

Abstract
The antischistosomal prodrug oxamniquine is activated by a sulfotransferase (SULT) in the parasitic flatworm Schistosoma mansoni. Of the three main human schistosome species, only S. mansoni is sensitive to oxamniquine therapy despite the presence of SULT orthologs in Schistosoma hematobium and Schistosoma japonicum. The reason for this species-specific drug action has remained a mystery for decades. Here we present the crystal structures of S. hematobium and S. japonicum SULTs, including S. hematobium SULT in complex with oxamniquine. We also examined the activity of the three enzymes in vitro; surprisingly, all three are active toward oxamniquine, yet we observed differences in catalytic efficiency that implicate kinetics as the determinant for species-specific toxicity. These results provide guidance for designing oxamniquine derivatives to treat infection caused by all species of schistosome to combat emerging resistance to current therapy.
Keywords: drug resistance, enzyme kinetics, enzyme structure, parasite, structural biology, sulfotransferase, helminth, oxamniquine, schistosome, schistosomiasis
Introduction
Schistosomes, commonly known as blood flukes, infected 261 million people in 78 countries in 2013 with up to 200,000 deaths caused by the associated disease (1). The parasite is acquired from freshwater snail hosts found in Africa, Asia, the Middle East, South America, and the West Indies. Schistosomiasis, the chronic illness resulting from schistosome infection, is classified by the World Health Organization as a neglected tropical disease. Three species of the human blood fluke, Schistosoma mansoni, Schistosoma hematobium, and Schistosoma japonicum, account for almost all cases of human schistosomiasis (2). Symptoms vary according to the infecting species, and chronic infection can damage the liver, spleen, lungs, intestines, and urogenital tract (3). Effective drug treatment is available using the drug praziquantel, currently the recommended drug for schistosome infection. Praziquantel intervention provides enormous health benefits, but the selective pressure the monotherapy applies to the parasite has the potential to drive resistance to the drug. Schistosomes with decreased susceptibility to praziquantel have already been documented in patients in Egypt, Senegal, and Tanzania (4); therefore, combination treatments are needed.
Oxamniquine was the first-line antischistosomal drug in Brazil until the 1980s when it was gradually replaced by praziquantel (5) because of its lower cost and the threat of emerging oxamniquine resistance (6–9). We recently identified a sulfotransferase (SULT)4 enzyme in S. mansoni (SmSULT) as the oxamniquine target, leading to the X-ray crystal structure of the enzyme in a complex with oxamniquine and the reaction product 3′-phosphoadenosine 5′-phosphate (PAP) (10). Oxamniquine is a species-specific treatment effective against S. mansoni but not S. hematobium or S. japonicum despite their SULTs sharing 71 and 58% sequence identity with SmSULT. The reason for the differences in oxamniquine efficacy between species is unknown. Here we describe the enzyme kinetics of SULT from all three sources and present the structures of S. hematobium SULT (ShSULT) in complexes with PAP with and without oxamniquine and of S. japonicum SULT (SjSULT) in complex with PAP. The results provide a framework for redesign of oxamniquine as a pan-species drug treatment to be used in conjunction with praziquantel.
Results
Structures of PAP complexes of schistosome sulfotransferases
Initial efforts to express full-length recombinant ShSULT were unsuccessful. Examination of the sequences of the three schistosome enzymes showed that ShSULT contains an additional N-terminal extension of 15 residues compared with SmSULT and SjSULT. Engineering this extension out of the ShSULT expression construct yielded a soluble protein amenable to crystallization. Although this modified ShSULT could be expressed using a construct containing a cleavable N-terminal His8 tag, little soluble protein was produced from N-terminally tagged constructs for SjSULT, and refolded protein failed to crystallize. The addition of a short Gly-Ser linker plus a C-terminal His8 tag to full-length SjSULT produced soluble protein that formed crystals. For both enzymes, the crystals were grown in the presence of PAP, yielding the structures of the enzyme–PAP complexes. Soaking the ShSULT crystals with racemic oxamniquine or the individual stereoisomers yielded the structures of the enzyme–PAP–oxamniquine complexes (Fig. 1 and Table 1).
Figure 1.
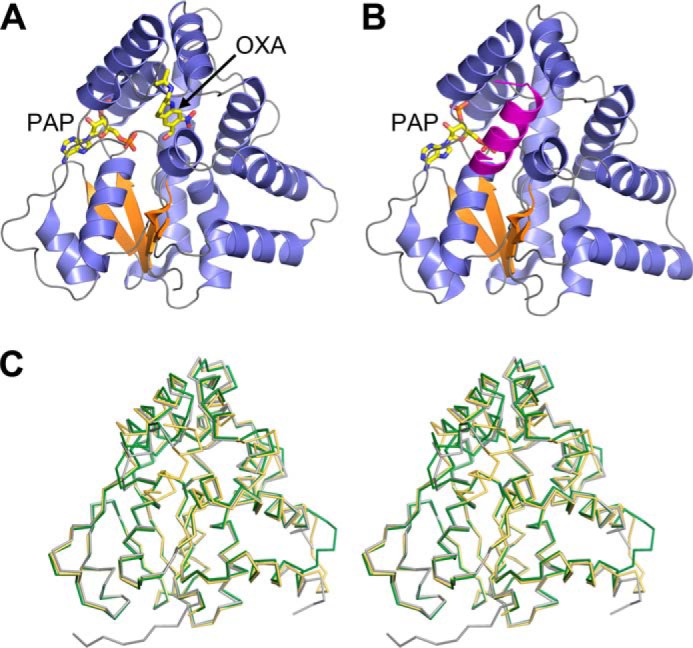
A, schematic representation of the ShSULT–PAP–oxamniquine (OXA) complex crystal structure. β strands are colored orange, and α helices are colored slate blue. B, schematic representation of the SjSULT–PAP complex crystal structure. Note that the predicted biological assembly is shown with a chain-swapped C-terminal α helix from the crystal symmetry-related molecule colored magenta. For clarity, the C-terminal Gly-Ser His8 affinity tag is not shown. C, walleyed stereoview of the superposition of the Cα traces of ShSULT (dark green) and SjSULT (light orange) onto SmSULT (gray).
Table 1.
Data collection and refinement statistics
UTHSCSA, University of Texas Health Science Center at San Antonio.
