

Abstract
Platinum-resistance is a major limitation to effective chemotherapy regimens in high-grade serous ovarian cancer (HGSOC). To better understand the mechanisms involved we characterized the proteome and phosphoproteome in cisplatin sensitive and resistant HGSOC primary cells using a mass spectrometry-based proteomic strategy. PCA analysis identified a distinctive phosphoproteomic signature between cisplatin sensitive and resistant cell lines. The most phosphorylated protein in cisplatin resistant cells was sequestosome-1 (p62/SQSTM1). Changes in expression of apoptosis and autophagy related proteins Caspase-3 and SQSTM1, respectively, were validated by Western blot analysis. A significant increase in apoptosis in the presence of cisplatin was observed in only the sensitive cell line while SQSTM1 revealed increased expression in the resistant cell line relative to sensitive cell line. Furthermore, site-specific phosphorylation on 20 amino acid residues of SQSTM1 was detected indicating a hyper-phosphorylation phenotype. This elevated hyper-phosphorylation of SQSTM1 in resistant HGSOC cell lines was validated with Western blot analysis. Immunofluoresence staining of s28-pSQSTM1 showed inducible localization to autophagosomes upon cisplatin treatment in the sensitive cell line while being constitutively expressed to autophagosomes in the resistant cell. Furthermore, SQSTM1 expression was localized in cancer cells of clinical high-grade serous tumors. Here, we propose hyper-phosphorylation of SQSTM1 as a marker and a key proteomic change in cisplatin resistance development in ovarian cancers by activating the autophagy pathway and influencing down-regulation of apoptosis.
Ovarian cancer is the leading cause of death among all other gynecologic malignancies, with high-grade serous ovarian carcinoma (HGSOC)1 as the predominant subtype (1, 2). Late diagnosis and chemoresistance are major factors in low survival outcomes. The standard treatment involves surgical removal of the tumor, associated with administration of platinum-based chemotherapy. Although this treatment is initially effective, it is often followed by relapse and subsequent chemoresistance. Cancer recurs in ∼25% of patients within six months and the overall five-year survival rate is 31% (3). Three main mechanisms for the cisplatin-resistant phenotype of tumor cells have been proposed: (1) decreased cellular drug accumulation, (2) altered detoxification mechanism, and (3) DNA repair (4). The involvement of one or more of these resistance mechanisms and alternations in other signaling pathways has been extensively studied in ovarian cancer models (5).
The inter- and intrastrand covalent adduction of DNA by cisplatin is generally accepted as the critical pharmacological target of cisplatin-induced cytotoxicity, triggering programmed cell death by induction of apoptosis (6, 7). However, a defect in the apoptosis pathway is associated with resistance in tumor cell lines (8). Autophagy is another signaling pathway that has been investigated for its role in cancer drug resistance upon cisplatin treatment (9–12). Autophagy has been shown to increase in cisplatin-resistant HGSOC cell lines in comparison to cisplatin-sensitive cell lines. Inhibition of autophagy by 3-methyladenine (3-MA) increases the rate of cell death with no effects on apoptosis (13). Furthermore, knockdown of autophagy inducer ERK by siRNA decreases autophagy and subsequently sensitizes ovarian cancer cells to cisplatin-induced apoptosis (14).
Circumventing cisplatin resistance remains a critical goal for chemotherapy strategies. Using an unbiased analysis platform, we describe the proteome and phosphoproteome of cisplatin-sensitive and resistant HGSOC-derived cells in the absence and presence of cisplatin. Our results suggest that hyper-phosphorylation of sequestosome-1 (p62/SQSTM1), a regulator of apoptosis and autophagy, is associated with development of cisplatin resistance.
EXPERIMENTAL PROCEDURES
Cell Lines
Primary cell lines M019i and OC002 originated from ascites of women with HGSOC. OC002 cells were derived from primary surgery while M019i were derived from interval surgery after neoadjuvant platinum-taxane chemotherapy. The patients had rapid progression of HGSOC with progression-free survival (PFS) of 2.4 and 10.1 months and overall survival of 34.3 and 12.0, respectively. Thus, for M019i a significant disease control was achieved with second line chemotherapy after relapse. Cisplatin-resistant variants (M019iCis and OC002Cis) of the original cells were generated using methods described previously (15). Briefly, the original cells were grown in stepwise increase of cisplatin concentrations up to 2.0 μg/ml (6.6 μmol/L). All cell lines were grown as spheroids in serum-free Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F12, Lonza, Basel, Switzerland) culture media supplemented with B-27® supplement (Life Technologies, NY), 20 ng/ml EGF (Sigma, St. Louis, MO), and 10 ng/ml bFGF (Invitrogen, Carlsbad, CA). Platinum resistant cells were treated with cisplatin in every third subculture and left to recover at least 3 days before plated for sample preparation. The cellular response to cisplatin was monitored with regular assays. Cells were plated on 96 well plates 2500 cells/well and cisplatin was added on the following day in final concentrations of 0.01, 0.1, 1.0, 10, and 100 μmol/L. Cell viability was measured 72 h after the first treatment using the ATP assay (CellTiter-Glo® Luminescent Cell Viability Assay, Promega, Madison, WI) in triplicate wells each. IC50 value for cisplatin increased from 0.6 μmol/L of M019i and 0.8 μmol/L of OC002 to 7.0 μmol/L of M019iCis and 5.0 of OC002Cis (Fig. 1A–1B and supplemental Fig. S1A). Three biological replicates from cell lysates were prepared from individual culture dishes and processed in parallel for cisplatin-sensitive M019i and resistant M019iCis cell lines in the absence and presence of IC50 dose of cisplatin for proteomic, phosphoproteomic, and validation analysis. To study the effect of autophagy inhibition the platinum resistant M019iCis cells were treated with 10 μmol/L cisplatin, 5 mmol/L 3-MA or both for 48 h after which cell viability was measured as above. OC002 cell lines were used to validate findings in MO19i cell lines. All experiments were performed according to institutional ethical guidelines.
Fig. 1.
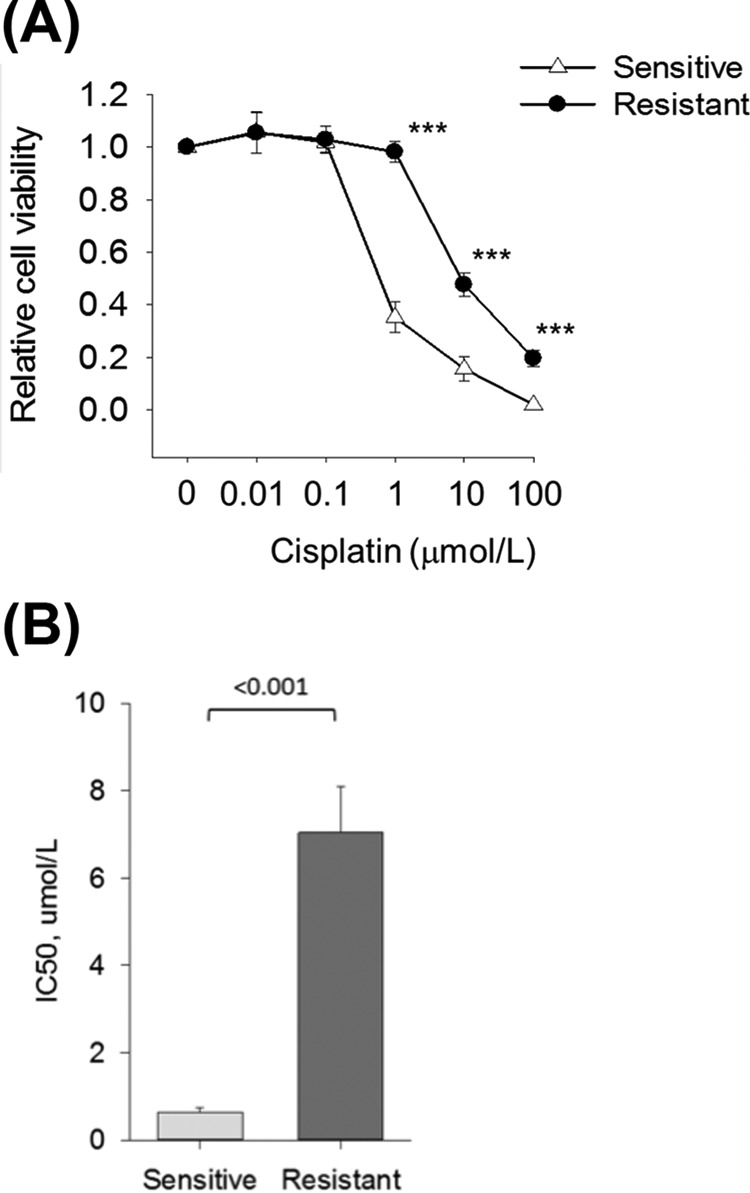
Cisplatin response in platinum sensitive M019i and resistant M019iCis cell lines. A, Relative cell viability as compared with untreated control. Statistical analysis between cell lines at different cisplatin concentrations were calculated using Student's t test (mean ± S.E., n = 5). *** p < 0.001. B, Half maximal inhibitory concentration (IC50) values of cisplatin for M019i (mean 0.630, S.E. 0.0476) and M019iCis (mean 7.038, S.E. 0.466) were calculated from five independent measurement. The IC50 concentrations of cisplatin were futher used to evaluate cells response to cisplatin at the molecular level.
