

Abstract
Flavin-containing monooxygenases (FMOs) are primarily studied as xenobiotic metabolizing enzymes with a prominent role in drug metabolism. In contrast, endogenous functions and substrates of FMOs are less well understood. A growing body of recent evidence, however, implicates FMOs in aging, several diseases, and metabolic pathways. The evidence suggests an important role for these well-conserved proteins in multiple processes and raises questions about the endogenous substrate(s) and regulation of FMOs. Here, we present an overview of evidence for FMOs' involvement in aging and disease, discussing the biological context and arguing for increased investigation into the function of these enzymes.
Keywords: aging, atherosclerosis, flavoprotein, metabolism, neurodegenerative disease, oxidation–reduction (redox), sulfur, xenobiotic, flavin-containing monooxygenase (FMO), trimethylamine–N-oxide (TMAO), xenobiotic metabolism, iron
Introduction to FMOs
Evolution and classification
Flavin-containing monooxygenases (FMOs)2 are ancient and widely conserved enzymes, being present in all kingdoms of life (1, 2). FMOs make up a subgroup of the Group B flavin-dependent monooxygenases (EC 1.14.13.8) along with Baeyer–Villiger monooxygenases (BVMOs) and N-hydroxylating monooxygenases (NMOs) (3). FMOs, BVMOs, and NMOs are each distinguished by variations in primary structural motifs, substrate preferences, and catalytic mechanisms. FMOs are also notably similar to the Group A enzymes dihydrolipoamide dehydrogenase (DLD), glutathione reductase (GR), and low-molecular-weight thioredoxin reductase (TRXR) (2, 4). These groups are distinguished from other flavin-dependent monooxygenases (Groups C–H) by their combined use of FAD and NAD(P)H. Any functional interplay between FMOs and Group A enzymes, perhaps as an NAD(P)H–thiol redox buffering system, is largely unexplored (5).
Catalytic cycle
The catalytic cycle of FMO enzymes is fairly well understood. FMOs utilize a tightly bound FAD prosthetic group, NAD(P)H, and molecular oxygen to monooxygenate or otherwise oxidize substrates, producing water and NAD(P)+ as by-products (6) (Fig. 1). Uncoupling has been observed in both the presence and the absence of substrate in vitro, resulting in “leakage” of hydrogen peroxide, and less frequently, superoxide (7). FMOs are notable for their “cocked and loaded” mechanism where they bind NAD(P)H and reduce FAD in the absence of substrate, creating an unusually stable C4a-hydroperoxyflavin ready to oxidize any substrate that accesses the active site (6). This mechanism is in contrast to that of cytochrome P450s and the Group A enzymes discussed above, which each require the presence of substrate to begin their catalytic cycles (6). The rate-limiting step of FMOs is thought to be the release of either H2O or NADP+. The significance of FMOs' reaction kinetics, reactive oxygen species leakage, and effects on cellular NAD(P)H is not known (7, 8).
Figure 1.
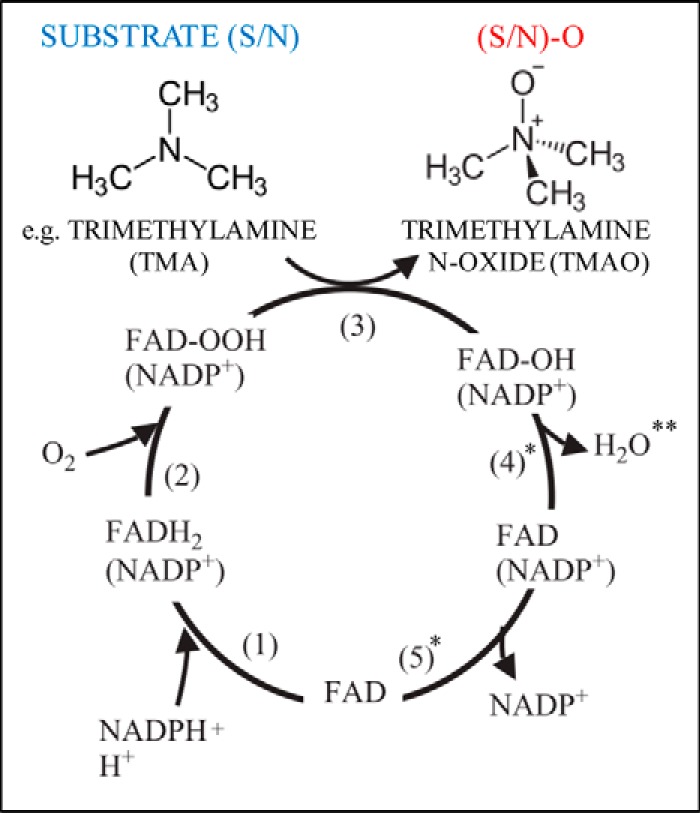
FMO catalytic cycle with canonical hFMO3 substrate TMA shown. * indicates candidate rate-limiting steps. ** indicates site where H2O2/O2− leakage may occur. Adapted from 6, Copyright © 2005 Elsevier B.V.
Substrates
FMOs typically monooxygenate the sulfur or nitrogen atoms of small, soft nucleophiles in a charge- and stereo-selective manner, but important exceptions exist. In addition to monooxygenation, FMOs can also catalyze oxidative decarboxylation (9), oxidative demethylation (10), and disulfide bond formation (11). FMOs act on a wide variety of sulfur- and nitrogen-containing compounds, but carbon, phosphorus, selenium, and other elements are also amenable to FMO-mediated oxidation (6). Pig FMO1 has a Km from 0.3 to 10 μm in vitro for several organic selenium-containing xenobiotics (6), and both hFMO1 and hFMO3 are capable of catalyzing the formation of pyruvate from selenocysteine (12), consistent with a role in endogenous selenium metabolism. Mammalian FMO5 does not metabolize typical FMO substrates, and may act more like a BVMO (8, 13).
The depth and width of the channel leading to the active site are thought to restrict overly large substrates' access (14), but this mechanism is not fully understood because no crystal structure has been solved for a mammalian FMO. The most evolutionarily related proteins with solved crystal structures are from bacterial trimethylamine monooxygenase (15–17) and yeast FMO (18), but their sequences are both shorter and significantly different from mammalian FMO sequences (19).
FMOs prefer uncharged substrates or substrates with a single positive charge. Neutral zwitterions are likely to be poor substrates (6), but the lone Saccharomyces cerevisiae FMO (yFMO1) can oxygenate free cysteine (20). FMOs' charge selectivity is the proposed mechanism by which they preferentially act on xenobiotics, as charged compounds cannot easily enter cells through the plasma membrane (6). Classically studied FMO substrates include xenobiotics such as imipramine, nicotine, clozapine, tamoxifen, and amphetamine (6). The oxygenation of these compounds increases their solubility and aids in their subsequent excretion, although it can occasionally also activate the compounds and increase their toxicity (21).
FMO metabolism of xenobiotics largely detoxifies compounds, whereas the role of FMO in endogenous metabolism is less characterized. FMOs can metabolize the sulfur amino acid pathway metabolites methionine, cysteine, and cysteamine, as well as the sulfur-containing co-factor lipoic acid (6, 20), but many related endogenous compounds remain untested.
