

Abstract
Cathepsin D is reportedly to be closely associated with tumor development, migration, and invasion, but its pathological mechanism is not fully elucidated. We aimed to evaluate phenotypic changes and molecular events in response to cathepsin D knockdown. Lowering endogenous cathepsin D abundance (CR) induced senescence in HeLa cells, leading to reduced rate of cell proliferation and impaired tumorigenesis in a mouse model. Quantitative proteomics revealed that compared with control cells (EV), the abundances of several typical lysosomal proteases were decreased in the lysosomal fraction in CR cells. We further showed that cathepsin D knockdown caused increased permeability of lysosomal membrane and reactive oxygen species accumulation in CR cells, and the scavenging of reactive oxygen species by antioxidant was able to rescue cell senescence. Despite the increased reactive oxygen species, the proteomic data suggested a global reduction of redox-related proteins in CR cells. Subsequent analysis indicated that the transcriptional activity of nuclear factor erythroid-related factor 2 (Nrf2), which regulates the expression of groups of antioxidant enzymes, was down-regulated by cathepsin D knockdown. Importantly, Nrf2 overexpression significantly reduced cell senescence. Although transient oxidative stress promoted the accumulation of Nrf2 in the nucleus, we showed that the Nrf2 protein exited nucleus if oxidative stress persisted. In addition, when cathepsin D was transiently knocked down, the cathepsin-related events followed a sequential order, including lysosomal leakage during the early stage, followed by oxidative stress augmentation, and ultimately Nrf2 down-regulation and senescence. Our results suggest the roles of cathepsin D in cancer cells in maintaining lysosomal integrity, redox balance, and Nrf2 activity, thus promoting tumorigenesis. The MS Data are available via ProteomeXchange with identifier PXD002844.
Cathepsin D, a member of the cathepsin superfamily, is a lysosome-residing aspartic protease. Because it was originally envisaged this enzyme requires acidic pH environment for its maximal catalytic activity (1, 2), its physiological role was initially studied in lysosomes. Lysosomes are the major apparatus for recycling damaged proteins and subcellular organelles through autophagosome-lysosome fusion and chaperone-mediated autophagy, whereas cathepsin D was found to facilitate protein homeostasis (3, 4). For instance, cathepsin D reportedly digests protein aggregates, like α-synuclein and β-amyloid, and it is the major protease for the generation of vesoinhibins (5–7).
Although numerous research support cathepsin D's role in maintaining tissue homeostasis and metabolism, the protein also has been shown to mediate stress-induced apoptosis in several cells. Diess et al. reported that a cathepsin D inhibitor, pepstatin A, constrained the apoptosis induced by interferon-γ and Fas/APO-1, while lowering the abundance of cathepsin D mRNA exerted the similar phenotype (8). The direct injection of cathepsin D into cytosol of mouse fibroblasts resulted in cytochrome c release and apoptosis, whereas this process was abrogated by pepstatin A or caspase-3 inhibitor, suggesting that the apoptosis mediated by cathepsin D was likely dependent upon caspase-3 activity (9). A mechanistic study showed that in response to TNF treatment, cathepsin D was colocalized with Bid in endosome, in which 9kDa tBid was generated through cleavage, and cytochrome c release and caspase-9 activity were augmented (10). Castino et al. revealed that cathepsin D activity facilitated the induction of apoptosis under oxidative stress through promoting Bax relocation to mitochondrial membrane and mitochondrial dysfunction (11). Thus, cathepsin D indeed plays a specific role in cell apoptosis, but its involvement in other cellular processes, like cellular senescence and autophagy, is not well studied yet.
In addition to its physiological role in lysosome and cytosol, cathepsin D is often found to be overexpressed in several cancer cells as well as tumor tissues (12–14). As mutated and noncatalytic cathepsin D could stimulate the growth of cancer cells (15), Vetvicka et al. hypothesized that procathepsin D could bind to a cell surface receptor, then resulted in the consequent signaling changes and enhancement of tumorigenesis (16). However, despite a significant effort during the past years, the potential cathepsin D receptor remains elusive. The mechanistic links of cathepsin D and tumorigenesis are far from well understood. It is generally accepted that the status changes of cathepsin D gene expression are likely to bring a network response within cells (9, 17–20), therefore, overall monitoring the initial and consequent molecular events is necessary for mechanism study. Some investigators attempted to profile the proteomic responses in the cells or tissues in response to cathepsin D abundance changes. Martin et al. employed a quantitative proteomics approach, iTRAQ, to measure the proteomic changes induced by cathepsin D inhibition in macrophage during bacterial infection and claimed that SOD2, a superoxide scavenging protein, and HSPA5, an ER stress marker, were positively regulated by the inhibition (21). Sabine et al. adopted similar approach to conduct proteomic study in crude synaptosomal fraction of brains in cathepsin D−/− mice, and revealed massive changes of the proteins related with cytoskeleton, which might account for the aberrant focal adhesion assembly in the cathepsin D knockout fibroblasts (22). Current information regarding proteomic response to perturbation of cathepsin D protein abundance, however, is still not good enough to abstract a mechanism figure that clearly clarifies how the cells or organelles cope with the micro-environmental changes induced by the abundance changes of cathepsin D.
Previous studies clearly showed that quantitative proteomics offered insights into the biologic issues related with cathepsin D, yet there are two concerns that we have to think of. First of all, because cathepsin D is frequently up-regulated in cancer cells or tumor tissues, majority of work in the field so far focused on the mechanistic study based on the cell model with cathepsin D up-regulation. What is happening once cancer cells are deprived of sufficient cathepsin D? Glondu et al. observed antisense-mediated down-regulation of cathepsin D did not change the in vitro invasion capacity of breast cancer cell MDA-MB-231 (23), whereas with the similar approach, Tedone et al. found that cathepsin D down-regulation reduced invasiveness of MCF-7, another breast cancer cell line (24). These limited literatures so far, nevertheless, could not provide systematic information regarding the phenotypic and molecular alterations in response to the lowered abundance of cathepsin D. Secondarily, as a major lysosomal protease, it is conceivable that lowering cathepsin D level would cause proteomic changes across different subcellular organelles in addition to a whole cell level. This additional layer of proteomic information would boost our understanding of cathepsin D-related phenotypic changes. Hence we hypothesized that an integration of subcellular proteomics survey and functional evaluation in a cell system with down-regulated cathepsin D benefits exploration toward the cathepsin D related mechanism. These considerations prompted us to initiate the experimental design as well as data analysis described in this communication.
Using a 2DE-based proteomics approach, we previously discovered cathepsin D as a potential biomarker for small cell lung cancer (SCLC) (25). The concentration of cathepsin D was found to be much higher in SCC tissues than in normal donors, suggesting that lysosomal protease cathepsin D is physiologically significant in promoting SCC. Based on previous reports and our study, a thorough investigation of cathepsin D's function in cancer progression is needed, which entails the characterization of both the phenotype alterations and molecular events induced by changes in cathepsin D levels. To this end, we constructed a stable cathepsin D knockdown HeLa cell line, designated as CR, in which the cathepsin D abundance was significantly attenuated because of shRNA. In contrast to empty vector transfected HeLa cells (EV), CR cells displayed impaired tumorigenicity and increased senescence. The CR cells were cultured in DMEM medium containing 13C6l-Lysine, mixed with EV, and three subcellular fractions, the cytosol, lysosome, and nucleus, were prepared followed by quantitative proteomics analysis. The quantitative proteomics revealed the abundances of a number of typical lysosomal proteins were reduced in the lysosome but augmented in the cytoplasm, with the abundances of redox proteins declining. We further showed that cathepsin D knockdown caused permeability changes in the lysosomal membrane and increased reactive oxygen species (ROS)1 in CR cells. Tracing regulation factors to expression of these redox genes, we found the transcriptional activity of nuclear factor erythroid factor-like 2 (Nrf2) was lower in CR cells. Moreover, overexpressing Nrf2 alleviated cellular senescence in these cells. Through time-course experiments, we observed a sequential occurrence of the cathepsin D-related events, including an increase in lysosomal membrane permeability during the early stage, ROS accumulation, and finally Nrf2 activity inhibition and enhanced cell senescence.
EXPERIMENTAL PROCEDURES
Plasmid Construction
The two siRNA sequences against CTSD were designed as 5′-GAATGGTACCTCGTTTGAC-3′ and 5′-GTATTACAAGGGTTCTCTG-3′, which were used to construct the small hairpin RNA (shRNA) according to Wetering et al. (26). The resultant shRNAs were introduced into the plasmid pTER+, and were named pTER-shCD1 and pTER-shCD2. The empty vector pTER+ was named pTER-EV. The entire coding sequence of human Nrf2 gene was cloned into the plasmid pCDNA3.1(+), which was named pCDNA3.1-Nrf2. The pGL3-Basic, pGL3-ARE and pRL-TK vectors were provided by Dr. Ningzhi Xu. The Antioxidant Response Element (ARE) on the pGL3-ARE plasmid was derived from the ARE sequence from NQO1, and the sequences of the sense strands of ARE was 5′-CGATCCAGTCACAGTGACTCAGCAGAATCTGGAGCT-3′.
Cell Culture, Stable Cell Line Establishment
HeLa cells were obtained from American Type Culture Collection. The cells were cultured in full DMEM supplemented with 10% FBS, penicillin, and streptomycin.
To establish the stable cells with/without the knockdown of cathepsin D, HeLa, or A549 cells were transfected with the vector pTER-CR1 and pTER-EV and then screened in complete DMEM medium with 100 μg/ml zeocin for ∼2 weeks. The stable cells with the knockdown of cathepsin D were named CR (for HeLa) and A549-CR, and the control cells, cells transfected with pTER-EV, were designated as EV (for HeLa) and A549-EV. A similar approach was adopted to generate Nrf2 overexpressing cells.
Metabolic Labeling, Subcellular Fractionation, and Peptide Preparation
To differentially label the CR and EV cells, the SILAC Protein Quantitation Kit (Thermo Fisher Scientific, Rockfield, IL) was used according to the manufacturer's instruction. Cells were centrifuged at 300 × g and the cell pellets were resuspended in isotonic buffer followed by the treatment with the syringe to break up the cells. Organelle preparation for nucleus, cytosol, and lysosome was implemented according to a protocol described previously (1, 38). The protein concentration was determined using the BCA assay. Equal amounts of EV and CR proteins from each fraction were mixed and further separated with 12% SDS-PAGE. The proteins in the gel were cut into 15 small slices per lane and tryptic digestion was then performed according the in-gel digestion protocol (44). The tryptic peptides were extracted with acetonitrile, dried in a vacuum and reconstituted with 0.1% FA.
Mass Spectrometry, Data Processing, and Functional Analysis
Peptides were delivered onto a C18 reverse phase column using a linear 65 min gradient of 0–80% ACN in 0.05% FA in a prominence nano HPLC (Shimazu, Tokyo, Japan). The eluted fractions directly entered the LTQ-Orbitrap-Velos mass spectrometer (Thermo Fisher Scientific), in which full MS scan from 300–2000 m/z was acquired in the Orbitrap. The following MS/MS scans were collected in the ion trap for the top eight most intense ions using collision-induced dissociation (CID). Two biological replicates were analyzed independently.