Protein Preparation
M019i and M019iCis cells were suspended in 50 mm Ammonium biocarbonate, 8 m Urea (pH6–7), and protease inhibitor mixture (Roche Applied Science, Indianapolois, IN). The lysates were vortexed and sonicated in three cycles for 5 mins. Proteins were precipitated overnight by ice cold acetone. Samples were centrifuged for 15 min at 3500RPM in 4°C. Samples were prepared in triplicate. Total protein measurements were determined using the Bicinchoninic acid protein assay (Bio-Rad, Hercules, CA). 100 μg and 1 mg of protein extracts were used for protein and phosphoprotein analysis, respectively. Ten micrograms of α-casein (Sigma) was added to 1 mg of protein extracts for phosphoproteomics. Both protein extracts were denatured with 8 m urea in 50 mm Tris-HCl (pH 8.5) before reduction with 10 mm DTT at 37 °C for 1 h and alkylation with 50 mm iodoacetamide in the dark for 1 h. Alkylation was stopped by addition of 50 mm DTT. The samples were digested with a .02 μg/μl modified trypsin (Promega) concentration in 50 mm Tris-HCl (pH8.5) at 37 °C for 20 h. Tryptic digests were slightly acidified with 10% TFA and desalted with an Empore C18 cartridge and eluted with 1 ml of 6% TFA/80% ACN. Peptides produced by this protocol are referred to as whole proteome samples.
Phosphopeptide Enrichment
For phosphopeptide enrichment, following the desalting protocol (vide supra), the peptides were enriched on 5 mg of TiO2 (GL Science, Saitama, Japan) placed in a pipette tip and positioned in a 1.5 ml Eppendorf tube. Phosphopeptides were eluted with 200 μl of 5% NH4OH and desalted immediately with a C18 column prepared in house. Phosphopeptides were eluted manually with 50 μ of 0.1% FA/80% ACN and evaporated to dryness in a SpeedVac. The dried peptides were reconstituted with 0.5% FA in 2% ACN. Peptides produced by this protocol are referred to as TiO2 enriched samples.
Mass Spectrometry Analysis
Proteins were identified from the whole proteome peptide samples by tandem mass spectra (MS/MS) of peptides on an Orbitrap Fusion™ Tribrid™ mass spectrometer (Thermo Scientific Corp., San Jose, CA) coupled to a NanoAquity UPLC system (Waters Corporation, Milford, MA). Peptides were trapped on a 100 μm i.d. x 20 mm long precolumn in-house packed with 200 Å (5 μm) Magic C18 particles (C18AQ; Michrom Bioresources Inc., Auburn, CA). Subsequent peptide separation was on an in-house constructed 75 μm i.d. x 180 mm long analytical column pulled using a Sutter Instruments P-2000 CO2 laser puller (Sutter Instrument Company, Novato, CA) and packed with 100 Å (5 μm) C18AQ particle. For each liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis, an estimated amount of 1 μg of peptides (0.2 μg/μl) was loaded on the precolumn at 4 μl/min in water/acetonitrile (95/5) with 0.1% (v/v) formic acid. Peptides were eluted using an acetonitrile gradient flowing at 250 nL/min using mobile phase gradient of 5–35% acetonitrile over 60 min with a total gradient time of 95 min. The eluting peptides were interrogated with an Orbitrap Fusion mass spectrometer using data-dependent acquisition method with the Top Speed decisions selection. FTMS1 spectra were collected using the following parameters: scan range 350–1800 m/z, resolving power 120 k, AGC target 4E5, and maximum injection time of 50ms. ITMS2 spectra were collected using the following parameters: rapid scan rate, CID NCE 35, 1.6 m/z isolation window, AGC target 1E4, and maximum injection time of 50 ms. MS2 precursors were selected for a 3 s cycle. Precursors with an assigned monoisotopic m/z and a charge state of 2–7 were interrogated. Precursors were filtered using a 60 s dynamic exclusion window. Samples were injected three times and analyzed as technical replicates.
Peptides enriched by TiO2 capture for phosphoprotein identification were analyzed in a similar manner using a Q Exactive quadrupole-orbitrap mass spectrometer (Thermo Fisher Scientific). The Q Exactive was coupled to an EASY-nLC II nanoflow LC instrument (Thermo Fisher Scientific). A 100 μm × 3 cm trap column and a 75 μm × 15 cm analytical column were in-house packed with Magic C18AQ resin (5 μm, 200-Å pore size; Michrom Bioresources). After injecting about 45% (v/v) of the sample onto the column, phosphopeptides were eluted using a 3-step linear gradient at a flow rate of 300 nl/ml using solvent A (0.2% FA in 2% ACN) and solvent B (0.2% FA in 95% ACN). The initial linear gradient was 2 to 20% B for 30 min, then 40% B until 40 min, and finally 100% B for 45–50 min. Data dependent acquisition was performed in positive ion mode. MS spectra were acquired from 300–2000 m/z in the orbitrap with resolution of 70,000, an AGC target value of 1e6 ions, and a maximal injection time of 120 ms. The 10 most abundant ions of charge states 2+ or higher were selected for subsequent fragmentation by HCD. The MS/MS spectra were acquired in the orbitrap with a resolution of 17,500, an AGC target value of 5e4 ions, maximal injection time of 250ms, lowest mass fixed at 100 m/z, and dynamic exclusion duration set to 20s.
Protein Identification
Data was searched using Mascot (v2.4.0; Matrix Science, London, UK) against the UniProt database (v2014–04, H. sapiens, 40,612 sequences), common contaminants, and reverse sequences, via Proteome Discoverer (v1.3.0.339, Thermo Fischer Scientific). Search parameters in analysis allowed modified residues (fixed cysteine carbamidomethylation and variable methionine oxidation, phosphorylation of serine/threonine/tyrosine, and acetylation on protein N terminus) and one missed cleavage site. A protein was considered with a confidence threshold of >95% and the identification of more than one unique peptide. Mass tolerance of mass spectra acquired on the Orbitrap Fusion™ Tribrid™ mass spectrometer from the whole proteome samples used a tolerance set to 5 ppm and 0.6 Da in MS and MS/MS mode, respectively. For the peptides enriched by TiO2 analyzed on a Q Exactive quadrupole-orbitrap mass spectrometer, a mass tolerance of 5 ppm and 0.02 Da in MS and MS/MS mode was used, respectively. Expectation value of ≤0.05 indicated peptide sequence identifications. An FDR of 1% was estimated using the target-decoy strategy separately for phosphorylated and nonphosphorylated peptides, as described by Marx et al. (16). PhosphoRS (17) v3.0 implemented in Proteome Discoverer (neutral loss option disabled) was used to validate phosphorylation sites. The MS proteomic data have been deposited to the Proteome Xchange Consortium (http://proteomecentral.org/) with data set identifier: PXD002394 and be accessed with the username: reviewer43047@ebi.ac.uk and password: K6aTjHQR.
Protein Quantification
Nonlabeled quantification was performed using Progenesis LC-MS (v4.1, Nonlinear). Here, extracted peptide intensity (MS1) features were generated for the evaluation of differential protein expressions. Selection of the best reference run was selected and an automatic alignment was performed for all data based on RP-HPLC retention times of each analysis. Detected features represent peptide ions. Proteins from the whole proteome samples were normalized to the sum of ion abundances of unique peptides, whereas phosphopeptides enriched by TiO2 capture were normalized to the abundance of phosphopeptides detected from the α-casein standard. All features that had no associated MS/MS spectra were deleted. Differentiation of proteins and phosphopeptides was determined using analysis of variance (ANOVA). Proteins and phosphopeptides with a p value ≤0.05 were considered significant with a power ≥0.8. The total cumulative abundance was calculated by averaging the individual abundance across the triplicate samples.
Functional Analysis
Functional annotation of the sensitive and resistance proteome and phosphoproteome was conducted using database for annotation, visualization, and integrated discovery (DAVID) software (18). Overrepresented functional categories among proteins enriched in each sample population was relative to a background of all identified proteins in study using a permutation-based p value <1e−3. Experimentally verified and published protein-protein interactions from several resources including Ingenuity's knowledgebase (www.Ingenuity.com) and STRING (19) were assessed.
