

Abstract
The dopamine transporter (DAT) regulates dopamine (DA) neurotransmission by recapturing DA into the presynaptic terminals and is a principal target of the psychostimulant cocaine. The sigma-1 receptor (σ1R) is a molecular chaperone, and its ligands have been shown to modulate DA neuronal signaling, although their effects on DAT activity are unclear. Here, we report that the prototypical σ1R agonist (+)-pentazocine potentiated the dose response of cocaine self-administration in rats, consistent with the effects of the σR agonists PRE-084 and DTG (1,3-di-o-tolylguanidine) reported previously. These behavioral effects appeared to be correlated with functional changes of DAT. Preincubation with (+)-pentazocine or PRE-084 increased the Bmax values of [3H]WIN35428 binding to DAT in rat striatal synaptosomes and transfected cells. A specific interaction between σ1R and DAT was detected by co-immunoprecipitation and bioluminescence resonance energy transfer assays. Mutational analyses indicated that the transmembrane domain of σ1R likely mediated this interaction. Furthermore, cysteine accessibility assays showed that σ1R agonist preincubation potentiated cocaine-induced changes in DAT conformation, which were blocked by the specific σ1R antagonist CM304. Moreover, σ1R ligands had distinct effects on σ1R multimerization. CM304 increased the proportion of multimeric σ1Rs, whereas (+)-pentazocine increased monomeric σ1Rs. Together these results support the hypothesis that σ1R agonists promote dissociation of σ1R multimers into monomers, which then interact with DAT to stabilize an outward-facing DAT conformation and enhance cocaine binding. We propose that this novel molecular mechanism underlies the behavioral potentiation of cocaine self-administration by σ1R agonists in animal models.
Keywords: conformational change, dopamine transporter, ligand-binding protein, oligomerization, sigma receptor, Sigma-1 receptor, cocaine binding, cocaine self-administration, cysteine accessibility
Introduction
Upon synaptic release of dopamine (DA),2 the dopamine transporter (DAT) recaptures DA into presynaptic terminals and regulates the intensity and duration of DA neurotransmission (1, 2). The DAT is a principal target of the abused psychostimulants, cocaine and methamphetamine. The abuse of and addiction to these drugs stem from their ability to inhibit DAT and elevate extracellular DA levels. Extensive characterization has shown that DAT function can be regulated by posttranslational modifications such as phosphorylation and by multiple interacting proteins (3–6). Recent breakthroughs in the X-ray crystal structure of the Drosophila DAT reveal that cocaine occupies the substrate-binding pocket in DAT while trapping the transporter in an outward-facing conformation (7).
The sigma receptor (σR) was initially proposed as a subtype of opioid receptors (8). After the molecular cloning of the sigma-1 receptor (σ1R) subtype as a 25-kDa membrane protein (9), studies from several groups have shown that it is a molecular chaperone that can interact with and modulate the activity of a variety of client proteins, including ankyrin (10), potassium channels (11, 12), BiP (13), DA D1 and D2 receptors (14, 15), and opioid receptors (16). The crystal structure of σ1R has recently been solved, showing a homotrimer with each protomer containing a single transmembrane domain (TM) and a cytoplasmic domain mediating ligand-binding and subunit multimerization (17).
σRs were shown to regulate midbrain DA neuronal firing and modulate DA release in earlier pharmacological studies (18, 19). Several σR ligands have been explored for their therapeutic potential in treating stimulant abuse (20, 21). A recent behavioral study showed that σ1R agonists such as PRE-084 and 1,3-di-o-tolylguanidine (DTG), but not σ1R antagonists, increased the potency of cocaine in a self-administration procedure (22). Furthermore, cocaine self-administration experience induced self-administration of σ1R agonists, an effect not likely mediated by changes of DA receptors (23, 24). In the present study we investigated the effects of σ1R ligands on DAT function using behavioral, pharmacological, and biochemical methods. We present several lines of evidence supporting the hypothesis that interaction of σ1R with DAT modulates the outward-facing conformation of DAT and facilitates cocaine binding to DAT. This molecular mechanism underscores a novel modulatory role of σ1R on DA neurotransmission and suggests therapeutic potentials of σ1R ligands in treating cocaine addiction.
Results
Rats with indwelling venous catheters were trained to self-administer cocaine using an established procedure in which each fifth press on a lever produced an injection. Lever-press responding maintained by cocaine injections was similar to that reported previously with cocaine or other more conventional reinforcers under similar schedules. A brief pause was followed by a sequence of five responses made in rapid succession producing the injection (Fig. 1A; top panel, see the inset for a magnified display of the micro-temporal pattern). Few if any responses were emitted on the inactive lever (vertical marks on the line below cumulative curve) or during the 2-min timeout periods (lower event line displaced upward) between successive components. In the extinction (EXT) component no injections were delivered, and response rates were low. As the dose of cocaine increased in successive components, response rates also increased. The highest rate of responding was obtained in the fourth component in which injections of 0.32 mg/kg were available (Fig. 1A, top record). When saline injections were available in the second through fifth components (data not shown), responses were never emitted at rates greater than those maintained in EXT. The average response rates were a bell-shaped function of cocaine dose (Fig. 1B, filled symbols). The maximum response rate averaging 0.339 (± S. E. 0.073) responses/s was obtained with a dose of 0.32 mg/kg/injection and was significantly greater than the rates averaging 0.023 (± S. E. 0.003) responses/s during EXT.
Figure 1.

The σ1R agonist (+)-pentazocine dose-dependently potentiated cocaine self-administration in rats. A, representative cumulative records of an individual subject responding showing patterns of self-administration in real time maintained by intravenous cocaine when each fifth-response produced an injection (fixed-ratio, or FR 5-response schedule). Ordinates show cumulative responses. Abscissae show time. The five 20-min self-administration components of each session are indicated by a downward displacement of the lowest line below each record. At the end of each component the cumulative-response curve reset to base. The preceding 2-min timeout periods are indicated by the upward position of the lowest line of each record. In the first component, each fifth response turned off the light-emitting diodes for 20 s but did not activate the infusion pump (EXT); in subsequent components, injections were also delivered with each fifth response (diagonal marks on the cumulative record) with doses (in mg/kg/injection) indicated. Vertical marks on the event line below the cumulative curve indicate responses on the left (inactive) lever. The encircled portion of the record in the third component is magnified to better show the temporal pattern of responding. Note the dose-dependent increase in responding up to the cocaine dose of 0.32 mg/kg/injection in the top record and the increases in responding at lower doses of cocaine after pretreatment with 3.2 mg/kg of (+)-pentazocine in the bottom record. In general, (+)-pentazocine affected overall rates without changes in the temporal pattern of responding. B, dose-effect curves of cocaine self-administration and the effect of (+)-pentazocine treatment. Each point represents the mean ± S.E. (n = 6) of response rates maintained during extinction (EXT, responses had no scheduled consequences) or under the FR5 schedule at the indicated cocaine doses/injection. Asterisks indicate significant effects of the doses of i.p. (+)-pentazocine (○, ▵, ▿) compared with saline pretreatment (●) as determined by two-way repeated measures ANOVA and Tukey post-hoc tests (*, p < 0.05; **, p < 0.01; ***, p < 0.001). Note that (+)-pentazocine rendered the subject more sensitive to cocaine dose.
Pre-session intraperitoneal (i.p.) injection with (+)-pentazocine dose-dependently shifted the cocaine self-administration dose-effect curve leftward, without affecting maximum response rate (Fig. 1B). The lowest dose of (+)-pentazocine (1.0 mg/kg) was inactive, whereas doses of 3.2 and 10 mg/kg produced leftward shifts that approximated 3- and 10-fold, respectively. These changes were obtained without appreciable effects on the temporal patterns of responding (Fig. 1A, bottom record). The major difference in the performances after (+)-pentazocine or vehicle pretreatment were that the highest response rates were obtained at lower doses of cocaine after (+)-pentazocine. Two-way repeated measures analysis of variance (ANOVA) indicated a significant effect on response rate (F4,60 = 5.60; p = 0.003), with drug pretreatment dose and component (EXT and cocaine dose) as factors. Post-hoc Tukey comparisons indicated that response rates maintained at 0.32 mg/kg/injection of cocaine were significantly decreased by 3.2 and 10 mg/kg of (+)-pentazocine (q = 5.78, 6.25, respectively; p values <0.001). However, the low response rates at 0.032 mg/kg/injection of cocaine were increased by 10.0 mg/kg of (+)-pentazocine (q = 4.66; p = 0.009). Overall, after (+)-pentazocine pretreatment lower doses of cocaine maintained the highest responses rates compared with vehicle pretreatment. Such potentiating effects on cocaine self-administration by (+)-pentazocine were consistent with those produced by other σ1R agonists (PRE-084 and DTG) in a previous report (22).