The raw mass spectrometry files acquired from all the fractions in each of replication were combined and processed with MaxQuant 1.5.0.28 (Maxplank Institute, Munich, Germany). Peak lists were extracted and searched against the database of ipi.human.v.3.68 (87,083 entries). The search parameters were set as follows: carbamidomethyl as the fixed modification, pyro-Glu/Gln (N-term), and Oxidation (M) as variable modifications, 20ppm for peptide tolerance and 0.5 Da for MS/MS tolerance. The enzyme was set as trypsin and the maximum missed cleavage was set to two. The false discovery rate for both proteins and peptides was set as 1%. Proteins with at least two ratio counts, as generated from either unique or razor peptides, were considered as quantified proteins for further analysis. The median values of the SILAC ratios H/L were taken as protein abundance (HeLa-CR/HeLa-EV) ratios to minimize the effects of outlier values. Contaminations were removed from the final list of identification and quantification. The p values for each quantification value were calculated as previously described (27). The iBAQ (Intensity based accurate quantification) values (43) were calculated as implemented in the MAXQUANT software and the relative iBAQ ranking of a protein in a specific fraction was derived by dividing the ranking of the iBAQ of that protein by the total number of proteins. The result was then decreased by one such that the proteins with higher iBAQ values had a higher relative iBAQ ranking. The differential proteins in each fraction were delivered into the DAVID database (NIAID, National Institute of Health) for functional enrichment analysis (21, 22). Regression analyses of SILAC quantification results were evaluated through comparison of the quantitative results to the differential proteins between the biological duplicates. At x axis, the SILAC ratios for differential proteins in log2 form from one of the duplicated samples were plotted, and at y axis, all the corresponding ratios in the other sample were paired. The regression analysis was performed with Microsoft Excel 2007 for the values of slope and regression coefficient.
Quantitative PCR Analysis
Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA), and reversely transcribed to generate cDNA libraries. The primers were designed against Nrf2 (F: 5′-TTCCCGGTCACATCGAGAG-3′, R: 5′-TCCTGTTGCATAC CGTCTAAATC-3′), Nqo1 (F: 5′-GATATTCCAGTTCCCCCTGCA, R: 5′-AAAGCACTGC CTTCTTACTCCG-3′), SOD2 (F: 5′-TTTCAATAAGGAACGGGGACAC, R: 5′-GTGCTCCCACACATCAATCC-3′), GCLM (F: 5′-TGTCTTGGAATGCACTGTATCTC-3′, R: 5′-CCCAGTAAGGCTGTAAATGCTC-3′), AKR1C1 (F: 5′-ATGGTCACTTCATGCCTGTC-3′, R: 5′-AAGCCAGCTTCAATTGCC-3′), and β-actin (F: 5′-CAAGGCCAACCGCGAGAAGA-3′, R: 5′-GGATAGCACAGCCTGGATAG-3′). The mRNA abundances were quantitatively determined using a PRISM 7300 system (ABI, Foster City, CA). The results were normalized with β-actin abundance.
Western-blot Analysis
Cells were harvested in lysis buffer (50 mm HEPES, pH 7.4, 2% SDS) supplemented with protease inhibitor mixture set I (Calbiochem, San Diego, CA). Proteins were separated through 10–15% SDS-PAGE and was detected by semiquantitative measurements of protein abundance among different samples were achieved using antibodies against cathepsin D, β-actin, aldose reductase, lamin-A, lamp1, and sod2 from Santa Cruz Biotechnology, Inc. (Dallas, TX), nqo1 and gclm from Abcam, Nrf2 from Cell Signaling Technology, Boston, MA, AKR1C1/2 and GSTP1 from BPI (Beijing, China).
Cell Growth, Cellular Senescence, and Tumorigenesis Assay
For cell growth estimation, HeLa cells with or without cathepsin D knockdown were seeded into 96-well plates. For each time point, three replicate wells were measured using the MTT Assay Kit (Beyotime, Beijing, China). Using the Senescence Cells Histochemical Staining Kit (Sigma-Aldrich), the cells were fixed for 7 min and then incubated with the staining solution for 14 h at 37 °C. The levels of cell senescence are represented by the ratio of senescent cells in the total cell population. The animal protocol described in this article was approved by the Animal Care and Welfare Committee at the Beijing Institute of Genomics, CAS. EV and CR cells (2 × 106) were bilaterally injected subcutaneously into ten 4–6 week old nude mice. Six weeks after injection, the tumor weight was measured.
Cell Cycle Analysis
Cells at 70–80% confluency were harvested, washed and then fixed in 70% ethanol overnight at 4 °C, the cells were then washed and centrifuged at 1000 × g. Propidium iodide was added to the cells with RNase at 100 μg/ml and 0.1% Triton X-100 in 200 μl PBS, and the cells were incubated at room temperature for 30 min in the dark. Finally, the cells were washed with 1 × PBS and analyzed by a flow cytometer (FACSCalibur™, BDIS, San Jose, CA). The results were analyzed by ModfitLTTM (Verity Software House, Topsham, ME).
Measurement of Intracellular ROS, Labile Iron and Mitochondrial Membrane Potential
Cells were incubated with complete DMEM medium containing 20 μm H2DCFDA at 37 °C, 5% CO2 for 40 min and then washed, followed by green fluorescence observation under an inverted fluorescence microscope (Olympus IX 70) or measurement with a 96-well plate fluorescence reader (FlexstationII®, Molecular Devices, Sunnyvale, CA) at an excitation wavelength of 488 nm and a barrier wavelength of 530 nm. Alternatively, trypsinized HeLa cells were loaded with 20 μm H2DCFDA, washed, and their fluorescence signals were recorded using a flow cytometer (FACSCalibur™) in the FL1 channel. For the measurement of labile iron pool, the cells were harvested and loaded with 1 μm Calcein-AM for 30 min at 37 °C, 5% CO2, washed, resuspended in PBS and were delivered to the flow cytometer mentioned above to record the FL1 signals. The ΔMMP was measured using the mitochondrial membrane potential measurement kit (JC-1, Beyotime, Beijing, China) according to the manufacturer's instructions. The fluorescence intensities were recorded using the flow cytometer mentioned above.
Lysosomal Membrane Stability Assay
For acridine orange staining, cells were harvested and incubated with 5 μg/ml AO in DMEM medium at 37 °C for 20 min. The cells were washed and the fluorescence intensity was measured by the flow cytometer mentioned above.
For the lysosomal pH measurement, the Lysosensor Yellow/Blue DND-160 (Life Technologies, Carlsbad, CA) was used as a lysosomal pH indicator according to a previous report (54). Briefly, EV and CR cells were trypsinized and labeled with 5 μm dye in DMEM medium, washed, and treated with monensin and nigericin in MES calibration buffer (pH 3.5–6.5). The labeled cells were transferred to a 96-well plate and their light emission intensities at 535 nm in response to 340 nm and 380 nm excitation were measured in a 96-well plate fluorescence reader. The ratios of the light emission intensity in response to the two excitation wavelengths were plotted against the pH value to generate the pH calibration curve. To measure the lysosomal pH of the EV and CR cells, the cells were stained with the dye and resuspended in MES buffer (pH 7.7). The lysosomal pH values for the two cell lines were estimated using the ratios of the emission intensities at 340/380 nm excitation and their respective calibration curves of the fluorescence probe.
Dual Luciferase Reporter Assay
Both EV and CR cells were seeded and transfected with reporter vector, either pGL3-Basic or pGL3-ARE, and pRL-TK using LipofectamineTM 2000. At 24 h post-transfection, the cells were washed once with PBS and 100 μl passive lysis buffer was added to lyse the cells with gentle shaking. Then the cell lysate was centrifuged and the luciferase activities were determined using a Dual luciferase Reporter Assay System (Promega, Madison, WI) with luminescence measurement with single-tube luminometer (Optocomp1, MGM Instruments). The relative firefly luciferase activities were normalized to the Renilla luciferase activities of the cotransfected pRL-TK vector.
Statistical Analysis
All of the experiments were performed with at least three biological replicates and significance was calculated using unpaired Student's t test, unless mentioned elsewhere.
RESULTS
Cathepsin D Knockdown Leads to a Series of Changes of Cellular Behavior in HeLa Cells
We generated CR in HeLa cells, in which cathepsin D abundance was reduced by 80% compared with EV cells, as shown in Fig. 1A. Thereafter, we conducted a systematic evaluation of the biologic characteristics of the EV and CR cells. The MTT assay was used to monitor cell growth, and the results clearly showed that the CR cells grew more slowly compared with the EV cells (Fig. 1B). The evaluation of senescence using senescence-associated β-gal (SA-β-gal) staining presented in Fig. 1C and 1D revealed the ratio of the senescent CR cells was about 44.9 ± 2.3%, significantly higher than that in the EV cells (3.4 ± 1.2%). In addition, another cathepsin D knockdown construct, pTER-shCD2, was used to transfect HeLa cells and establish HeLa-CR2 (CR2) cells, in which cathepsin D abundance was down-regulated (supplemental Fig. S1A). When CR2 cells were subjected to SA-β-gal assay, they also exhibited cellular senescence that was quite similar with CR cells (supplemental Fig. S1B). Besides, as depicted in Fig. 1E, ∼88.1 ± 2.3% of the CR cells were at the G2/M phase, whereas only 21.9 ± 2.2% of the EV cells were at this stage. The cellular behavior of apoptosis and autophagy was examined as well, but no consistent changes were found in the two cell lines (supplemental Fig. S2). Taken together, we deduced that lowering the cathepsin D abundance in the HeLa cells resulted in decreased proliferation, which was likely to be associated with cellular senescence and an alteration in cell cycle distribution. We further inquired whether the changes of cathepsin D abundance in the HeLa cells could affect tumorigenesis using the xenograft mouse model. For each mouse, the EV and CR cells were inoculated subcutaneously in the two sides. As depicted in Fig. 1F, in 10 xenografted mice, the tumors induced by the EV and CR cells were 1.56 ± 0.61 g and 0.20 ± 0.14 g, respectively, indicating that the tumors with the implanted CR cells were generally smaller than those with the EV cells. This suggests that cathepsin D knockdown reduces in vivo tumorigenesis.
Fig. 1.