Western Blot Analysis
For Western blotting, cell pellets were lysed in RIPA buffer (150 MM Tris-HCL, pH 7.4, 1% Nonidet P-40, 150 mm NaCl, 0.5% sodium deoxycholate, 1 mm EDTA and 1 mm SDS) with protease inhibitor mixture (Roche) and 1 mm orthovanadate (Sigma). Each sample containing 20 μg of total protein was separated by 10–15% polyacrylamide gel and blotted onto nitrocellulose membranes. Primary antibodies from Autophagy Antibody Sampler Kit (Cell Signaling Technology, Beverly, MD) as well as rabbit polyclonal anti-human SQSTM1 (Pierce, Carlsbad, CA, PA5–12092), rabbit polyclonal anti-human phospho-SQSTM1 (pSer28, Pierce, PA5–35409), rabbit polyclonal anti-human phospho-SQSTM1 (pThr269/Ser272, Cell Signaling Technology, #13121) mouse monoclonal anti-human PSMD9 (Abcam, ab58115), mouse monoclonal anti-human ubiquitin (PFD1, Cell Signaling Technology, Beverly, MD, #3936) and rabbit monoclonal anti-human Cleaved Caspase-3 (Asp175) (5A1E, Cell Signaling Technology, #9664) were used to validate protein expression in cell samples. Horseradish peroxidase-conjugated secondary antibodies were purchased from Dako. The signals were visualized by enhanced chemiluminescence (Thermo Scientific, Waltham, MA) and quantitated using ImageJ (version 1.47v). Statistical analyses were performed using the statistical software Sigma Stat 3.11 (Systat Software Inc., Chicago, IL).
Patient Samples
To study SQSTM1 expression in clinical tumor specimens we used a prospective ovarian cancer cohort of HGS-OvCa patients treated at the Department of Obstetrics and Gynecology, Turku University Hospital (Turku, Finland; ClinicalTrials.gov Id: NCT01276574 (20, 21)). The study and the use of all clinical material have been approved by (1) The Ethics Committee of the Hospital District of Southwest Finland (ETMK): ETMK 53/180/2009 × 238 and (2) National Supervisory Authority for Welfare and Health (Valvira): DNRO 6550/05.01.00.06/2010 and STH507A. Patients were treated with either primary surgery followed by six cycles of platinum- and taxane-based chemotherapy or three cycles of neoadjuvant chemotherapy (NACT) followed by interval debulking surgery and three to six chemotherapy cycles. The treatment modality for each patient was determined based on preoperative imaging studies and diagnostic laparoscopy. Tumor samples were collected during operation.
Quantitative Real-time RT-PCR
Fresh frozen tumor specimens were collected before and after neoadjuvant chemotherapy from eleven women with HGSOC. RNA was extracted with RNeasy kit (Qiagen, Hilden, Germany) and reverse transcribed to cDNA using Tetro cDNA synthesis kit (Bioline, London, UK) and oligo dT primers from tumor specimens and cell lines. Expression of selected markers was determined in triplicate samples using TaqMan qRT-PCR with Applied Biosystems 7900HT instrument (FinnishDNAMicroarray Centre, Turku Centre for Biotechnology, University of Turku, Turku, Finland). The primers and probes were designed using Universal ProbeLibrary Assay Design Center (Roche Applied Science). Raw qRT-PCR Ct values were normalized against the geometric mean of PPIA and TBP (21).
Immunofluorescence Staining and Microscopy
For immunofluorescence, cells were grown adherently in RPMI medium supplemented with 10% serum, ultra-glutamine and antibiotics. Cells were allowed to attach on gelatin-coated coverslips for 6 h, treated with or without 10 μmol/L cisplatin for 18 h and fixed with 4% paraformaldehyde. For localization of S28-pSQSTM1 and LC3, sections were stained with rabbit anti-human S28-pSQSTM1 antibody (1:250; Pierce, PA5–35409) and rabbit anti-human LC3 antibody (1:100; Cell Signaling Technology, #4108) followed by Alexa Fluor 568 or 488 goat anti-rabbit IgG (1:500; Invitrogen). Stainings was performed in phosphate buffered saline (PBS) supplemented with 5% bovine serum albumin and 0.25% Triton X-100. Epifluorescence images were taken with confocal microscope (LSM780 microscope, Zeiss Microscopy GmbH Germany) and analyzed with ZEN 2 software (Zeiss Microscopy GmbH, Oberkochen, Germany).
Immunohistochemistry
Sections of paraffin-embedded high-grade serous ovarian cancer specimens were stained with rabbit anti-human SQSTM1 polyclonal antibody (1:75; Pierce) using a Benchmark XT autostainer device (Roche Tissue Diagnostics/Ventana Medical Systems, Tucson, AZ) by pretreatment with standard CC1 (pH 8.5), antibody incubation at 37C for 44 min followed by counterstaining with Hematoxylin for 4 min. For detection, an UltraView Universal DAB Detection Kit was used (Roche Tissue Diagnostics/Ventana Medical Systems).
RNA In Situ Hybridization
Five micrometer tissue sections from paraffin-embedded tumor specimens were subjected for RNA in situ hybridization. Chromogenic target mRNA detection using RNAscope 2.5 HD detection kit -BROWN (#322300) was performed according to the manual. In short, tissue sections were baked for 1 h at 60 °C, deparaffinized, and treated with hydrogen peroxide for 10 min at room temperature (RT). Target retrieval was performed for 15 min at 100 °C, followed by protease treatment for 30 min at 40 °C. The probe hs-SQSTM1 (#415881), as well as positive and negative control probes Hs-PPIB (#313901) and dihydrodipicolinate reductase (dapB) (#310043), respectively, were hybridized for 2 h at 40 °C followed by RNAscope amplification steps. The samples were incubated for 60 min with AMP 5 reagent. The samples were treated with DAB for 10 min at room temperature followed by counterstaining with 50% hematoxylin. The sections were dipped in ammonium water and dehydrated before mounting.
RESULTS
Overview of M019i HGSOC Cellular Proteome and Phosphorylated Proteome
From the whole proteome and TiO2 enriched peptide samples of sensitive M019i (IC50 = 0.6 μmol/L) and resistant M019iCis (IC50 = 7.0μmol/L) cell lines (Fig. 1A–1B), 2790 and 1361 quantifiable proteins were identified in both the absence and presence of cisplatin, respectively. A total of 2905 unique phosphorylation sites were mapped from the TiO2 enriched samples with 644 proteins identified in both the whole proteome and TiO2 enriched sample populations with 87 of the proteins being phosphorylated in both populations. Supplemental Tables S1 and S2 contain lists of identified proteins and phosphorylation sites. Proteins identified in the non-enriched samples were functionally categorized to cytosol, RNA, mitochondrion, and ribosome relative to the entire human proteome. Proteins in the TiO2 enriched samples were categorized to nuclear, cytoskeleton, cell cycle, and mitosis (p value <1 × 10−10); see supplemental Tables S3 and S4 for details. The results are in agreement with >70% of phosphorylation estimated to occur on nuclear proteins (22). All identified proteins were quantified and used as background in functional categorization by differential statistics. Principle component analysis (PCA) of differentially expressed proteins (i.e. proteins whose relative abundance changed between pair-wise comparisons) segregated the M019i and M019iCis cell lines; however, phosphorylation of a few proteins provided better classification of the cellular subtypes (Fig. 2A). Furthermore, a larger flux in identifications between the M019i and M019iCis cell lines in the presence and absence of cisplatin was only seen in the phosphoproteome data (Fig. 2B).
Fig. 2.

HGSOC landscape in the absence and presence of cisplatin. Whole proteome and TiO2 enriched peptides. A, Comparative principle component analysis of all samples. B, Protein identifications for platinum sensitive M019i and resistant M019iCis cells for untreated samples and with cisplatin treatment IC50 (0.6 μmol/L). (mean ± S.D., n = 3).
Proteome of M019i HGSOC Cell Lines in Absence and Presence of Cisplatin
In the Absence of Cisplatin
A total of 312 differentially expressed proteins were identified between sensitive M019i and resistant M019iCis HGSOC cell lines in the absence of cisplatin in the whole proteome samples. The majority of differentially expressed proteins (n = 237) were more abundant in the cisplatin-resistant M019iCis cell line, while 75 proteins were more abundant in the cisplatin-sensitive M019i cell line; see supplemental Table S5. Functional pathway analysis revealed an enrichment of proteins involved in transcription in the cisplatin sensitive cell line while enrichment in proteins involved in glutathione metabolism and caspase activity was indicated in the cisplatin resistant cell line.
In the Presence of Cisplatin
Both cell lines showed an enrichment of proteins associated with the mitochondria, endoplasmic reticulum, and protein transport. Supplemental Tables S6 and S7 contain a list of differentially expressed proteins in the absence and presence of cisplatin treated M019i and M019iCis cells, respectively. The proteasome complex was, however, only enriched in the resistant cell line (Fig. 3). Functional mapping of over-expressed proteins in the presence versus the absence of cisplatin indicated proteasomal protein catabolic processes only in the platinum resistant M019iCis cell line (supplemental Table S8).
