8, 2017
Article in press: May 13, 2017
P- Reviewer: Friedman EA, Navarro-Gonzalez JF, Zuo L S- Editor: Song XX L- Editor: A E- Editor: Lu YJ
References
- 1.Gunay-Aygun M, Font-Montgomery E, Lukose L, Tuchman Gerstein M, Piwnica-Worms K, Choyke P, Daryanani KT, Turkbey B, Fischer R, Bernardini I, et al. Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology. 2013;144:112–121.e2. doi: 10.1053/j.gastro.2012.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sweeney W, Avner ED. Polycystic Kidney Disease, Autosomal Recessive. Gene Reviews [Internet] In: Pagon RA, Adam MP, Ardinger HH, editors. Seattle (WA): University of Washington; 2001. p. [updated 2016]. Available from: http://www.ncbi.clm.nih.gov/books/NBK1326. [Google Scholar]
- 3.Sweeney WE, Avner ED. Pathophysiology of childhood polycystic kidney diseases: new insights into disease-specific therapy. Pediatr Res. 2014;75:148–157. doi: 10.1038/pr.2013.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cramer MT, Guay-Woodford LM. Cystic kidney disease: a primer. Adv Chronic Kidney Dis. 2015;22:297–305. doi: 10.1053/j.ackd.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Sweeney WE, Gunay-Aygun M, Patil A, Avner ED. Childhood Polycystic Kidney Disease. In: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, Emma F, Goldstein S, editors. Pediatr Nephrol. 2016;31:1–58. [Google Scholar]
- 6.Gunay-Aygun M, Tuchman M, Font-Montgomery E, Lukose L, Edwards H, Garcia A, Ausavarat S, Ziegler SG, Piwnica-Worms K, Bryant J, et al. PKHD1 sequence variations in 78 children and adults with autosomal recessive polycystic kidney disease and congenital hepatic fibrosis. Mol Genet Metab. 2010;99:160–173. doi: 10.1016/j.ymgme.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guay-Woodford LM, Muecher G, Hopkins SD, Avner ED, Germino GG, Guillot AP, Herrin J, Holleman R, Irons DA, Primack W, et al. D. T, Waldo FB, Lunt PW, Zerres K. The severe perinatal form of autosomal recessive polycystic kidney disease maps to chromosome 6p21.1-p12: Implications for genetic counseling. Am J Hum Genet. 1995;56:1101–1107. [PMC free article] [PubMed] [Google Scholar]
- 8.Zerres K, Rudnik-Schoneborn S, Steinkamm C, Becker J, Mucher G. Autosomal recessive polycystic kidney disease. J Mol Med. 1998;76:303–309. doi: 10.1007/s001090050221. [DOI] [PubMed] [Google Scholar]
- 9.Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70:1305–1317. doi: 10.1086/340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 11.Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Prospective change in renal volume and function in children with ADPKD. Clin J Am Soc Nephrol. 2009;4:820–829. doi: 10.2215/CJN.02810608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ong AC, Harris PC. Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int. 2005;67:1234–1247. doi: 10.1111/j.1523-1755.2005.00201.x. [DOI] [PubMed] [Google Scholar]
- 13.Streets AJ, Magayr TA, Huang L, Vergoz L, Rossetti S, Simms RJ, Harris PC, Peters DJ, Ong AC. Parallel microarray profiling identifies ErbB4 as a determinant of cyst growth in ADPKD and a prognostic biomarker for disease progression. Am J Physiol Renal Physiol. 2017;312:F577–F588. doi: 10.1152/ajprenal.00607.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60:321–337. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest. 2014;124:2315–2324. doi: 10.1172/JCI72272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sweeney WE, Avner ED. Diagnosis and management of childhood polycystic kidney disease. Pediatr Nephrol. 2011;26:675–692. doi: 10.1007/s00467-010-1656-1. [DOI] [PubMed] [Google Scholar]
- 17.Wilson PD. Polycystic kidney disease: new understanding in the pathogenesis. Int J Biochem Cell Biol. 2004;36:1868–1873. doi: 10.1016/j.biocel.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 18.Zhang MZ, Mai W, Li C, Cho SY, Hao C, Moeckel G, Zhao R, Kim I, Wang J, Xiong H, et al. PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc Natl Acad Sci USA. 2004;101:2311–2316. doi: 10.1073/pnas.0400073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y, Dai XQ, Li Q, Chen CX, Mai W, Hussain Z, Long W, Montalbetti N, Li G, Glynne R, et al. Kinesin-2 mediates physical and functional interactions between polycystin-2 and fibrocystin. Hum Mol Genet. 2006;15:3280–3292. doi: 10.1093/hmg/ddl404. [DOI] [PubMed] [Google Scholar]