Genomic organization and expression
Humans have five protein-encoding FMO genes (hFMO1–5) on chromosome 1 (22), each of which displays one-to-one orthology with FMO1–5 across mammalian species (23). Humans and other mammals also have several Fmo pseudogenes (24). Alternative splicing of hFMO1–5, especially hFMO4, is possible, but its function is unknown (25). hFMO1–4 are tightly clustered, likely the result of a gene duplication event that predates mammals, whereas the ancestral hFMO5 is ∼20 Mb away (22, 26). Consistent with this hypothesis, mouse Fmo5 (mFmo5) is on chromosome 3, whereas mFmo1–4 are clustered together on chromosome 1 (22). Subfunctionalization of mammalian FMOs probably followed duplication, possibly in response to new xenobiotics encountered in terrestrial environments (26).
hFMO1–5 show distinct developmental and tissue-specific patterns of expression (27, 28). Briefly, hFMO1 predominates in the fetal liver, whereas hFMO3 and hFMO5 are the major isoforms expressed in the postnatal liver (27, 28). In contrast, mFmo1 and mFmo5 are the major adult mouse liver FMOs, whereas mFmo3 is much more highly expressed in female mice (29). There are conflicting reports regarding conservation in humans of this sex-dependent differential expression, but it is clear that the conserved differences reported in humans are of a smaller magnitude than those in mice (30–32).
Several factors that regulate FMO transcription are known (33–37). Estrogen (29) and insulin (34) activate FMO transcription, whereas testosterone (29) and glucagon (37) are repressors of FMO transcription, but FMO5 is again an exception (38). Several hormone receptors have been placed both upstream and downstream of FMOs (39, 40). Large differences can exist between FMO mRNA abundance, FMO protein levels, and FMO functional activity (39, 41, 42), indicating multiple levels of regulation that require further study.
FMOs and disease
Although FMOs have been causally linked to only one disease, trimethylaminuria, evidence is accumulating that FMOs affect the pathology of multiple major diseases (Table 1). The nature of FMOs' involvement in these diseases, however, remains largely undefined.
Table 1.
FMOs in human disease and disease models
| Diseases | Nature of association |
|---|---|
| Fmo1 | |
| Sporadic ALS | • 3′-UTR SNPs associated with increased disease risk (62) |
| Fmo-2 | |
| No data | No data |
| Fmo3 | |
| TMAU | • Fmo3 mutations cause TMAU (43) |
| Atherosclerosis | • Increased FMO3 activity increases TMAO, in turn increasing atherosclerosis risk (45) |
| Chronic kidney disease | • Minor alleles at hFMO3 residue 158 were associated with increased circulating TMAO and faster epidermal growth factor receptor (eGFR) decline (95) |
| Diabetes | • Fmo3 downregulated in rodent models of diabetes |
| Sideroblastic anemia | • Single study with small sample size found Fmo3 dysfunction in cases of sideroblastic anemia (67) |
| Hemochromatosis | • Most differentially expressed hepatic transcript in mouse model of hemochromatosis (68) |
| Fmo4 | |
| No data | No data |
| Fmo5 | |
| Diabetes | • Hepatic FMO5 expressed at ratio of 0.41 in diabetic:non-diabetic patients (55) |
| Sporadic ALS | • FMO5 SNP found associated with ALS, especially in female patients (96) |
| Locus 1q24.3 | |
| Neurodegeneration (multiple diseases) | • Genome-wide association study (GWAS): Fmo polymorphisms affect lentiform nucleus volume, itself associated with multiple neurodegenerative diseases (63) |
| Parkinson's | • Decreased expression of FMO1 in rotenone model of Parkinson's in cultured primary midbrain dopaminergic neurons (64) |
Trimethylaminuria (TMAU)
Much of the FMO-related literature is focused on the only disease known to be caused by altered FMO activity, TMAU (43). TMAU, or “fish odor syndrome,” is a disorder in which the volatile compound trimethylamine (TMA) cannot be converted to the soluble trimethylamine–N-oxide (TMAO), leading to excretion of TMA through the skin. TMA is a small metabolite derived from dietary intake or produced by gut bacteria that has a distinctive “fishy” smell. hFMO3 mutations are the main cause of TMAU, but other causes exist including variations in hFMO3 expression and microbiome overproduction of TMA. There is no cure for TMAU, so treatment consists of limiting dietary intake of TMA and its precursors such as choline and carnitine.
Scant epidemiological data exist on whether TMAU alters risk for other diseases. An early study observed that TMAU patients have a high incidence of hypertension (43), and there are at least two reported cases of TMAU co-presenting with neurological disorders unlikely to be related to patients' social stress (44). These cases are interesting in relation to the other diseases recently connected to FMOs.
Atherosclerosis and cardiovascular disease (CVD)
Beginning in 2011, several studies concluded, based on both mouse and human data, that FMO3-dependent production of TMAO increases the risk for atherosclerosis and general CVD (45–47). These studies shed light on the importance of gut microbiota in determining disease risk and demonstrated a clear association between elevated TMAO and CVD. In the proposed model, gut bacteria produce TMA from dietary precursors, hepatic FMO3 converts TMA to TMAO, and TMAO increases the risk for atherosclerosis and CVD. Proposed mechanisms for TMAO increasing CVD risk include effects on cholesterol (45), a prolongation of the pressor effects of angiotensin II (48), and platelet hyperreactivity and thrombosis potential (49).
These mechanistic explanations are not fully convincing, however, and contrast with other data more consistent with TMAO production being a protective response to CVD. First, seafood rich in TMA and TMAO is widely thought to lower CVD risk (50). Second, as mentioned, an early study of TMAU patients found a high incidence of hypertension in the TMAU-afflicted group (43). The proposed mechanism was that FMO3 could metabolize tyramine, an endogenous pressor molecule, thereby reducing its pressor effects and lowering blood pressure (43). A follow-up study found that none of three examined FMO3 polymorphisms predispose to hypertension in a sample of several hundred Caucasian patients, but also noted that severe, highly penetrant loss-of-function mutations could “unmask pressor effects of variation in other drug metabolizing enzymes previously buffered by FMO3” (51). At least two more recent studies are consistent with TMAO production having a reactive, protective function in response to CVD pathology (52, 53), with one of these studies asserting that TMAO is, in fact, protective against CVD risk (53). What is clear from these publications is that TMAO is a molecule of great interest due to its diverse functions including osmolyte, chemical chaperone, reactive oxygen species scavenger, and now, potential risk factor (54).
Diabetes and metabolic disorders
FMO expression is altered in human diabetic patients and rodent models of diabetes (55, 56), and recent reports have revealed that FMOs can alter carbohydrate and lipid metabolism (40, 56–60). Two studies using streptozotocin-induced diabetes in rats show altered FMO expression in diabetic states (56). A third study comparing liver biopsy samples from Type 2 diabetes mellitus patients with samples from non-diabetic patients found hFMO5 down-regulated (55). A study examining expression of FMOs in diabetes found a trend that FMO3 is up-regulated and FMO5 is down-regulated in the disease state (61). As in CVD, it is not clear whether these FMO transcriptional changes are causative, protective, or have little effect on the disease process.