Fig. 3.

Functional analysis of differentially expressed proteins of cisplatin sensitive and resistant cells in the absence and presence of treatment.
Phosphorylated Proteome of MO19i HGSOC Cells in Absence and Presence of Cisplatin
In the Absence of Cisplatin
A total of 27 differentially expressed phosphorylation sites were identified between the platinum sensitive M019i and resistant M019iCis HGSOC cell lines in the absence of cisplatin in the TiO2 enriched samples. Phosphorylation sites were identified on 13 proteins, with over half of the phosphorylation sites mapped to the sequestosome-1 (SQSTM1) protein (Fig. 4A). SQSTM1 has been proposed to have many putative phosphorylation sites (23). The seven phosphorylation sites identified as more abundant in the M019i cells are cited as being regulators in mitotic cells (22) and pluripotency of human embryonic cells (24). Among the six phosphorylated proteins more abundant in the M019iCis cells, two of the other top three most changed proteins (ubiquitin-40S ribosomal protein and neighbor of BRCA1 gene (NBR1)) are hypothesized to interact with SQSTM1 in selective autophagy clearance of ubiquitinated proteins (25). Cisplatin-sensitive ovarian cancer cells have been shown to accumulate ubiquitinated proteins in the presence of cisplatin while resistant cell lines have not (26). SQSTM1 is suggested to shuttle polyubiquitinated proteins for degradation by autophagy reducing apoptosis induced by ER stress (26) and mediation of caspases (27, 28), but the functional relationship of these processes is still being elucidated.
Fig. 4.

Phosphoproteome fold change results. A, Fold change in resistant and sensitive cell lines in absence of cisplatin treatment (mean ± S. D. of ratio, n = 3). B, Fold change in sensitive cell lines in absence and presence of cisplatin treatment (mean ± S.D. of ratio, n = 3).
In the Presence of Cisplatin
This same phosphoproteomic signature was observed in the comparison of M019i cell line in the absence and presence of cisplatin (Fig. 4B). There, eight phosphorylation sites were found to be increased in M019iCis cells upon exposure to cisplatin involved in acetylation (p value 2.8 × 10−2); see supplemental Table S9. These proteome and phosphoproteome results led to further investigation of the apoptotic pathway, autophagy, and proteasome to elucidate their role in these cell lines.
Apoptosis is Induced Only in the Platinum Sensitive Cell Lines Upon Exposure to Cisplatin
Because of the known effects of cisplatin to induce apoptosis, the protein expression levels of proteins associated with apoptosis was investigated. The summation of the label-free relative quantities of proteins (GRP78 (31), CH60 (32), CH10 (32), CYC (33), and STAT1 (34)) was significantly increased in the M019i cell line upon exposure to cisplatin (p value = .05, t test), which was not observed in the M019iCis cell line (p value = .14, t test) (supplemental Fig. S2). Western blot analysis of caspase 3, responsible for apoptosis execution, further confirmed an increase upon cisplatin treatment in the sensitive M019i cell line not seen in the M019iCis cell line (p value = 2.0 × 10−3) (Fig. 5). Similar result was observed in cell line OC002 (supplemental Fig. S1B).
Fig. 5.
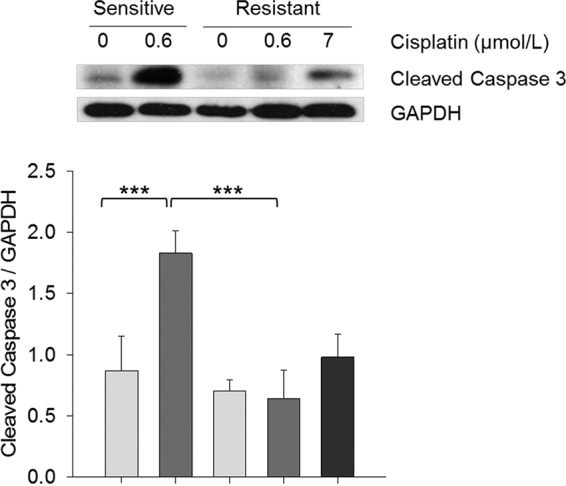
Quantitative results of cleaved caspase 3 associated with apoptosis. Western blot analysis of cleaved Caspase 3. Statistical differences were calculated using Two-Way ANOVA with multiple comparisons using posthoc Tukey test for paired treatment groups (- and +) of sensitive and resistant cells and using One-Way ANOVA with multiple comparisons using posthoc Holm-Sidak method within treatment groups of resistant cell line. * p < 0.05, ** p < 0.01, *** p < 0.001.
Autophagy is Induced in the Platinum Resistant Cell Line
Six proteins were identified in the whole proteome samples to be involved with autophagy (35): SQSTM1, MAP1B, Cathepsin B, Cathepsin D, Rab 7, and ATG3. The first four proteins were more abundant in M019iCis relative to M019i cell lines in the absence of cisplatin; see supplemental Table S5. None of these proteins were found to be differentially expressed between the absence and presence of cisplatin in both cell lines; see Supporting supplemental Tables S6 and S7. Western blot analysis of proteins involved in autophagy showed a significant increase in expression in the resistant M019iCis and OC002Cis cell lines. Specifically, the abundance of SQSTM1 (Fig. 6A and supplemental Fig. S1B), Atg7(B), Atg5 bound to Atg12(C), LC3–1(D), and LC3-II(E) was more highly expressed compared with the sensitive M019i and OC002 cell lines in absence of cisplatin.
Fig. 6.

Western blot analysis of autophagy markers in platinum sensitive M019i and resistant M019iCis cells. In addition to untreated samples (0 μmol/L) M019i cells were treated with cisplatin at IC50 (0.6 μmol/L) while M019iCis cells were treated with IC50 of both cell lines (0.6 μmol/L or 7 μmol/L; IC50 of M019iCis). Two-Way ANOVA with multiple comparisons using posthoc Tukey test was used to evaluate statistical differences between paired treatment groups of sensitive and resistant cells. One-Way ANOVA with multiple comparisons using posthoc Holm-Sidak method was used to compare treatment groups of resistant cell line. Each bar shows mean ± S.E. of three separate experiments. *p < 0.05, ** p < 0.01, *** p < 0.001. A, SQSTM1, (B) Atg7, (C) Atg5 bound to atg12, (D) LC3-I, (E) LC3-II, (F) LC3-II/LC3-I, (G) Relative viability of platinum resistant cells in presens of cisplatin (10 μmol/L), 3-MA (5 mmol/L) or both.
The LC3-II/LC3-I ratio was measured as an indicator for autophagy activation. Upon autophagy activation, LC3-I is cleaved to proteolytic-derived LC3-II which then aggregates to autophagosomal membranes (36). The LC3-II/LC3-I ratio (Fig. 6F) indicated that autophagy is decreased by cisplatin in M019i cells but increased in the M019iCis cells. A significant change of the LC3-II/LC3–1 ratio in sensitive OC002 cells was not observed in presence of cisplatin. Autophagy markers SQSTM1, Atg7, LC3-II, and the ratio of LC3-II/LC3-I in the resistant M019iCis and OC002Cis cell lines maintained an elevated expression in the presence of cisplatin compared with the sensitive cell line. Furthermore, inhibition of autophagy by 3-MA, a specific Pi3K inhibitor, reduced viability of the platinum resistant M019iCis cells (Fig. 6G), the effect being even more potent when combined with cisplatin. In contrast to the MS analysis, no significant change in a proteasome marker PSMD9 expression or ubiquitinated proteins was observed by Western blot analysis (supplemental Fig. S3).
Hyper-phosphorylation of Sequestosome-1 (SQSTM1) Increases During Resistance Development
To correlate hyper-phosphorylation on SQSTM1 to the increased expression of autophagy in the resistant M019iCis cell line, the distribution of phosphorylation on 20 individual amino acids where phosphorylation was differentially expressed were plotted from the TiO2 enriched samples. Ratios for the sensitive M019i cell line in the presence and absence of cisplatin, (Sensitive), and M019iCis to M019i cell lines in the absence of cisplatin, (− cisplaton), identified in the phosphoproteomic signature (Fig. 4) were compared. For peptides containing more than one phosphorylationite, the share of phosphorylation signal was equally divided among the sites. Hyper-phosphorylation at these sites was more prominent in the (−cisplaton) than (Sensitive), with 80% to 20% of phosphorylation sites having a fold change >2, respectively (Fig. 7A). It is important to note that SQSTM1 expression at the unmodified protein level is increased in the M019iCis cell line compared with the M019i cell line in absence of cisplatin by a fold change of 18 (supplemental Tables S5). 7/20 sites showed an increase of phosphorylation by a FC of over 40. While the increase of hyper-phosphorylation from (Sensitive) to (− cisplaton) is a potential response to cisplatin that leads to resistance, it should be noted that much of the observed phosphorylation increase was located toward the C terminus of SQSTM1. The percent distribution of phosphorylation in SQSTM1 showed that most of the signal of phosphorylation was observed in the PBI (3–102) (37, 38), LC3 (321–342)(39), and ubiquitin binding(387–436)(40) domains as well as domains shown to allow transit through mitosis (269–272)(41) upon phosphorylation (Fig. 7B).