| ShSULT–PAP | ShSULT–PAP–racemic OXA | ShSULT–PAP–(R)-OXA | ShSULT–PAP–(S)-OXA | SjSULT–PAP | |
|---|---|---|---|---|---|
| PDB code | 5TIV | 5TIW | 5TIX | 5TIY | 5TIZ |
| Data collection | |||||
| X-ray Source | Advanced Photon Source 24-ID-E | Advanced Photon Source 24-ID-C | UTHSCSA X-ray Crystallography Core Laboratory | UTHSCSA X-ray Crystallography Core Laboratory | Advanced Photon Source 24-ID-E |
| Space group | C2221 | P21212 | P212121 | C2221 | P4122 |
| Cell dimensions | |||||
| a, b, c (Å) | 52.6, 73.6, 139.6 | 69.7, 139.4, 49.6 | 51.7, 72.6, 138.7 | 52.5, 73.4, 139.0 | 57.3, 57.3, 181.5 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 0.9792 | 0.9795 | 1.54178 | 1.54178 | 0.9792 |
| Resolution (Å) | 139.6–1.43 (1.51–1.43)a | 139.42–1.66 (1.75–1.66) | 46.23–1.78 (1.88–1.78) | 46.34–1.76 (1.86–1.76) | 54.65–2.87 (3.03–2.87) |
| Rsym | 0.055 (0.901) | 0.056 (0.914) | 0.086 (0.645) | 0.055 (0.714) | 0.158 (0.667) |
| Rpim | 0.025 (0.395) | 0.024 (0.382) | 0.045 (0.329) | 0.028 (0.376) | 0.115 (0.493) |
| Mean I/σI | 18.3 (2.0) | 21.9 (2.1) | 13.9 (2.0) | 15.5 (2.0) | 7.7 (2.0) |
| Completeness (%) | 99.7 (100) | 99.8 (99.9) | 99.2 (98.4) | 99.6 (100) | 96.9 (98.8) |
| Redundancy | 7.0 (7.2) | 7.2 (7.4) | 4.5 (4.6) | 4.6 (4.6) | 2.8 (2.8) |
| Wilson value (Å2) | 17.6 | 26.5 | 22.9 | 27.3 | 39.0 |
| Refinement | |||||
| Resolution (Å) | 36.48–1.43 | 69.71–1.66 | 39.00–1.78 | 40.84–1.76 | 54.65–2.87 |
| No. reflections | 50,234 | 57,773 | 50,286 | 26,838 | 7,107 |
| Rwork/Rfree | 0.159/0.191 | 0.175/0.203 | 0.188/0.230 | 0.167/0.214 | 0.225/0.276 |
| No. atoms | |||||
| Protein | 2,073 | 4,092 | 4,048 | 2,036 | 2,059 |
| Ligand/ion | 35 (1 PAP, 1 TRS)b | 140 (2 PAP, 2 (S)-OXA, 5 Ca2+, 1 PEG, 1BCN) | 94 (2 PAP, 2 (R)-OXA) | 47 (1 PAP, 1 (S)-OXA) | 27 (1 PAP) |
| Solvent | 240 | 290 | 433 | 184 | |
| B-factors (Å2) | |||||
| Protein | 22.5 | 29.8 | 28.6 | 32.3 | 31.5 |
| Ligand | 18.5 | 33.5 | 31.7 | 30.0 | 16.8 |
| Solvent | 33.0 | 37.5 | 38.6 | 40.6 | |
| r.m.s.d. bond lengths (Å) | 0.008 | 0.013 | 0.003 | 0.004 | 0.010 |
| r.m.s.d. bond angles (°) | 1.003 | 1.193 | 0.608 | 0.690 | 0.848 |
| Ramachandran statistics: favored, allowed, outliers (%) | 96.4, 3.6, 0.0 | 97.6, 2.4, 0.0 | 96.7, 3.3, 0.0 | 97.2, 2.8, 0.0 | 95.6, 4.0, 0.4 |
a Highest resolution shell is shown in parentheses.
b Ligand abbreviations: OXA, oxamniquine; BCN, Bicine (N,N-bis(2-hydroxyethyl)glycine); TRS, Tris buffer.
The overall structures of both ShSULT and SjSULT are similar to that of SmSULT (Protein Data Bank (PDB) code 4MUA). The ShSULT molecule superimposes onto SmSULT with an r.m.s.d. of 0.8 Å over 243 of 253 residues with no significant deviations other than a fully ordered loop (ShSULT residues 76–80) in the α3-loop-α4 region that is disordered in the SmSULT crystal structure. The N-terminal 17–20 residues are disordered. The SjSULT molecule superimposes onto SmSULT with an r.m.s.d. of 1.0 Å over 232 of 253 residues with one significant deviation in a C-terminal α helix (α12) caused by its domain swap into a neighboring molecule related by 2-fold crystal symmetry. The swapped α12 does not superimpose on the C-terminal α helices of SmSULT or ShSULT associated with α11 in a helix-turn-helix motif. Rather, SjSULT α12 is displaced and packs against α2 (supplemental Fig. 1, A and B) with half of the C-terminal tag (Gly-Ser-His-His-His) visible in the electron density map. The tag region makes van der Waals interactions with the protein surface formed by α1-β1-α2 secondary structures. We speculate that this arrangement is an artifact caused by the domain swap. Threading the SjSULT C-terminal sequence onto the C-terminal structure of either SmSULT or ShSULT reveals that the SjSULT sequence is compatible with the ShSULT fold (supplemental Fig. 1C) provided small adjustments are made such as selecting common rotamers.
All three schistosome SULTs adopt an α/β fold characteristic to the PAPS-dependent SULT family of enzymes with a five-stranded parallel β sheet sandwiched between two clusters of five and seven α helices. A search of the Protein Data Bank (11) for structural homologs using the PDBeFold server (12) yields the human integral membrane tyrosylprotein sulfotransferase 2 (TPST2) (13) as the top match to SmSULT (Z score, 7.2; r.m.s.d., 2.8 Å for 157 of 253 residues), ShSULT (Z score, 6.8; r.m.s.d., 2.8 Å for 156 of 246 residues), and SjSULT (Z score, 7.9; r.m.s.d., 2.7 Å for 161 of 253 residues). Fig. 2A presents a secondary structure topology diagram of the schistosome SULT fold indicating elements in common with the mammalian SULTs. Schistosome SULTs have an additional three α helices in common with TPST2 that are absent from the well characterized canonical mammalian SULTs in the SULT1, SULT2, and SULT4 families with known structures. Five α helices in the schistosome SULT do not have a mammalian enzyme counterpart (Fig. 2, A–C).
Figure 2.

A, diagram of secondary structure topology shared between the three schistosome SULTs illustrating the variation in fold as compared with mammalian SULTs. The β strands and α helices in yellow are common between the known schistosome and mammalian SULT protein structures, including SULTs 1A1, 1A3, 1B1, 1C1, 1C2, 1C3, 1D1, 1E1, 2A1, 2A3, and 4A1 and TPST2. Additionally, the α helices in cyan are common between schistosome SULT and TPST2, but α helices in red do not share a mammalian enzyme counterpart. N and C termini are labeled. B, diagram of ShSULT, TPST2 (PDB code 3AP1), and SULT1E1 (PDB code 1G3M) for comparison. The color scheme of the secondary structure elements pictured is as in A. Secondary structures of TPST2 and SULT1E1 without a schistosome counterpart are shown in gray. C, superposition of common secondary structure elements (shown in isolation) between schistosome and mammalian SULTs. Mammalian secondary structures are colored as in B, and ShSULT secondary structures are colored dark green.
The PAPS-binding site in the schistosome SULTs is almost identical to that seen in other members of the PAPS-dependent SULT family (14, 15). A conserved arginine residue in the β1-loop-α1 bridges the 3′- and 5′-phosphates of PAP, whereas a conserved serine residue in the same loop forms a hydrogen bond to N7 of the adenine (Fig. 3A). Separate conserved arginine and serine residues from the β4-loop-α6 interact with the negative charge of the 3′-phosphate. The lysine residue that forms a hydrogen bond with a sulfate oxygen in PAPS in mammalian SULTs is replaced by a histidine in all three schistosome enzymes (Table 2 and Fig. 3, A and B).
Figure 3.