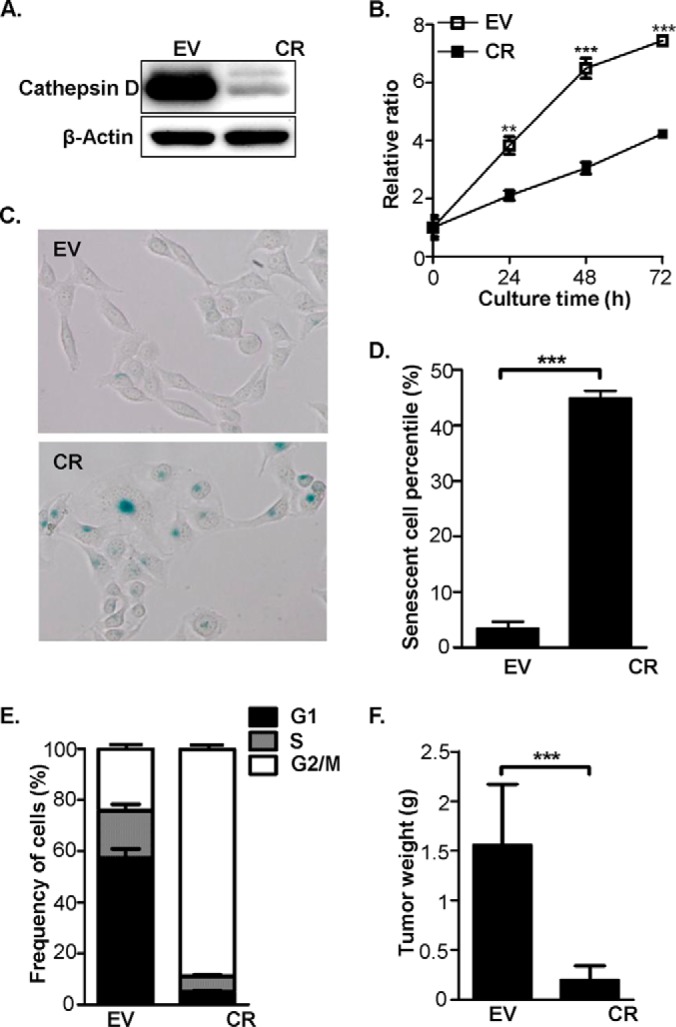
Cathepsin D knockdown induced multiple phenotype alterations in HeLa cells. A, The cathepsin D abundance was evaluated by Western-blot in EV and CR cells. B, The proliferation curves of EV and CR cells were drawn based on the data of MTT assay. The y axis data represents the ratios of the absorbance at 570 nm in CR against EV cells. C, Cell senescence was evaluated by SA-β-gal staining in EV and CR cells. D, Comparison of the positive ratios of SA-β-gal staining between EV and CR cells, which were derived from the numbers of SA-β-gal positive staining divided by the total cell numbers in each cell type. E, The cell cycle distribution patterns of EV and CR cells were estimated by a flowcytometer followed by software analysis with Modfit LT. F, The tumor cell growth in vivo was estimated by the nude mouse model, in which EV and CR cells were bilaterally and intravenously injected into 10 mice followed by weighing the tumor tissue after 4 weeks. In the above figures, the sign of ** represents p < 0.01 and *** as p < 0.001.
The Proteomic Response to Cathepsin D Knockdown Follows an Organelle Specific Manner
Cathepsin D is a typical lysosomal protease that also distributes in the cytosol (28). As knockdown of cathepsin D in HeLa cells resulted in significant changes in cellular behavior, an integrated investigation of proteomic responses of the cytosolic and lysosomal fractions to cathepsin D knockdown should help reveal the mechanisms underlying the phenotypic changes observed in HeLa cells. Moreover, as key regulators of the cell cycle are generally considered to reside in the nucleus, proteomic information of the CR nucleus is assumed to offer useful information toward understanding cell cycle regulation. We therefore analyzed the proteomic responses to cathepsin D knockdown in the three organelles, cytosol, lysosome, and nucleus.
EV and CR cells were cultured in the light (l-Lysine 6C12) or heavy isotopes (l-Lysine 6C13) incorporated DMEM medium for at least five passages to ensure that the isotopic amino acids were fully incorporated. After the cells were harvested and the organelles were enriched by ultracentrifugation, the organelle enrichment quality was examined by Western-blot analysis using antibodies against several typical markers (supplemental Fig. S3A). The results revealed that these organelle markers were significantly enriched in the corresponding fractions, indicating acceptable organelle enrichment. For each fraction, the same amount of protein from the CR and EV cells was mixed, denatured, and separated using SDS-PAGE. After tryptic digestion of the gel fractions, the peptides were extracted, separated, and identified through reverse phase liquid chromatography coupled with an Orbitrap-based mass spectrometer. In the cytosolic, lysosomal, and nuclear fractions, a total of 28,229, 12,026, and 7081 peptides were identified from 323,291, 61,851, and 48,222 MS/MS spectra, and resulted in the identification of 3116, 2237, and 1523 unique proteins, respectively (Fig. 2A and supplemental Table S1). The mass deviations of the 95% peptides were less than 0.8 ppm (supplemental Fig. S3B). We also assessed the quality of organelle preparation using mass spectrometric signals. The iBAQ values, which represent the relative abundance of each protein, were calculated for the three subcellular fractions and were transformed into relative iBAQ rankings decribed in the “Experimental Procedures” section. As shown in Fig. 2B, all of the iBAQ rankings for the three markers appeared at high levels in the corresponding organelle. Thus, the iBAQ evidence was in accordance with western-blot analysis (supplemental Fig. S3A) to confirm the quality of the organelle enrichment.
Fig. 2.

Quantitative proteomic analysis revealed that cathepsin D knockdown resulted in a global change in subcellular protein profiles. A, Overlap analysis of the proteins identified in each subcellular fraction (blue circle for cytosol, green circle for lysosome and red circle for nucleus). The numbers of identified proteins were shown on the Venn diagram. B, The relative iBAQ rankings for the marker proteins in three subcellular fractions, AKR1B1 for cytosol in blue, Lamp1 for lysosome in green, and Lamin A for nucleus in red. The relative iBAQ rankings were estimated as described in “Materials and Methods,” in which a protein with the highest iBAQ value was defined the relative iBAQ ranking as 1. C, Biological reproducibilities between two independent SILAC experiments were evaluated in the cytosol, the lysosome and the nucleus. SILAC ratios for each differential protein were log2 transformed, and the slope and correlation coefficency of linear fitting curve for each subcellular fraction were indicated. D, Evaluation of correlation efficiency of SILAC ratios among “lysosome” proteins between cytosol and lysosomal fraction. In proteins that were both quantified in cytosol and lysosomal fractions, the “lysosome” proteins, as indicated by Gene Ontology, were selected, and their SILAC ratios in cytosol and lysosomal fractions were log2 transformed and plotted on the dot plot. Specifically, the SILAC ratios of cathepsin D were removed. Several typical lysosome-residing proteins, such as cathepsin A and C, β-galactosidase and prosaposin, were indicated by arrows. E, The frequency distribution of selected protein subsets in cytosol and nuclear fractions were charted against their log2-transformed SILAC ratios. Note that proteins constituting hnRNP complex tend to increase abundance in cytosol, and proteins that were related with chromosome accumulate in nucleus of CR cells.
All of the identified peptides were further analyzed using MaxQuant to select qualified peptides in a quantitative evaluation. To make sure the SILAC quantitative data were reproducible, a biological duplication was conducted through all experiments, in which the two cell batches were separately cultured and harvested followed by treatments with same experimental protocol, and only proteins assigned with quantitation values in both replicates were considered quantified proteins. After MaxQuant filtration, 1537, 476, and 472 proteins were quantified in the cytosolic, lysosomal, and nuclear fractions, respectively. The limited number of quantified proteins in the lysosome and nuclear fraction mainly stemmed from a much lower numbers of identified proteins in one of the biological replicates. We adopted a set of criteria, balanced between sensitivity and reproducibility, to define the differential proteins as follows: (1) the fold changes of the differential proteins should be larger than 1.3 and have a p value less than 0.05 in at least one biological replicate, (2) the fold changes of the two biological replicates for the same protein should be in the same direction. According to these criteria, 425, 145, and 119 proteins were defined as the differential proteins in the cytosol, lysosome, and nucleus, respectively (supplemental Table S4). Among them, only 29, 18, and 26 proteins overlapped between the cytosol-lysosome, cytosol-nuclear, and lysosome-nuclear fractions (supplemental Fig. S3C). Therefore, the proteome changes induced by cathepsin D knockdown appear to follow an organelle-specific manner. The regression analyses to the differential proteins in two biological replicates were performed in each subcellular organelle. The acceptable correlation efficiency was obtained, R2 = 0.70 in the cytosol, 0.62 in lysosome, and 0.63 in nucleus (Fig. 2C), respectively.
The Cathepsin D-related Proteins are Tightly Associated with the Organelle Functions
DAVID analyses were performed for understanding of the biological processes, cellular components, and molecular functions of the differential proteins in response to cathepsin D knockdown (supplemental Table S5). The most prominent feature is the prevalent reduction of oxido-reductases in all of the three organelles. Twenty six out of 192 proteins were down-regulated in the cytosol; 14 out of 88 proteins were down-regulated in the lysosome; nine out of 38 down-regulated proteins in the nucleus participate in the process of oxidation reduction (Table I and supplemental Table S5). Similarly, proteins with oxidoreductase activity were also enriched in the down-regulated cytosol and lysosomal fractions (supplemental Table S5), suggesting that cathepsin D knockdown might compromise redox balance. Additionally, 29 out of 233 proteins up-regulated in cytosol participate in RNA splicing, and eight of the 29 proteins are constituents of heterogeneous nuclear ribonucleoproteins (Fig. 2E and supplemental Table S4), suggesting cathepsin D knockdown induced cellular senescence were accompanied by massive alterations in RNA processing (supplemental Table S2). Intriguingly, 12 “lysosome” proteins, as annotated by Gene Ontology, were significantly up-regulated in the cytosol. In contrast, eight “lysosome” proteins were down-regulated in the lysosomal fraction (Table II). For the 14 “lysosome” proteins that were quantified both in lysosomal and cytosol fraction, most of them had SILAC ratios >1 in cytosol and <1 in lysosome, and the log2 ratio of these proteins in the two organelles were negatively correlated with correlation coefficiency of 0.38 (Fig. 2D). The above analysis led us to the hypothesis that cathepsin D knockdown somehow redistributes proteins between the cytosol and lysosome. Regarding the nucleus, as many as 11 proteins that are components of chromosomes were significantly up-regulated, including members of the histone H1, H2, and H3 families (Fig. 2E and supplemental Table S5). This is in accordance with our findings that more than 80% of the CR cell population was in the G2/M phase, where the DNA contents is amplified and the amount of chromosome-binding proteins are increased proportionally. On the basis of the proteomic evidence above, such as the down-regulation of redox proteins in the cytosol, diagonal changes of “lysosome” proteins in the cytosolic and lysosomal fractions, and augmentation of chromosome-located proteins in nucleus, we proposed that cathepsin D knockdown led to impaired lysosome stability and disturbance of the redox balance within cells, ultimately resulting in cell senescence and cell cycle arrest.