Fig. 7.

Comparative phosphorylation site analysis of SQSTM1 in (1) resistant versus sensitive cell lines in absence of cisplatin, and (2) presence of cisplatin versus absence in sensitive cell line. A, Fold change increase. B, Percent distribution of phosphorylation in resistant cell lines in absence of cisplatin and sensitive cell lines in presence of cisplatin.
Hyper-phosphorylation of SQSTM1 is Increased in the Platinum Resistant Cell Lines
Our analysis revealed that both SQSTM1 mRNA and protein expression, and protein phosphorylation were significantly elevated in platinum resistant M019Cis cells relative to M019i cells (Fig. 8A–8C). Cisplatin treatment increased SQSTM1 phosphorylation in M019i cells (S28-pSQSTM1, p value<0.001) (Fig. 8B) with an observable increase at T269/272-p-SQSTM1 of > 3-fold (Fig. 8C). In the presence of cisplatin, M019iCis cells maintained the level of phosphorylation at the IC50 concentrations of cisplatin of both cell lines. A ratio of revealed over-expression of the phosphorylated form relative to protein expression in the M019iCis cell lines (Fig. 8D–8E). The result was confirmed in OC002 cells using Western blot analysis (supplemental Fig. S1D). In an additional cell line OC016, which initially showed similar results, the platinum resistance was lost with parallel reduction in both SQSTM1 protein phosphorylation and mRNA expression (supplemental Fig. S4). It was observed that cisplatin in presence of inhibitors to potential upstream regulators of autophagy (e.g. AKT, mTOR, ikk, ERK, and Pi3K pathways) altered the expression of S28-pSQSTM1 in M019i and M019iCis cell lines (supplemental Fig. S5).
Fig. 8.

Messenger RNA and protein expression of SQSTM1 and phosphorylated SQSTM1 on serine residue on amino acid position 28 and threonine 269/serine 272 in platinum sensitive M019i and resistant M109iCis cells using qRT-PCR and Western blot analysis. Statistical differences were calculated using Two-Way ANOVA with multiple comparisons using posthoc Tukey test for paired treatment groups of sensitive and resistant cells and using One-Way ANOVA with multiple comparisons using posthoc Holm-Sidak method within treatment groups of resistant cell line. Each bar shows mean ± S.E. of three separate experiments. A, SQSTM1 mRNA expression, (B) Serine 28 phosphorylated SQSTM1, (C) Threonine 269/serine 272 phosphorylated SQSTM1, (D) Serine 28 phosphorylated SQSTM1 to SQSTM1, (E) Threonine 269/serine 272 phosphorylated SQSTM1 to SQSTM1.
Expression and Localization of SQSTM1 in the Platinum Resistant Cell Line and Clinical Tumor Specimens
In the platinum sensitive M019i cells S28-pSQSTM1 was localized in the cytoplasm (Fig. 9A). Upon cisplatin treatment, an inducible localization of S28-pSQSTM1 to autophagosomes was observed. In the resistant M019iCis cells, S28-pSQSTM1 was constitutively expressed in the activated autophagosomes marked with strong LC3 localization without platinum treatment. In sections of cancer tissue, both RNA in situ hybridization and immunohistochemical staining demonstrated SQSTM1 expression in the high-grade serous tumor cells, typically in the cytoplasm (Fig. 9B), whereas stromal fibroblasts and lymphocytes remained negative. Interestingly, the SQSTM1 mRNA levels were increased during chemotherapy, and the increase appeared to be highest in tumors in which the chemotherapy response was poor (Fig. 9C). The change was not statistically significant, possibly due to a small number of sample pairs.
Fig. 9.

Expression and localization of SQSTM1 in cell lines and human tumor specimens. A, Subcellular localization of S28-pSQSTM1 in platinum sensitive M019i and resistant M019iCis cells using immunocytochemical staining. DAPI is represented with blue stain. S28-pSQSTM1 (red) is localized in cytoplasm of the sensitive cell line while it partially localizes into autophagosomes when treated with cisplatin. In resistant cells S28-pSQSTM1 is observed mainly in autophagosomes marked with LC3 staining (green) even without cisplatin treatment. B, Representative SQSTM1 RNA in situ hybridization (left) and immunohistochemical staining (right) shows SQSTM1 expression in the tumor cells but not in surrounding stroma. C, The fold difference in SQSTM1 mRNA expression in eleven HGSOC tumor pairs before and after neoadjuvant chemotherapy classified by histological response to chemotherapy. SQSTM1 mRNA levels (mean, S.E., n = 3) were increased during chemotherapy as presented with the fold difference over 1.0 especially in tumors in which the chemotherapy response was poor. The change was not statistically significant, possibly due to a small number of sample pairs.
DISCUSSION
Platinum-based chemotherapy (e.g. cisplatin) remains the first line treatment for HGSOC. Over 70% of patients initially respond to cisplatin treatment but most relapse as resistance develops (42). Thus, we sought to develop a better understanding of the mechanism of cisplatin resistance. Patient derived cisplatin-sensitive M019i and generated cisplatin-resistant M019iCis spheroidal HGSOC cells were used to model in vivo disease, especially in metastatic spread in ascites. Proteomic analysis revealed enrichment in glutathione metabolism, apoptosis regulation, and proteasome complex in the resistant cell line. Knockdown experiments to Glutathione S-transferase P1, one of the first enzymes to metabolize platinum drugs, have been shown to re-sensitize ovarian tumor cell lines to cisplatin treatment (43) and intense investigations of how glutathione metabolism inactivates and detoxifies cells of cisplatin have been conducted (6, 44) without elucidating mechanisms to avoid resistance. However, it is known that induction of apoptosis is the primary target for cisplatin anticancer activity (45) and that development of resistance to cisplatin is correlated to a defective apoptotic response (45–48). We found that caspase-3 expression levels confirmed that apoptosis significantly increases upon cisplatin exposure only in the platinum sensitive cell line while exposure to cisplatin had no significant effect on caspase-3 expression levels in the resistant cell lines, a finding that agrees with previous studies.
In the presence of cisplatin, mitochondrial components were enriched in both the sensitive and resistant cell lines. Although the accepted pharmacological cytotoxic effects of cisplatin have been credited to cross-linked adducts of DNA, only ∼1% of intracellular platinum is bound to DNA with the great majority of the intracellular drug available to interact with available nucleophilic sites such as are present in mitochondria (51, 52). For example, cisplatin was shown to preferentially bind to mitochondrial DNA in head and neck squamous cells (53) and to induce mitochondrial-dependent reactive oxygen species (ROS) response in Saccharomyces cerevisiae (54), contributing to the cytotoxic effect caused by DNA damage. Recent interest has developed in the role of “metabolic reprogramming” in development of ovarian cancer resistance by studying the mitochondrial function and metabolism in the presence of cisplatin (55–58), but a detailed discussion is beyond the scope of our findings.
As expected the proteasome complex, a well-known death mediated pathway involved with the degradation of ubiquitinated proteins, was found to be over-expressed in proteomic analysis of platinum resistant cells relative to sensitive cells in the presence of cisplatin. In addition, the most abundant proteins in the phosphoproteomic signature of resistance were SQSTM1-an integral part of the sequestosome complex, ubiquitin-40S ribosomal protein, and neighbor of BRCA1 gene (NBR1). These results imply the importance of degradation of ubiquitinated proteins through autophagy or the proteasome complex. Both these pathways were highlighted in both proteome and phosphoproteome analysis. Although ∼1400 phosphorylated proteins were identified in this study, there were only a total of 20 phosphorylated proteins among the cellular subtypes whose expression changed significantly enough to be measured. Nonetheless, phosphorylation of the most abundant proteins revealed phosphorylation controlled signaling in the autophagy pathway to play a potential role in cisplatin resistance development in HGOSC cell lines. Increased expression of autophagy markers in the resistant cell line were validated by Western blot analysis.