The relation between cocaine self-administration and DAT function suggests that potentiation of cocaine self-administration by σ1R agonists may involve modulation of DAT by these drugs. Because cocaine inhibits the DAT by direct binding, we tested the effects of σ1R ligands on the binding of [3H]WIN35428, a radiolabeled cocaine analog, in rat brain tissues with a high density of DAT. Freshly harvested rat striatal slices were incubated with σ1R agonists, washed multiple times to remove residual drugs, then homogenized to measure binding the of [3H]WIN35428. Specific binding was adjusted for variations of protein concentrations among treatment groups. After preincubation with 10 μm (+)-pentazocine or PRE-084, Bmax values of [3H]WIN35428 binding in striatal homogenates were significantly increased (mean ± S.E.: 187 ± 30% and 166 ± 15% of vehicle, respectively; Fig. 2, A and B), whereas Ki values were not substantially changed.
Figure 2.

Regulation of DAT function by σ1R agonists in native tissues and transfected cells. A and B, rat striatal slices were incubated with σ1R agonists, washed, and homogenized to measure [3H]WIN35428 binding. Shown are representative binding curves with triplicate samples (mean ± S.D.) and summarized Bmax values (mean ± S.E., n = 4–5 experiments) in the bar graph. C and D, HEK293 cells transfected with DAT and σ1R were incubated with σ1R agonists, washed, and measured for [3H]WIN35428 binding. Shown are representative binding curves with triplicate samples (mean ± S.D.) and summarized Bmax values (mean ± S.E., n = 3 experiments) in the bar graph. E and F, HEK293 cells transfected with DAT and σ1R were incubated with σ1R agonists, washed, and measured for [3H]DA uptake. Shown are representative results with triplicate samples (mean ± S.D.) and summarized Vmax values (mean ± S.E., n = 3 - 5 experiments) in the bar graph. *, p < 0.05; **, p < 0.01, one-way ANOVA and post-hoc Dunnett's test, compared with vehicle. (+)pent, (+)-pentazocine.
We further examined the effects of σ1R agonists on DAT function in HEK293 cells co-transfected with DAT and σ1R. Because cocaine preferentially binds to DAT in the outward-facing conformation (7), we reasoned that potential changes of DAT conformation and cocaine binding might be more easily unmasked under conditions of low extracellular Na+, in which the conformational equilibrium of DAT is shifted toward the inward-facing state. We found that [3H]WIN35428 binding was correlated with increasing extracellular Na+ in a dose-dependent manner in these cells, and that although binding at 50 mm Na+ was reduced to half that in normal Na+ (150 mm), it still could be measured reliably (data not shown). Thus, cells were incubated with σ1R ligands and subsequently assayed for DAT binding or substrate uptake in a buffer with 50 mm NaCl and 100 mm N-methyl-d-glucamine. Similar to results obtained using striatal tissues, there was a significant increase of [3H]WIN35428 binding Bmax in these cells pretreated with (+)-pentazocine or PRE-084 (130 ± 7% or 125 ± 6% of vehicle, respectively; Fig. 2, C and D) without substantial changes in Ki values. Similar effects of σ1R agonists on DAT binding were observed in another cell line expressing HA-tagged DAT and Myc-tagged σ1R (data not shown). Additionally, there was a significant increase of [3H]DA uptake Vmax values in cells preincubated with (+)-pentazocine or PRE-084 (133 ± 8% or 145 ± 12% of vehicle, respectively; Fig. 2, E and F) without substantial changes of Km values.
As σ1R has been shown to be a versatile molecular chaperone that can interact with multiple membrane proteins (25), we examined whether σ1R could interact with neurotransmitter transporters. In HEK293 cells GST-tagged-σ1R was co-expressed together with Myc-tagged-σ1R, DAT, or the excitatory amino acid transporter-2 (EAAT2). When GST-σ1R was affinity-purified from cell lysates by glutathione beads, a strong signal of Myc-σ1R was observed, suggesting the existence of a robust constitutive interaction that facilitated the formation of multimers. Substantial signals from the DAT were also detected, whereas no signal of EAAT2 was detected despite its higher expression level (Fig. 3A). The bands detected for DAT corresponded to unglycosylated (∼55 kDa), glycosylated (∼80–90 kDa), and high molecular weight (Mr) oligomeric forms, suggesting that σ1R likely interacts with DAT directly or indirectly in various cellular compartments, including those mature, glycosylated DAT on the cell surface. Furthermore, in cells co-transfected with σ1R and DAT, σ1R could be co-immunoprecipitated with DAT using DAT-specific antibody MAB369 but not with control IgG (Fig. 3B).
Figure 3.

Interaction between DAT and σ1R. A, DAT, but not EAAT2, was pulled down with GST-σ1R in transfected HEK293 cells. Representative blots from five experiments are shown. Note in the pull-down sample the presence of unglycosylated, glycosylated, and oligomeric forms of DAT, with a possible partial degradation product (∼45 kDa). B, co-immunoprecipitation of DAT and σ1R in transfected HEK293 cells. DAT was immunoprecipitated (IP) by rat monoclonal antibody (MAB369) but not by normal rat IgG as control. Left, detection of σ1R; right, membrane was reblotted to confirm enrichment of DAT. Antibodies raised from different species were used to minimize cross-reactivity to IgG proteins. C, molecular interaction between σ1R and DAT measured by BRET. HEK 293T cells were transfected with a constant amount of the RLuc-fusion construct and increasing amounts of the Venus-fusion construct. Molecular interactions between σ1R and DAT and between σ1R and EAAT2 are compared by measuring the energy transfer between the two fusion protein partners: σ1R-Rluc-Venus-EAAT2 (●) and σ1R-Rluc-Venus-DAT (▴). Homomeric pairs for DAT, EAAT2, and σ1R were used as controls: Rluc-EAAT2-Venus-EAAT2 (○), Rluc-DAT-Venus-DAT (▵), σ1R-Rluc - σ1R-Venus (□). All data points were performed in triplicate (S.E. error bars were obscured by symbols). The BRETmax values were calculated by nonlinear regression using a single-site saturation binding model. D, analysis of interaction domains on σ1R with DAT. GST-tagged full-length (FL) σ1R or C-terminal deletion variants (σ1R-xs and σ1R-Δ3) were co-transfected with Myc-tagged DAT. Stronger DAT signals were co-enriched with GST-σ1 xs or Δ3 compared with GST-σ1 FL (upper panel). The membrane was then blotted with antibodies against GST to confirm GST pulldown (lower panel).
The interaction between σ1R and DAT was further verified in live cells using bioluminescence resonance energy transfer (BRET) method. Consistent with the results using GST pulldown assays, co-expression of σ1R and DAT resulted in substantially higher BRETmax signals than co-expression of σ1R and EAAT2 (Fig. 3C, ▴ and ●, respectively), whereas DAT-DAT and EAAT2-EAAT2 interactions (Fig. 3C, inset, ▵ and ○, respectively) showed similar BRETmax values, indicating that the difference between the BRETmax between σ1R-DAT and σ1R-EAAT2 was significant. The σ1R-σ1R BRET pair exhibited the most robust signals (Fig. 3C, inset, □), confirming the multimerization of σ1R, as shown in Fig. 3A.