Recently, mammalian FMOs have been shown to affect intermediary carbon metabolism. Researchers who first linked FMO3 to atherosclerosis also found that mFMO3 activity correlated with hepatic and/or plasma lipids and glucose levels (40) and that FMO3 inhibition can divert cholesterol away from biliary excretion (57). The former study suggested that mFMO3's effects were peroxisome proliferator-activated receptor α (PPARα)- and KLF15-mediated, whereas the latter study concluded that mFMO3 affected cholesterol balance through TMAO production, but that mFMO3's effects on lipids and inflammation were mediated by another substrate. Two other reports examining mFmo1 and mFmo5 knock-out mice, respectively, found that both were capable of altering metabolism and energy balance sufficiently to cause gross alterations in body size (58, 59). A recent review discusses these mFMO-related effects on carbohydrate and lipid metabolism and energy balance (60).
Neurodegeneration and neurological disease
There is substantial evidence connecting FMO expression to neurodegenerative diseases including sporadic amyotrophic lateral sclerosis (ALS), Parkinson's disease (PD), and schizophrenia. hFMO1 expression is consistently decreased in the spinal cord of ALS patients (62), and single nucleotide polymorphisms in the hFMO1 3′-untranslated region occur more frequently in female patients with sporadic ALS (62). Additionally, mFmo1 is up-regulated in a mouse model of ALS, which, although in the opposite direction of human findings, may be explained by different stages of the disease affecting expression differently (62).
The role of FMOs in other neurodegenerative diseases is also intriguing but largely correlative. The FMO gene cluster containing FMO1–4 is associated with lentiform nucleus volume, a physiological marker associated with PD, schizophrenia, and other neurological disorders (63). Additionally, hFMO1 and Parkin are down-regulated in a rotenone model of PD carried out in cell culture (64). The same study showed up-regulation of caspase 3, and that hFMO1 inhibition was sufficient to activate caspase 3, an executor of apoptotic cell death implicated in the loss of dopaminergic neurons in PD (64). These data suggest a possible role for FMO1 in protecting against multiple neurodegenerative disorders.
Iron dyshomeostasis
Studies by a single research group support FMOs acting in iron homeostasis. Building on a known FMO–calreticulin complex (65), a ferrireductase role for FMO in an iron import complex termed “paraferritin” was described (66). The complex was further described as including FMO, calreticulin, DMT1, and other proteins (66). Its suggested roles include serving as an alternative, non-transferrin mechanism for cellular iron uptake and as a means to deliver iron to mitochondrial ferrochelatase for incorporation into heme. While acknowledging that larger studies would be necessary to determine prevalence, this group also suggested diminished FMO activity as a risk factor for sideroblastic anemia based on four cases (67).
Mouse models of hereditary hemochromatosis, a disease characterized by excessive intestinal iron absorption and subsequent iron overload throughout bodily tissues, alter hepatic mFmo transcription. Mutation of the hemochromatosis gene Hfe (high iron Fe) and high dietary iron both cause liver iron loading, but they do so via secondary and primary iron overload, respectively. Hepatic mFmo3 transcription was highly up-regulated by genetic hereditary hemochromatosis, but counterintuitively, heavily down-regulated in mice fed high dietary iron (68). By magnitude, mFmo3 was the most altered transcript across both conditions. In a separate study, mFmo3 was up-regulated in Hfe-deficient D2 mice, and although the data were not shown, mFmo3 was described not to change in WT mice fed a high-iron diet (69). These studies, in addition to those describing paraferritin, suggest that FMOs act in iron homeostatic pathways in the liver or elsewhere, and that this novel role for FMOs requires further exploration.
FMOs and aging
Published data from the past decade provide increasing evidence that FMOs play an important role during aging. Specific Fmo genes are transcriptionally activated in numerous mouse longevity models including dietary restriction (DR), growth hormone/insulin-like growth factor 1 (GH/IGF1) signaling disruption, and rapamycin treatment (70–73). These are among the most robustly conserved longevity–promoting interventions (74). The correlation between increased mFmo expression and longevity suggests that FMOs could play a causal role in promoting longevity. Further supporting this, Caenorhabditis elegans (nematode worm) fmo-2 is up-regulated by DR and is necessary for lifespan extension from solid DR, a form of DR (75). Nematode FMO-2 is also sufficient to extend lifespan and improve healthspan and stress resistance when ubiquitously overexpressed (75).
FMOs and mammalian aging
Multiple gene expression analyses from mouse models of delayed aging show that mFmo gene expression is often increased in long-lived mice (Table 2). For example, a 2007 meta-analysis of published liver microarray data found that mFmo3 is consistently up-regulated in a variety of long-lived knock-out models and in response to longevity-promoting interventions (73). Similarly, a 2008 study of XME gene expression in liver found mFmo3 and mFmo4 to be up-regulated in long-lived male mice following DR, GH/IGF1 mutation, or rapamycin treatment (71). Of particular interest, an independent study showed that growth hormone receptor mutant mice have increased levels of both mFmo3 and TMAO, further correlating mFmo3 expression and activity with longevity (76).
Table 2.
FMOs and aging in model systems.
| FMO genes | Lifespan/healthspan effects | Expression changes in longevity interventions |
|---|---|---|
| Mus musculus (mouse) | ||
| Fmo1 | No data | No data |
| Fmo2 | No data | • Caloric restriction ↑ (70) |
| Fmo3 | • Regulates whole-body cholesterol balance in mice (57) | • Caloric restriction ↑ (70, 71) |
| • ΔIIS ↑ (71, 73) | ||
| • Rapamycin ↑ (71) | ||
| • Methionine restriction ↑ | ||
| Fmo4 | No data | • Caloric restriction ↑ (71) |
| • ΔIIS (growth hormone receptor knock-out (GHRKO), Little, Snell) | ||
| ↑ (71) | ||
| • Rapamycin ↑ (71) | ||
| Fmo5 | • Suggested to be a metabolic regulator of aging (59) | • Caloric restriction ↑ (70) |
| D. melanogaster (fly) | ||
| Fmo1 | No data | No data |
| Fmo2 | • RNAi shortens adult lifespan (86) | No data |
| C. elegans (worm) | ||
| fmo-1 | No data | • Dietary restriction ↑ (82, 83) |
| fmo-2 | • Overexpression sufficient for LS extension (75, 97) and stress resistance (75) | • Dietary restriction ↑ (75, 82) |
| • Necessary for solid DR longevity (75) | • Hypoxic response ↑ (79, 80) | |
| • Necessary for hypoxic response longevity (75) | • Mitochondrial disruption ↑ (81) | |
| • Long-telomere worms ↑ (97) | ||
| fmo-3 | No data | No data |
| fmo-4 | No data | • Dietary restriction ↑ (75, 82) |
| • Hypoxic response ↑ (79, 80) | ||
| fmo-5 | No data | No data |
| C46H11.2 | No data | • Dietary restriction ↑ (83) |
| C01H6.4 | No data | No data |
| S. cerevisiae (yeast) | ||
| fmo1 | No data | • Responds to altered sulfur availability (possibly related to methionine restriction) (98, 99) |
| A. thaliana (plant) | ||
| YUC6 | • OE sufficient for longevity and stress resistance (87) | No data |
Dietary restriction and treatment with the drug rapamycin are the two best-documented and most effective interventions for delaying diseases of aging and increasing lifespan in mice (77), and several studies, including those listed above, have observed increased FMO gene expression in animals subjected to both interventions (Table 2). For example, a comparison of mouse DR and gene expression over time found multiple FMOs up-regulated by DR (70), with mFmo3 and mFmo5 among the most significantly up-regulated liver transcripts, along with heart mFmo3. Overall, mFmo1 and mFmo2 were among the most significantly elevated genes when all 17 tested tissues were considered as a group. Interestingly, the same study found that mFmo1 was significantly down-regulated with age in animals fed a normal diet, suggesting that reduced FMO1 expression could be a biomarker of normative aging or even causally involved in the aging process. The most extensive analysis to date of gene expression changes associated with rapamycin treatment in mice found that hepatic mFMO levels are consistently elevated by both DR and rapamycin in both male and female mice (72).