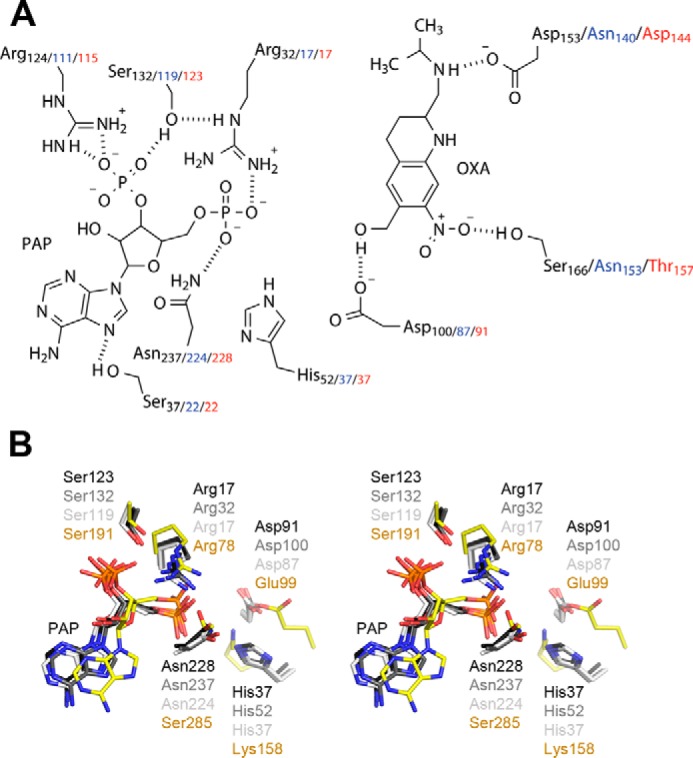
A, schematic of the ShSULT active site (SjSULT residues in blue and SmSULT residues in red) showing oxamniquine (OXA) in position for sulfate transfer from PAPS (PAP shown). Hydrogen bonds are indicated as dotted lines. B, walleyed stereoview of the superposition of TPST2 and schistosome SULT active-site residues. SmSULT, dark gray; ShSULT, medium gray; SjSULT, light gray; TPST2, yellow.
Table 2.
Structurally equivalent active site residues in schistosomal and mammalian SULTs
| TPST2 | SULT1E1 | SmSULT | ShSULT | SjSULT |
|---|---|---|---|---|
| Arg-78 | Lys-47 | Arg-17 | Arg-32 | Arg-17 |
| Thr-81 | Thr-50 | Thr-20 | Thr-35 | Thr-20 |
| Glu-99 | His-107 | Asp-91 | Asp-100 | Asp-87 |
| Lys-158 | Lys-105 | His-37 | His-52 | His-37 |
| Ser-191 | Ser-137 | Ser-123 | Ser-132 | Ser-119 |
| Arg-183 | Arg-129 | Arg-115 | Arg-124 | Arg-111 |
| Ser-285 | Asn-228 | Asn-237 | Asn-224 |
Structures of S. hematobium sulfotransferase with oxamniquine plus PAP
To investigate the interaction of oxamniquine with ShSULT, the ShSULT–PAP crystals were soaked overnight with the individual stereoisomers or racemic oxamniquine. Difference electron density maps (Fo − Fc) clearly indicated the presence of oxamniquine in the predicted substrate-binding region in all cases. In the crystals soaked with (S)-oxamniquine, the drug is in a similar position to that seen in the SmSULT–PAP–(S)-oxamniquine complex (PDB code 5BYK) (Fig. 4A). ShSULT Asp-100 accepts a hydrogen bond from the putative reactive hydroxyl group. Ser-166 donates a hydrogen bond to the nitro group, and Asp-153 accepts a hydrogen bond from the amine in the isopropylaminomethyl group of oxamniquine. In the crystals soaked with the (R)-oxamniquine, the drug is in a position that can be described as a possibly unproductive binding mode. It is oriented similarly along the longest axis of the molecule when compared with (S)-oxamniquine, but its hydroxyl group is not in hydrogen-bonding distance to Asp-100. Rather, an intervening solvent molecule is positioned in hydrogen-bonding distance to both Asp-100 and the oxamniquine hydroxyl group (Fig. 4B). In both complexes, the pucker of the oxamniquine piperidine ring differs from that seen in SmSULT. In the (S)-oxamniquine and (R)-oxamniquine complexes of ShSULT, carbon 3 is on the same side of the piperidine plane as the isopropylaminomethyl group. In both complexes of SmSULT, carbon 3 is on the opposite side of the plane (supplemental Fig. 2).
Figure 4.
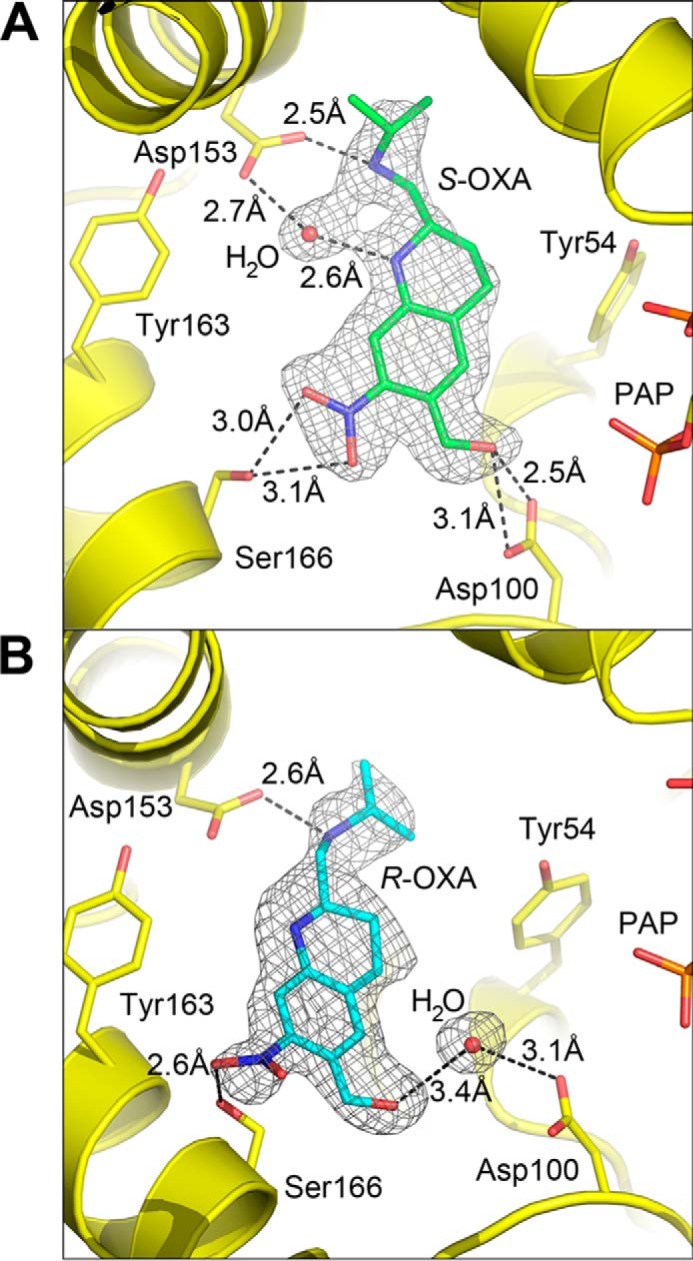
A, composite omit map (2mFo − DFc) calculated for the ShSULT–PAP–(S)-oxamniquine (OXA) complex contoured at 1. 0 σ. B, composite omit map (2mFo −DFc) calculated for the ShSULT–PAP–(R)-oxamniquine complex contoured at 1.0 σ.