Several splice variants of σ1R display different lengths in their cytoplasmic domains, including one variant (GenBankTM NM_147157.2) lacking exon 3 (σ1-Δ3) that encodes amino acids 119–149 and another (GenBankTM BC007839.2) that has a frameshift-induced early termination and deletion of amino acids 103–223 (σ1R-xs). When GST-tagged σ1R-Δ3 and σ1R-xs were examined for interaction with DAT in transfected cells, both exhibited stronger association than the full-length (FL) σ1R, whereas σ1R-xs showed the strongest interaction with DAT (Fig. 3D). These surprising results suggested that the interaction between DAT and σ1R was likely mediated by the TM of σ1R, as deletion of its cytoplasmic domain did not impair but, instead, enhanced DAT-σ1R association.
We previously studied conformational changes of DAT using cysteine accessibility assays. Upon cocaine binding, the conformation of DAT was changed so that several residues exhibited altered accessibility, including cysteine 306 (Cys-306) and threonine 316 (Thr-316) on TM6a (Fig. 4A), a key domain forming a part of the extracellular vestibule in the outward-facing conformation of DAT (26). To understand why preincubation with σ1R agonists increased cocaine binding (Fig. 2), we examined whether σ1R drugs could modulate the DAT conformation. In HEK293 cells co-transfected with wild-type DAT and Myc-tagged σ1R, exposure of 1 μm cocaine substantially increased the accessibility of Cys-306 (253 ± 29%, mean ± S.E., compared with vehicle), as probed by maleimide-PEG2-biotin, a sulfhydryl-specific, membrane-impermeant reagent. Preincubation with PRE-084 increased the cocaine-induced effects in a dose-dependent manner, with 10 μm PRE-084 producing a significant enhancement to 364 ± 36% of vehicle (Fig. 4B). Similarly, 10 μm (+)-pentazocine enhanced cocaine-induced changes in Cys-306 accessibility from 273 ± 20% to 349 ± 29% of vehicle treatment (Fig. 4C). However, neither PRE-084 nor (+)-pentazocine pretreatment significantly altered Cys-306 accessibility in the absence of cocaine (Fig. 4, B and C). Moreover, neither σ1R agonists changed the cell-surface expression levels of DAT, as measured by cell-surface biotinylation using sulfo-NHS-SS-biotin, a membrane-impermeant probe that selectively reacts with primary amine moieties in proteins (Fig. 4D).
Figure 4.

Modulation of DAT conformation by σ1R agonists in the presence of cocaine. A, schematic of the Drosophila DAT structure with cocaine (yellow) bound (PDB 4XP4) (7). Highlighted positions corresponding to Cys-306 (red), Thr-316 (green), and TM6a (blue) in human DAT. Part of TM11 was omitted for clarity. B and C, potentiation of cocaine-induced accessibility changes of Cys-306 in DAT by σ1R agonists. Transfected HEK293 cells expressing DAT and epitope-tagged σ1R were preincubated with PRE-084 or (+)-pentazocine for 1 h at 37 °C, washed with PBSCM, and labeled with maleimide-PEG2-biotin at 4 °C for 45 min in the presence or absence of cocaine. Biotinylated proteins were enriched from cell lysates with NeutrAvidin beads, subjected to SDS-PAGE, and detected on immunoblot using DAT-specific antibodies (see details under “Experimental procedures”). D, σ1R agonists did not change cell expression levels of DAT. Transfected cells were incubated with σ1R drugs for 1 h at 37 °C, washed with PBSCM, and labeled with sulfo-NHS-SS-biotin at 4 °C for 45 min. E, σ1R antagonist CM304 blocked the effects of PRE-084 on cocaine-induced accessibility changes of Cys-306 in DAT. Cells were incubated with CM304 for 15 min before PRE-084 treatment for 1 h at 37 °C with CM304 also present. F, potentiation of cocaine-induced accessibility changes of T316C/C306A DAT by σ1R agonists. All bar graphs (B–F) show summarized results (mean ± S.E.) from multiple experiments (n) with representative immunoblots of labeled DAT and lysate DAT signals. *, p < 0.05; ***, p < 0.001, one-way ANOVA with post-hoc Bonferroni's multiple comparison test. NS, not significant. (+)pent, (+)-pentazocine.
Pretreatment with 2 μm CM304, a novel antagonist with subnanomolar affinity and a high selectivity for σ1R (27, 28), effectively blocked the potentiating effects of 10 μm PRE-084 (Fig. 4E), verifying that these effects were mediated specifically through σ1R. CM304 alone did not alter cocaine-induced changes in Cys-306 accessibility on DAT.
We then probed the cysteine accessibility at a position in closer proximity to the inhibitor-binding pocket of DAT, using a substituted cysteine construct T316C/C306A in which Thr-316 and Cys-306 were mutated to cysteine and alanine, respectively. In cells co-transfected with T316C/C306A DAT and Myc-tagged σ1R, cocaine binding significantly increased T316C thiol side-chain reactivity toward maleimide-PEG2-biotin to 387 ± 22% of vehicle (Fig. 4F). The effect of cocaine was significantly enhanced after preincubation with 20 μm (+)-pentazocine or PRE-084 to 487 ± 26% or 548 ± 49%, respectively (Fig. 4F). As in Fig. 4, B and C, neither PRE-084 nor (+)-pentazocine altered T316C accessibility in the absence of cocaine (Fig. 4F).
Together, these biochemical data indicated that pre-exposure to σ1R agonists enhanced cocaine-induced conformational changes of DAT, suggesting that the interaction with σ1R likely shifted the conformational equilibrium of DAT toward the outward-facing state which preferentially binds cocaine.
Because σ1R agonists did not directly alter DAT conformation or its cell-surface expression levels, their modulatory effects on DAT conformation are most likely mediated by σ1R-DAT interactions. The crystal structure of σ1R showed a homotrimeric assembly (17). Each monomer has a single TM and a cytoplasmic portion comprising a ligand-binding pocket and trimerization interface. Because oligomerization of membrane proteins may regulate their function, we devised a novel biochemical method to detect σ1R multimerization. HEK293 cells were transfected with σ1R containing N-terminal FLAG-2 × His8 tag (FH-σ1R), which has a predicted Mr of 32 kDa (25 kDa σ1R + 7 kDa tags with linker). After incubation with σ1R ligands, cells were solubilized using a mild detergent, glyco-diosgenin (GDN), and electrophoretically separated under non-denaturing conditions using another mild detergent, perfluorooctanoic acid (PFO). FLAG antibodies detected immunoreactive signals of two lower Mr bands (∼40 and 70 kDa) and high-Mr diffused bands (>150 kDa), likely corresponding in apparent Mr to the σ1R monomer, dimer, and oligomer (possibly larger than trimer). Compared with vehicle, the agonist (+)-pentazocine significantly decreased, whereas the antagonist CM304 significantly increased signals of σ1R oligomers (Fig. 5A), suggesting that ligand binding to σ1R has distinct effects on the multimeric states of σ1R. When GDN lysates of FH-σ1R were run under denaturing conditions in SDS-PAGE, only one band (∼30 kDa) was seen, similar to σ1R signals in Fig. 3, A and B.
Figure 5.

Effects of σ1R ligands on σ1R multimerization. A, cells expressing FH-σ1R were incubated with ligands in culture medium at 37 °C for 1 h, then lysed with GDN lysis buffer and subjected to PFO-PAGE. FLAG antibodies detected multiple bands, corresponding to monomer, dimer, and high-order oligomers of σ1R. Shown are quantified results of multimeric band signals (mean ± S.E., n = 3 experiments) with a representative blot. **, p < 0.01, one way ANOVA and post-hoc Dunnett's test, compared with vehicle. B, after ligand incubation, FH-σ1R cells were surface biotinylated with sulfo-NHS-biotin at 4 °C. Biotinylated proteins from GDN lysates of cells were enriched with NeutrAvidin beads, separated in PFO-PAGE or SDS-PAGE, and immunoblotted with FLAG antibody (representative blots shown). C, cell surface FH-σ1R bands from PFO-PAGE were quantified. The fraction of monomer out of total signals (including monomer, dimer, and multimer) in each treatment was calculated and normalized to that of vehicle. (+)-Pentazocine significantly increased monomeric FH-σ1R fractions (mean ± S.E., n = 4 experiments). *, p < 0.05, one way ANOVA and post-hoc Dunnett's test, compared with vehicle. D, cell surface FH-σ1R bands from SDS-PAGE were quantified. Drug treatment did not significantly alter the overall amount of FH-σ1R on the cell surface (mean ± S.E., n = 4 experiments).