Autophagic degradation of ubiquitinated protein aggregates is important for cell survival. The LC3B-II domain on autophagosomes interacts with aggregates via selective substrates to shuttle for degradation in autophagy (59). SQSTM1 is a well-characterized autophagy selective substrate in ubiquitinated protein aggregates (39, 60, 61). NBR1 is another independent autophagy selective substrate functioning in a similar manner to SQSTM1 (25, 62). A high expression of SQSTM1 in cytoplasm in comparison to the nucleus has recently been associated with poor prognosis in epithelial ovarian cancer (63). Inhibition of autophagy with class III phosphoinositide 3-kinase (PI3K) inhibitor 3-methyladenine (3-MA) and siRNA knockdown of SQSTM1 enhanced ubiquitinated protein accumulation and enhanced apoptosis determined by re-expression of ER stress chaperone protein glucose-regulated protein-78 (Grp78) (26). SiRNA knockdown of beclin 1, known to be in complex with PI3K and necessary for the initiation of autophagy (64), has been shown to increase the apoptotic rate in the presence of cisplatin not seen with 3-MA treatment determined by flow cytometry (13). Additionally, cisplatin has also been shown to induce extracellular signal kinase (ERK) by phosphorylation (14). SiRNA knockdown of ERK was shown to decrease in autophagy and cell proliferation upon cisplatin treatment. Furthermore, emerging evidence suggests that MAPK activation plays a possible role in the regulation of autophagy (65–68). These results indicate a protective role of autophagy through regulation by key kinases in resistant ovarian cancers. In our study, 0.25% and 4.7% of the proteins identified were kinases in the proteome and phosphoproteome, respectively. Full characterization of the kinome in these type cells using a kinase-enrichment protocol would likely be of value to the field.
Platinum resistant ovarian cancer cells are hypothesized to avoid cisplatin induced ER stressed apoptosis by shuttling ubiquitinated proteins to degradation via autophagy. Here, apoptosis was observed to increase in platinum sensitive cells in the presence of cisplatin whereas autophagy, based on LC3-II/LC3-I ratio, decreased as expected. In contrast, platinum resistant HGSOC cells expressed elevated levels of autophagy markers ATG7, ATG5, and LC3-II and LC3-II/LC3-I ratio with increasing cisplatin concentrations in the resistant cell line. Furthermore, our data also shows that inhibition of autophagy with 3-MA reduces the viability of platinum resistant cells, and this reduction strengthens in the presence of cisplatin. Our data is in agreement with previous studies (14)(24) supporting an important role of autophagy in platinum resistance of HGSOC. Damaged organelles and molecules are engulfed by autophagosomes and degraded by lysosomal hydrolase (69), providing a mechanism that enables cancer cells to maintain homeostasis, energy recycling, and the potential to develop drug resistance (70, 71). Interestingly, elevated expression and hyper-phosphorylation of SQSTM1 in platinum resistant HGSOC cells was observed in our study, a finding that contrasts with previous results where SQSTM1 was shown to decrease following cisplatin treatment and was correlated with an increase in autophagy (14, 26). We showed an inducible phosphorylated SQSTM1 localization in autophagosomes of platinum sensitive cells after cisplatin treatment whereas the same autophagosomal localization pattern was constitutively expressed in resistant cells before and after platinum treatment. Furthermore, native SQSTM1 was shown to be expressed in the cancer cells of clinical tumor specimens. Finally, it is known that as a scavenger protein SQSTM1 is induced by several pathways and stressors, such as oxidative stress (72) and starvation (73), which lends support to our finding. Indeed, our data from a small sample set (n = 11) of primary-interval sample pairs showed a 2-fold increase in SQSTM1 mRNA after neoadjuvant chemotherapy in tumors with poor histological response.
Targeting the autophagic pathway is regarded as a promising new strategy for cancer drug discovery. Elaiophylin, a recently developed novel autophagy inhibitor specific for autophagic flux, was shown to achieve significant in vivo antitumor effect upon administration with or without cisplatin in 4-week-old athymic mice bearing palpable SKOV3 ovarian tumors (74). Here we attribute cisplatin-induced autophagy to the hyper-phosphorylation of SQSTM1. We thus propose hyper-phosphorylation of SQSTM1 to be an inducer in cisplatin resistance development in HGOSC ovarian cancers by shuttling ubiquitinated proteins to the autophagy pathway. Our data showed that loss of resistance in OC016 cell lines was correlated to a parallel reduction in SQSTM1 phosphorylation. Phosphorylation at serine 24 has been shown to induce homo-polymerization of SQSTM1 required for autophagosome formation and to disrupt binding to NBR1, PKC, and MEK5 (75). Phosphorylation at serine 403 increases the affinity between its ubiquitin-associated(UBA) domain and polyubiquitin chains, stabilizing SQSTM1 with ubiquitinated proteins for autophagosome entry (23). Point mutation experiments attribute phosphorylation of serine 403 to regulation of autophagic degradation of ubiquitinated proteins that are poorly degraded by the proteasome. Mutations of acidic residues to non-polar alanine residues in the LC3B binding domain of SQSTM1 (amino acid 321–342) disrupted interaction between the two proteins, and the degradation of ubiquitinated proteins was significantly inhibited (39). The interruption in the interaction was attributed to the loss of the electrostatic interactions between the negative charged residues of SQSTM1 with the positive charged residues of LC3B. These findings suggest that the interaction of SQSTM1 to its targets is highly governed by phosphorylation and supports a vital role for hyper-phosphorylation of SQSTM1 in the induction of autophagy.
To conclude, we showed for the first time an elevated level of hyper-phosphorylated SQSTM1 in platinum-resistant HGSOC cells derived from patient primary cell lines produced by spheroidal growth. Hyper-phosphorylation of SQSTM1 in the sensitive cells was expressed in presence of cisplatin, but to a greater extent between the resistant and sensitive cells. Notably, we found that the levels of hyper-phosphorylated SQSTM1 were remarkably higher than the native protein and reported the distribution of phosphorylation site modifications on the detected phosphorylated amino acid residues as being like sites previously reported by others. Phosphorylation of SQSTM1 in different domains governs its interaction with binding partners such as ubiquitinated proteins, homo-polymerization, and the LC3 binding domain in autophagosomes. Indeed, upon platinum treatment phosphorylated SQSTM1 was shown to induce localization from the cytoplasm to autophagosomes in sensitive cell lines whereas constitutively being expressed in autophagosomes in the resistant cell lines. Inhibition of autophagy reduced the viability of platinum resistant cells and decreased phosphorylated SQSTM1 in sensitive and resistant cell lines upon cisplatin treatment. Thus, we propose that a hyper-phosphorylated phenotype of SQSTM1, and not the native SQSTM1, plays a vital role in resistance development by the induction of autophagy.
DATA AVAILABILITY
Data produced herein has been deposited in the PRIDE database as follows: ProteomeXchange title: Proteomic and phosphoproteomic analysis of cisplatin resistance in patient derived serous ovarian cancer; ProteomeXchange accession: PXD002394; PubMed ID: 28455291; Publication DOI: Not applicable; Project Webpage: http://www.ebi.ac.uk/pride/archive/projects/PXD002394; FTP Download: ftp://ftp.pride.ebi.ac.uk/pride/data/archive/2017/05/PXD002394.
Supplementary Material
Acknowledgments
We thank the Turku Proteomics Facility, in particular Susumu Imanishi and Pekka Haapaniemi for constructive criticism and technical assistance, respectively, the support of Biocenter Finland, and the University of Maryland School of Pharmacy Mass Spectrometry Center (SOP1841–IQB2014).
Footnotes
Author contributions: R.L., O.C., and D.R.G. designed research; E.V.N., K.H., Y.G., K.K., N.A., V.R., and J.H. performed research; K.H. and K.K. contributed new reagents or analytic tools; E.V.N. analyzed data; E.V.N. and K.H. wrote the paper.
* This work was conducted as part of the National Technology Agency of Finland and funded by the Finland Distinguished Professor Programme (grant 40398/11), the Academy of Finland, the Centre of Excellence in Molecular Systems Immunology and Physiology Research, 2012–2017 (grant 250114), the Sigrid Jusélius Foundation, The Cancer Society of Finland, The Paulo Foundation, and Turku University Hospital Research Funds.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- HGSOC
- High-grade serous ovarian cancer
- AGC
- Automatic gain control
- ANOVA
- Analysis of Variance
- bFGF
- basic fibroblast growth factor
- DAPI
- 4,6-diamidino-2-phenylindole
- DNA
- Deoxyribonucleic acid
- EGF
- Epidermal growth factor
- FA
- Formic acid
- HCL
- Hydrochloric acid
- IT
- Ion trap
- MS/MS
- tandem mass spectrometry
- NH4OH
- Ammonium hydroxide
- PCA
- Principal component analysis
- PFS
- Progression-free survival
- P62/SQSTM1
- Sequestosome
- ROS
- Reactive oxygen species
- siRNA
- Small interfering ribonucleic acid
- TiO2
- Titanium dioxide
- 3-MA
- 3-methyladenine.