Oxamniquine is a racemic mixture in the formulation given as treatment to patients, although the S-stereoisomer has higher potency against S. mansoni in vivo (16). When racemic oxamniquine is soaked into crystals of SmSULT, the active site contains only the S-isomer. The ShSULT–PAP–racemic oxamniquine complex contains two molecules per asymmetric unit that superimpose with an r.m.s.d. of 0.5 Å. The active sites in both chains superimpose closely with the exceptions of Tyr-54, Ser-166, and the oxamniquine molecule. In chain B, both Tyr-54 and Ser-166 have similar positions to those of their counterparts in SmSULT, Phe-39 and Thr-157 (Fig. 5A). Oxamniquine also binds in a position similar to that seen in SmSULT, and the electron density clearly shows that only (S)-oxamniquine is bound in ShSULT. In chain A, Tyr-54, Ser-166, and oxamniquine are in two conformations (Fig. 5B), and all refine to nearly 0.5 occupancy. For Tyr-54, one position is shifted away from the central cavity, and the other matches the single position adopted in chain B. One position of the side chain of Ser-166 matches its position in chain B, allowing formation of a hydrogen bond with the nitro moiety of oxamniquine. In the other position, the side chain is rotated so that the hydroxyl faces away from the oxamniquine molecule. For oxamniquine, one conformation matches the position of oxamniquine observed in chain B and in the crystal structure of ShSULT complexed with pure (S)-oxamniquine, but the other conformation is slightly rotated around an axis perpendicular to the piperidine plane. To summarize, in chain A, Tyr-54 positioned “in” toward the substrate pocket with the Ser-166 Oγ positioned “out” mimics the S. mansoni-like oxamniquine position, whereas Tyr-54 out and Ser-166 in promote an alternate oxamniquine position. Comparison with the ShSULT–PAP–(R)-oxamniquine and ShSULT–PAP–(S)-oxamniquine structures suggests that both oxamniquine conformations seen in chain A contain only (S)-oxamniquine. The electron density does not support modeling an (R)-oxamniquine as one of the two conformations depicted in Fig. 5B.
Figure 5.
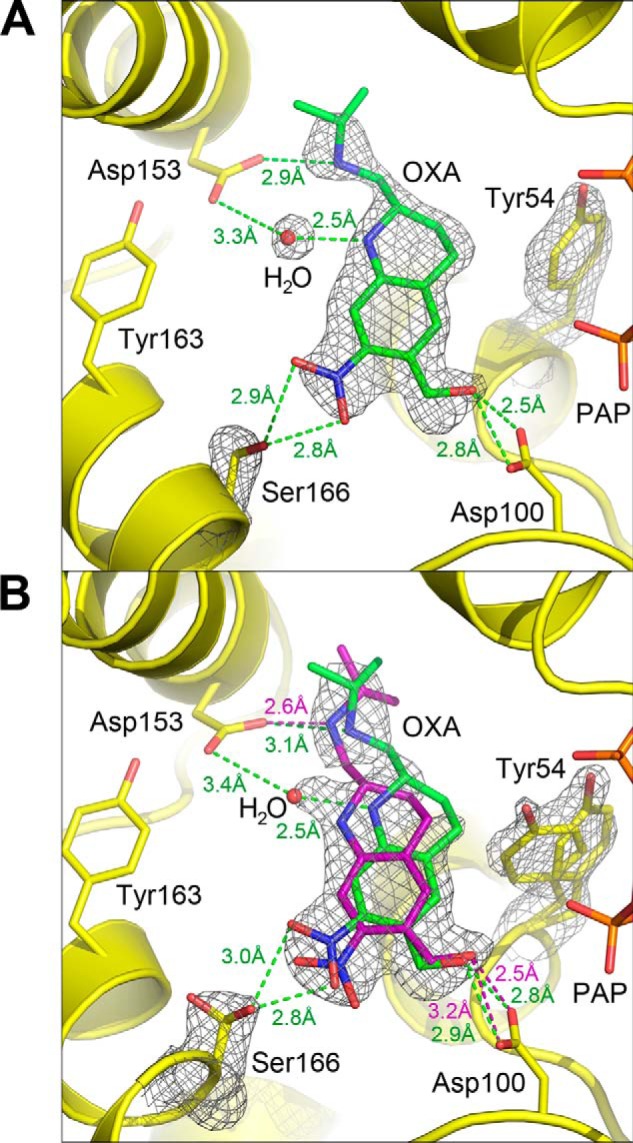
Crystal structure of ShSULT soaked with racemic oxamniquine. A, chain B shows a single conformation for bound (S)-oxamniquine. A composite omit map (2mFo − DFc) contoured at 1.0 σ is superimposed on the atoms shown. B, oxamniquine (OXA) binds in the ShSULT active site in two positions in chain A determined by the alternate conformations of Tyr-54 and Ser-166. A composite omit map (2mFo − DFc) contoured at 1.0 σ is superimposed on the atoms shown. Oxamniquine alternate confirmations and corresponding hydrogen bonding interactions are indicated in green and purple. A solvent atom is positioned between Asp-153 and oxamniquine when the alternate conformation shown in green is present (compare with A).
Schistosome sulfotransferase sulfation kinetics
The specific activities of all three recombinant enzymes were determined using the common sulfotransferase substrate quercetin (17). With this flavanol, SmSULT and SjSULT have similar specific activities, whereas ShSULT has a higher specific activity by a factor of 0.3 (Table 3). Alanine substitutions of the putative catalytic base in SmSULT (D91A) or ShSULT (D100A) decrease the activity of the enzymes 5- and 11-fold compared with the wild-type enzymes, respectively.
Table 3.
Activities of schistosomal SULTs with quercetin as substrate
The following conditions were used: 10 μm quercetin, 0.05 μm inositol monophosphatase 3, 50 mm MOPS, pH 7.0, 15 mm MgCl2 at 37 °C. Errors are standard deviations calculated for four independent experiments.
| Enzyme | Specific activity |
|---|---|
| mmol product/mmol enzyme/min | |
| Wild-type SmSULT | 3.5 ± 0.4 |
| D91A SmSULT | 0.71 ± 0.11 |
| Wild-type ShSULT | 4.6 ± 0.4 |
| D100A ShSULT | 0.41 ± 0.01 |
| Wild-type SjSULT | 3.5 ± 0.08 |
To determine which oxamniquine stereoisomer is preferred by each of the enzymes, kcat/Km values were determined. In preliminary analyses with the racemic mixtures, significant substrate inhibition, frequently observed with sulfotransferases (18–21), was seen with SmSULT and ShSULT but not SjSULT. This precluded determining reliable kcat and Km values for these two enzymes. For SmSULT, the highest activity with racemic oxamniquine of 9 min−1 was at 6–7 μm; ShSULT had the highest activity with racemic oxamniquine, 16 min−1, at ∼60 μm; the kcat and Km values for SjSULT with the racemic substrate were 1.2 ± 0.2 min−1 and 50 ± 19 μm. It was still possible to determine kcat/Km values in all cases by determining initial rates at low concentrations of each stereoisomer of oxamniquine (Table 4). The results show significant differences among the abilities of the three enzymes to utilize the two isomers of oxamniquine as substrates. SmSULT does not show a significant preference for either stereoisomer. In contrast, (S)-oxamniquine is a better substrate by an order of magnitude for ShSULT, whereas SjSULT shows about a 3-fold preference for (R)-oxamniquine.
Table 4.
Steady-state kinetic parameters for schistosomal SULTs with oxamniquine (OXA) as substrate
The following conditions were used: 0.05 μm inositol monophosphatase 3, 50 mm MOPS, pH 7.0, 15 mm MgCl2 at 37 °C.
| Enzyme |
kcat/Km |
|
|---|---|---|
| (S)-OXA | (R)-OXA | |
| μm−1 min−1 | ||
| SmSULT | 1.7 ± 0.4 | 1.3 ± 0.3 |
| ShSULT | 0.89 ± 0.22 | 0.067 ± 0.008 |
| SjSULT | 0.0071 ± 0.0016 | 0.021 ± 0.004 |
Characterization of the product of oxamniquine sulfation by schistosome SULTs
The sulfated product of the SULT reaction with oxamniquine is proposed to rapidly eliminate sulfate to form a highly reactive ethylene moiety (17, 22). Continuous-flow mass spectrometry was used to identify the product of the SULT reaction. Recombinant SmSULT in a volatile buffer was mixed with a solution containing PAPS and oxamniquine, and the mixture was injected into an OrbiTrap mass spectrometer. After a reaction of 6 s, the resulting mass spectrum showed a clear signal at m/z 280.1658 that corresponds to the [M + H]+ ion of oxamniquine (C14H22N3O3) (Fig. 6A). A smaller signal (1) was also detectable at m/z 382.1043. This mass is consistent with the formula C14H21N3O6SNa (predicted m/z, 382.1049) and can thus be identified as a sulfated oxamniquine with an associated sodium ion. This ion was not detectable when PAPS or enzyme was omitted from the reaction or when the D91A mutant enzyme was used in a complete reaction (Fig. 6B). The same ion was produced in reactions with recombinant ShSULT (Fig. 6C). Thus, the enzymes from both sources are able to catalyze the transfer of sulfate from PAPS to oxamniquine to produce a sulfated product. No ion corresponding to the proposed ethylene product was detected.