Crowded litter mice represent an alternative model of DR where animals experience nutrient restriction only during the first 3 weeks of life (78). Interestingly, even this early life restriction is sufficient to cause persistent induction of mFmo3 in liver up to 12 months later. This could suggest that epigenetic changes associated with DR, and perhaps other longevity interventions, induce persistent changes in FMO expression that contribute to healthy aging even after the intervention is discontinued.
In contrast to data supporting a role for FMOs in promoting longevity, loss of mFmo5 results in a blood profile of cholesterol, glucose/insulin, and other biomarkers that resembles a more youthful state (59). The authors suggest that this could mean that FMO5 itself promotes metabolic aging. However, because this study did not test the long-term health effects of the metabolic profile, it is unclear whether these results are representative of aging or metabolic reprogramming (59). FMO5 also has a unique substrate profile among mammalian FMOs (8), so it may not be representative of the majority of FMOs. Given their wide taxonomic distribution and numerous evolutionary modifications, there are likely to be exceptions to any broad claims about FMOs as a group.
FMOs and aging in non-mammalian species
Extensive data linking FMO function to aging have come from studies performed in the nematode C. elegans. Several independent studies show that fmo-2 is up-regulated by both genetic and environmental models of increased lifespan in this organism, including DR, hypoxia, mutation of the Von Hippel Lindau tumor suppressor, stabilization of the hypoxic response transcription factor, and developmental electron transport chain inhibition (79–83). Recently, a direct, causal role for FMO-2 as a longevity-promoting factor was uncovered by work showing that either ubiquitous or intestine-specific overexpression of FMO-2 in otherwise wild-type animals is sufficient to extend lifespan in worms (75). Consistent with this, deletion of fmo-2 prevented full lifespan extension following activation of the hypoxic response or DR, further supporting a model that activation of fmo-2 contributes to lifespan extension under these conditions (75).
Additional worm fmo genes are up-regulated by these same longevity interventions, but they have not yet been studied for their roles in aging. Of note, fmo-1 is up-regulated in a more lasting manner than fmo-2 by fasting (83). Also, fmo-4 is induced similarly to fmo-2 by hypoxia (79). Worm fmo-4 is expressed in the hypodermis, whereas fmo-2 is expressed in the intestine, pharynx, and excretory cells (84). Worm fmo-4 also displays a hypoosmotic sensitivity phenotype unique among worm fmo genes (85). Taken together, this evidence suggests that further valuable details can be learned about worm FMOs' roles in healthy aging, stress resistance, and normal physiological processes.
Evidence from flies and plants further suggests that FMOs promote longevity. RNAi knockdown of Drosophila melanogaster (fly) Fmo2 shortens adult lifespan (86). As with worm and mammalian FMOs, fly Fmo2 is not directly orthologous to worm fmo-2 or mammalian FMO2. In fact, both fly Fmo genes are more similar to the ancestral yeast Fmo genes (19). Plants have evolved a distinct group of FMOs termed “YUCCAs.” Arabidopsis thaliana YUC6 overexpression in potato plants results in increased height, erect stature, and longevity due to YUC6's role in auxin production (87). These phenotypes may be plant-specific, or there may be overlap with FMO functions conserved in animal species. YUCCAs also promote drought resistance, and some FMOs are involved in osmoregulation in worms (85) and fish (88), in addition to producing the osmolyte TMAO in humans (43). Interestingly, there is overlap between the DR and osmotic stress pathways (89, 90), and it is plausible that FMOs act at this intersection.
Concluding remarks
FMOs and disease: Unifying mechanisms?
Our understanding of FMO–disease relationships is nascent, but the data already suggest two common mechanisms. Altered sulfur amino acid (SAA) metabolism affects CVD (92), metabolic disease (92), and neurodegenerative disease pathology (93). Again, several SAA pathway metabolites that are credible FMO substrates remain untested as such. Iron metabolism is a second pathway that links FMOs, SAAs, CVD, metabolic disease, and neurological disease.
FMOs and aging: Evolutionary considerations
Data from non-mammalian model systems demonstrate that FMOs can play a direct, causal role in promoting longevity. In worms, fmo-2 is sufficient to extend lifespan and is required for lifespan extension from numerous interventions. FMO induction in response to multiple longevity-enhancing interventions in mice supports a conserved role, as do the primary structural similarities shared by worm and mammalian FMOs.
If FMOs promote longevity in a conserved manner, then the conditions shaping both the evolution of FMOs and aging will merit attention. Induction of FMOs in response to both DR and osmotic stress, for example, suggests that a “harsh times” survival strategy can underlie longevity. Model systems will continue to be of great utility in such investigations.
Future directions
FMOs are emerging as enzymes of considerable interest, with clear experimental and theoretical research directions identifiable. Two lines of experimentation will greatly solidify our foundational knowledge of FMOs. First, solving the crystal structures of hFMOs would conclusively answer structural questions and directly inform functional ones. Second, thorough testing for endogenous substrates, aging, disease, and basic cellular function would solidify the conserved endogenous role of these proteins. There are several candidate endogenous substrates of major biological interest that have not been tested in vitro or otherwise. Metabolites in the sulfur amino acid metabolism pathway, including S-adenosyl methionine, homocysteine (91), and homocysteine adducts, are potential targets for prioritization.
Evidence points to FMO involvement in multiple major diseases and the aging process (Fig. 2). There is an ongoing debate whether aging should be reclassified as a disease, but it is clearly established that aging is the biggest risk factor for the major causes of death including CVD, cancer, and neurodegeneration (94). The risk for each of these increases exponentially with age, independent of other risk factors. Taking everything into consideration, FMOs are an exciting, undercharacterized subgroup of well-conserved enzymes that may play central roles in basic biological processes affecting human health, and there are clear first steps to characterizing them more fully.
Figure 2.