Because cell surface DAT is vital in its function, and σ1R can interact with glycosylated DAT, which is likely in the plasma membrane (Fig. 3, A and D), we further examined the multimeric status of σ1R on the cell surface. The FLAG epitope (DYKDDDDK) and enterokinase cleavage sequence (DDDDK) in FH-σ1R has three lysine residues whose primary amine side chain can react with sulfo-NHS-biotin, a membrane-impermeant cell-surface biotinylation reagent. FH-σ1R-expressing cells were treated with σ1R ligands at 37 °C for 1 h in culture medium, washed with cold PBSCM (PBS containing 0.10 mm CaCl2, 1.0 mm MgCl2, pH 7.1), and biotinylated at 4 °C. After cell lysis with GDN, biotinylated proteins were enriched with NeutrAvidin beads, then eluted with sample buffer containing 4% PFO to preserve the multimeric states of σ1R. Although the majority of σ1R was intracellular, a portion of σ1R was present on the cell surface and labeled by sulfo-NHS-biotin. In PFO-PAGE three different bands (∼40, ∼70, and >150 kDa) of FH-σ1R were observed, likely corresponding to the monomer, dimer, and multimer (Fig. 5B). With (+)-pentazocine pretreatment, the fraction of σ1R monomers on the cell surface was significantly increased compared with vehicle (Fig. 5C). However, if the NeutrAvidin beads PFO eluates were then incubated with SDS sample buffer and run in SDS-PAGE, only one band of ∼30 kDa was seen, which represented the denatured σ1R monomer derived from monomer, dimer, and multimer under native conditions. No statistical difference in σ1R band densities in SDS-PAGE was seen with σ1R drug treatment (Fig. 5D). These results demonstrate that (+)-pentazocine does not change the overall amount of FH-σ1R on the cell surface but specifically alters its quaternary organization to induce more monomer formation.
Discussion
In the present study we explored molecular mechanisms underlying behavioral potentiation of cocaine self-administration by σ1R agonists. Our data in native tissues and transfected cells show that pretreatment of σ1R agonists shifts DAT conformation toward an outward-facing conformation (Fig. 4), which facilitates cocaine binding (Fig. 2) and enhances cocaine self-administration potency (Fig. 1). Furthermore, σ1R agonists did not directly affect DAT conformation or cell-surface expression levels but likely exert their effects by altering multimeric states of σ1R, thus modulating its dynamic interaction with DAT. The agonist (+)-pentazocine dissociated σ1R multimers to monomers and dimers (Fig. 5) and potentiated cocaine-induced changes in DAT conformation (Fig. 4), whereas the antagonist CM304 had the opposite effects on σ1R oligomerization and no effects on DAT conformation. Taken together, our results support the hypothesis that the dissociation of σ1R multimers to monomers by σ1R agonists facilitates the interaction of σ1R with DAT and promotes an outward-facing conformation of DAT, thus enhancing cocaine binding and potentiating the response to cocaine (Fig. 6).
Figure 6.
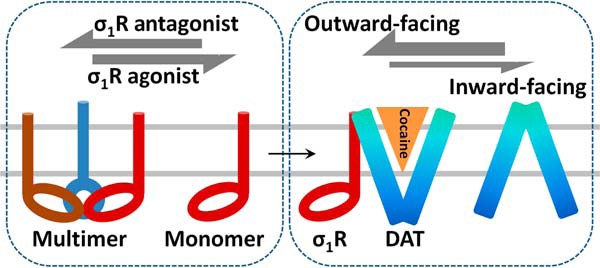
Hypothesis schematic summarizing the modulation of DAT conformation and cocaine binding by σ1R. Binding of σ1R agonists facilitates dissociation of σ1R multimers to monomers, which then dynamically interact with DAT to promote an outward-facing conformation of DAT, thus enhancing cocaine binding and potentiating cocaine's behavioral response.
The transport cycle of DAT involves coordinated movement of key domains that enables DAT to switch between the outward-facing and inward-facing conformations. Pharmacological studies (26, 29) and recent atomic structures of Drosophila DAT (7) have confirmed that cocaine binds to DAT in the outward-facing conformation. Based on biochemical, pharmacological and behavioral data presented in this study, we speculate that interaction with σ1R shifts the DAT conformation toward an outward-facing state in which cocaine binding is facilitated, although detailed molecular mechanisms involved in such modulation requires further elucidation. Our cysteine accessibility assays were optimized to examine cocaine-induced conformational changes of DAT at Cys-306 and T316C, but they did not detect significant effects of σ1R agonists in the absence of cocaine (Fig. 4). It is possible that such modulation may involve conformational changes of other domains on DAT. Because cocaine binding to DAT is promoted by DAT-σ1R interaction, modulatory effects by σ1R agonists were eventually unmasked as potentiation of cysteine accessibility changes at Cys-306 or T316C. In other words, conformational changes of DAT induced by cocaine binding are apparently enhanced after σ1R agonists treatment.
The σ1R agonist (+)-pentazocine dose-dependently shifted the cocaine self-administration dose-effect curve to the left (Fig. 1). A similar effect was obtained previously with other σ1R agonists, PRE-084 and DTG (22). Additionally, the effects of the σ1R agonists on cocaine self-administration were similar to the effects of DA uptake inhibitors, such as methylphenidate or WIN35428 (22), suggesting that σ1R agonists may act through DAT to potentiate the reinforcing effects of cocaine. In contrast, σ1R antagonists at doses effective in blocking σ1R agonist effects had no effects on cocaine self-administration when administered alone, suggesting a crucial modulatory role of σ1R on the DA signaling that is critically involved in cocaine self-administration. In the present study we identified a novel modulatory mechanism of σ1R on DAT, the key element initiating DA signaling due to cocaine administration. Data presented here suggest that the interaction with σ1R likely stabilizes a conformation of DAT, which is preferable for cocaine binding. This correlates with the observations that lower doses of cocaine were sufficient to produce reinforcing effects, i.e. a leftward shift of cocaine dose response.
We previously found that membrane cholesterol promotes the outward-facing conformation of DAT and increases [3H]WIN35428 binding Bmax in native tissues and transfected cells (26). Indeed, cholesterol has been shown to be associated with Drosophila DAT bound with inhibitors such as cocaine in X-ray crystal structures (7, 30). As cholesterol binding hinders the movement of TM1a, which is necessary for DAT to transition into the inward-facing conformation, it is speculated that cholesterol plays a critical role in stabilizing outward-facing conformation of DAT (30).
Interestingly, the effects of σ1R agonists on DAT binding resemble those exerted by increasing the membrane cholesterol content. Both treatments increased Bmax values of [3H]WIN35428 binding (Fig. 2, B and D) without changing cell-surface expression levels of DAT (Fig. 4D). Furthermore, σ1R has been suggested to bind cholesterol (31) and contain steroid-binding domains like-I and II (32, 33). It is reasonable to speculate that interaction with σ1R may increase the membrane cholesterol content in the microdomains surrounding DAT proteins, thus shifting the equilibrium of DAT conformation toward the outward-facing direction. This conjecture will require further studies to resolve several important questions. It remains to be elucidated whether cholesterol can occupy the ligand-binding pocket of σ1R or associate with σ1R in another manner and how multimeric status of σ1R can impact its ability to bind cholesterol. Nevertheless, cumulative data from the work reported here and by others appear to support the hypothesis that as a cholesterol carrier σ1R may interact with and modulate the function of various membrane proteins.
Alternatively, association with σ1R may affect the quaternary structure of DAT. It is worth noting that high-Mr DAT bands appeared to be preferentially enriched with GST-σ1R (Fig. 3, A and D), suggesting direct or indirect association of σ1R with oligomeric DAT proteins. Earlier studies have shown that DAT likely exists as multimers in native membrane environment (34–37), and its oligomeric states can affect [3H]WIN35428 binding through protomer cooperativity (38). Thus, the dynamic interaction between DAT and σ1R at molecular levels will require future investigation employing more sophisticated techniques.