REFERENCES
- 1. Siegel R., Naishadham D., and Jemal A. (2013) Cancer statistics, 2013. CA Cancer J. Clin. 63, 11–30 [DOI] [PubMed] [Google Scholar]
- 2. Seidman J. D., Horkayne-Szakaly I., Haiba M., Boice C. R., Kurman R. J., and Ronnett B. M. (2004) The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int. J. Gynecol. Pathol. 23, 41–44 [DOI] [PubMed] [Google Scholar]
- 3. Miller D. S., Blessing J. A., Krasner C. N., Mannel R. S., Hanjani P., Pearl M. L., Waggoner S. E., and Boardman C. H. (2009) Phase II evaluation of pemetrexed in the treatment of recurrent or persistent platinum-resistant ovarian or primary peritoneal carcinoma: a study of the Gynecologic Oncology Group. J. Clin. Oncol. 27, 2686–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galluzzi L., Senovilla L., Vitale I., Michels J., Martins I., Kepp O., Castedo M., and Kroemer G. (2012) Molecular mechanisms of cisplatin resistance. Oncogene 31, 1869–1883 [DOI] [PubMed] [Google Scholar]
- 5. Jin L., Huo Y., Zheng Z., Jiang X., Deng H., Chen Y., Lian Q., and Ge R. (2014) Down-regulation of Ras-related protein Rab 5C-dependent endocytosis and glycolysis in cisplatin-resistant ovarian cancer cell lines. Mol. Cell. Proteomics 13, 3138–3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Perez R. P. (1998) Cellular and molecular determinants of cisplatin resistance. Eur. J. Cancer 34, 1535–1542 [DOI] [PubMed] [Google Scholar]
- 7. E. A. (1999) The mechanism of action of cisplatin: From adducts to apoptosis, in Cisplatin., Bernhard Lippert ed edn. Wiley-VCH, Switzerland: pp. 111–134 [Google Scholar]
- 8. Reed J. C. (1997) Bcl-2 family proteins: regulators of apoptosis and chemoresistance in hematologic malignancies. Semin. Hematol. 34, 9–19 [PubMed] [Google Scholar]
- 9. Zou Z., Wu L., Ding H., Wang Y., Zhang Y., Chen X., Zhang C. Y., Zhang Q., and Zen K. (2012) MicroRNA-30a sensitizes tumor cells to cis-platinum via suppressing beclin 1-mediated autophagy. J. Biol. Chem. 287, 4148–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ren J. H., He W. S., Nong L., Zhu Q. Y., Hu K., Zhang R. G., Huang L. L., Zhu F., and Wu G. (2010) Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. Cancer Biother. Radiopharm. 25, 75–80 [DOI] [PubMed] [Google Scholar]
- 11. O'Donovan T. R., O'Sullivan G. C., and McKenna S. L. (2011) Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 7, 509–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sirichanchuen B., Pengsuparp T., and Chanvorachote P. (2012) Long-term cisplatin exposure impairs autophagy and causes cisplatin resistance in human lung cancer cells. Mol. Cell Biochem. 364, 11–18 [DOI] [PubMed] [Google Scholar]
- 13. Bao L., Jaramillo M. C., Zhang Z., Zheng Y., Yao M., Zhang D. D., and Yi X. (2015) Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol. Med. Rep. 11, 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang J. and Wu G. S. (2014) Role of autophagy in cisplatin resistance in ovarian cancer cells. J. Biol. Chem. 289, 17163–17173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsai L. L., Yu C. C., Chang Y. C., Yu C. H., and Chou M. Y. (2011) Markedly increased Oct4 and Nanog expression correlates with cisplatin resistance in oral squamous cell carcinoma. J. Oral. Pathol. Med. 40, 621–628 [DOI] [PubMed] [Google Scholar]
- 16. Marx H., Lemeer S., Schliep J. E., Matheron L., Mohammed S., Cox J., Mann M., Heck A. J., and Kuster B. (2013) A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat. Biotechnol. 31, 557–564 [DOI] [PubMed] [Google Scholar]
- 17. Taus T., Kocher T., Pichler P., Paschke C., Schmidt A., Henrich C., and Mechtler K. (2011) Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362 [DOI] [PubMed] [Google Scholar]
- 18. Dennis G. Jr, Sherman B. T., Hosack D. A., Yang J., Gao W., Lane H. C., and Lempicki R. A. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3. [PubMed] [Google Scholar]
- 19. Jensen L. J., Kuhn M., Stark M., Chaffron S., Creevey C., Muller J., Doerks T., Julien P., Roth A., Simonovic M., Bork P. and von Mering C. (2009) STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37, D412–D6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vallius T., Peter A., Auranen A., Carpen O., Kemppainen J., Matomaki J., Oksa S., Roering P., Seppanen M., Grenman S., and Hynninen J. (2016) 18F-FDG-PET/CT can identify histopathological non-responders to platinum-based neoadjuvant chemotherapy in advanced epithelial ovarian cancer. Gynecol. Oncol. 140, 29–35 [DOI] [PubMed] [Google Scholar]
- 21. Chen P., Huhtinen K., Kaipio K., Mikkonen P., Aittomaki V., Lindell R., Hynninen J., Auranen A., Grenman S., Lehtonen R., Carpen O., and Hautaniemi S. (2015) Identification of prognostic groups in high-grade serous ovarian cancer treated with platinum-taxane chemotherapy. Cancer Res. 75, 2987–2998 [DOI] [PubMed] [Google Scholar]
- 22. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., and Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. [DOI] [PubMed] [Google Scholar]
- 23. Matsumoto G., Wada K., Okuno M., Kurosawa M., and Nukina N. (2011) Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell 44, 279–289 [DOI] [PubMed] [Google Scholar]
- 24. Rigbolt K. T., Prokhorova T. A., Akimov V., Henningsen J., Johansen P. T., Kratchmarova I., Kassem M., Mann M., Olsen J. V., and Blagoev B. (2011) System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci. Signal. 4, rs3. [DOI] [PubMed] [Google Scholar]
- 25. Kirkin V., Lamark T., Johansen T., and Dikic I. (2009) NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 5, 732–733 [DOI] [PubMed] [Google Scholar]
- 26. Yu H., Su J., Xu Y., Kang J., Li H., Zhang L., Yi H., Xiang X., Liu F., and Sun L. (2011) p62/SQSTM1 involved in cisplatin resistance in human ovarian cancer cells by clearing ubiquitinated proteins. Eur. J. Cancer 47, 1585–1594 [DOI] [PubMed] [Google Scholar]
- 27. Huang S., Okamoto K., Yu C., and Sinicrope F. A. (2013) p62/sequestosome-1 up-regulation promotes ABT-263-induced caspase-8 aggregation/activation on the autophagosome. J. Biol. Chem. 288, 33654–33666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Korolchuk V. I., Mansilla A., Menzies F. M., and Rubinsztein D. C. (2009) Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 33, 517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deleted in proof.
- 30. Deleted in proof.