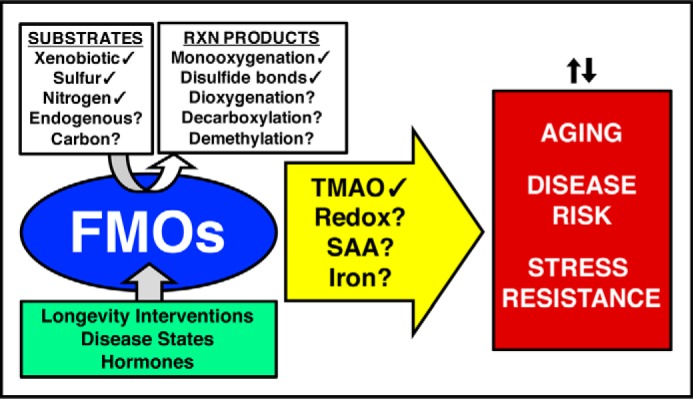
Summary of current knowledge about the regulation and broader role of FMOs. The green box (lower left) depicts known regulators of FMO transcription. The white box (upper left) and arrow (center) show both observed (✓) and hypothetical (?) reactions and mechanisms of FMOs, leading to the broader effects in the red box (right). Redox refers to changes in oxidation/reduction balance resulting from changes to the NADPH/NADP+ ratio and/or increased production of hydrogen peroxide.
This work was supported by National Institutes of Health Grants R00AG045200 and P30AG024824 (to S. F. L.), National Institutes of Health Grant R01AG038518 (to M. K.), and National Institutes of Health Training Grant T32AG000057 (to R. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- FMO
- flavin-containing monooxygenase
- BVMO
- Baeyer–Villiger monooxygenase
- NMO
- N-hydroxylating monooxygenase
- TMA
- trimethylamine
- TMAU
- trimethylaminuria
- TMAO
- trimethylamine–N-oxide
- CVD
- cardiovascular disease
- PD
- Parkinson's disease
- ALS
- amyotrophic lateral sclerosis
- DR
- dietary restriction
- SAA
- sulfur amino acid
- h
- human
- m
- mouse
- y
- yeast.
References
- 1. Mascotti M. L., Lapadula W. J., and Juri Ayub M. (2015) The origin and evolution of Baeyer–Villiger monooxygenases (BVMOs): an ancestral family of flavin monooxygenases. PLoS ONE 10, e0132689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mascotti M. L., Juri Ayub M., Furnham N., Thornton J. M., and Laskowski R. A. (2016) Chopping and changing: the evolution of the flavin-dependent monooxygenases. J. Mol. Biol. 428, 3131–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huijbers M. M. E., Montersino S., Westphal A. H., Tischler D., and Van Berkel W. J. H. (2014) Flavin dependent monooxygenases. Arch. Biochem. Biophys. 544, 2–17 [DOI] [PubMed] [Google Scholar]
- 4. Ojha S., Meng E. C., and Babbitt P. C. (2007) Evolution of function in the “two dinucleotide binding domains” flavoproteins. PLoS Comput. Biol. 3, e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ziegler D. M., Duffel M. W., and Poulsen L. L. (1979) Studies on the nature and regulation of the cellular thiol:disulphide potential. Ciba Found. Symp. 191–204 [DOI] [PubMed] [Google Scholar]
- 6. Krueger S. K., and Williams D. E. (2005) Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol. Ther. 106, 357–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Siddens L. K., Krueger S. K., Henderson M. C., and Williams D. E. (2014) Mammalian flavin-containing monooxygenase (FMO) as a source of hydrogen peroxide. Biochem. Pharmacol. 89, 141–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fiorentini F., Geier M., Binda C., Winkler M., Faber K., Hall M., and Mattevi A. (2016) Biocatalytic characterization of human FMO5: unearthing Baeyer–Villiger reactions in humans. ACS Chem. Biol. 11, 1039–1048 [DOI] [PubMed] [Google Scholar]
- 9. Mashiguchi K., Tanaka K., Sakai T., Sugawara S., Kawaide H., Natsume M., Hanada A., Yaeno T., Shirasu K., Yao H., McSteen P., Zhao Y., Hayashi K., Kamiya Y., and Kasahara H. (2011) The main auxin biosynthesis pathway in Arabidopsis. Proc. Natl. Acad. Sci. 108, 18512–18517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gut I., and Conney A. H. (1993) Trimethylamine N-oxygenation and N-demethylation in rat liver microsomes. Biochem. Pharmacol. 46, 239–244 [DOI] [PubMed] [Google Scholar]
- 11. Suh J. K., Poulsen L. L., Ziegler D. M., and Robertus J. D. (1999) Yeast flavin-containing monooxygenase generates oxidizing equivalents that control protein folding in the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 96, 2687–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rooseboom M., Commandeur J. N. M., Floor G. C., Rettie A. E., and Vermeiden N. P. E. (2001) Selenoxidation by flavin-containing monooxygenases as a novel pathway for β-elimination of selenocysteine Se-conjugates. Chem. Res. Toxicol. 14, 127–134 [DOI] [PubMed] [Google Scholar]
- 13. Neuhoff C., Gunawan A., Farooq M. O., Cinar M. U., Große-Brinkhaus C., Sahadevan S., Frieden L., Tesfaye D., Tholen E., Looft C., Schellander K., and Uddin M. J. (2015) Preliminary study of FMO1, FMO5, CYP21, ESR1, PLIN2 and SULT2A1 as candidate gene for compounds related to boar taint. Meat Sci. 108, 67–73 [DOI] [PubMed] [Google Scholar]
- 14. Ziegler D. M. (2002) An overview of the mechanism, substrate specificities, and structure of FMOs. Drug Metab. Rev. 34, 503–511 [DOI] [PubMed] [Google Scholar]
- 15. Alfieri A., Malito E., Orru R., Fraaije M. W., and Mattevi A. (2008) Revealing the moonlighting role of NADP in the structure of a flavin-containing monooxygenase. Proc. Natl. Acad. Sci. U.S.A. 105, 6572–6577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cho H. J., Cho H. Y., Kim K. J., Kim M. H., Kim S. W., and Kang B. S. (2011) Structural and functional analysis of bacterial flavin-containing monooxygenase reveals its ping-pong-type reaction mechanism. J. Struct. Biol. 175, 39–48 [DOI] [PubMed] [Google Scholar]
- 17. Li C. Y., Chen X. L., Zhang D., Wang P., Sheng Q., Peng M., Xie B. B., Qin Q. L., Li P. Y., Zhang X. Y., Su H. N., Song X. Y., Shi M., Zhou B. C., Xun L. Y., et al. (2017) Structural mechanism for bacterial oxidation of oceanic trimethylamine into trimethylamine N-oxide. Mol. Microbiol. 103, 992–1003 [DOI] [PubMed] [Google Scholar]
- 18. Eswaramoorthy S., Bonanno J. B., Burley S. K., and Swaminathan S. (2006) Mechanism of action of a flavin-containing monooxygenase. Proc. Natl. Acad. Sci. U.S.A. 103, 9832–9837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. UniProt Consortium (2015) UniProt: A hub for protein information. Nucleic Acids Res. 43, D204–D212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suh J. K., Poulsen L. L., Ziegler D. M., and Robertus J. D. (1996) Molecular cloning and kinetic characterization of a flavin-containing monooxygenase from Saccharomyces cerevisiae. Arch. Biochem. Biophys. 336, 268–274 [DOI] [PubMed] [Google Scholar]
- 21. Henderson M. C., Krueger S. K., Stevens J. F., and Williams D. E. (2004) Human flavin-containing monooxygenase form 2 S-oxygenation: sulfenic acid formation from thioureas and oxidation of glutathione. Chem. Res. Toxicol. 17, 633–640 [DOI] [PubMed] [Google Scholar]
- 22. Hernandez D., Janmohamed A., Chandan P., Phillips I. R., and Shephard E. A. (2004) Organization and evolution of the flavin-containing monooxygenase genes of human and mouse: identification of novel gene and pseudogene clusters. Pharmacogenetics 14, 117–130 [DOI] [PubMed] [Google Scholar]