Table I. The SILAC ratios of down-regulated proteins involved in oxidation reduction in cytosol, lysosome and nucleus.
| Accession No. | Gene symbols | Mean SILAC Ratio |
Cytosol |
Lysosome |
Nucleus |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cytosol | Lysosome | Nucleus | 1 | 2 | 1 | 2 | 1 | 2 | ||
| IPI00152981 | ACAD9 | 0.76 | 0.55 | 0.76 | 0.76 | 0.55 | ||||
| IPI00028031 | ACADVL | 1.08 | 0.45 | 1.10 | 1.20 | 0.95 | 0.08 | 0.82 | 1.08 | 1.13 |
| IPI00651738 | ADI1 | 0.48 | 0.41 | 0.54 | ||||||
| IPI00413641 | AKR1B1* | 0.77 | 0.97 | 0.84 | 0.71 | 1.35 | 0.58 | |||
| IPI00029733 | AKR1C1* | 0.67 | 0.48 | 0.73 | 0.75 | 0.60 | 0.39 | 0.57 | 1.06 | 0.40 |
| IPI00005668 | AKR1C2 | 0.26 | 0.27 | 0.25 | ||||||
| IPI00291483 | AKR1C3* | 0.61 | 0.47 | 0.65 | 0.57 | 0.47 | ||||
| IPI00295386 | CBR1* | 0.90 | 0.57 | 1.28 | 0.83 | 0.96 | 0.71 | 0.44 | 1.75 | 0.81 |
| IPI00010810 | ETFA | 1.36 | 0.42 | 1.19 | 1.38 | 1.35 | 0.39 | 0.46 | 1.36 | 1.03 |
| IPI00026781 | FASN | 0.78 | 0.75 | 0.71 | 0.76 | 0.80 | 0.54 | 0.97 | 0.84 | 0.59 |
| IPI00554521 | FTH1* | 0.76 | 0.52 | 0.71 | 0.82 | 0.83 | 0.21 | |||
| IPI00216008 | G6PD* | 0.71 | 0.77 | 0.24 | 0.65 | 0.78 | 0.66 | 0.87 | 0.24 | |
| IPI00219018 | GAPDH | 0.74 | 0.58 | 0.60 | 0.69 | 0.79 | 0.42 | 0.74 | 0.48 | 0.73 |
| IPI00010090 | GCLM* | 0.82 | 0.99 | 0.65 | ||||||
| IPI00219025 | GLRX | 0.78 | 0.64 | 0.92 | ||||||
| IPI00017726 | HSD17B10 | 0.67 | 0.32 | 0.65 | 0.59 | 0.75 | 0.18 | 0.45 | 0.70 | 0.60 |
| IPI00604598 | JMJD6 | 0.83 | 0.70 | 0.96 | ||||||
| IPI00947127 | LDHA | 0.95 | 0.93 | 0.77 | 0.93 | 0.97 | 0.71 | 1.15 | 0.90 | 0.65 |
| IPI00008215 | ME1* | 0.49 | 0.42 | 0.56 | ||||||
| IPI00218342 | MTHFD1* | 0.64 | 1.15 | 0.61 | 0.68 | 1.15 | ||||
| IPI00889777 | MTRR | 0.54 | 0.54 | 0.53 | ||||||
| IPI00419266 | NDUFA6 | 0.70 | 0.70 | 0.70 | ||||||
| IPI00947328 | NDUFB5 | 0.78 | 0.80 | 0.75 | ||||||
| IPI00926925 | OGDH | 0.74 | 1.07 | 0.94 | 0.54 | 0.98 | 1.16 | |||
| IPI00384280 | PCYOX1 | 0.25 | 0.09 | 0.41 | ||||||
| IPI00219525 | PGD* | 0.76 | 0.55 | 0.68 | 0.85 | 0.55 | ||||
| IPI00000874 | PRDX1* | 0.65 | 0.83 | 1.91 | 0.60 | 0.70 | 0.64 | 1.02 | 1.72 | 2.11 |
| IPI00027350 | PRDX2 | 0.77 | 0.93 | 2.58 | 0.69 | 0.85 | 0.73 | 1.13 | 3.26 | 1.89 |
| IPI00024919 | PRDX3 | 1.15 | 0.37 | 1.03 | 1.06 | 1.25 | 0.23 | 0.52 | 1.21 | 0.85 |
| IPI00024915 | PRDX5 | 0.81 | 0.71 | 0.85 | 0.77 | 0.85 | 0.75 | 0.67 | 0.85 | 0.84 |
| IPI00013871 | RRM1 | 0.19 | 0.35 | 0.37 | 0.14 | 0.24 | 0.35 | 0.36 | 0.38 | |
| IPI00011118 | RRM2* | 0.73 | 0.86 | 0.60 | ||||||
| IPI00016443 | SDHAF2 | 1.10 | 0.64 | 1.18 | 1.03 | 0.66 | 0.62 | |||
| IPI00022314 | SOD2 | 0.90 | 0.21 | 0.71 | 0.78 | 1.03 | 0.18 | 0.24 | 0.54 | 0.88 |
| IPI00384643 | TP53I3 | 0.62 | 0.59 | 0.64 | ||||||
| IPI00216298 | TXN* | 0.87 | 0.85 | 0.49 | 0.84 | 0.90 | 0.85 | 0.79 | 0.20 | |
| IPI00642032 | TXNL1 | 0.66 | 0.67 | 0.67 | 0.64 | 0.67 | ||||
| IPI00156689 | VAT1 | 0.85 | 0.87 | 0.88 | 0.81 | 0.99 | 0.75 | |||
* Represents the proteins known to be regulated by Nrf2.
Table II. SILAC ratios for proteins with GO “lysosome” annotation that are up-regulated in cytosol and down-regulated in lysosome by cathepsin D knockdown.
| Accession | Protein name | Cytosol |
Lysosome |
||
|---|---|---|---|---|---|
| 1 | 2 | 1 | 2 | ||
| IPI00002255 | Lipopolysaccharide-responsive and beige-like anchor protein | 1.46 | 1.16 | ||
| IPI00005107 | Niemann-Pick C1 protein | 0.47 | 0.37 | ||
| IPI00012585 | Beta-hexosaminidase subunit beta | 2.02 | 1.83 | ||
| IPI00016342 | Ras-related protein Rab-7a | 0.77 | 0.75 | ||
| IPI00018871 | ADP-ribosylation factor-like protein 8B | 0.54 | 0.44 | ||
| IPI00022810 | Dipeptidyl-peptidase 1 precursor | 2.59 | 2.72 | ||
| IPI00023728 | Gamma-glutamyl hydrolase precursor | 4.21 | 2.80 | ||
| IPI00027253 | Gamma-aminobutyric acid receptor-associated protein | 1.50 | 1.07 | ||
| IPI00027851 | Beta-hexosaminidase subunit alpha precursor | 2.32 | 2.60 | ||
| IPI00215998 | CD63 antigen | 0.31 | 0.44 | ||
| IPI00217766 | Lysosome membrane protein 2 | 0.67 | 0.64 | ||
| IPI00219825 | Prosaposin | 3.53 | 2.27 | ||
| IPI00296441 | Adenosine deaminase | 1.43 | 2.08 | ||
| IPI00301579 | Epididymal secretory protein E1 precursor | 1.60 | 2.39 | ||
| IPI00384280 | Prenylcysteine oxidase 1 precursor | 0.09 | 0.41 | ||
| IPI00640525 | cathepsin A isoform a precursor | 2.05 | 2.62 | ||
| IPI00644708 | Nucleolysin TIAR | 1.15 | 1.45 | ||
| IPI00796919 | β-galactosidase | 4.96 | 2.95 | ||
| IPI00884105 | Lysosome-associated membrane glycoprotein 1 precursor | 0.41 | 0.43 | ||
Enhancement of Oxidative Stress by Cathepsin D Knockdown Leads to Cell Senescence
The intracellular ROS levels in EV and CR cells were estimated using H2DCFDA, which emits green fluorescence signals after being oxidized by ROS, and analyzed by either a flow cytometer (Fig. 3A) or a fluorescence 96-well plate reader (Fig. 3B). Data obtained using both approaches showed the level of ROS was much higher in CR than in EV cells. Besides, the ROS in CR2 cells was determined and its level was also higher than control cells (supplemental Fig. S1C). Importantly, an antioxidant, N-acetyl cysteine (NAC), was able to reduce ROS level and partially rescue cell senescence in CR cells (Fig. 3C and 3D), suggesting that oxidative stress induced by cathepsin D knockdown is tightly correlated with cell senescence.
Fig. 3.

ROS increment was correlated with the cell senescence induced by cathepsin D knockdown. A and B, Levels of ROS in EV and CR cells were measured by H2DCF-DA staining coupled with flowcytometry (A) and 96-well plate fluorescence reader (B). C, The ROS responses in the HeLa cells that were treated with antioxidant, NAC. EV and CR cells were seeded in a 96-well plate, treated with mock or N-acetyl cysteine (NAC) at indicated concentrations for 48 h, and their ROS levels were monitored using H2DCF-DA staining. Results were expressed as relative mean fluorescence intensities (MFI) using mock treated EV cells as the reference. D, Comparison of senescence ratios of EV cells, CR cells, and CR cells treated with NAC for 48 h followed by SA-β-gal staining. The sign of * represents p < 0.05, ** as p < 0.01 and *** as p < 0.001.
How ROS was enhanced by cathepsin D knockdown? We first sought the answer based on the proteomic data. One of the most prominent features of our data sets was the overall increase of lysosome-residing proteins in the cytosol (Table II). To verify proteomic data, the colocalization of cathepsin B, a lysosomal protease, and lamp2, a lysosomal membrane protein, was examined by confocal microscope with immunofluorescence. In EV cells, the fluorescence signals of two proteins were colocalized very well with the colocalization M1 score more than 0.95, whereas those two proteins were not evidently colocalized in CR and CR2 cells with the M1 coefficient less than 0.8 (Supplemental Fig. S1E and S1F). Because the lysosomal membrane permeability (LMP) increase induced by hydroxychloroquine promoted ROS generation (29, 30), we propose that LMP augmentation might mediate the ROS increase in the CR cells. Acridine orange (AO), a cell-permeable dye that emits red fluorescence when trapped and aggregated in the lysosome (31), was adopted to probe the integrity of the lysosomes. As illustrated in Fig. 4A, the fluorescence signals for the EV cells were mainly retained at the Q2 phase (97.1%), whereas the CR cells were distributed within two phases, the Q2 phase (82.9%) and Q3 phase (14.3%), suggesting that the cathepsin D knockdown increased the fraction of cells with “pale” lysosomes that could not retain AO. As a positive control, the EV cells were treated by bafilomycin A1 (BA), a lysosomal membrane H+ pump inhibitor, and only 43.3% of cells were at the Q2 phase because of BA induced LMP increase (Fig. 4A, right panel). To support the idea that cathepsin D knockdown specifically induced LMP, the acridine orange retention assay was performed on EV, CR, and CR2 cells, and the two cell lines with cathepsin D knockdown had significant proportion that fell in Q2 phase (2.09% in CR and 1.75% in CR2) while nearly none of EV cells (0.452%) were in the Q2 phase, when they were analyzed together (supplemental Fig. S1D). Cathepsin D knockdown somehow impaired lysosomes in HeLa cells. Once the integrity of the lysosomal membrane is impaired, a reasonable deduction is that the lysosomal pH will increase because of H+ leakage. Lysosensor Yellow/Blue DND-160, a ratiometric probe that is used to measure pH in acidic organelles (32, 33), was used to test the pH values of the lysosomes. Using this probe, the lysosome pH values in the EV and CR were estimated at 5.0 ± 0.3 and 5.7 ± 0.2, respectively (Fig. 4B). Thus, the additional evidences, such as mismatch of cathepsin B and lamp2, compromised AO retention and lysosome alkylation, strongly supported that the LMP increase was induced by cathepsin D knockdown.