Figure 6.

Continuous-flow mass spectra of SULT reactions after 6 s. A, mass spectrum of the reaction of 10 μm SmSULT with 20 μm oxamniquine (OXA) and 50 μm PAPS in 50 mm ethanediamine acetate, 12.5 mm ammonium acetate, 5% methanol, pH 8.0. B, mass spectrum of the reaction of 10 μm SmSULT D91A with 20 μm oxamniquine and 50 μm PAPS in 50 mm ethanediamine acetate, 12.5 mm ammonium acetate, 5% methanol, pH 8.0. C, mass spectrum of the reaction of 10 μm ShSULT with 20 μm oxamniquine and 50 μm PAPS in 50 mm ethanediamine acetate, 12.5 mm ammonium acetate, 5% methanol, pH 8.0. D, mass spectrum of the reaction of 10 μm SmSULT with 20 μm oxamniquine and 50 μm PAPS in 12.5 mm ammonium acetate, 5% methanol, pH 7.0. The spectra represent averages of 200 scans at a flow rate of 1.0 μl/min at 25 °C. Ion signals are normalized to the most intense ion. E, proposed mechanism for formation of the products of the SULT reaction.
The mass spectra from the reactions of both enzymes also show a major species (2) with m/z 322.2240; expansion of the spectra shows that a second smaller ion (3) with m/z 322.1762 is also produced by both enzymes. Neither ion is formed by the inactive mutant enzyme, establishing that they are products of the SULT reaction. The m/z values of the two ions correspond to ions with the formulae C16H28N5O2 (predicted m/z, 322.2238) and C16H24N3O4 (predicted m/z, 322.1761), respectively. The formation of both 2 and 3 was time-dependent with the ion intensities being 2–3-fold greater at 6 s than at 1.2 s (supplemental Table 1). The formation of 2 can be attributed to nucleophilic displacement of the sulfate from oxamniquine sulfate by ethanediamine in the buffer (Fig. 6E), whereas 3 can be attributed to similar displacement of the sulfate by acetate. Consistent with this proposal, if the SmSULT reaction was carried out in ammonium acetate buffer without ethanediamine, 2 was not detectable, and significantly more oxamniquine sulfate was formed (Fig. 6D). If the ethanediamine was replaced with Tris as buffer at pH 7.0, 2 was similarly not detectable, but a new peak with m/z 383.2291 was seen, and the amount of oxamniquine sulfate increased, consistent with nucleophilic displacement of sulfate from oxamniquine sulfate by Tris (predicted m/z, 383.2289).
Discussion
The SULTs from the three main species responsible for almost all human blood fluke infections have the hallmark characteristics of PAPS-dependent SULTs, including the β strand-loop-α helix PAPS-binding motif. Structurally, the schistosome enzymes are closer to the membrane-bound mammalian tyrosine protein sulfotransferases such as TPST2 than to cytosolic mammalian sulfotransferases such as SULTE1 (Table 2), although they lack obvious sequences for membrane binding. The schistosome SULTs contain five α helices without a counterpart in mammalian SULT structures; these helices enclose the active site and are likely to be important to the specificity of the schistosome enzymes. The enclosed active sites of the schistosome enzymes would be expected to prevent access of residues on protein surfaces in the absence of significant conformational changes, suggesting that the schistosome enzymes sulfate small molecule substrates.
The oxamniquine-binding sites of all three schistosome enzymes are located within cavities surrounded by α helices 2, 6, 7, 8, 11, and 12. The cavities have the capacity to accommodate substrates of various sizes considering the internal volumes of 984, 985, and 1189 Å3 for SmSULT, ShSULT, and SjSULT, respectively (note that the cavity volume for SjSULT is based on the arrangement of the domain-swapped C-terminal helix, α12, in the crystal structure). For comparison, the calculated molecular volume of oxamniquine is 212 Å3. The active sites of the schistosome enzymes are consistent with their utilizing a different set of substrates from the mammalian enzymes (Figs. 2 and 3). The mammalian SULTs have structural elements that would appear to impede productive oxamniquine binding (supplemental Fig. 3), suggesting that oxamniquine is not a substrate. Indeed, a study of metabolism of oxamniquine in several mammals, including humans, showed that the major metabolite excreted in urine has a carboxylate substitution for the hydroxymethyl group, consistent with metabolism by a cytochrome P450 rather than a SULT (23). Thus, the selective toxicity of oxamniquine toward S. mansoni and not humans is likely attributable to the structural variation between the SULT homologs and the availability of an alternative detoxification pathway in humans.
The mechanism of SULTs is proposed to involve nucleophilic attack of the deprotonated substrate hydroxyl group on the sulfur of PAPS, resulting in sulfuryl transfer from PAPS to the substrate (13, 14, 24–26). Mutational analyses of conserved active-site residues in TPST2 support catalytic functions for Arg-78, Glu-99, Lys-158, and Ser-285 (13). Superposition of the structures of SmSULT, ShSULT, and SjSULT onto that of TPST2 identifies the counterparts of these residues in the schistosome enzymes (Fig. 2B and Table 2). All three contain an active-site aspartate in place of Glu-99 appropriately positioned to act as the catalytic base to abstract a proton from the hydroxyl group of oxamniquine and other substrates (Fig. 2, A and B). The finding that substitution of this residue to alanine in SmSULT and ShSULT reduces activity supports this functional assignment. The schistosome enzymes also contain a conserved arginine in the same position as Arg-78 in TPST2. This residue was proposed to donate a proton to the bridging oxygen of the phosphosulfate moiety in PAPS (13), but the computational analysis of Marforio et al. (25) is more consistent with this residue acting as a shuttle for the SO3− as it is transferred from donor to acceptor. The latter role is also more consistent with the high pKa of arginine. The asparagine found in the schistosome enzymes in place of Ser-285 is positioned to play a similar role of stabilizing the transition state by forming a strong hydrogen bond with a non-bridging oxygen on the phosphate (13, 25). Lys-158, proposed to stabilize the transition state, is replaced with histidine in the schistosome SULTs. This residue could readily serve as a positive charge to stabilize the transition state if the imidazole ring were protonated.
When racemic oxamniquine is soaked into crystals of SmSULT, the active site contains only the S-isomer, establishing that this stereoisomer binds more tightly to that enzyme. One possible explanation for oxamniquine inefficacy in S. hematobium is that ShSULT preferentially binds (R)-oxamniquine. However, soaking racemic oxamniquine into ShSULT crystals similarly shows (S)-oxamniquine bound. (R)-Oxamniquine does bind but adopts a potentially unproductive binding mode where its hydroxyl group is separated from the catalytic aspartate by an intervening solvent molecule (Fig. 4B). The propensity for this binding mode may correlate to the observation that kcat/Km for (R)-oxamniquine is an order of magnitude lower than for (S)-oxamniquine as an ShSULT substrate.