- 23. Phillips I. R., Dolphin C. T., Clair P., Hadley M. R., Hutt A. J., McCombie J. R. R., Smith R. L., and Shephard E. A. (1995) The molecular biology of the flavin-containing monooxygenases of man. Chem. Biol. Interact. 96, 17–32 [DOI] [PubMed] [Google Scholar]
- 24. Hines R. N., Hopp K. A., Franco J., Saeian K., and Begun F. P. (2002) Alternative processing of the human FMO6 gene renders transcripts incapable of encoding a functional flavin-containing monooxygenase. Mol. Pharmacol. 62, 320–325 [DOI] [PubMed] [Google Scholar]
- 25. Lattard V., Zhang J., and Cashman J. R. (2004) Alternative processing events in human FMO genes. Mol. Pharmacol. 65, 1517–1525 [DOI] [PubMed] [Google Scholar]
- 26. Hao D. C., Chen S. L., Mu J., and Xiao P. G. (2009) Molecular phylogeny, long-term evolution, and functional divergence of flavin-containing monooxygenases. Genetica 137, 173–187 [DOI] [PubMed] [Google Scholar]
- 27. Koukouritaki S. B., Simpson P., Yeung C. K., Rettie A. E., and Hines R. N. (2002) Human hepatic flavin-containing monooxygenases 1 (FMO1) and 3 (FMO3) developmental expression. Pediatr. Res. 51, 236–243 [DOI] [PubMed] [Google Scholar]
- 28. Zhang J., and Cashman J. R. (2006) Quantitative analysis of FMO gene mRNA levels in human tissues. Drug Metab. Dispos. 34, 19–26 [DOI] [PubMed] [Google Scholar]
- 29. Falls J. G., Blake B. L., Cao Y., Levi P. E., and Hodgson E. (1995) Gender differences in hepatic expression of flavin-containing monooxygenase isoforms (FMO1, FMO3, and FMO5) in mice. J. Biochem. Toxicol. 10, 171–177 [DOI] [PubMed] [Google Scholar]
- 30. Cashman J. R., Yang Z., Yang L., and Wrighton S. A. (1993) Stereo- and regioselective N- and S-oxidation of tertiary amines and sulfides in the presence of adult human liver microsomes. Drug Metab. Dispos. 21, 492–501 [PubMed] [Google Scholar]
- 31. Sadeque A. J., Thummel K. E., and Rettie A. E. (1993) Purification of macaque liver flavin-containing monooxygenase: a form of the enzyme related immunochemically to an isozyme expressed selectively in adult human liver. Biochim. Biophys. Acta 1162, 127–134 [DOI] [PubMed] [Google Scholar]
- 32. Bennett B. J., de Aguiar Vallim T. Q., Wang Z., Shih D. M., Meng Y., Gregory J., Allayee H., Lee R., Graham M., Crooke R., Edwards P. A., Hazen S. L., and Lusis A. J. (2013) Trimethylamine–N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 17, 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Luo Z., and Hines R. N. (2001) Regulation of flavin-containing monooxygenase 1 expression by ying yang 1 and hepatic nuclear factors 1 and 4. Mol. Pharmacol. 60, 1421–1430 [DOI] [PubMed] [Google Scholar]
- 34. Borbás T., Benko B., Dalmadi B., Szabó I., and Tihanyi K. (2006) Insulin in flavin-containing monooxygenase regulation: flavin-containing monooxygenase and cytochrome P450 activities in experimental diabetes. Eur. J. Pharm. Sci. 28, 51–58 [DOI] [PubMed] [Google Scholar]
- 35. Klick D. E., Shadley J. D., and Hines R. N. (2008) Differential regulation of human hepatic flavin containing monooxygenase 3 (FMO3) by CCAAT/enhancer-binding protein β (C/EBPβ) liver inhibitory and liver activating proteins. Biochem. Pharmacol. 76, 268–278 [DOI] [PubMed] [Google Scholar]
- 36. Zhang J., Chaluvadi M. R., Reddy R., Motika M. S., Richardson T. A., Cashman J. R., and Morgan E. T. (2009) Hepatic flavin-containing monooxygenase gene regulation in different mouse inflammation models. Drug Metab. Dispos. 37, 462–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miao J., Ling A. V., Manthena P. V., Gearing M. E., Graham M. J., Crooke R. M., Croce K. J., Esquejo R. M., Clish C. B., Morbid Obesity Study Group, Vicent D., and Biddinger S. B. (2015) Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat. Commun. 6, 6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Houseman L. (2008) Flavin-containing monooxygenases: Regulation, endogenous roles and dietary supplements. Ph.D. thesis, University of London [Google Scholar]
- 39. Celius T., Roblin S., Harper P. A., Matthews J., Boutros P. C., Pohjanvirta R., and Okey A. B. (2008) Aryl hydrocarbon receptor-dependent induction of flavin-containing monooxygenase mRNAs in mouse liver. Drug Metab. Dispos. 36, 2499–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shih D. M., Wang Z., Lee R., Meng Y., Che N., Charugundla S., Qi H., Wu J., Pan C., Brown J. M., Vallim T., Bennett B. J., Graham M., Hazen S. L., and Lusis A. J. (2015) Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J. Lipid Res. 56, 22–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Celius T., Pansoy A., Matthews J., Okey A. B., Henderson M. C., Krueger S. K., and Williams D. E. (2010) Flavin-containing monooxygenase-3: induction by 3-methylcholanthrene and complex regulation by xenobiotic chemicals in hepatoma cells and mouse liver. Toxicol. Appl. Pharmacol. 247, 60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Novick R. M., Vezina C. M., and Elfarra A. A. (2010) Isoform distinct time-, dose-, and castration-dependent alterations in flavin-containing monooxygenase expression in mouse liver after 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment. Biochem. Pharmacol. 79, 1345–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Treacy E. P., Akerman B. R., Chow L. M. L., Youil R., Bibeau C., Lin J., Bruce A. G, Knight M., Danks D. M., Cashman J. R., and Forrest S. M. (1998) Mutations of the flavin-containing monooxygenase gene (FMO3) cause trimethylaminuria, a defect in detoxication. Hum. Mol. Genet. 7, 839–845 [DOI] [PubMed] [Google Scholar]
- 44. McConnell H. W., Mitchell S. C., Smith R. L., and Brewster M. (1997) Trimethylaminuria associated with seizures and behavioural disturbance: a case report. Seizure 6, 317–321 [DOI] [PubMed] [Google Scholar]
- 45. Wang Z., Klipfell E., Bennett B. J., Koeth R., Levison B. S., Dugar B., Feldstein A. E., Britt E. B., Fu X., Chung Y.-M., Wu Y., Schauer P., Smith J. D., Allayee H., Tang W. H. W., et al. (2011) Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tang W. H. W., Wang Z., Levison B. S., Koeth R. A., Britt E. B., Fu X., Wu Y., and Hazen S. L. (2013) Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 368, 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Koeth R. A., Wang Z., Levison B. S., Buffa J. A., Org E., Sheehy B. T., Britt E. B., Fu X., Wu Y., Li L., Smith J. D., DiDonato J. A., Chen J., Li H., Wu G. D., et al. (2013) Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 19, 576–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ufnal M., Jazwiec R., Dadlez M., Drapala A., Sikora M., and Skrzypecki J. (2014) Trimethylamine–N-oxide: a carnitine-derived metabolite that prolongs the hypertensive effect of angiotensin II in rats. Can. J. Cardiol. 30, 1700–1705 [DOI] [PubMed] [Google Scholar]