Fig. 4.

Cathepsin D knockdown induced the ROS-related events in HeLa cells. A, The LMP was estimated by AO staining in the HeLa cells, in which the fluorescence intensities on FL1-H (∼525 nm) and FL3-H (>670 nm) were measured by flowcytometry for EV cells (left panel), CR cells (middle panel), and EV cells treated bafilomycine A1 (BA) (right panel) as a positive control. B, The pH values of EV and CR lysosomes were estimated according to the calibration curve using the method described in “Experimental Procedures.” C and D, The LMP was evaluated by leakage of labile iron from lysosome. 1 to 3 days after transfection, both shEV and shCD cells were stained with calcein-AM, followed by measurement of their fluorescence intensities with flowcytometry at FL1-H channel. For desferoxamine (DFO) treatment, at 2 days after transfection, HeLa cells were treated with 0.25 μm DFO for additional 24h. Representative fluorescence intensity histograms were shown in (C). The gray line represents shEV cells and the black line stands for shCD cell. The ratios of cells with FL1-H fluorescence intensity value of less than 65 were calculated and shown in (D). E, Evaluation of ΔMMP on the HeLa cells using on flowcytometry at channel of FL1-H and FL2-H (∼575 nm). The EV cells (left panel), CR cells (middle panel), and EV cells treated with CCCP (right panel), were stained with JC-1, a dye for ΔMMP measurement, as described in “Experimental Procedures.” The sign of * represents p < 0.05 and *** as p < 0.001.
How was the LMP augmentation correlated with oxidative stress? In eukaryotic cells, iron is usually present in its chelated form in the cytosol, whereas unchelated, labile iron promotes ROS generation through the Fenton reaction (34). The lysosome constitutes one of the largest pools of labile iron in human cells (35). We hypothesized that ROS generation was promoted, at least in part, through the leakage of labile iron from the lysosome to the cytosol. Calcein-AM is a membrane-permeable ester, which is processed into membrane impermeable, fluorescent calcein in the cytosol, whose fluorescence is quenched by cytosolic low mass labile iron (35). Because the leaking of labile iron is a dynamic process, we transfected HeLa cells with either the pTER-EV or pTER-shCD1, and monitored the alterations of cytosolic labile iron pool during the course of cathepsin D knockdown using calcein-AM. As shown in supplemental Fig. S4A, the abundance of cathepsin decreased at 2–4 days post-transfection in the HeLa cells transfected with pTER-shCD1 (shCD cells) compared with those transfected with pTER-shEV (shEV cells). Meanwhile, the flow cytometric measurement (Fig. 4C and 4D) showed that the ratios of the fluorescence negative cells began declining 2 days after transfection and the trend continued at 3 days in the shCD cells as compared with shEV cells. In addition, at 2 days after transfection, shEV and shCD cells were further treated with Desferoxamine (DFO), an iron chelator, for 1 day, and the treatment resulted in an overall increase of calcein fluorescence in both cells, indicating the decrease of fluorescence intensity indeed reflected the increased labile iron pool in the cytosol. This result implied cathepsin D knockdown induced the leakage of labile iron from the lysosome to the cytosol which possibly resulted in ROS generation.
In some cells lysosomal leakage is reportedly accompanied by changes in the mitochondrial membrane potential (ΔMMP), leading to an increase in ROS. Hence, we used the dye JC-1 coupled with a flow cytometer to test whether the ΔMMP was alternated in the CR cells. JC-1 is cell-permeable and emits red fluorescence in normal mitochondria, but emits green fluorescence in depolarized mitochondria and cytosol. As shown in Fig. 4E, in EV cells (left panel), the fluorescence signals were mainly retained at the Q2 phase (96.5%), while in the CR cells (middle panel), the percentile of fluorescence signals at the Q2 declined to 75.9%, suggesting that cathepsin D knockdown impaired the ΔMMP. As a positive control, when the EV cells were treated with the mitochondrial inhibitor (right panel), CCCP, the Q2 phase percentile dropped to 77.4%, indicating that alterations in the JC-1 fluorescence reflected the ΔMMP reduction.
All above observations related with cathepsin D knockdown in HeLa cells, including increased LMP and cytosolic labile iron pool and decreased ΔMMP, were well correlated with ROS generation. Our observations therefore support the postulate that cathepsin D knockdown leads to cell senescence through ROS accumulation.
The Oxidative Stress-Related Events Induced by Cathepsin D Knockdown Follow a Sequential Order
Because several cathepsin D abundance-related events occurred in the HeLa cells as mentioned above, we questioned if those events occurred simultaneously or in a certain order in response to cathepsin D knockdown. Similar to the calcein-AM assay, to dynamically monitor the occurrences of these events during decreasing cathepsin D abundance, the LMP, ROS, and cell senescence were measured in the shEV and shCD cells at 1 day to 4 days after transfection. For LMP, both the shEV and shCD cells were stained with AO and analyzed by flow cytometry. Cathepsin D knockdown did not significantly affect the shEV cell fluorescence intensity, though for the shCD cells, the fraction of “pale cells” (Q3 phase) increased from 11% to 15% and to 24% within 3 days of transfection (Fig. 5A). Impressively, this finding was in parallel with the decrease in calcein fluorescence (Fig. 4C and 4D), suggesting an increase in the cytosolic labile iron pool occurred simultaneously with LMP augmentation. For ROS, the FACS data showed its levels in the shCD cells remained similar to that in the shEV cells at 1 to 2 days post-transfection, whereas the ROS level was significantly higher than in the shEV cells during 3 to 4 days after cathepsin D knockdown (Fig. 5B and 5C). In addition, the FACS data was consistent with the fluorescence microscopic observation of the ROS dye (supplemental Fig. S4B and S4C, left panel), which indicated that ROS began to accumulate from 3 days post pTER-shCD transfection. For cell senescence, although no significant changes were observed during the initial 3 days, 31.0 ± 2.6% of the shCD cells senesced at 4 days post-transfection, whereas only 11.9 ± 1.1% shEV cells were positive for SA-β-gal staining (Fig. 5C and supplemental Fig. S4C, right panel). The dynamic data sets recapitulated that the relevant changes in the biological events because of cathepsin D knockdown were time-dependent and sequential3ly-ordered: After shCD transfection, the reduction of cathepsin D protein abundance and the augment of LMP occurred at day 2, the ROS accumulation became evident at day 3 and cell senescence occurred at day 4. Therefore, we deduced that cathepsin D knockdown first impairs the lysosomal membrane and causes labile iron to release into cytosol, subsequently induces ROS accumulation and finally results in cell senescence.
Fig. 5.

Time-course analysis to shCD transfected HeLa cells revealed sequential order of the ROS-related phenotype changes. A, Measurement of LMP with AO staining for the HeLa cells transfected for 1–3 days with either pTER-shEV (shEV, in gray dots) or pTER-shCD1 (shCD, in black dots). The method was given in the “Experimental Procedure.” B and C, Measurement of ROS accumulation because of transient knockdown of cathepsin D. 1 to 4 days after transfection, the shEV and shCD cells were stained with H2DCF-DA and the fluorescence intensities were measured using flow cytometry. The histogram for H2DCF-DA fluorescence under each condition were shown in B, and the mean values and standard deviations of fluorescence intensity were shown as in C. D, Measurement of cell senescence ratios for the shEV and shCD cells. 1 to 4 days after transfection, SA-β-gal staining was implemented for counting the senescent cells. The sign of * represents p < 0.05 and *** as p < 0.001.
Cathpesin D Knockdown Inhibits Nrf2 Transcriptional Activity in HeLa Cells but not A549 Cells
ROS can activate the expression of antioxidant genes, which help cells combat oxidative stress (36–38). In contrast to the common principle, our proteomic data revealed the opposite scenario. More than 40% of the down-regulated redox proteins (11 in 26) were previously shown to be positively regulated by Nrf2 (39–41), a main transcription factor to regulate antioxidant gene expression (Table I). The Western-blot results showed that the protein abundance of Nrf2 and several of its targets, including NQO1, GCLM, AKR1C1, GSTP1, and SOD2, were significantly down-regulated in the CR compared with the EV cells (Fig. 6A). In addition, the Nrf2 abundance was also observed down-regulation in CR2 (supplemental Fig. S1A). Using real-time PCR with β-actin as a loading control, we showed that the mRNA levels of the Nrf2 targets were also lowered in the CR cells by ∼twofold (Fig. 6B). In addition, the nuclear level of Nrf2 protein was decreased in CR cells (Fig. 6C). The good agreement of mRNA and protein levels for Nrf2 and its target genes thus prompted us further to inquire to if the Nrf2 transcriptional activity was repressed by cathepsin D knockdown.
Fig. 6.

Cathepsin D knockdown inhibited Nrf2-ARE transcriptional activity and down-regulated expression of the Nrf2 target genes. A, Comparison of the abundance of the typically Nrf2 transcriptional targets in the lysates of EV and CR cells using Western-blot. B, Comparison of the mRNA abundances of four Nrf2 targets in EV and CR cells using Real-time PCR. C, Nrf2 protein abundance in nuclear lysates of EV and CR cells was evaluated by Western-blot with lamin A as a loading control. D, The Nrf2-ARE transcriptional activities in EV and CR cells were assayed using the dual-luciferase reporter system as described in the “Experimental Procedures.” E, The abundance of Nrf2 protein in Nrf2-transfected CR cells (NRF2 cells) was evaluated using Western-blot, alongside with empty vector-transfected CR cells (pCDNA3.1 cells). β-actin was used a loading control. F, Comparison of senescent cell ratios between the pCDNA3.1 cells and with Nrf2 cells. The sign *represents p < 0.05, ** as p < 0.01 and *** as p < 0.001.