One of the two molecules in the asymmetric unit in the ShSULT crystals with racemic oxamniquine shows (S)-oxamniquine bound in more than one conformation, whereas in the other subunit the (S)-oxamniquine position is comparable with the oxamniquine position in the (S)-oxamniquine crystal soak of both SmSULT and ShSULT (Figs. 4A and 5A). The combination of side chain rotamers of Tyr-54 and Ser-166 in ShSULT appears to select the position for bound (S)-oxamniquine (Fig. 5B). In SmSULT, the equivalent residue to Ser-166 is Thr-157, and its methyl group may stabilize the side chain where it borders a hydrophobic patch comprising Leu-92, Val-128, Leu-129, and Leu-161. The combination of a stable Thr-157 rotamer and a Phe-39 unable to donate a hydrogen bond to its neighboring asparagine likely imposes a more rigid active site for oxamniquine binding in SmSULT than in ShSULT. The crystal structures of oxamniquine-bound SmSULT and ShSULT also differ in that ShSULT-bound oxamniquine adopts a piperidine ring pucker opposite to SmSULT-bound oxamniquine (supplemental Fig. 2), although it is not clear that this plays a significant role in oxamniquine affinity.
A number of residues in contact with oxamniquine in the ShSULT–PAP–oxamniquine and SmSULT–PAP–oxamniquine crystal structures are different in SjSULT (supplemental Fig. 4). Although SjSULT still has the aspartate that serves as the active-site base (Asp-87), an asparagine (Asn-140) replaces an aspartate seen in the other two enzymes (Asp-153 in ShSULT and Asp-144 in SmSULT). In addition, SjSULT contains Asn-153 in place of Ser-166 (ShSULT) and Thr-157 (SmSULT). It is not clear that the two asparagine substitutions would prevent oxamniquine binding to SjSULT as the asparagine side chain is still capable of forming a hydrogen bond with the oxamniquine nitro and amine groups. The nearby substitution of Val-139 for glycine (SmSULT Gly-143 and ShSULT Gly-152) likely has a more significant impact on binding. The additional bulk of SjSULT Val-139 appears to create steric clash with the isopropylaminomethyl group of oxamniquine according to structural superpositions with SmSULT–PAP–oxamniquine (supplemental Fig. 4) and ShSULT–PAP–oxamniquine complexes.
The continuous-flow mass spectral analyses establish directly that schistosome SULTs catalyze a sulfotransferase reaction with oxamniquine as substrate. The observation of additional products resulting from nucleophilic displacement of sulfate by buffer components is consistent with the oxamniquine sulfate generated by the enzymes being highly susceptible to nucleophilic attack. Oxamniquine has been proposed to exert its schistosomicidal activity upon the decay of the sulfated product of the SULT reaction to a reactive ethylene with toxic alkylating activity within the parasite (17, 22). Assays using tritiated oxamniquine show the formation of labeled macromolecules in worm extracts, consistent with activation of oxamniquine by a SULT generating a reactive species (10, 17, 22). However, the variation in the amount of oxamniquine sulfate when the buffer is changed is more consistent with an SN2-like mechanism in which a nucleophile displaces the sulfate in a concerted reaction than with an SN1-like mechanism in which loss of sulfate generates a discrete ethylene intermediate that is subsequently attacked by a nucleophile. Thus, the present data suggest an alternative hypothesis for oxamniquine toxicity in which macromolecules in the parasite participate directly in the nucleophilic displacement of sulfate from activated oxamniquine.
The kinetic analyses confirm the activity of all three schistosome enzymes with oxamniquine as substrate and allow direct comparison of their enzymatic activities with oxamniquine. Based on the kcat/Km values, SmSULT has the highest activity with oxamniquine as substrate and shows no significant preference for either stereoisomer. ShSULT is about one-half as active as SmSULT with (S)-oxamniquine but has much lower activity with the R-isomer. SjSULT is less active with oxamniquine as substrate by about an order of magnitude compared with SmSULT and shows a preference for the R-isomer. These differences may account for the efficacy of oxamniquine as a drug against S. mansoni infection and its failure against S. hematobium and S. japonicum.
The reduction in ShSULT catalytic efficiency toward oxamniquine may be related to its ability to bind oxamniquine in multiple positions (Figs. 4 and 5) because the active sites of the two enzymes are quite similar. In the case of SjSULT, the substitution of Gly-143 with Val-139 and the substitution of Asp-153 with Asn-140 (supplemental Fig. 4) provide a structural rationale for the significant decrease in catalytic efficiency toward oxamniquine. If this is correct, small modifications in the structure of oxamniquine that avoid the clash with Val-139 may generate a more effective drug.
In summary, the present results provide a rationale for the lack of efficacy of oxamniquine with S. hematobium and S. japonicum and provide structural and kinetic information that will be critical to develop oxamniquine derivatives with efficacy against these two species. We speculate that efficient turnover of oxamniquine in S. mansoni creates a large enough pool of toxic product to kill the blood fluke, whereas oxamniquine is not effective against S. hematobium and S. japonicum due to its diminished turnover. The finding that ShSULT is almost as active as SmSULT with a very similar active site suggests that small structural changes to oxamniquine may be sufficient to develop an improved derivative. In addition, we have identified a possible structural basis for the low enzymatic activity of SjSULT with oxamniquine that will be useful in developing a derivative targeting that enzyme. The structures of all three schistosome SULTs show that their active sites are similar to but not identical to their mammalian counterparts, providing a rationale for the different effects in humans and worms. On the whole, the crystal structures of the schistosome SULTs and the evidence for in vitro oxamniquine binding and turnover in both ShSULT and SjSULT support the initiative for developing an oxamniquine derivative as a pan-species antischistosomal drug.
Experimental procedures
Cloning, expression, and purification
DNA encoding the ShSULT gene was subcloned into the pAG8H vector derived from pKM260 (27), which contains an inducible lacZ promoter and an N-terminal His8 tag that can be released from the target protein via a tobacco etch virus (TEV) protease cleavage site. The plasmid was transformed into Escherichia coli strain BL21 pLysS (Promega, Madison, WI), and the cells were grown at 37 °C. When the A600 reached 0.7, the temperature was decreased to 18 °C, and expression was induced by the addition of isopropyl β-d-thiogalactoside to a final concentration of 1 mm. Cells from 6 liters of cell culture were washed and resuspended in 50 ml of 50 mm Tris, pH 8.0, 500 mm NaCl (column buffer) containing 250 μl of Sigma protease inhibitor mixture (Sigma-Aldrich)/liter of cultured cells and lysed by sonication on ice. The full-length ShSULT localized in inclusion bodies, and little soluble protein was expressed. Sequence analysis indicated that ShSULT contained an N-terminal extension compared with SmSULT; thus, to attempt to alleviate the solubility problem, a more compact ShSULT construct was designed with 16 residues truncated at the N terminus. The truncated ShSULT was successfully overexpressed and localized in the soluble fraction. The clarified supernatant was loaded onto a Ni2+-nitrilotriacetic acid affinity chromatography column (GE Healthcare), washed with 5 volumes of column buffer, and eluted using a 10–500 mm imidazole gradient. Fractions identified as ShSULT by SDS-PAGE were pooled, and His-tagged TEV protease was added in a protein to protease ratio of 15:1. The resulting solution was dialyzed overnight at 4 °C against 50 mm Tris, pH 8.0, 100 mm NaCl, 2 mm dithiothreitol. The dialysate was passed over the Ni2+-nitrilotriacetic acid column again to remove the His tag and the His-tagged TEV protease, and the cleaved target protein flowed through. The sample was loaded onto a GE Healthcare prepacked Q anion-exchange column and eluted with a 0.1–1.0 m NaCl gradient. Fractions containing ShSULT as identified by SDS-PAGE were pooled and dialyzed overnight at 4 °C in 25 mm Tris, pH 8.5, 50 mm NaCl, 2 mm tris(carboxyethyl)phosphine. The product was ∼98% pure as estimated by SDS-PAGE. When attempts to produce sufficient soluble N-terminal His8-tagged SjSULT failed, DNA encoding the SjSULT gene was subcloned into the pNC8H vector derived from pKM260, which contains an inducible lacZ promoter and a non-cleavable C-terminal His8 tag with a two-residue Gly-Ser linker. SjSULT was prepared as described above for ShSULT omitting the affinity tag cleavage step. Purified ShSULT and SjSULT proteins were concentrated to 10 mg/ml for crystallization trials. PAP was added in a 4:1 stoichiometric ratio over protein, and the sample was incubated for 1 h on ice immediately prior to crystallization screening. The D100A mutation was introduced into the pAG8H-ShSULT plasmid using the Phusion (Thermo Fisher Scientific, Waltham, MA) site-directed mutagenesis protocol, and the protein was prepared as described above. The D91A mutation was introduced into the pAG8H-SmSULT vector in the same way, and wild-type SmSULT and SmSULT D91A proteins were prepared as described previously (10).