A one-TM topology of σ1R was proposed when it was originally cloned (9). Subsequent studies suggested that σ1R possessed two TMs (11, 13). Recently the crystal structure of σ1R showed a homotrimer with each protomer containing a single TM and a cytoplasmic C terminus (17). Our data support this one-TM model, with an extracellular N terminus and a cytoplasmic C terminus when σ1R is expressed on the cell surface, because N-terminal FLAG-tagged σ1R was efficiently labeled at lysine residues by the membrane-impermeant sulfo-NHS-biotin (Fig. 5B). In addition, a substituted cysteine construct of σ1R containing W9C at its N terminus was labeled by the membrane-impermeant maleimide-PEG2-Biotin. In contrast, another construct with M170C at its C terminus was not labeled (data not shown).
We devised a novel biochemical method to examine the multimeric status of σ1R. First, σ1R-expressing cells were solubilized by GDN, a mild detergent that has been shown to be superior to dodecylmaltoside in stabilizing solubilized membrane proteins (39). Second, GDN-solubilized proteins were electrophoretically separated using buffer containing PFO, another mild detergent shown to preserve the oligomerization status of membrane proteins (40–43). It appeared that, at least in transfected cells, the bulk of the epitope-tagged σ1R exists as high-order multimers, because only high-Mr bands were observed when GDN lysates were incubated with PFO sample buffer at room temperature. The apparent Mr (>150 kDa) of these bands suggested a quaternary assembly possibly larger than trimers (Fig. 5A). This is consistent with results from early purification studies using [3H](+)SKF10047 or [3H]azido-DTG as affinity ligands in which the labeled protein complex under non-denaturing conditions appeared to be >150 kDa (44–46).
High- and low-Mr σ1R bands were revealed after optimizing the incubation temperature of GDN lysates and PFO sample buffer to 50 °C. Although in non-denaturing gels the migration rate of a protein might not accurately correlate with its Mr as in SDS-PAGE, the apparent Mr (∼40 and 70 kDa) of low-Mr bands most likely corresponded to monomeric and dimeric σ1R. The absence of signals between 70 and ∼150 kDa suggested that larger assemblies than trimers were predominant in these cells.
Treatment with (+)-pentazocine or CM304 had opposite effects on the multimeric states of σ1R (Fig. 5, A and B). Several agonists or antagonists had effects similar to those of (+)-pentazocine or CM304, respectively.3 Additionally, differential effects of ligands on σ1R multimerization were observed using HA- or Myc-tagged σ1R, ruling out possibly spurious results from the FLAG epitope.3 These biochemical results are consistent with recent findings that Förster resonance energy transfer signals between cyan or yellow fluorescent protein-tagged σ1R were decreased by agonist (+)-pentazocine but increased by antagonist haloperidol in transfected cells (47). It has also been reported that multimeric states of purified σ1R in detergent micelles were stabilized by some ligands (48).
Our biochemical data indicate that through direct or indirect manners σ1R associates with unglycosylated, glycosylated, and oligomeric DAT (Fig. 3, A and D), suggesting their interactions occur in various intracellular compartments and on the plasma membrane. Although the majority of σ1R reside on the ER, we detected the presence of σ1R in the plasma membrane using cell-surface biotinylation assays (Fig. 5B). Furthermore, there were distinct effects of σ1R agonists or antagonists on multimeric states of σ1R on the cell surface (Fig. 5C). It is plausible that the portion of σ1R in the plasma membrane plays a more important role in modulating the function of various interacting membrane proteins such as the channels and receptors identified so far and the DAT in this study. We hypothesize that σ1R agonists promote σ1R-DAT interactions by increasing σ1R monomers. Previous studies employing atomic force microscopy have demonstrated that σ1R associates with a trimeric acid-sensing channel with 3-fold symmetry (49) and with a tetrameric sodium channel with 4-fold symmetry (50). Although we cannot rule out the possibility that DAT associates with oligomeric σ1R, such interactions may be energetically unfavorable due to steric hindrance. Furthermore, the σ1R-xs splice variant, which retains the TM sequence but lacks cytoplasmic ligand binding and trimerization domains, exhibited stronger association with DAT (Fig. 3D). Therefore, current data favor the model that σ1R monomers preferentially interact with DAT.
In summary, in this study we present biochemical, pharmacological, and behavioral evidence to demonstrate that σ1R interacts with DAT and modulates DAT function. Our data support the hypothesis that agonists of σ1R affect its oligomerization and interaction with DAT, inducing an outward-facing conformation of DAT and facilitating cocaine binding to DAT. This study represents a first step to decipher the mechanisms underlying potentiation of cocaine self-administration by σ1R agonists, although further work is necessary to fully elucidate the molecular and cellular underpinnings of these neural adaptations. As σ1R ligands have been shown to indirectly modulate DA signaling, exploration of their therapeutic potentials may open a new avenue in treating cocaine addiction.
Experimental procedures
Chemicals, radioligands, and antibodies
Sources of reagents are as follows: (−)-cocaine HCl, (+)-pentazocine succinate, WIN35428, National Institute on Drug Abuse (NIDA) Drug Supply Program; PRE-084, Tocris (Minneapolis, MN); maleimide-PEG2-biotin, sulfo-NHS-SS-biotin, sulfo-NHS-biotin, NeutrAvidin-agarose beads, BCA protein kit, Pierce; [3H]DA (NET673, 16.9 to 30.6 Ci/mmol) and [3H]WIN35428 (NET1033, 85.9 Ci/mmol), PerkinElmer Life Sciences; PFO, TCI America (Portland, OR); GDN, Anatrace (Maumee, OH); all other chemicals, Sigma or Fisher. Antibodies were from: anti-DAT MAB369, Chemicon (Temecula, CA); rabbit antisera against DAT N terminus (26); σ1R rabbit antisera (Su laboratory); anti-Myc 9E10 mAb, protein A/G beads, Santa Cruz Biotechnology (Santa Cruz, CA); anti-FLAG rat mAb L5, anti-GST rabbit poly9248, Biolegend (San Diego, CA); glutathione-conjugated Sepharose beads, GE Healthcare; HRP-conjugated secondary antibodies, Biolegend or Jackson ImmunoResearch (West Grove, PA).
Behavioral pharmacology
Adult male Sprague-Dawley rats (Taconic Farms, Germantown, NY) were surgically prepared with indwelling venous catheters and trained to self administer cocaine under a fixed-ratio (FR) five-response schedule of reinforcement in standard operant conditioning chambers using previously described methods (51). Experimental sessions were conducted daily with subjects enclosed within chambers containing two response levers (with stimulus lights above each) and a food tray. The catheter was attached via tubing to an external infusion pump via a ceiling-mounted swivel. After recovery from surgery, subjects were allowed to self administer cocaine (0.32 mg/kg/injection, intravenous) and trained over a series of sessions. Under the final conditions the emission of 5 responses produced a cocaine injection during 20-min components of the session separated by 2-min timeout periods in which cocaine was not available. Unit dose of cocaine increased from 0 mg/kg/injection in the first component to 1.0 mg/kg/injection in the fifth component. Test sessions before which σ1R agonists were administered were interspersed between training sessions and separated by a minimum of 72 h. The response rates were calculated by dividing the total responses by the elapsed time during components, excluding responses and time during time-out periods. Med Associates Inc. (St. Albans, VT) software was used to control the experiments and to create cumulative records of responding. Effects on response rates were assessed by ANOVA, with post hoc Tukey tests using SigmaPlot software (Systat Software, Inc., San Jose, CA) with statistical significance set at p < 0.05.