- 49. Zhu W., Gregory J. C., Org E., Buffa J. A., Gupta N., Wang Z., Li L., Fu X., Wu Y., Mehrabian M., Sartor R. B., McIntyre T. M., Silverstein R. L., Tang W. H., DiDonato J. A., et al. (2016) Gut microbial metabolite TMAO enhances platelet article gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. 165, 111–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tomasova L., Konopelski P., and Ufnal M. (2016) Gut bacteria and hydrogen sulfide: the new old players in circulatory system homeostasis. Molecules 21, E1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dolan C., Shields D. C., Stanton A., O'Brien E., Lambert D. M., O'Brien J. K., and Treacy E. P. (2005) Polymorphisms of the flavin containing monooxygenase 3 (FMO3) gene do not predispose to essential hypertension in Caucasians. BMC Med. Genet. 6, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fukami K., Yamagishi S., Sakai K., Kaida Y., Yokoro M., Ueda S., Wada Y., Takeuchi M., Shimizu M., Yamazaki H., and Okuda S. (2015) Oral l-carnitine supplementation increases trimethylamine–N-oxide but reduces markers of vascular injury in hemodialysis patients. J. Cardiovasc. Pharmacol. 65, 289–295 [DOI] [PubMed] [Google Scholar]
- 53. Collins H. L., Drazul-Schrader D., Sulpizio A. C., Koster P. D., Williamson Y., Adelman S. J., Owen K., Sanli T., and Bellamine A. (2016) l-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE−/− transgenic mice expressing CETP. Atherosclerosis 244, 29–37 [DOI] [PubMed] [Google Scholar]
- 54. Ufnal M., Zadlo A., and Ostaszewski R. (2015) TMAO: a small molecule of great expectations. Nutrition 31, 1317–1323 [DOI] [PubMed] [Google Scholar]
- 55. Takamura T., Sakurai M., Ota T., Ando H., Honda M., and Kaneko S. (2004) Genes for systemic vascular complications are differentially expressed in the livers of Type 2 diabetic patients. Diabetologia 47, 638–647 [DOI] [PubMed] [Google Scholar]
- 56. Vahabzadeh M., and Mohammadpour A.-H. (2015) Effect of diabetes mellitus on the metabolism of drugs and toxins. J. Clin. Toxicol. 5, 233 [Google Scholar]
- 57. Warrier M., Shih D. M., Burrows A. C., Ferguson D., Gromovsky A. D., Brown A., Marshall S., McDaniel A., Schugar R. C., Wang Z., Sacks J., Rong X., Vallim T., Chou J., Ivanova P. T., et al. (2015) The TMAO-generating enzyme flavin monooxygenase 3 is a central regulator of cholesterol balance. Cell Rep. 10, 326–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Veeravalli S., Omar B. A., Houseman L., Hancock M., Gonzalez Malagon S. G., Scott F., Janmohamed A., Phillips I. R., and Shephard E. A. (2014) The phenotype of a flavin-containing monooyxgenase knockout mouse implicates the drug-metabolizing enzyme FMO1 as a novel regulator of energy balance. Biochem. Pharmacol. 90, 88–95 [DOI] [PubMed] [Google Scholar]
- 59. Gonzalez Malagon S. G., Melidoni A. N., Hernandez D., Omar B. A., Houseman L., Veeravalli S., Scott F., Varshavi D., Everett J., Tsuchiya Y., Timms J. F., Phillips I. R., and Shephard E. A. (2015) The phenotype of a knockout mouse identifies flavin-containing monooxygenase 5 (FMO5) as a regulator of metabolic ageing. Biochem. Pharmacol. 96, 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Petriello M. C., Hoffman J. B., Morris A. J., and Hennig B. (2017) Emerging roles of xenobiotic detoxification enzymes in metabolic diseases. Rev. Environ. Health. 32, 105–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Motika M. S., Zhang J., and Cashman J. R. (2007) Flavin-containing monooxygenase 3 and human disease. Expert Opin. Drug Metab. Toxicol. 3, 831–845 [DOI] [PubMed] [Google Scholar]
- 62. Bozzoni V., Pansarasa O., Diamanti L., Nosari G., Cereda C., and Ceroni M. (2016) Amyotrophic lateral sclerosis and environmental factors. Funct. Neurol. 31, 7–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hibar D. P., Stein J. L., Ryles A. B., Kohannim O., Jahanshad N., Medland S. E., Hansell N. K., McMahon K. L., de Zubicaray G. I., Montgomery G. W., Martin N. G., Wright M. J., Saykin A. J., Jack C. R. Jr., Weiner M. W., et al. (2013) Genome-wide association identifies genetic variants associated with lentiform nucleus volume in N=1345 young and elderly subjects. Brain Imaging Behav. 7, 102–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li B., Yuan Y., Zhang W., He W., Hu J., and Chen N. (2014) Flavin-containing monooxygenase, a new clue of pathological proteins in the rotenone model of parkinsonism. Neurosci. Lett. 566, 11–16 [DOI] [PubMed] [Google Scholar]
- 65. Guan S. H., Falick A. M., Williams D. E., and Cashman J. R. (1991) Evidence for complex formation between rabbit lung flavin-containing monooxygenase and calreticulin. Biochemistry 30, 9892–9900 [DOI] [PubMed] [Google Scholar]
- 66. Umbreit J. N., Conrad M. E., Hainsworth L. N., and Simovich M. (2002) The ferrireductase paraferritin contains divalent metal transporter as well as mobilferrin. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G534–G539 [DOI] [PubMed] [Google Scholar]
- 67. Barber M., Conrad M. E., Umbreit J. N., Barton J. C., and Moore E. G. (2000) Abnormalities of flavin monooxygenase as an etiology for sideroblastic anemia. Am. J. Hematol. 65, 149–153 [DOI] [PubMed] [Google Scholar]
- 68. Muckenthaler M., Roy C. N., Custodio A. O., Miñana B., deGraaf J., Montross L. K., Andrews N. C., and Hentze M. W. (2003) Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hemochromatosis. Nat. Genet. 34, 102–107 [DOI] [PubMed] [Google Scholar]
- 69. Coppin H., Darnaud V., Kautz L., Meynard D., Aubry M., Mosser J., Martinez M., and Roth M.-P. (2007) Gene expression profiling of Hfe−/− liver and duodenum in mouse strains with differing susceptibilities to iron loading: identification of transcriptional regulatory targets of Hfe and potential hemochromatosis modifiers. Genome Biol. 8, R221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Swindell W. R. (2009) Genes and gene expression modules associated with caloric restriction and aging in the laboratory mouse. BMC Genomics 10, 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Steinbaugh M. J., Sun L. Y., Bartke A., and Miller R. A. (2012) Activation of genes involved in xenobiotic metabolism is a shared signature of mouse models with extended lifespan. Am. J. Physiol. Endocrinol. Metab. 