- 20.Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, Leonhard WN, van de Wal A, Ward CJ, Verbeek S, Deruiter MC, Breuning MH, de Heer E, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet. 2004;13:3069–3077. doi: 10.1093/hmg/ddh336. [DOI] [PubMed] [Google Scholar]
- 21.Gallagher AR, Germino GG, Somlo S. Molecular advances in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:118–130. doi: 10.1053/j.ackd.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan HF, Gainullin VG, Rossetti S, Torres VE, Harris PC. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest. 2012;122:4257–4273. doi: 10.1172/JCI64313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fedeles SV, Gallagher AR, Somlo S. Polycystin-1: a master regulator of intersecting cystic pathways. Trends Mol Med. 2014;20:251–260. doi: 10.1016/j.molmed.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris PC, Bae KT, Rossetti S, Torres VE, Grantham JJ, Chapman AB, Guay-Woodford LM, King BF, Wetzel LH, Baumgarten DA, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2006;17:3013–3019. doi: 10.1681/ASN.2006080835. [DOI] [PubMed] [Google Scholar]
- 25.Orskov B, Christensen KB, Feldt-Rasmussen B, Strandgaard S. Low birth weight is associated with earlier onset of end-stage renal disease in Danish patients with autosomal dominant polycystic kidney disease. Kidney Int. 2012;81:919–924. doi: 10.1038/ki.2011.459. [DOI] [PubMed] [Google Scholar]
- 26.Chapman AB. The fetal environment: a critical phase that determines future renal outcomes in autosomal dominant polycystic kidney disease. Kidney Int. 2012;81:814–815. doi: 10.1038/ki.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cadnapaphornchai MA. Hypertension in children with autosomal dominant polycystic kidney disease (ADPKD) Curr Hypertens Rev. 2013;9:21–26. doi: 10.2174/1573402111309010004. [DOI] [PubMed] [Google Scholar]
- 28.Selistre L, de Souza V, Ranchin B, Hadj-Aissa A, Cochat P, Dubourg L. Early renal abnormalities in children with postnatally diagnosed autosomal dominant polycystic kidney disease. Pediatr Nephrol. 2012;27:1589–1593. doi: 10.1007/s00467-012-2192-y. [DOI] [PubMed] [Google Scholar]
- 29.Hartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics. 2014;134:e833–e845. doi: 10.1542/peds.2013-3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoyer PF. Clinical manifestations of autosomal recessive polycystic kidney disease. Curr Opin Pediatr. 2015;27:186–192. doi: 10.1097/MOP.0000000000000196. [DOI] [PubMed] [Google Scholar]
- 31.Grantham JJ. Rationale for early treatment of polycystic kidney disease. Pediatr Nephrol. 2015;30:1053–1062. doi: 10.1007/s00467-014-2882-8. [DOI] [PubMed] [Google Scholar]
- 32.Trowe T, Boukouvala S, Calkins K, Cutler RE, Fong R, Funke R, Gendreau SB, Kim YD, Miller N, Woolfrey JR, et al. EXEL-7647 inhibits mutant forms of ErbB2 associated with lapatinib resistance and neoplastic transformation. Clin Cancer Res. 2008;14:2465–2475. doi: 10.1158/1078-0432.CCR-07-4367. [DOI] [PubMed] [Google Scholar]
- 33.Gendreau SB, Ventura R, Keast P, Laird AD, Yakes FM, Zhang W, Bentzien F, Cancilla B, Lutman J, Chu F, et al. Inhibition of the T790M gatekeeper mutant of the epidermal growth factor receptor by EXEL-7647. Clin Cancer Res. 2007;13:3713–3723. doi: 10.1158/1078-0432.CCR-06-2590. [DOI] [PubMed] [Google Scholar]
- 34.Sweeney WE, von Vigier RO, Frost P, Avner ED. Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol. 2008;19:1331–1341. doi: 10.1681/ASN.2007060665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nauta J, Ozawa Y, Sweeney WE, Jr Rutledge JC, Avner ED. Renal and biliary abnormalities in a new murine model of autosomal recessive polycystic kidney disease. Pediatr Nephrol. 1993;7:163–172. doi: 10.1007/BF00864387. [DOI] [PubMed] [Google Scholar]