Radioligand binding assay
Male Sprague-Dawley rats (150–250 g, Charles River) were euthanized, and striatal tissues were dissected immediately. Tissues were quickly rinsed with artificial cerebrospinal fluid (aCSF, 125 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 1 mm MgCl2, 26 mm NaHCO3, 11 mm glucose, 2.4 mm CaCl2) and then chopped into 0.5-mm slices using a McIlwain tissue chopper, washed 3 times with aCSF, and divided into approximately equal portions in 20 ml of aCSF containing σ1R ligands or vehicle. Tissues were incubated at 35 °C with continuous oxygenation for 1 h and then washed 3 times with ice-cold sucrose-phosphate buffer (SPB, 0.32 m sucrose, 7.74 mm Na2HPO4, 2.26 mm NaH2PO4, pH 7.4). Afterward tissues were resuspended in 5 ml of SPB, homogenized using a Brinkman Polytron, and centrifuge at 10,000 × g for 10 min at 4 °C. The pellet was resuspended in 5 ml of SPB and centrifuged again at 10,000 × g for 10 min at 4 °C. The resulting pellet was resuspended in 5 ml of SPB and divided into 0.2-ml aliquots in glass test tubes. Aliquots were also saved for measuring protein concentrations. The binding reactions were initiated by adding competing ligands and 0.5 nm [3H]WIN35428 in a total volume of 0.5 ml, then incubated on ice for 2 h and terminated by rapid filtration through Whatman GF/B filters (presoaked in 0.05% polyethyleneimine (PEI)) using a cell harvester (Brandel Instruments, Gaithersburg, MD). The filters were washed twice with 5.0 ml of cold buffer, soaked with scintillation mixture overnight, and measured for radioactivity using a Tri-Carb 2910TR liquid scintillation counter (PerkinElmer Life Sciences) at 50% efficiency. Nonspecific binding measured in the presence of 100 μm cocaine was subtracted from total counts to obtain specific binding. Data were analyzed using GraphPad Prism (San Diego, CA) for non-linear regression to derive Bmax and Ki values. All animal-related protocols were approved by the National Institute on Drug Abuse Intramural Research Program Animal Care and Use Committee.
DNA subcloning and stable expression cell lines
The coding sequences of human DAT or human σ1R cDNA were subcloned into CMV promoter-based mammalian expression vectors: pcDNA3.1(+) (Invitrogen), pCMV-tag5A (Stratagene, La Jolla, CA), or plasmids expressing N-terminal fusion of Myc, HA, GST, or FLAG-2 × His8 tags that were custom-made in the laboratory and verified by standard DNA sequencing procedures. Plasmids were linearized with PvuI, MluI, or ApaL1 and transfected into HEK293 cells with TransIT LT1 reagent (Mirus Bio, Madison, WI). Cells were then cultured in DMEM (Sigma) with 10% FBS (Invitrogen), penicillin-streptomycin, and 0.5 mg/ml G418 in humidified incubators with 5% CO2 at 37 °C. Expression of DAT and σ1R in G418-resistant pools or clones was verified by immunoblot using antibodies against DAT, σ1R, or epitope tags. To overcome potential limitations of cell-line specific effects, multiple lines of HEK293 cells were constructed to stably co-express DAT and σ1R. The following cell lines were used in uptake, binding, or cysteine accessibility assays, including “D-S” (expressing DAT + σ1R), “D-SM” (DAT + C-Myc-tagged σ1R), “HD-MS” (HA-tagged DAT + N-Myc tagged σ1R), and “T316C-MS” (T316C/C306A DAT + N-Myc tagged σ1R).
[3H]DA uptake and [3H]WIN35428 binding in transfected cells
D-S cells were seeded into PEI-coated 96-well plates and cultured to confluency. After cultured medium was aspirated using an ELx50 plate washer (BioTek, Winooski, VT), cells were washed twice and then incubated in low-Na+ buffer (NaCl 50 mm, 100 mm N-methyl-d-glucamine, 2 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 5 mm glucose, 5 mm HEPES, pH 7.4) for 2 h at 37 °C in the absence or presence of σ1R ligands.
For uptake assays, after drug incubation cells were washed twice and then incubated at room temperature for 5 min with 100 μl of low-Na+ buffer containing 25 nm [3H]DA and various concentrations of unlabeled DA and 5 μm catechol-O-methyl transferase inhibitor Ro 41–0960. Cells were then washed with cold PBSCM using a plate washer and lysed with 0.2 ml of scintillation mixture overnight. Retained radioactivity was determined by a Wallac MicroBeta2 liquid scintillation counter (PerkinElmer Life Sciences). Data were analyzed using the Michaelis-Menten kinetic equation with GraphPad Prism.
For binding assays, after drug incubation D-S cells or HD-MS cells were then washed twice and incubated with low-Na+ buffer containing 7.5 nm [3H]WIN35428 and various concentrations of unlabeled WIN35428 at 4 °C for 1.5 h. Cells were then washed with cold PBSCM using a plate washer, and bound radioactivity was determined as described above. Data were analyzed with GraphPad Prism for non-linear curve-fitting.
Co-immunoprecipitation and GST pulldown
HEK293 cells in 60-mm culture dishes were transfected to co-express GST-σ1R and Myc-σ1R, DAT, or EAAT2. Two days after transfection, cells were collected and incubated with TNE lysis buffer (1% Triton X-100, NaCl 150 mm, EDTA 1 mm, Tris 10 mm, pH 7.5, and protease inhibitors) at 4 °C for 1 h followed by centrifugation at 18,000 × g for 20 min at 4 °C. Aliquots of the supernatants were saved as lysate input and for protein concentration measurement; the remaining portions were incubated with 40 μl of 50% slurry of glutathione-conjugated Sepharose beads (prewashed with TNE buffer) on a rotator for 4 h at 4 °C. After washing beads three times with TNE lysis buffer, proteins were eluted with SDS sample buffer and separated by SDS-PAGE, transferred to PVDF membranes, and detected by immunoblotting and chemiluminescence methods.
Confluent D-S cells in 60-mm culture dishes were incubated with TNE lysis buffer at 4 °C for 1 h and centrifugation at 18,000 × g for 20 min at 4 °C. Supernatants were incubated with 1 μg of anti-DAT MAB369 (rat mAb) or normal rat IgG at 4 °C for 2 h followed by the addition of 40 μl of protein A/G beads and incubated for 2 h. Afterward beads were washed three times with TNE lysis buffer. Proteins were eluted and detected as described above.
BRET assays
The cDNAs encoding Renilla luciferase (RLuc) or monomeric Venus were fused in-frame to the C terminus of σ1R and N terminus of DAT or EAAT2 in the pcDNA3.1 vector. The BRET assays were performed as described previously (52). Briefly, HEK293T cells were transfected using a constant amount of plasmid DNA but various ratios of plasmids encoding the fusion protein partners. A control corresponding to mock-transfected cells was included in order to subtract raw basal luminescence and fluorescence from the data. Expression of monomeric Venus fusion proteins was estimated by measuring fluorescence at 535 nm after excitation at 485 nm. Expression of RLuc fusion proteins was estimated by measuring the luminescence of the cells after incubation with 5 μm coelenterazine-h. In parallel, BRET was measured as the fluorescence of the cells at 535 nm at the same time points using a Mithras LB940 reader (Berthold Technologies, Bad Wildbad, Germany).
DAT cysteine accessibility assays and cell-surface biotinylation
D-SM cells or T316C-MS cells were seeded into PEI-coated 6-well or 12-well plates and cultured to confluency. Cells were washed 3 time with PBSCM + 10 mm glucose (PBSCMG) and then incubated with σ1R ligands in PBSCMG at 37 °C for 1 h. Afterward cells were washed 3 times with cold PBSCM, then incubated with 0.5 mg/ml maleimide-PEG2-biotin in the presence or absence of cocaine for 45 min at 4 °C with gentle shaking. The labeling reaction was quenched with PBSCM containing 2 mm DTT or 50 mm cysteine for 15 min at 4 °C. Cells were then washed, harvested, and lysed in TNE lysis buffer for 1 h at 4 °C followed by a 20-min centrifugation at 18,000 × g. Aliquots of supernatants were saved for protein concentration measurement. Remaining supernatants were incubated with 60 μl of NeutrAvidin-agarose beads overnight at 4 °C. After washing beads three times with TNE buffer, biotinylated proteins were eluted with SDS sample buffer, separated by SDS-PAGE, transferred to PVDF membranes, and probed with MAB369 or rabbit antisera for DAT. Chemiluminescent signals were captured with a MultiImage III device (Alpha Innotech, San Leandro, CA) as digital TIFF images without pixel saturation. Mean densities of DAT bands were quantified using the NIH ImageJ software and normalized to percent of vehicle. DAT or σ1R expression levels on the cell surface were measured by procedures similar to those outlined above, except that cells were incubated with 0.5 mg/ml sulfo-NHS-SS-biotin or sulfo-NHS-biotin in PBSCM at 4 °C for 45 min followed by quenching with 100 mm glycine.