303, E488–E95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Miller R. A., Harrison D. E., Astle C. M., Fernandez E., Flurkey K., Han M., Javors M. A., Li X., Nadon N. L., Nelson J. F., Pletcher S., Salmon A. B., Sharp Z. D., Van Roekel S., Winkleman L., and Strong R. (2014) Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell 13, 468–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Swindell W. R. (2007) Gene expression profiling of long-lived dwarf mice: longevity-associated genes and relationships with diet, gender and aging. BMC Genomics 8, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pitt J. N., and Kaeberlein M. (2015) Why is aging conserved and what can we do about it? PLOS Biol. 13, e1002131; Correction (2015) PLOS Biol. 13, e1002176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Leiser S. F., Miller H., Rossner R., Fletcher M., Leonard A., Primitivo M., Rintala N., Ramos F. J., Miller D. L., and Kaeberlein M. (2015) Cell nonautonomous activation of flavin-containing monooxygenase promotes longevity and health span. Science 350, 1375–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schirra H. J., Anderson C. G., Wilson W. J., Kerr L., Craik D. J., Waters M. J., and Lichanska A. M. (2008) Altered metabolism of growth hormone receptor mutant mice: a combined NMR metabonomics and microarray study. PLoS ONE 3, e2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kaeberlein M., Rabinovitch P. S., and Martin G. M. (2015) Healthy aging: the ultimate preventative medicine. Science 350, 1191–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sun L., Sadighi Akha A. A., Miller R. A., and Harper J. M. (2009) Life-span extension in mice by preweaning food restriction and by methionine restriction in middle age. J. Gerontol. A Biol. Sci. Med. Sci. 64, 711–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shen C., Nettleton D., Jiang M., Kim S. K., and Powell-Coffman J. A. (2005) Roles of the HIF-1 hypoxia-inducible factor during hypoxia response in Caenorhabditis elegans. J. Biol. Chem. 280, 20580–20588 [DOI] [PubMed] [Google Scholar]
- 80. Bishop T., Lau K. W., Epstein A. C. R., Kim S. K., Jiang M., O'Rourke D., Pugh C. W., Gleadle J. M., Taylor M. S., Hodgkin J., and Ratcliffe P. J. (2004) Genetic analysis of pathways regulated by the von Hippel-Lindau tumor suppressor in Caenorhabditis elegans. PLoS Biol. 2, e289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lee S.-J., Hwang A. B., and Kenyon C. (2010) Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 20, 2131–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Honjoh S., Yamamoto T., Uno M., and Nishida E. (2009) Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature 457, 726–730 [DOI] [PubMed] [Google Scholar]
- 83. Uno M., Honjoh S., Matsuda M., Hoshikawa H., Kishimoto S., Yamamoto T., Ebisuya M., Yamamoto T., Matsumoto K., and Nishida E. (2013) A fasting-responsive signaling pathway that extends life span in C. elegans. Cell Rep. 3, 79–91 [DOI] [PubMed] [Google Scholar]
- 84. Petalcorin M. I. R., Joshua G. W., Agapow P. M., and Dolphin C. T. (2005) The fmo genes of Caenorhabditis elegans and C. briggsae: characterisation, gene expression and comparative genomic analysis. Gene 346, 83–96 [DOI] [PubMed] [Google Scholar]
- 85. Hirani N., Westenberg M., Seed P. T., Petalcorin M. I., and Dolphin C. T. (2016) C. elegans flavin-containing monooxygenase-4 is essential for osmoregulation in hypotonic stress. Biol. Open 5, 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Attrill H., Falls K., Goodman J. L., Millburn G. H., Antonazzo G., Rey A. J., and Marygold S. J., FlyBase Consortium (2016) FlyBase: establishing a Gene Group resource for Drosophila melanogaster. Nucleic Acids Res. 44, D786–D792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cheol Park H., Cha J.-Y., and Yun D.-J. (2013) Roles of YUCCAs in auxin biosynthesis and drought stress responses in plants. Plant Signal. Behav. 8, e24495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schlenk D. (1993) A comparison of endogenous and exogenous substrates of the flavin-containing monooxygenases in aquatic organisms. Aquat. Toxicol. 26, 157–162 [Google Scholar]
- 89. Matzkin L. M., and Markow T. A. (2009) Transcriptional regulation of metabolism associated with the increased desiccation resistance of the cactophilic Drosophila mojavensis. Genetics 182, 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chandler-Brown D., Choi H., Park S., Ocampo B. R., Chen S., Le A., Sutphin G. L., Shamieh L. S., Smith E. D., and Kaeberlein M. (2015) Sorbitol treatment extends lifespan and induces the osmotic stress response in Caenorhabditis elegans. Front. Genet. 6, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. McCully K. S. (2015) Homocysteine metabolism, atherosclerosis, and diseases of aging. Compr. Physiol. 6, 471–505 [DOI] [PubMed] [Google Scholar]
- 92. Poloni S., Blom H. J., and Schwartz I. V. (2015) Stearoyl-CoA desaturase-1: Is it the link between sulfur amino acids and lipid metabolism? Biology (Basel) 4, 383–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gibrat C., and Cicchetti F. (2011) Potential of cystamine and cysteamine in the treatment of neurodegenerative diseases. Prog Neuropsychopharmacol. Biol. Psychiatry 35, 380–389 [DOI] [PubMed] [Google Scholar]
- 94. Kaeberlein M. (2013) Longevity and aging. F1000Prime Rep. 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Robinson-Cohen C., Newitt R., Shen D. D., Rettie A. E., Kestenbaum B. R., Himmelfarb J., and Yeung C. K. (2016) Association of FMO3 variants and trimethylamine N-oxide concentration, disease progression, and mortality in CKD patients. PLoS ONE 11, e0161074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gagliardi S., Gallo A., Policicchio S., La Salvia S., Diamanti L., Bernuzzi S., Pansarasa O., and Cereda C. (2016) Environmental and genetic factors in ALS: positive correlation of Snps in flavin-containing monooxygenase 5 gene. Ann. Neurodegener. Disord. 1, 1014 [Google Scholar]
- 97. Park M. C., Park D., Lee E. K., Park T., and Lee J. (2010) Genomic analysis of the telomeric length effect on organismic lifespan in Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 396, 382–387 [DOI] [PubMed] [Google Scholar]
- 98. Brauer M. J., Huttenhower C., Airoldi E. M., Rosenstein R., Matese J. C., Gresham D., Boer V. M., Troyanskaya O. G., and Botstein D. (2008) Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol. Biol. Cell 19, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kumar A., John L., Maity S., Manchanda M., Sharma A., Saini N., Chakraborty K., and Sengupta S. (2011) Converging evidence of mitochondrial dysfunction in a yeast model of homocysteine metabolism imbalance. J. Biol. Chem. 286, 21779–21795 [DOI] [PMC free article] [PubMed] [Google Scholar]