We established the system for Nrf2 activity determination with a dual-luciferase reporter, in which pGL3-NQO1 was constructed with the plasmid, pGL3, and the Antioxidant Response Element (ARE) promoter sequence of NQO1. After transfection of pGL3-NQO1 into EV and CR cells, the luminescence intensity ratios in the EV cells were almost twofold higher than that in the CR cells (Fig. 6D). To identify whether the Nrf2 activity down-regulation contributed to cell senescence, CR cells were transfected with either pCDNA3.1-Nrf2 or pCDNA3.1 empty vector. The cells overexpressing Nrf2 were selected and cell senescence was measured using the SA-β-gal assay. As depicted in Fig. 6E, two stable cell lines were established and denoted as CR-Nrf2 and CR-pCDNA3.1, and the senescence ratios, 35.2 ± 2.3% in CR-pCDNA3.1 and 13.0 ± 2.8% in CR-Nrf2, were significantly different (Fig. 6F and Supplemental Fig. S5). These data hence implicated that the up-regulation of Nrf2 might rescue the cell senescence induced by cathepsin D knockdown.
Because A549 cells have been reported to harbor a critical point mutation on the Kelch domain of Keap1, which is an Nrf2 inhibitor, the Nrf2 in these cells are constitutively active (42). We were interested in whether the senescence-related events could be reproduced in such cells with cathepsin D knockdown. We constructed A549 stable cells with cathepsin D knockdown, termed A549-CR, and the protein abundance of cathepsin D was ∼40% in A549-CR cells as it was in control cells, A549-EV (supplemental Fig. S6A). Similar with HeLa cells, the lysosomal protease cathepsin B was observed to release from lysosome to cytosol in A549-CR, indicating lowering the endogenous abundance of cathepsin D also resulted in increased LMP in A549 cells (supplemental Fig. S6E and S6F). However, the ROS level in A549-CR cells was not increased (supplemental Fig. S6D), and the protein abundance of Nrf2 remained unchanged as expected (supplemental Fig. S6A). Intriguingly, cathepsin D knockdown in A549 cells did induce senescence, although the senescent rate is not as high as that in HeLa-CR cells (supplemental Fig. S6B and S6C). Maybe LMP increase alone is sufficient to induce cell senescence in A549 cells, through alternative pathways other than ROS/Nrf2.
Nrf2 Gene Expression Responds to Oxidative Stress in a Two-Phase Manner
Next we tested how the activity of Nrf2 was down-regulated in CR cells. We tested the effect of oxidative stress on Nrf2 activity by incubating the cells with sublethal amounts of H2O2 for different time spans. We determined that HeLa cells were capable of withstanding a 200 μm H2O2 treatment for as long as 24 h, by which time the cell senescence ratios were increased, but with minimal cell death (data not shown). As illustrated in supplemental Fig. S7, after the HeLa cells were treated with H2O2 with the final concentration of 200 μm, the cellular ROS level remained relatively stable at the first 4 h, started to increase after 12 h and continued to increase until 24 h post-treatment. During this period, the nuclear abundance of the Nrf2 protein showed an increase at 0.5 and 1 h after H2O2 treatment and declined thereafter (Fig. 7A and 7B). By 24 h after H2O2 treatment, Nrf2 abundance dropped by ∼40% in the nucleus. In good agreement with Nrf2 protein level in the nucleus, the dynamic responses of Nrf2 mRNA and its transcriptional targets after the treatment of 200 μm H2O2 appeared to have two clear cut-off phases, whereby oxidative stress evoked dramatic up-regulation of their mRNA abundance within the period of 0.5–1 h, and caused significant down-regulation of their mRNA abundance after 12 h (Fig. 7C). Taking the above observation together, we postulate that the inhibition of Nrf2 gene expression and the down-regulation of Nrf2 target genes are likely to be the consequences of ROS increase at late phase. Furthermore, Nrf2 abundance in HeLa cells was monitored in shEV and shCD cells. The data in Fig. 7D and 7E reveal that the ratios of Nrf2 abundance of shCD/shEV cells were not significantly different within 3 days of transfection even though obvious ROS accumulation was observed during the same period (Fig. 5B), whereas the ratios were significantly down-regulated at 4 days after transfection. Because ROS began accumulating 3 days after transfection in the shCD cells, our data suggested Nrf2 down-regulation might be a downstream event of ROS augmentation. As the CR cells were selected and cultured for many passages, the cells were tolerant to the higher oxidative stress induced by cathepsin D knockdown. The chronic oxidative stress in such HeLa cells, thus, was likely to block Nrf2 gene expression as seen the results in Fig. 7A–7C.
Fig. 7.

Chronic oxidative stress led to turning down Nrf2 expression and activity. A, Time-dependent changes of Nrf2 abundance responding to treatment of 200 μm H2O2 in nucleus of the HeLa cells were evaluated by Western-blot, with Lamin A as a loading control. A representative blot was shown. B, Relative changes of Nrf2 protein abundance in nucleus responding to 200 μm H2O2 treatment were estimated based on band intensities on the three independent Western-blots with lamin A as a reference. C, The dynamic changes of relative mRNA abundance for Nrf2 and its targets responding to 200 μm H2O2, with varying length of duration. The mRNA abundances were monitored by Real-time PCR and the corresponding mRNA levels without H2O2 treatment were employed as controls. D, Protein abundance changes of Nrf2 and cathespin D during transfection of pTER-shEV or pTER-shCD1 were evaluated by Western-blot with β-actin as the loading control. A representative blot was shown. E, Time-course of relative protein abundance changes for Nrf2 and cathepsin D during transfection. The relative protein abundances were calculated based on band intensities on the three independent Western-blots, and were normalized by the β-actin abundance at each time point. The sign of *represents p < 0.05, ** as p < 0.01 and *** as p < 0.001.
DISCUSSION
Based on a gene-knockdown approach, we characterized the phenotypic alterations and molecular changes at the protein level induced by cathepsin D knockdown. The down-regulation of cathepsin D abundance in HeLa cells had dramatic effects on the cancer cell phenotype, including a reduction of cell proliferation rate and induction of cell senescence. More importantly, the cathepsin D protein abundance in tumor is of physiological importance — the injection of cancer cells with lower cathepsin D level into nude mice resulted in a significant reduction of tumor development. Cell senescence is an irreversible state of cell growth and cell cycle arrest that could be induced by various exogenous stimuli, including oxidative stress, DNA damage, and oncogene activation. The induction of cellular senescence in cancer has been proposed to be the first barrier against tumorigenesis. Thus cathepsin D knockdown is likely to inhibit tumor growth by promoting cellular senescence. Inspired by these observations, we believed the combination of the phenotypic characterization and molecular events would deepen our understanding of cathepsin D's unique role in tumor development and cellular senescence.
We carried out a systematic investigation of the proteomic changes induced by cathepsin D knockdown. SILAC was the prime choice of quantification methodology because the systematic errors introduced during, e.g. protein digestion and peptide labeling are eliminated, likely yielding more accurate quantitative results. Three subcellular fractions, cytosolic, lysosomal, and nuclear, were selected to study these proteomic changes. Two important features of the proteomic changes induced by cathepsin D knockdown were revealed, one of them being the reciprocal changes of the lysosome-residing proteins in the cytosol and lysosome: their abundance was increased in the cytosol yet decreased in the lysosome. Our proteomic data, combined with the acridine orange release assay, lysosomal pH assay, and immunofluorescence assay by confocal microscopy, clearly showed that cathepsin D knockdown induced lysosomal membrane permeabilization. Although several previous studies reported an increase in the abundance of other cathepsin proteases when cathepsin D was inhibited or knocked out, a lack of subcellular proteomic information masked the proteomic changes occurred in lysosome. Although few reports provided the direct evidence that indicated the protein abundance of cathepsin D could affect LMP, there were some indirect evidences showing that lowering this protease destabilized lysosome. For instance, accumulation of β-amyloid in lysosome resulted in LMP and cell death, possibly acting as a lysosomotropic reagent (43, 44). Hamazaki et al. found cathepsin D was responsible for the clearance of β-amyloid (7). It is thus possible that cathepsin D knockdown leads to accumulation/aggregation of its catalytic substrates, which exerts detrimental effect on lysosome integrity.
Another important feature of CR proteomics is that many redox proteins, especially antioxidant enzymes, exhibited reduced abundance in the cytosol and lysosomal fractions, indicating an imbalanced redox status within these cells. Either transient or stable knockdown of cathepsin D in the HeLa cells increased ROS level, which mediated cellular senescence. Several groups have paid attention to cellular senescence induced by ROS. Chen et al. found that the diploid fibroblasts appeared senescence after treatment with hydrogen peroxide (45), whereas Lee et al. showed that the senescence induced by an oncogene, Ras, in fibroblasts was mediated by ROS, and was rescued by ROS scavenger (46). Regarding the regulatory mechanism of ROS to senescence, except that the classic senescence pathway involved with p21 could be adjusted by ROS (47), Rai et al. proposed that ROS could cause DNA damage and telomere shortening, subsequently leading to cellular senescence (48). Thus cathepsin D abundance is possibly crucial for maintaining a relatively low level of ROS in cancer cells. Because the CR cells displayed many senescence-related events, including lysosomal membrane permeabilization and oxidative stress induction, we felt it necessary to delineate the temporal sequence of these events. Using a cathepsin D transient knockdown system, we validated that the ROS increase occurred after lysosomal membrane permeabilization (evidenced by acridine orange retention assay) and earlier than cellular senescence. The ferrous iron released from the lysosome to the cytosol possibly initiates the ROS accumulation upon cathepsin D knockdown, but how are ROS maintained at a high level in HeLa cells?
Nrf2 is a transcription factor of many antioxidant proteins. In response to oxidative stress generators, Nrf2 translocates from the cytosol to the nucleus, promoting the expression of many antioxidant proteins, such as NQO1, HO-1, and SOD1, etc. Despite the increased oxidative stress, the intrinsic ROS-scavenging system is inhibited rather than activated: the protein abundance, as well as the transcriptional activity of Nrf2 is decreased in the HeLa-CR cells. How is this phenomenon explained? As oxidative stress induced by cathepsin D knockdown was not easily controlled, we introduced an in vitro system to monitor the Nrf2 abundance changes and its transcriptional activity, in which the HeLa cells were incubated with a sublethal concentration of H2O2 for different periods. As illustrated in Fig. 7, H2O2 stimulated the expression of Nrf2 and its target genes within a short period of time, yet inhibited Nrf2 transcriptional activity after 12 h. Importantly, the Nrf2 abundance was significantly enriched in the nucleus at 0.5 and 1 h after H2O2 treatment, indicating its transcriptional activity was increased. However, the Nrf2 protein was gradually eliminated in the nucleus after 4 h of H2O2 exposure. This result suggests that the Nrf2 response to oxidative stress could be divided into two phases. During the early phase, oxidative stress activates Nrf2, which up-regulates the antioxidant proteins to protect cells from oxidative stress; during at the late phase, it may inactivate Nrf2 because the consistent stress may modify the transcriptional factors or other regulators, resulting in dysfunction of the global antioxidant network. Although many studies have shown that Nrf2 is activated in response to oxidative stress within short periods (from 10 min to 6 h) (36, 38), studies regarding the effect of long-term oxidative stress on Nrf2 activity and its related mechanisms are lacking. Several reports, however, have consistently shown the co-occurrence of increased oxidative stress and decreased expression of Nrf2 down-stream target genes in tissues. Kim et al. reported that the rats with chronic renal failure exhibited signs of oxidative stress, such as increased lipid peroxidation and depleted glutathione, meanwhile the Nrf2 activity and its down-stream target genes were down-regulated (49). Safdar et al. found that oxidative stress in the aged muscle, indicated by protein carbonylation and 4-HNE content, was increased as compared with that in the young muscle, whereas activation of Nrf2-ARE signaling in the aged samples failed, because the expression of Nrf2 downstream targets, such as HMOX1 and γ-GCLC, was down-regulated (50). Our study hence provided evidence in a cell model that distinct from acute oxidative stress, persistent oxidative stress could inhibit Nrf2 activity, and paved ways for exploring the molecular mechanism how the Nrf2 protein is regulated by chronic oxidative stress.