Crystallization, structure determination, and refinement
Automated screening for crystallization was carried out using the sitting-drop vapor-diffusion method with an Art Robbins Instruments (Sunnyvale, CA) Phoenix system in the X-ray Crystallography Core Laboratory at the University of Texas Health Science Center at San Antonio. ShSULT–PAP crystals were obtained from the Molecular Dimensions (Suffolk, UK) Morpheus screen condition number 1-12. ShSULT–PAP–oxamniquine complex crystals were prepared by soaking PAP-containing crystals overnight in the mother liquor saturated with racemic oxamniquine or the individual (R)- and (S)-oxamniquine enantiomers prepared as described previously (16). SjSULT–PAP crystals were obtained from the Rigaku (Tokyo, Japan) Precipitant Synergy 2 screen condition number 151. The SjSULT–PAP crystals grew as polycrystalline clusters with an epitaxially twinned core branching into single crystal rods, and only an isolated rod produced a useful single crystal diffraction pattern. Attempts to soak oxamniquine into isolated SjSULT–PAP crystals returned damaged crystals and unproductive X-ray diffraction. All crystals were mounted in undersized nylon loops with excess mother liquor wicked off and flash-cooled in liquid nitrogen prior to data collection. The structures of ShSULT and SjSULT were determined by the molecular replacement method implemented in Phaser (28) using the SmSULT structure (PDB code 4MUA) as the search model. Coordinates for all models were refined using PHENIX (29), including simulated annealing with torsion angle dynamics, and alternated with manual rebuilding using Coot (30). The occupancies for oxamniquine coordinates were held at 1.0 during refinement with the exception of the racemic oxamniquine soaked into ShSULT crystals where occupancies for alternate conformations were refined independently. Data were collected using our home source Rigaku MicroMax 007-HF equipped with R-AXIS-HTC detectors and at the Advanced Photon Source Northeastern Collaborative Access Team (NE-CAT) beamlines 24-ID-C and 24-ID-E and integrated and scaled using XDS (31). Data collection and refinement statistics are shown in Table 1. Structural pocket volume calculations were performed using the Computed Atlas of Surface Topography of Proteins (CASTp) server (32). Figures were generated using PyMOL (Schrödinger, LLC), TopDraw (33), and ChemDraw (PerkinElmer Life Sciences).
Sulfotransferase assay
Standard assay conditions were 10 μm quercetin, 0.05 μm inositol monophosphatase 3, 50 mm MOPS, pH 7.0, 15 mm MgCl2 at 37 °C. Protein concentrations in the assays were 0.2 μm for SmSULT, ShSULT, and SjSULT and 1.2 μm for SmSULT D91A and ShSULT D100A. Assays were started by adding 25 μl of 100 μm PAPS in MOPS/MgCl2 buffer to 25 μl of the reaction mixture. For standard assays, 10-μl aliquots were taken at 5-min intervals and quenched with 6 μl of malachite green reagent A from the Universal Sulfotransferase Activity kit (R&D Systems Inc., Minneapolis, MN). Water (20 μl) and 6 μl of malachite green reagent B were then added; after 20 min, the absorbance at 620 nm was determined. To obtain the kcat/Km values for oxamniquine stereoisomers, the concentration of each was varied over a concentration range where the rate was linearly dependent on the substrate concentration (1–5 μm for SmSULT and 10–50 μm for ShSULT and SjSULT). Aliquots were taken from the reactions each minute for 4 min to determine the initial rate of product formation. Protein concentrations in the assays were 0.1–0.2 μm for SmSULT and ShSULT and 0.8–1.2 μm for SjSULT. All assays were corrected for background hydrolysis of PAPS based on controls from which enzyme was omitted. The respective kcat/Km values were determined from weighted linear fits of the initial rates as a function of substrate concentration. Data from multiple experiments were combined to obtain the values in Table 4.
Continuous-flow mass spectrometry
The products of the reaction of SmSULT and ShSULT with oxamniquine were detected by continuous-flow mass spectrometry using a Thermo Scientific LTQ OrbiTrap Discovery interfaced with a New Objectives (Woburn, MA) PicoView nanospray source and a Peltier-controlled nanovolume continuous-flow mixer (Eksigent, Dublin, CA) (34). A solution of 20 μm SULT in buffer in one line was continuously mixed with an equal volume of 40 μm oxamniquine, 100 μm PAPS, 10% methanol, 5 mm ammonium acetate, pH 7.0, from a second line at 25 °C and injected into the mass spectrometer after 1.2 or 6 s. Reactions were monitored in positive-ion spectral mode from m/z 110 to 600.
Author contributions
A. B. T., K. M. R., N. E. C., P. T. L., P. F. F., and P. J. H. conceived and designed the experiments. A. B. T, K. M. R., X. C., S. P. H., E. D., C. M. P., L. P.-M., D. C., R. S. T., and S. F. M. performed the experiments and analyzed the data. A. B. T., P. T. L., P. F. F., and P. J. H. analyzed the data and wrote the paper. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
Support for the X-ray Crystallography Core Laboratory by the University of Texas Health Science Center at San Antonio Office of the Vice President for Research and the Cancer Therapy and Research Center (National Institutes of Health Grant P30 CA054174) is gratefully acknowledged. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by National Institute of General Medical Sciences, National Institutes of Health Grant P41 GM103403. The Pilatus 6M detector on 24-ID-C beam line is funded by National Institutes of Health Office of Research Infrastructure Programs High-End Instrumentation Grant S10 RR029205. This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357.
This work was supported by National Institutes of Health Grant R01AI115691 (to P. J. H. and P. T. L.) and Welch Foundation Grants AQ-1245 (to P. F. F.) and AQ-1399 (to P. J. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5TIV, 5TIW, 5TIX, 5TIY, and 5TIZ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains supplemental Figs. 1–4 and Table 1.
- SULT
- sulfotransferase
- PAP
- 3′-phosphoadenosine 5′-phosphate
- PAPS
- 3′-phosphoadenosine 5′-phosphosulfate
- ShSULT
- S. haematobium sulfotransferase
- SjSULT
- S. japonicum sulfotransferase
- SmSULT
- S. mansoni sulfotransferase
- r.m.s.d.
- root mean square deviation
- TPST2
- tyrosylprotein sulfotransferase 2
- TEV
- tobacco etch virus.