Analysis of σ1R multimeric states by PFO-PAGE
Confluent FH-σ1R cells in 12-well plates were incubated with σ1R ligands at 37 °C for 1 h in culture medium. Cells were then washed with cold PBSCM, harvested, and incubated with lysis buffer (0.1% GDN, NaCl 150 mm, EDTA 1 mm, Tris 10 mm, pH 7.5, and protease inhibitors) for 1 h at 4 °C. After lysates were centrifuged at 20,000 × g for 20 min, supernatants were mixed with an equal volume of 2× PFO sample buffer (8% PFO, 40% glycerol, bromphenol blue 0.005%, Tris 100 mm, pH 7.5) to a final concentration of 4% PFO and heated at 50 °C for 10 min. Samples were run in 5–15% polyacrylamide Tris-glycine gels (running buffer: 0.1% PFO, 25 mm Tris, 192 mm glycine, pH 8.3). Proteins were transferred to PVDF membranes and immunoblotted with rat monoclonal anti-FLAG L5 antibodies. Because commercial protein markers containing SDS were not suitable in this system, the following proteins were mixed with PFO sample buffer and used as molecular standards: egg white lysozyme (14 kDa), ovalbumin (45 kDa), BSA (66 kDa monomer, 132-kDa dimer), and equine apoferritin (443 kDa).
Author contributions
W. C. H. designed, conducted, and analyzed the experiments in Figs. 2–5 and wrote most of the paper. T. H. and J. L. K. designed, performed, and analyzed the experiments Fig. 1. H. Y. designed, conducted, and analyzed the experiments in Fig. 3. C. R. M. and F. T. C. provided novel reagents. S. G. A., T.-P. S., and all other authors contributed to designing experiments, reviewing the results, and writing the paper. All approved the final version of the manuscript.
Acknowledgments
The National Institute on Drug Abuse Drug Supply Program kindly provided (−)-cocaine HCl, (+)-pentazocine succinate, and WIN35428. We thank Dr. Linda Werling for providing human σ1R cDNA and critical reading of this manuscript.
This work was supported by Butler University Faculty Startup Fund and Holcomb Research Award (to W. C. H.), National Institutes of Health Intramural Research Programs (to H. Y., T. H., T. P. S., S. G. A., and J. L. K.), Grant R37 DA007595 (to S. G. A.), and in part by Grants R01 DA023205 (to C. R. M.) and P20 GM104932 (to C. R. M.). C. R. M. and F. T. C. are co-inventors and patent holders of CM304. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
W. C. Hong, manuscript in preparation.
- DA
- dopamine
- DAT
- DA transporter
- σR
- sigma receptor
- σ1R
- sigma-1 receptor
- TM
- transmembrane domain
- DTG
- 1,3-di-o-tolylguanidine
- EXT
- extinction
- EAAT2
- excitatory amino acid transporter-2
- BRET
- bioluminescence resonance energy transfer
- GDN
- glyco-diosgenin
- PFO
- perfluorooctanoic acid
- ANOVA
- analysis of variance
- SPB
- sucrose-phosphate buffer
- FR
- fixed-ratio
- RLuc
- Renilla luciferase
- FH-σ1R
- FLAG-2 × His8-σ1R.
References
- 1. Amara S. G., and Kuhar M. J. (1993) Neurotransmitter transporters: recent progress. Annu. Rev. Neurosci. 16, 73–93 [DOI] [PubMed] [Google Scholar]
- 2. Sulzer D., Cragg S. J., and Rice M. E. (2016) Striatal dopamine neurotransmission: regulation of release and uptake. Basal Ganglia 6, 123–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. German C. L., Baladi M. G., McFadden L. M., Hanson G. R., and Fleckenstein A. E. (2015) Regulation of the dopamine and vesicular monoamine transporters: pharmacological targets and implications for disease. Pharmacol. Rev. 67, 1005–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kristensen A. S., Andersen J., Jørgensen T. N., Sørensen L., Eriksen J., Loland C. J., Strømgaard K., and Gether U. (2011) SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 63, 585–640 [DOI] [PubMed] [Google Scholar]
- 5. Vaughan R. A., and Foster J. D. (2013) Mechanisms of dopamine transporter regulation in normal and disease states. Trends Pharmacol. Sci. 34, 489–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bermingham D. P., and Blakely R. D. (2016) Kinase-dependent regulation of monoamine neurotransmitter transporters. Pharmacol. Rev. 68, 888–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang K. H., Penmatsa A., and Gouaux E. (2015) Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 521, 322–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martin W. R., Eades C. G., Thompson J. A., Huppler R. E., and Gilbert P. E. (1976) The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 197, 517–532 [PubMed] [Google Scholar]
- 9. Hanner M., Moebius F. F., Flandorfer A., Knaus H. G., Striessnig J., Kempner E., and Glossmann H. (1996) Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc. Natl. Acad. Sci. U.S.A. 93, 8072–8077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hayashi T., and Su T. P. (2001) Regulating ankyrin dynamics: Roles of sigma-1 receptors. Proc. Natl. Acad. Sci. U.S.A. 98, 491–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aydar E., Palmer C. P., Klyachko V. A., and Jackson M. B. (2002) The sigma receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron 34, 399–410 [DOI] [PubMed] [Google Scholar]
- 12. Kourrich S., Hayashi T., Chuang J. Y., Tsai S. Y., Su T. P., and Bonci A. (2013) Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell 152, 236–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hayashi T., and Su T. P. (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 131, 596–610 [DOI] [PubMed] [Google Scholar]
- 14. Navarro G., Moreno E., Aymerich M., Marcellino D., McCormick P. J., Mallol J., Cortés A., Casadó V., Canela E. I., Ortiz J., Fuxe K., Lluís C., Ferré S., and Franco R. (2010) Direct involvement of sigma-1 receptors in the dopamine D1 receptor-mediated effects of cocaine. Proc. Natl. Acad. Sci. U.S.A. 107, 18676–18681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Navarro G., Moreno E., Bonaventura J., Brugarolas M., Farré D., Aguinaga D., Mallol J., Cortés A., Casadó V., Lluís C., Ferre S., Franco R., Canela E., and McCormick P. J. (2013) Cocaine inhibits dopamine D2 receptor signaling via sigma-1-D2 receptor heteromers. PLoS ONE 8, e61245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim F. J., Kovalyshyn I., Burgman M., Neilan C., Chien C. C., and Pasternak G. W. (2010) Sigma 1 receptor modulation of G-protein-coupled receptor signaling: potentiation of opioid transduction independent from receptor binding. Mol. Pharmacol. 77, 695–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schmidt H. R., Zheng S., Gurpinar E., Koehl A., Manglik A., and Kruse A. C. (2016) Crystal structure of the human sigma1 receptor. Nature 532, 527–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walker J. M., Bowen W. D., Walker F. O., Matsumoto R. R., De Costa B., and Rice K. C. (1990) Sigma receptors: biology and function. Pharmacol. Rev. 42, 355–402 [PubMed] [Google Scholar]
- 19. Gonzalez-Alvear G. M., and Werling L. L. (1994) Regulation of [3H]dopamine release from rat striatal slices by sigma receptor ligands. J. Pharmacol. Exp. Ther. 271, 212–219 [PubMed] [Google Scholar]
- 20. Narayanan S., Mesangeau C., Poupaert J. H., and McCurdy C. R. (2011) Sigma receptors and cocaine abuse. Curr. Top. Med. Chem. 11, 1128–1150 [DOI] [PubMed] [Google Scholar]
- 21. Robson M. J., Noorbakhsh B., Seminerio M. J., and Matsumoto R. R. (2012) Sigma-1 receptors: potential targets for the treatment of substance abuse. Curr. Pharm. Des. 18, 902–919 [DOI] [PubMed] [Google Scholar]