According to previous publications, the abundance of Nrf2 is an important parameter to determine whether cells go to senescence. Caveolin-1 (Cav-1) is a scaffolding protein located within caveolar membrane. Recently, it has been found to be a negative regulator of Nrf2 signaling through physical interaction, and the overexpression of Nrf2 mutant, which does interact with Cav-1, inhibited cellular senescence induced by H2O2 stress in MEFs. Similarly, inhibiting Nrf2 transcriptional activity by Cav-1 overexpression promoted senescence and reduces growth on soft agar in HCT-116 cells (51). Yang et al. showed that the H2S donor could promote Nrf2 transcriptional activity and reverse cell senescence in mouse embryonic fibroblasts (MEFs) (52). Interestingly, Volonte et al. found subcytotoxic levels of hydrogen peroxide induced the expression and transcriptional activity of Cav-1, which promoted cell senescence in diploid fibroblasts (53), thus it is possible that mild and chronic oxidative stress, induced by cathepsin D knockdown, might inhibit Nrf2 through promoting cav-1 sequestration of Nrf2 on caveolar. In our study, we found Nrf2 inhibition, caused by increased ROS, promoted cell senescence in HeLa: Nrf2 overexpression could abolish cell senescence. Nevertheless, further efforts are needed in detailed study of the regulation of Nrf2 activity by oxidative stress, especially in long-term.
The data we obtained from A549 cells suggested alternative pathways to senescence exist. Because A549-CR cells possess hyperactivity of Nrf2, we were interested in whether the senescence-related events could be reproduced in such cells. Similar with HeLa cells, cathepsin D knockdown induced increased LMP and cell senescence, although to a lesser extent compared with in HeLa cells. However, the ROS level in A549-CR cells was not increased, and the protein abundance of Nrf2 remained unchanged. It seemed that in A549 cells, ROS increase and Nrf2 inhibition is not required for cathepsin D knockdown to induce senescence as long as lysosomal content is released and the mechanistic links await exploration. Proteomic investigation in A549 cells would certainly aid in exploration of underlying causes.
In conclusion, our study revealed a novel role of cathepsin D to maintain redox balance in HeLa cells. The knockdown of cathepsin D initiates a series of the LMP-associated consequences, like accumulation of ROS, inhibition of Nrf2 transcriptional activity and augmentation of cell senescence. Thus, we propose cathepsin D could play three unique biological roles that have been not reported yet. First of all, cathepsin D protein level serves to maintain lysosomal membrane integrity and redox balance. Secondarily, different from the exogenous ROS inducers that are likely to bring cellular apoptosis, cathepsin D knockdown can mildly and chronically increase oxidative stress within cells, which drives cell senescence rather than apoptosis. Thirdly, the late phase responses to oxidative stress, instead of Nrf2 activation at the early phase response, were the inhibition of Nrf2 activity and global attenuation of redox protein expression. Our study therefore provides a new angle to survey the biological functions of cathepsin D. A schematic diagram of cathepsin D knockdown inducing cell senescence is shown in Fig. 8.
Fig. 8.
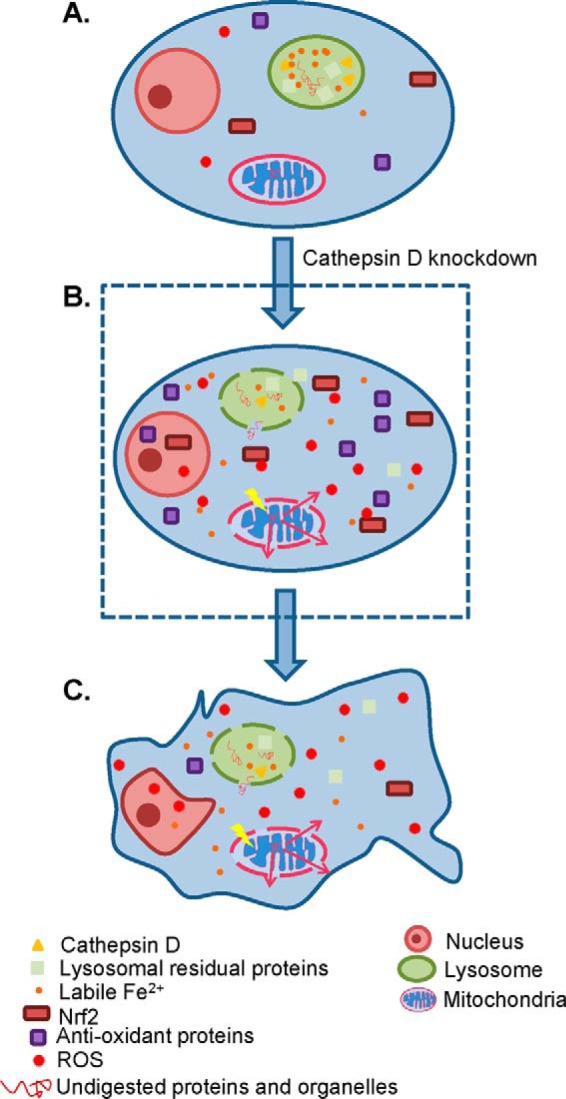
Schematic diagram of cathepsin D knockdown induced biological events. A, A normal HeLa cell is depicted, and the lysosomal contents, including cathepsin D, other lysosomal residual proteins, Fe2+, and undigested proteins and organelles, are well retained within lysosome. B, The knockdown of cathepsin D increases LMP and decreases ΔMMP, followed by ROS accumulation. The elevation of ROS transiently activates Nrf2 and promotes the transcription of many antioxidant proteins. C, Persistent oxidative stress because of lysosome rupture and mitochondria leakage, however, serves to inhibit the Nrf2 transcriptional activity and reduces the amount of antioxidant proteins, thus further aggravates oxidative stress within cells and finally leads to cellular senescence.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (54) via the PRIDE partner repository with the data set identifier PXD002844.
Supplementary Material
Footnotes
Author contributions: S.S., X.L., and S.L. designed research; S.S., D.X., and X.L. performed research; L.L., J. Zi, Y.D., J. Zhang, J. Zhu, and N.X. contributed new reagents or analytic tools; S.S., X.Z., Y.W., Y.Z., X.L., and S.L. analyzed data; S.S., X.C., Y.X., X.L., and S.L. wrote the paper.
* This work was supported by the National Natural Science Foundation of China (30800568) and the National High Technology Research and Development Program of China (2012AA020206).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- ROS
- Reactive oxygen species
- AKR1B1
- Aldo-keto reductase family 1 member B1
- AKR1C1
- Aldo-keto reductase family 1 member C1
- DMEM DMEM
- Dulbecco's Modified Eagle Medium
- GCLM
- Glutamate-cysteine ligase regulatory subunit
- GSTP1
- Glutathione S transferase P
- H2DCFDA
- 2′,7′-dichlorodihydrofluorescein diacetate
- iBAQ
- Intensity-based absolute quantification
- LMP
- Lysosomal membrane permeability
- NQO1
- NAD(P)H dehydrogenase [quinone] 1
- Nrf2
- Nuclear factor erythroid 2-related factor 2
- SILAC
- Stable isotope labeling by amino acids in cell culture.