- 36.Nauta J, Sweeney WE, Rutledge JC, Avner ED. Biliary epithelial cells from mice with congenital polycystic kidney disease are hyperresponsive to epidermal growth factor. Pediatr Res. 1995;37:755–763. doi: 10.1203/00006450-199506000-00014. [DOI] [PubMed] [Google Scholar]
- 37.Sweeney WE, Hamahira K, Sweeney J, Garcia-Gatrell M, Frost P, Avner ED. Combination treatment of PKD utilizing dual inhibition of EGF-receptor activity and ligand bioavailability. Kidney Int. 2003;64:1310–1319. doi: 10.1046/j.1523-1755.2003.00232.x. [DOI] [PubMed] [Google Scholar]
- 38.Nemo R, Murcia N, Dell KM. Transforming growth factor alpha (TGF-alpha) and other targets of tumor necrosis factor-alpha converting enzyme (TACE) in murine polycystic kidney disease. Pediatr Res. 2005;57:732–737. doi: 10.1203/01.PDR.0000159513.51898.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katsuyama M, Masuyama T, Komura I, Hibino T, Takahashi H. Characterization of a novel polycystic kidney rat model with accompanying polycystic liver. Exp Anim. 2000;49:51–55. doi: 10.1538/expanim.49.51. [DOI] [PubMed] [Google Scholar]
- 40.Talbot JJ, Song X, Wang X, Rinschen MM, Doerr N, LaRiviere WB, Schermer B, Pei YP, Torres VE, Weimbs T. The cleaved cytoplasmic tail of polycystin-1 regulates Src-dependent STAT3 activation. J Am Soc Nephrol. 2014;25:1737–1748. doi: 10.1681/ASN.2013091026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sato Y, Harada K, Kizawa K, Sanzen T, Furubo S, Yasoshima M, Ozaki S, Ishibashi M, Nakanuma Y. Activation of the MEK5/ERK5 cascade is responsible for biliary dysgenesis in a rat model of Caroli’s disease. Am J Pathol. 2005;166:49–60. doi: 10.1016/S0002-9440(10)62231-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int. 2000;57:33–40. doi: 10.1046/j.1523-1755.2000.00829.x. [DOI] [PubMed] [Google Scholar]
- 43.Nakanishi K, Sweeney WE, Jr, Zerres K, Guay-Woodford LM, Avner ED. Proximal tubular cysts in fetal human autosomal recessive polycystic kidney disease. J Am Soc Nephrol. 2000;11:760–763. doi: 10.1681/ASN.V114760. [DOI] [PubMed] [Google Scholar]
- 44.Wilson PD. Polycystic kidney disease. N Engl J Med. 2004;350:151–164. doi: 10.1056/NEJMra022161. [DOI] [PubMed] [Google Scholar]
- 45.Bello-Reuss E, Holubec K, Rajaraman S. Angiogenesis in autosomal-dominant polycystic kidney disease. Kidney Int. 2001;60:37–45. doi: 10.1046/j.1523-1755.2001.00768.x. [DOI] [PubMed] [Google Scholar]
- 46.Yamaguchi T, Nagao S, Wallace DP, Belibi FA, Cowley BD, Pelling JC, Grantham JJ. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003;63:1983–1994. doi: 10.1046/j.1523-1755.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- 47.Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004;279:40419–40430. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- 48.Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, Helmkamp GM, Grantham JJ. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int. 2004;66:964–973. doi: 10.1111/j.1523-1755.2004.00843.x. [DOI] [PubMed] [Google Scholar]
- 49.Huang JL, Woolf AS, Long DA. Angiogenesis and autosomal dominant polycystic kidney disease. Pediatr Nephrol. 2013;28:1749–1755. doi: 10.1007/s00467-012-2305-7. [DOI] [PubMed] [Google Scholar]
- 50.Martins DP, Souza MA, Baitello ME, Nogueira V, Oliveira CI, Pinhel MA, Caldas HC, Filho MA, Souza DR. Vascular endothelial growth factor as an angiogenesis biomarker for the progression of autosomal dominant polycystic kidney disease. Genet Mol Res. 2016;15 doi: 10.4238/gmr.15017623. [DOI] [PubMed] [Google Scholar]
- 51.Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med. 2003;9:1323–1326. doi: 10.1038/nm935. [DOI] [PubMed] [Google Scholar]
- 52.Wallace DP. Cyclic AMP-mediated cyst expansion. Biochim Biophys Acta. 2011;1812:1291–1300. doi: 10.1016/j.bbadis.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014;25:18–32. doi: 10.1681/ASN.2013040398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rastogi A, Rosner MH, Barash I, Steinman TI, El-Zoghby ZM, Nurko S, Goldberg S, El-Meanawy A, El Ters M, Roche M, et al. Tyrosine Kinase Inhibitor Tesevatinib for Patients with Autosomal Dominant Polycystic Kidney Disease. J. Am Soc Nephrol. 2015:830A. [Google Scholar]