References
- 1. World Health Organization (2015) http://www.who.int/mediacentre/factsheets/fs115/en
- 2. Hotez P. J., Bundy D. A. P., Beegle K., Brooker S., Drake L., de Silva N., Montresor A., Engels D., Jukes M., Chitsulo L., Chow J., Laxminarayan R., Michaud C., Bethony J., Correa-Oliveira R., et al. (2006) Helminth infections: soil-transmitted helminth infections and schistosomiasis, in Disease Control Priorities in Developing Countries (Jamison D. T., Breman J. G., Measham A. R., Alleyne G., Claeson M., Evans D. B., Jha P., Mills A., and Musgrove P., eds) pp. 467–482, 2nd Ed., The World Bank, Washington, D. C. [PubMed] [Google Scholar]
- 3. Centers for Disease Control and Prevention (2012) http://www.cdc.gov/parasites/schistosomiasis
- 4. Greenberg R. M. (2013) New approaches for understanding mechanisms of drug resistance in schistosomes. Parasitology 140, 1534–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beck L., Favre T. C., Pieri O. S., Zani L. C., Domas G. G., and Barbosa C. S. (2001) Replacing oxamniquine by praziquantel against Schistosoma mansoni infection in a rural community from the sugar-cane zone of Northeast Brazil: an epidemiological follow-up. Mem. Inst. Oswaldo Cruz 96, (suppl.) 165–167 [DOI] [PubMed] [Google Scholar]
- 6. Cioli D., Pica-Mattoccia L., and Moroni R. (1992) Schistosoma mansoni: hycanthone/oxamniquine resistance is controlled by a single autosomal recessive gene. Exp. Parasitol. 75, 425–432 [DOI] [PubMed] [Google Scholar]
- 7. Gentile R., and Oliveira G. (2008) Brazilian studies on the genetics of Schistosoma mansoni. Acta Trop. 108, 175–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Katz N., Dias E. P., Araujo N., and Souza C. P. (1973) Study of a human strain of Schistosoma mansoni resistant to schistosomicidal agents. Rev. Soc. Brasil. Med. Trop. 7, 381–387 [Google Scholar]
- 9. Rogers S. H., and Bueding E. (1971) Hycanthone resistance: development in Schistosoma mansoni. Science 172, 1057–1058 [DOI] [PubMed] [Google Scholar]
- 10. Valentim C. L., Cioli D., Chevalier F. D., Cao X., Taylor A. B., Holloway S. P., Pica-Mattoccia L., Guidi A., Basso A., Tsai I. J., Berriman M., Carvalho-Queiroz C., Almeida M., Aguilar H., Frantz D. E., et al. (2013) Genetic and molecular basis of drug resistance and species-specific drug action in schistosome parasites. Science 342, 1385–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., and Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 13. Teramoto T., Fujikawa Y., Kawaguchi Y., Kurogi K., Soejima M., Adachi R., Nakanishi Y., Mishiro-Sato E., Liu M. C., Sakakibara Y., Suiko M., Kimura M., and Kakuta Y. (2013) Crystal structure of human tyrosylprotein sulfotransferase-2 reveals the mechanism of protein tyrosine sulfation reaction. Nat. Commun. 4, 1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kakuta Y., Pedersen L. G., Carter C. W., Negishi M., and Pedersen L. C. (1997) Crystal structure of estrogen sulphotransferase. Nat. Struct. Biol. 4, 904–908 [DOI] [PubMed] [Google Scholar]
- 15. Kakuta Y., Pedersen L. G., Pedersen L. C., and Negishi M. (1998) Conserved structural motifs in the sulfotransferase family. Trends Biochem. Sci. 23, 129–130 [DOI] [PubMed] [Google Scholar]
- 16. Taylor A. B., Pica-Mattoccia L., Polcaro C. M., Donati E., Cao X., Basso A., Guidi A., Rugel A. R., Holloway S. P., Anderson T. J., Hart P. J., Cioli D., and LoVerde P. T. (2015) Structural and functional characterization of the enantiomers of the antischistosomal drug oxamniquine. PLoS Negl. Trop. Dis. 9, e0004132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pica-Mattoccia L., Carlini D., Guidi A., Cimica V., Vigorosi F., and Cioli D. (2006) The schistosome enzyme that activates oxamniquine has the characteristics of a sulfotransferase. Mem. Inst. Oswaldo Cruz 101, Suppl. 1, 307–312 [DOI] [PubMed] [Google Scholar]
- 18. Gulcan H. O., and Duffel M. W. (2011) Substrate inhibition in human hydroxysteroid sulfotransferase SULT2A1: studies on the formation of catalytically non-productive enzyme complexes. Arch. Biochem. Biophys. 507, 232–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stjernschantz E., Reinen J., Meinl W., George B. J., Glatt H., Vermeulen N. P., and Oostenbrink C. (2010) Comparison of murine and human estrogen sulfotransferase inhibition in vitro and in silico—implications for differences in activity, subunit dimerization and substrate inhibition. Mol. Cell. Endocrinol. 317, 127–140 [DOI] [PubMed] [Google Scholar]
- 20. Lu L. Y., Hsieh Y. C., Liu M. Y., Lin Y. H., Chen C. J., and Yang Y. S. (2008) Identification and characterization of two amino acids critical for the substrate inhibition of human dehydroepiandrosterone sulfotransferase (SULT2A1). Mol. Pharmacol. 73, 660–668 [DOI] [PubMed] [Google Scholar]
- 21. Barnett A. C., Tsvetanov S., Gamage N., Martin J. L., Duggleby R. G., and McManus M. E. (2004) Active site mutations and substrate inhibition in human sulfotransferase 1A1 and 1A3. J. Biol. Chem. 279, 18799–18805 [DOI] [PubMed] [Google Scholar]
- 22. Cioli D., Pica-Mattoccia L., and Archer S. (1995) Antischistosomal drugs: past, present, and future? Pharmacol. Ther. 68, 35–85 [DOI] [PubMed] [Google Scholar]
- 23. Kaye B., and Woolhouse N. M. (1976) The metabolism of oxamniquine—a new schistosomicide. Ann. Trop. Med. Parasitol. 70, 323–328 [DOI] [PubMed] [Google Scholar]
- 24. Kakuta Y., Petrotchenko E. V., Pedersen L. C., and Negishi M. (1998) The sulfuryl transfer mechanism. Crystal structure of a vanadate complex of estrogen sulfotransferase and mutational analysis. J. Biol. Chem. 273, 27325–27330 [DOI] [PubMed] [Google Scholar]
- 25. Marforio T. D., Giacinto P., Bottoni A., and Calvaresi M. (2015) Computational evidence for the catalytic mechanism of tyrosylprotein sulfotransferases: a density functional theory investigation. Biochemistry 54, 4404–4410 [DOI] [PubMed] [Google Scholar]
- 26. Teramoto T., Sakakibara Y., Liu M. C., Suiko M., Kimura M., and Kakuta Y. (2009) Snapshot of a Michaelis complex in a sulfuryl transfer reaction: crystal structure of a mouse sulfotransferase, mSULT1D1, complexed with donor substrate and accepter substrate. Biochem. Biophys. Res. Commun. 383, 83–87 [DOI] [PubMed] [Google Scholar]
- 27. Melcher K. (2000) A modular set of prokaryotic and eukaryotic expression vectors. Anal. Biochem. 277, 109–120 [DOI] [PubMed] [Google Scholar]
- 28. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., and Liang J. (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, W116–W118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bond C. S. (2003) TopDraw: a sketchpad for protein structure topology cartoons. Bioinformatics 19, 311–312 [DOI] [PubMed] [Google Scholar]
- 34. Roberts K. M., Tormos J. R., and Fitzpatrick P. F. (2014) Characterization of unstable products of flavin- and pterin-dependent enzymes by continuous-flow mass spectrometry. Biochemistry 53, 2672–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.