- 31. Lee A. S. (2001) The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem. Sci. 26, 504–510 [DOI] [PubMed] [Google Scholar]
- 32. Samali A., Cai J., Zhivotovsky B., Jones D. P., and Orrenius S. (1999) Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J. 18, 2040–2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang X. and Wang X. (2004) Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 73, 87–106 [DOI] [PubMed] [Google Scholar]
- 34. Thomas M., Finnegan C. E., Rogers K. M., Purcell J. W., Trimble A., Johnston P. G., and Boland M. P. (2004) STAT1: a modulator of chemotherapy-induced apoptosis. Cancer Res. 64, 8357–8364 [DOI] [PubMed] [Google Scholar]
- 35. Glick D., Barth S., and Macleod K. F. (2010) Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. González-Polo R. A., Niso-Santano M., Ortíz-Ortíz M. A., Gómez-Martín A., Morán J. M., García-Rubio L., Francisco-Morcillo J., Zaragoza C., Soler G., and Fuentes J. M. (2007) Inhibition of paraquat-induced autophagy accelerates the apoptotic cell death in neuroblastoma SH-SY5Y cells. Toxicol. Sci. 97, 448–458 [DOI] [PubMed] [Google Scholar]
- 37. Ren J., Wang J., Wang Z., and Wu J. (2014) Structural and biochemical insights into the homotypic PB1-PB1 complex between PKCζ and p62. Sci. China Life Sci. 57, 69–80 [DOI] [PubMed] [Google Scholar]
- 38. Wilson M. I., Gill D. J., Perisic O., Quinn M. T., and Williams R. L. (2003) PB1 domain-mediated heterodimerization in NADPH oxidase and signaling complexes of atypical protein kinase C with Par6 and p62. Mol. Cell 12, 39–50 [DOI] [PubMed] [Google Scholar]
- 39. Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J. A., Outzen H., Øvervatn ABjørkøy G., and Johansen T. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 [DOI] [PubMed] [Google Scholar]
- 40. Ciani B., Layfield R., Cavey J. R., Sheppard P. W., and Searle M. S. (2003) Structure of the ubiquitin-associated domain of p62 (SQSTM1) and implications for mutations that cause Paget's disease of bone. J. Biol. Chem. 278, 37409–37412 [DOI] [PubMed] [Google Scholar]
- 41. Linares J. F., Amanchy R., Greis K., Diaz-Meco M. T., and Moscat J. (2011) Phosphorylation of p62 by cdk1 controls the timely transit of cells through mitosis and tumor cell proliferation. Mol. Cell Biol. 31, 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Agarwal R. and Kaye S. B. (2003) Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 3, 502–516 [DOI] [PubMed] [Google Scholar]
- 43. Sawers L., Ferguson M. J., Ihrig B. R., Young H. C., Chakravarty P., Wolf C. R., and Smith G. (2014) Glutathione S-transferase P1 (GSTP1) directly influences platinum drug chemosensitivity in ovarian tumour cell lines. Br. J. Cancer 111, 1150–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Johnson S. W., Ozols R. F., and Hamilton T. C. (1993) Mechanisms of drug resistance in ovarian cancer. Cancer 71, 644–649 [DOI] [PubMed] [Google Scholar]
- 45. Henkels K. M., and Turchi J. J. (1997) Induction of apoptosis in cisplatin-sensitive and -resistant human ovarian cancer cell lines. Cancer Res. 57, 4488–4492 [PubMed] [Google Scholar]
- 46. Segal-Bendirdjian E., and Jacquemin-Sablon A. (1995) Cisplatin resistance in a murine leukemia cell line is associated with a defective apoptotic process. Exp. Cell Res. 218, 201–212 [DOI] [PubMed] [Google Scholar]
- 47. Diebold J., Baretton G., Felchner M., Meier W., Dopfer K., Schmidt M., and Löhrs U. (1996) bcl-2 expression, p53 accumulation, and apoptosis in ovarian carcinomas. Am. J. Clin. Pathol. 105, 341–349 [DOI] [PubMed] [Google Scholar]
- 48. Li J., Feng Q., Kim J. M., Schneiderman D., Liston P., Li M., Vanderhyden B., Faught W., Fung M. F., Senterman M., Korneluk R. G., and Tsang B. K. (2001) Human ovarian cancer and cisplatin resistance: possible role of inhibitor of apoptosis proteins. Endocrinology 142, 370–380 [DOI] [PubMed] [Google Scholar]
- 49. Deleted in proof.
- 50. Deleted in proof.
- 51. Gonzalez V. M., Fuertes M. A., Alonso C., and Perez J. M. (2001) Is cisplatin-induced cell death always produced by apoptosis? Mol. Pharmacol. 59, 657–663 [DOI] [PubMed] [Google Scholar]
- 52. Fuertes M. A., Castilla J., Alonso C., and Pérez J. M. (2003) Cisplatin biochemical mechanism of action: from cytotoxicity to induction of cell death through interconnections between apoptotic and necrotic pathways. Curr. Med. Chem. 10, 257–266 [DOI] [PubMed] [Google Scholar]
- 53. Yang Z., Schumaker L. M., Egorin M. J., Zuhowski E. G., Guo Z., and Cullen K. J. (2006) Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: possible role in apoptosis. Clin. Cancer Res. 12, 5817–5825 [DOI] [PubMed] [Google Scholar]
- 54. Marullo R., Werner E., Degtyareva N., Moore B., Altavilla G., Ramalingam S. S., and Doetsch P. W. (2013) Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 8, e81162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chappell N. P., Teng P. N., Hood B. L., Wang G., Darcy K. M., Hamilton C. A., Maxwell G. L., and Conrads T. P. (2012) Mitochondrial proteomic analysis of cisplatin resistance in ovarian cancer. J. Proteome Res. 11, 4605–4614 [DOI] [PubMed] [Google Scholar]
- 56. Dai Z., Yin J., He H., Li W., Hou C., Qian X., Mao N., and Pan L. (2010) Mitochondrial comparative proteomics of human ovarian cancer cells and their platinum-resistant sublines. Proteomics 10, 3789–3799 [DOI] [PubMed] [Google Scholar]
- 57. Montopoli M., Bellanda M., Lonardoni F., Ragazzi E., Dorigo P., Froldi G., Mammi S., and Caparrotta L. (2011) “Metabolic reprogramming” in ovarian cancer cells resistant to cisplatin. Curr. Cancer Drug Targets 11, 226–235 [DOI] [PubMed] [Google Scholar]
- 58. Hirama M., Isonishi S., Yasuda M., and Ishikawa H. (2006) Characterization of mitochondria in cisplatin-resistant human ovarian carcinoma cells. Oncol. Rep. 16, 997–1002 [PubMed] [Google Scholar]
- 59. Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., and Johansen T. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ichimura Y., Kominami E., Tanaka K., and Komatsu M. (2008) Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy 4, 1063–1066 [DOI] [PubMed] [Google Scholar]
- 61. Komatsu M., Waguri S., Koike M., Sou Y. S., Ueno T., Hara T., Mizushima N., Iwata J., Ezaki J., Murata S., Hamazaki J., Nishito Y., Iemura S., Natsume T., Yanagawa T., Uwayama J., Warabi E., Yoshida H., Ishii T., Kobayashi A., Yamamoto M., Yue Z., Uchiyama Y., Kominami E., and Tanaka K. (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163 [DOI] [PubMed] [Google Scholar]
- 62. Kirkin V., Lamark T., Sou Y. S., Bjørkøy G., Nunn J. L., Bruun J. A., Shvets E., McEwan D. G., Clausen T. H., Wild P., Bilusic I., Theurillat J. P., Øvervatn Ishii A. T., Elazar Z., Komatsu M., Dikic I., and Johansen T. (2009) A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 33, 505–516 [DOI] [PubMed] [Google Scholar]
- 63. Iwadate R., Inoue J., Tsuda H., Takano M., Furuya K., Hirasawa A., Aoki D., and Inazawa J. (2014) High Expression of SQSTM1/p62 Protein Is Associated with Poor Prognosis in Epithelial Ovarian Cancer. Acta Histochem. Cytochem. 47, 295–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kihara A., Kabeya Y., Ohsumi Y., and Yoshimori T. (2001) Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2, 330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Comes F., Matrone A., Lastella P., Nico B., Susca F. C., Bagnulo R., Ingravallo G., Modica S., Lo Sasso G., Moschetta A., Guanti G., and Simone C. (2007) A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ. 14, 693–702 [DOI] [PubMed] [Google Scholar]
- 66. Corcelle E., Nebout M., Bekri S., Gauthier N., Hofman P., Poujeol P., Fénichel P., and Mograbi B. (2006) Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res. 66, 6861–6870 [DOI] [PubMed] [Google Scholar]
- 67. Pattingre S., Bauvy C., and Codogno P. (2003) Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J. Biol. Chem. 278, 16667–16674 [DOI] [PubMed] [Google Scholar]
- 68. Wang J., Whiteman M. W., Lian H., Wang G., Singh A., Huang D., and Denmark T. (2009) A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol. Chem. 284, 21412–21424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Eskelinen E. L., and Saftig P. (2009) Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 1793, 664–673 [DOI] [PubMed] [Google Scholar]
- 70. White E. (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 12, 401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sui X., Chen R., Wang Z., Huang Z., Kong N., Zhang M., Han W., Lou F., Yang J., Zhang Q., Wang X., He C., and Pan H. (2013) Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 4, e838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jain A., Lamark T., Sjottem E., Larsen K. B., Awuh J. A., Overvatn A., McMahon M., Hayes J. D., and Johansen T. (2010) p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 285, 22576–22591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sahani M. H., Itakura E., and Mizushima N. (2014) Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 10, 431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhao X., Fang Y., Yang Y., Qin Y., Wu P., Wang T., Lai H., Meng L., Wang D., Zheng Z., Lu X., Zhang H., Gao Q., Zhou J., and Ma D. (2015) Elaiophylin, a novel autophagy inhibitor, exerts antitumor activity as a single agent in ovarian cancer cells. Autophagy 0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Christian F., Krause E., Houslay M. D., and Baillie G. S. (2014) PKA phosphorylation of p62/SQSTM1 regulates PB1 domain interaction partner binding. Biochim. Biophys. Acta 1843, 2765–2774 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data produced herein has been deposited in the PRIDE database as follows: ProteomeXchange title: Proteomic and phosphoproteomic analysis of cisplatin resistance in patient derived serous ovarian cancer; ProteomeXchange accession: PXD002394; PubMed ID: 28455291; Publication DOI: Not applicable; Project Webpage: http://www.ebi.ac.uk/pride/archive/projects/PXD002394; FTP Download: ftp://ftp.pride.ebi.ac.uk/pride/data/archive/2017/05/PXD002394.