REFERENCES
- 1. Benes P., Vetvicka V., and Fusek M. (2008) Cathepsin D – many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 68, 12–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ferguson J. B., Andrews J. R., Voynick I. M., and Fruton J. S. (1973) The specificity of cathepsin D. J. Biol. Chem. 248, 6701–6708 [PubMed] [Google Scholar]
- 3. Hah Y. S., Noh H. S., Ha J. H., Ahn J. S., Hahm J. R., Cho H. Y., and Kim D. R. (2012) Cathepsin D inhibits oxidative stress-induced cell death via activation of autophagy in cancer cells. Cancer Lett. 323, 208–214 [DOI] [PubMed] [Google Scholar]
- 4. Redmann M., Ouyang X., Benavides G. a., Liang Q., Darley-Usmar V. M., and Zhang J. (2013) Novel contributions of the lysosomal pathway to autophagy, neuronal bioenergetics, oxidative stress, and neurodegeneration; the role of cathepsin D. Free Rad. Biol. Med. 65, S48 [Google Scholar]
- 5. Cruz-Soto M. E., Cosio G., Jeziorski M. C., Vargas-Barroso V., Aguilar M. B., Carabez A., Berger P., Saftig P., Arnold E., Thebault S., Martinez de la Escalera G., and Clapp C. (2009) Cathepsin D is the primary protease for the generation of adenohypophyseal vasoinhibins: cleavage occurs within the prolactin secretory granules. Endocrinology 150, 5446–5454 [DOI] [PubMed] [Google Scholar]
- 6. Sevlever D., Jiang P., and Yen S. H. (2008) Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 47, 9678–9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hamazaki H. (1996) Cathepsin D is involved in the clearance of Alzheimer's beta-amyloid protein. FEBS Lett. 396, 139–142 [DOI] [PubMed] [Google Scholar]
- 8. Deiss L. P., Galinka H., Berissi H., Cohen O., and Kimchi A. (1996) Cathepsin D protease mediates programmed cell death induced by interferon-gamma, Fas/APO-1, and TNF-alpha. EMBO J. 15, 3861–3870 [PMC free article] [PubMed] [Google Scholar]
- 9. Roberg K., Kagedal K., and Ollinger K. (2002) Microinjection of cathepsin D induces caspase-dependent apoptosis in fibroblasts. Am. J. Pathol. 161, 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heinrich M., Neumeyer J., Jakob M., Hallas C., Tchikov V., Winoto-Morbach S., Wickel M., Schneider-Brachert W., Trauzold A., Hethke A., and Schutze S. (2004) Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ. 11, 550–563 [DOI] [PubMed] [Google Scholar]
- 11. Castino R., Bellio N., Nicotra G., Follo C., Trincheri N. F., and Isidoro C. (2007) Cathepsin D-Bax death pathway in oxidative stressed neuroblastoma cells. Free Rad. Biol. Med. 42, 1305–1316 [DOI] [PubMed] [Google Scholar]
- 12. Montcourrier P., Mangeat P. H., Salazar G., Morisset M., Sahuquet A., and Rochefort H. (1990) Cathepsin D in breast cancer cells can digest extracellular matrix in large acidic vesicles. Cancer Res. 50, 6045–6054 [PubMed] [Google Scholar]
- 13. Johnson M. D., Torri J. A., Lippman M. E., and Dickson R. B. (1993) The role of cathepsin D in the invasiveness of human breast cancer cells. Cancer Res. 53, 873–877 [PubMed] [Google Scholar]
- 14. Berchem G., Glondu M., Gleizes M., Brouillet J. P., Vignon F., Garcia M., and Liaudet-Coopman E. (2002) Cathepsin-D affects multiple tumor progression steps in vivo: proliferation, angiogenesis, and apoptosis. Oncogene 21, 5951–5955 [DOI] [PubMed] [Google Scholar]
- 15. Glondu M., Coopman P., Laurent-Matha V., Garcia M., Rochefort H., and Liaudet-Coopman E. (2001) A mutated cathepsin-D devoid of its catalytic activity stimulates the growth of cancer cells. Oncogene 20, 6920–6929 [DOI] [PubMed] [Google Scholar]
- 16. Vetvicka V., Benes P., and Fusek M. (2002) Procathepsin D in breast cancer: what do we know? Effects of ribozymes and other inhibitors. Cancer Gene Therap. 9, 854–863 [DOI] [PubMed] [Google Scholar]
- 17. Walls K. C., Klocke B. J., Saftig P., Shibata M., Uchiyama Y., Roth K. A., and Shacka J. J. (2007) Altered regulation of phosphatidylinositol 3-kinase signaling in cathepsin D-deficient brain. Autophagy 3, 222–229 [DOI] [PubMed] [Google Scholar]
- 18. Ollinger K. (2000) Inhibition of cathepsin D prevents free-radical-induced apoptosis in rat cardiomyocytes. Arch. Biochem. Biophys. 373, 346–351 [DOI] [PubMed] [Google Scholar]
- 19. Follo C., Ozzano M., Montalenti C., Santoro M. M., and Isidoro C. (2013) Knockdown of cathepsin D in zebrafish fertilized eggs determines congenital myopathy. Biosci. Rep. 33, e00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shacka J. J., Klocke B. J., Young C., Shibata M., Olney J. W., Uchiyama Y., Saftig P., and Roth K. A. (2007) Cathepsin D deficiency induces persistent neurodegeneration in the absence of Bax-dependent apoptosis. J. Neurosci. 27, 2081–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bewley M. A., Pham T. K., Marriott H. M., Noirel J., Chu H. P., Ow S. Y., Ryazanov A. G., Read R. C., Whyte M. K., Chain B., Wright P. C., and Dockrell D. H. (2011) Proteomic evaluation and validation of cathepsin D regulated proteins in macrophages exposed to Streptococcus pneumoniae. Mol. Cell. Proteomics 10, M111.008193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koch S., Scifo E., Rokka A., Trippner P., Lindfors M., Korhonen R., Corthals G. L., Virtanen I., Lalowski M., and Tyynela J. (2013) Cathepsin D deficiency induces cytoskeletal changes and affects cell migration pathways in the brain. Neurobiol. Dis. 50, 107–119 [DOI] [PubMed] [Google Scholar]
- 23. Glondu M., Liaudet-Coopman E., Derocq D., Platet N., Rochefort H., and Garcia M. (2002) Down-regulation of cathepsin-D expression by antisense gene transfer inhibits tumor growth and experimental lung metastasis of human breast cancer cells. Oncogene 21, 5127–5134 [DOI] [PubMed] [Google Scholar]
- 24. Tedone T., Correale M., Barbarossa G., Casavola V., Paradiso A., and Reshkin S. J. (1997) Release of the aspartyl protease cathepsin D is associated with and facilitates human breast cancer cell invasion. FASEB J. 11, 785–792 [DOI] [PubMed] [Google Scholar]
- 25. Lou X., Xiao T., Zhao K., Wang H., Zheng H., Lin D., Lu Y., Gao Y., Cheng S., Liu S., and Xu N. (2007) Cathepsin D is secreted from M-BE cells: its potential role as a biomarker of lung cancer. J. Proteome Res. 6, 1083–1092 [DOI] [PubMed] [Google Scholar]
- 26. van de Wetering M., Oving I., Muncan V., Pon Fong M. T., Brantjes H., van Leenen D., Holstege F. C., Brummelkamp T. R., Agami R., and Clevers H. (2003) Specific inhibition of gene expression using a stably integrated, inducible small-interfering-RNA vector. EMBO Rep. 4, 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cui X., and Churchill G. A. (2003) Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 4, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khalkhali-Ellis Z., and Hendrix M. J. C. (2014) Two Faces of Cathepsin D: Physiological Guardian Angel and Pathological Demon. Biol. Med. 6, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boya P., Gonzalez-Polo R. A., Casares N., Perfettini J. L., Dessen P., Larochette N., Metivier D., Meley D., Souquere S., Yoshimori T., Pierron G., Codogno P., and Kroemer G. (2005) Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 25, 1025–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boya P., and Kroemer G. (2008) Lysosomal membrane permeabilization in cell death. Oncogene 27, 6434–6451 [DOI] [PubMed] [Google Scholar]
- 31. Moriyama Y., Takano T., and Ohkuma S. (1982) Acridine orange as a fluorescent probe for lysosomal proton pump. J. Biochem. 92, 1333–1336 [DOI] [PubMed] [Google Scholar]
- 32. Lin H. J., Herman P., Kang J. S., and Lakowicz J. R. (2001) Fluorescence lifetime characterization of novel low-pH probes. Anal. Biochem. 294, 118–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Z., Zhang J., Wang Y., Xing R., Yi C., Zhu H., Chen X., Guo J., Guo W., Li W., Wu L., Lu Y., and Liu S. (2013) Matrine, a novel autophagy inhibitor, blocks trafficking and the proteolytic activation of lysosomal proteases. Carcinogenesis 34, 128–138 [DOI] [PubMed] [Google Scholar]
- 34. Dixon S. J., and Stockwell B. R. (2014) The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17 [DOI] [PubMed] [Google Scholar]
- 35. Tenopoulou M., Kurz T., Doulias P. T., Galaris D., and Brunk U. T. (2007) Does the calcein-AM method assay the total cellular “labile iron pool” or only a fraction of it? Biochem. J. 403, 261–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Purdom-Dickinson S. E., Sheveleva E. V., Sun H., and Chen Q. M. (2007) Translational control of nrf2 protein in activation of antioxidant response by oxidants. Mol. Pharmacol. 72, 1074–1081 [DOI] [PubMed] [Google Scholar]
- 37. Fratta Pasini A., Albiero A., Stranieri C., Cominacini M., Pasini A., Mozzini C., Vallerio P., Cominacini L., and Garbin U. (2012) Serum oxidative stress-induced repression of Nrf2 and GSH depletion: a mechanism potentially involved in endothelial dysfunction of young smokers. PloS One 7, e30291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Covas G., Marinho H. S., Cyrne L., and Antunes F. (2013) Activation of Nrf2 by H2O2: de novo synthesis versus nuclear translocation. Methods Enzymol. 528, 157–171 [DOI] [PubMed] [Google Scholar]
- 39. Mitsuishi Y., Taguchi K., Kawatani Y., Shibata T., Nukiwa T., Aburatani H., Yamamoto M., and Motohashi H. (2012) Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79 [DOI] [PubMed] [Google Scholar]
- 40. Lee J. M., Calkins M. J., Chan K., Kan Y. W., and Johnson J. A. (2003) Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 278, 12029–12038 [DOI] [PubMed] [Google Scholar]
- 41. Li J., Lee J. M., and Johnson J. A. (2002) Microarray analysis reveals an antioxidant responsive element-driven gene set involved in conferring protection from an oxidative stress-induced apoptosis in IMR-32 cells. J. Biol. Chem. 277, 388–394 [DOI] [PubMed] [Google Scholar]
- 42. Singh A., Misra V., Thimmulappa R. K., Lee H., Ames S., Hoque M. O., Herman J. G., Baylin S. B., Sidransky D., Gabrielson E., Brock M. V., and Biswal S. (2006) Dysfunctional KEAP1-NRF2 interaction in nonsmall-cell lung cancer. PLoS Med. 3, e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Johansson A. C., Appelqvist H., Nilsson C., Kagedal K., Roberg K., and Ollinger K. (2010) Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 15, 527–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zheng L., Terman A., Hallbeck M., Dehvari N., Cowburn R. F., Benedikz E., Kågedal K., Cedazo-Minguez A., and Marcusson J. (2014) Macroautophagy-generated increase of lysosomal amyloid β-protein mediates oxidant-induced apoptosis of cultured neuroblastoma cells. Autophagy 7, 1528–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Q., and Ames B. N. (1994) Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc. Natl. Acad. Sci. U.S.A. 91, 4130–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee A. C., Fenster B. E., Ito H., Takeda K., Bae N. S., Hirai T., Yu Z. X., Ferrans V. J., Howard B. H., and Finkel T. (1999) Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 274, 7936–7940 [DOI] [PubMed] [Google Scholar]
- 47. Macip S., Igarashi M., Fang L., Chen A., Pan Z. Q., Lee S. W., and Aaronson S. A. (2002) Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 21, 2180–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rai P. (2010) Oxidation in the nucleotide pool, the DNA damage response and cellular senescence: defective bricks build a defective house. Mutat. Res. 703, 71–81 [DOI] [PubMed] [Google Scholar]
- 49. Kim H. J., and Vaziri N. D. (2010) Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Renal Physiol. 298, F662–F671 [DOI] [PubMed] [Google Scholar]
- 50. Safdar A., deBeer J., and Tarnopolsky M. A. (2010) Dysfunctional Nrf2-Keap1 redox signaling in skeletal muscle of the sedentary old. Free Rad. Biol. Med. 49, 1487–1493 [DOI] [PubMed] [Google Scholar]
- 51. Volonte D., Liu Z., Musille P. M., Stoppani E., Wakabayashi N., Di Y. P., Lisanti M. P., Kensler T. W., and Galbiati F. (2013) Inhibition of nuclear factor-erythroid 2-related factor (Nrf2) by caveolin-1 promotes stress-induced premature senescence. Mol. Biol. Cell 24, 1852–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang G., Zhao K., Ju Y., Mani S., Cao Q., Puukila S., Khaper N., Wu L., and Wang R. (2013) Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxidants Redox Signal. 18, 1906–1919 [DOI] [PubMed] [Google Scholar]
- 53. Volonte D., Zhang K., Lisanti M. P., and Galbiati F. (2002) Expression of caveolin-1 induces premature cellular senescence in primary cultures of murine fibroblasts. Mol. Biol. Cell 13, 2502–2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vizcaino J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Rios D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., and Gatto L. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (54) via the PRIDE partner repository with the data set identifier PXD002844.