- 22. Hiranita T., Soto P. L., Tanda G., and Katz J. L. (2010) Reinforcing effects of sigma-receptor agonists in rats trained to self-administer cocaine. J. Pharmacol. Exp. Ther. 332, 515–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hiranita T., Mereu M., Soto P. L., Tanda G., and Katz J. L. (2013) Self-administration of cocaine induces dopamine-independent self-administration of sigma agonists. Neuropsychopharmacology 38, 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Katz J. L., Hong W. C., Hiranita T., and Su T. P. (2016) A role for sigma receptors in stimulant self-administration and addiction. Behav. Pharmacol. 27, 100–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kourrich S., Su T. P., Fujimoto M., and Bonci A. (2012) The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci. 35, 762–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hong W. C., and Amara S. G. (2010) Membrane cholesterol modulates the outward facing conformation of the dopamine transporter and alters cocaine binding. J. Biol. Chem. 285, 32616–32626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. James M. L., Shen B., Zavaleta C. L., Nielsen C. H., Mesangeau C., Vuppala P. K., Chan C., Avery B. A., Fishback J. A., Matsumoto R. R., Gambhir S. S., McCurdy C. R., and Chin F. T. (2012) New positron emission tomography (PET) radioligand for imaging sigma-1 receptors in living subjects. J. Med. Chem. 55, 8272–8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Katz J. L., Hiranita T., Kopajtic T. A., Rice K. C., Mesangeau C., Narayanan S., Abdelazeem A. H., and McCurdy C. R. (2016) Blockade of cocaine or sigma receptor agonist self-administration by subtype-selective sigma receptor antagonists. J. Pharmacol. Exp. Ther. 358, 109–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reith M. E., Berfield J. L., Wang L. C., Ferrer J. V., and Javitch J. A. (2001) The uptake inhibitors cocaine and benztropine differentially alter the conformation of the human dopamine transporter. J. Biol. Chem. 276, 29012–29018 [DOI] [PubMed] [Google Scholar]
- 30. Penmatsa A., Wang K. H., and Gouaux E. (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Palmer C. P., Mahen R., Schnell E., Djamgoz M. B., and Aydar E. (2007) Sigma-1 receptors bind cholesterol and remodel lipid rafts in breast cancer cell lines. Cancer Res. 67, 11166–11175 [DOI] [PubMed] [Google Scholar]
- 32. Fontanilla D., Hajipour A. R., Pal A., Chu U. B., Arbabian M., and Ruoho A. E. (2008) Probing the steroid binding domain-like I (SBDLI) of the sigma-1 receptor binding site using N-substituted photoaffinity labels. Biochemistry 47, 7205–7217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pal A., Chu U. B., Ramachandran S., Grawoig D., Guo L. W., Hajipour A. R., and Ruoho A. E. (2008) Juxtaposition of the steroid binding domain-like I and II regions constitutes a ligand binding site in the sigma-1 receptor. J. Biol. Chem. 283, 19646–19656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hastrup H., Karlin A., and Javitch J. A. (2001) Symmetrical dimer of the human dopamine transporter revealed by cross-linking Cys-306 at the extracellular end of the sixth transmembrane segment. Proc. Natl. Acad. Sci. U.S.A. 98, 10055–10060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sitte H. H., and Freissmuth M. (2003) Oligomer formation by Na+-Cl-coupled neurotransmitter transporters. Eur. J. Pharmacol. 479, 229–236 [DOI] [PubMed] [Google Scholar]
- 36. Sorkina T., Doolen S., Galperin E., Zahniser N. R., and Sorkin A. (2003) Oligomerization of dopamine transporters visualized in living cells by fluorescence resonance energy transfer microscopy. J. Biol. Chem. 278, 28274–28283 [DOI] [PubMed] [Google Scholar]
- 37. Torres G. E., Carneiro A., Seamans K., Fiorentini C., Sweeney A., Yao W. D., and Caron M. G. (2003) Oligomerization and trafficking of the human dopamine transporter: mutational analysis identifies critical domains important for the functional expression of the transporter. J. Biol. Chem. 278, 2731–2739 [DOI] [PubMed] [Google Scholar]
- 38. Zhen J., Antonio T., Cheng S. Y., Ali S., Jones K. T., and Reith M. E. (2015) Dopamine transporter oligomerization: impact of combining protomers with differential cocaine analog binding affinities. J. Neurochem. 133, 167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chae P. S., Rasmussen S. G., Rana R. R., Gotfryd K., Kruse A. C., Manglik A., Cho K. H., Nurva S., Gether U., Guan L., Loland C. J., Byrne B., Kobilka B. K., and Gellman S. H. (2012) A new class of amphiphiles bearing rigid hydrophobic groups for solubilization and stabilization of membrane proteins. Chemistry 18, 9485–9490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ramjeesingh M., Huan L. J., Garami E., and Bear C. E. (1999) Novel method for evaluation of the oligomeric structure of membrane proteins. Biochem. J. 342, 119–123 [PMC free article] [PubMed] [Google Scholar]
- 41. Kedei N., Szabo T., Lile J. D., Treanor J. J., Olah Z., Iadarola M. J., and Blumberg P. M. (2001) Analysis of the native quaternary structure of vanilloid receptor 1. J. Biol. Chem. 276, 28613–28619 [DOI] [PubMed] [Google Scholar]
- 42. Penna A., Demuro A., Yeromin A. V., Zhang S. L., Safrina O., Parker I., and Cahalan M. D. (2008) The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature 456, 116–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xu J., Liu Y., Yang Y., Bates S., and Zhang J. T. (2004) Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J. Biol. Chem. 279, 19781–19789 [DOI] [PubMed] [Google Scholar]
- 44. McCann D. J., and Su T. P. (1991) Solubilization and characterization of haloperidol-sensitive (+)-[3H]SKF-10,047 binding sites (sigma sites) from rat liver membranes. J. Pharmacol. Exp. Ther. 257, 547–554 [PubMed] [Google Scholar]
- 45. Kavanaugh M. P., Tester B. C., Scherz M. W., Keana J. F., and Weber E. (1988) Identification of the binding subunit of the sigma-type opiate receptor by photoaffinity labeling with 1-(4-azido-2-methyl[6-3H]phenyl)-3-(2-methyl[4,6-3H]phenyl)guanidine. Proc. Natl. Acad. Sci. U.S.A. 85, 2844–2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schuster D. I., Arnold F. J., and Murphy R. B. (1995) Purification, pharmacological characterization, and photoaffinity labeling of sigma receptors from rat and bovine brain. Brain Res. 670, 14–28 [DOI] [PubMed] [Google Scholar]
- 47. Mishra A. K., Mavlyutov T., Singh D. R., Biener G., Yang J., Oliver J. A., Ruoho A., and Raicu V. (2015) The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem. J. 466, 263–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gromek K. A., Suchy F. P., Meddaugh H. R., Wrobel R. L., LaPointe L. M., Chu U. B., Primm J. G., Ruoho A. E., Senes A., and Fox B. G. (2014) The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J. Biol. Chem. 289, 20333–20344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Carnally S. M., Johannessen M., Henderson R. M., Jackson M. B., and Edwardson J. M. (2010) Demonstration of a direct interaction between sigma-1 receptors and acid-sensing ion channels. Biophys. J. 98, 1182–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Balasuriya D., Stewart A. P., Crottès D., Borgese F., Soriani O., and Edwardson J. M. (2012) The sigma-1 receptor binds to the Nav1.5 voltage-gated Na+ channel with 4-fold symmetry. J. Biol. Chem. 287, 37021–37029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hiranita T., Soto P. L., Newman A. H., and Katz J. L. (2009) Assessment of reinforcing effects of benztropine analogs and their effects on cocaine self-administration in rats: comparisons with monoamine uptake inhibitors. J. Pharmacol. Exp. Ther. 329, 677–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sohy D., Yano H., de Nadai P., Urizar E., Guillabert A., Javitch J. A., Parmentier M., and Springael J. Y. (2009) Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protean effects of “selective” antagonists. J. Biol. Chem. 284, 31270–31279 [DOI] [PMC free article] [PubMed] [Google Scholar]