

Abstract
Rev-erbβ is a heme-responsive transcription factor that regulates genes involved in circadian rhythm maintenance and metabolism, effectively bridging these critical cellular processes. Heme binding to Rev-erbβ indirectly facilitates its interaction with the nuclear receptor co-repressor (NCoR1), resulting in repression of Rev-erbβ target genes. Fe3+-heme binds in a 6-coordinate complex with axial His and Cys ligands, the latter provided by a heme-regulatory motif (HRM). Rev-erbβ was thought to be a heme sensor based on a weak Kd value for the Rev-erbβ·heme complex of 2 μm determined with isothermal titration calorimetry. However, our group demonstrated with UV-visible difference titrations that the Kd value is in the low nanomolar range, and the Fe3+-heme off-rate is on the order of 10−6 s−1 making Rev-erbβ ineffective as a sensor of Fe3+-heme. In this study, we dissected the kinetics of heme binding to Rev-erbβ and provided a Kd for Fe3+-heme of ∼0.1 nm. Loss of the HRM axial thiolate via redox processes, including oxidation to a disulfide with a neighboring cysteine or dissociation upon reduction of Fe3+- to Fe2+-heme, decreased binding affinity by >20-fold. Furthermore, as measured in a co-immunoprecipitation assay, substitution of the His or Cys heme ligands in Rev-erbβ was accompanied by a significant loss of NCoR1 binding. These results demonstrate the importance of the Rev-erbβ HRM in regulating interactions with heme and NCoR1 and advance our understanding of how signaling through HRMs affects the major cellular processes of circadian rhythm maintenance and metabolism.
Keywords: DNA-binding protein, enzyme kinetics, heme, ligand-binding protein, nuclear receptor, pre-steady-state kinetics, redox regulation, transcription repressor, ultraviolet-visible spectroscopy (UV-visible spectroscopy)
Introduction
Heme (Fe-protoporphyrin IX) is a versatile protein cofactor that can adopt a wide range of redox potentials and reactivities depending on its environment (1). As a result, heme plays a critical role in many cellular processes, including electron transport during oxidative phosphorylation, binding oxygen in hemoglobin and myoglobin, and generating high valence and reactive iron-oxo species in enzymes such as cytochrome P450s (2). In contrast to its archetypal role as a cofactor, recent studies have implicated heme as an important signaling molecule regulating diverse pathways ranging from heme biosynthesis to circadian rhythm maintenance (3–8).
A small fraction of total cellular heme (∼6%) comprises a regulatory heme pool that changes in concentration based on the metabolic condition of the cell (9, 10). Proteins that respond to and reversibly bind regulatory heme contain a conserved sequence consisting of a Cys-Pro dipeptide followed by a hydrophobic residue known as the heme-regulatory motif (HRM),2 where the Cys residue is a ligand to Fe3+-heme (11). HRM-containing proteins regulate key metabolic pathways; for example, the rate-limiting enzyme in heme biosynthesis, aminolevulinic acid synthase-1, contains three HRMs that mediate its mitochondrial import, maturation, and facilitate its degradation (4, 12). BACH1, a mammalian transcription factor and repressor of heme-oxygenase 1 and α/β globin genes, harbors six HRMs, several of which bind heme and inhibit DNA binding and target the protein for degradation (13, 14). Other HRM-containing proteins, including p53 (a tumor suppressor protein), iron response regulator, and iron regulatory protein 2 (regulators of heme and iron homeostasis, respectively), are degraded in response to heme binding suggesting that modulation of protein stability is a major role of the HRM (15–17). Ultimately, interplay between regulatory heme and HRMs represents an important signal transduction circuit that regulates energy and iron metabolism.
Another cellular process inextricably linked to heme and metabolism is circadian rhythm maintenance (18–22). In higher eukaryotes, cells are entrained to a diurnal cycle by factors, including light and feeding. At the molecular level, these signals are integrated into a set of core clock proteins that form a transcription-translation feedback loop (23). Two of these proteins, Bmal1 and CLOCK (or NPAS2), form a heterodimeric complex that activates transcription of clock-controlled genes. Although NPAS2 does not harbor an HRM, it binds heme and CO, which regulate its DNA-binding activity (24); NPAS2 is also a redox sensor that integrates energy metabolism into the circadian cycle by sensing changes in the NAD+/NADH couple (25). Genes activated by Bmal1·CLOCK include per1, per2, and per3 (encoding Period 1, 2, and 3), and cry1 and cry2 (encoding Cryptochromes 1 and 2), Rev-erbα, and Rev-erbβ. Although Period and Cryptochrome proteins heterodimerize and feedback to inhibit the activator function of the Bmal1·CLOCK complex, Rev-erbs are potent repressors of both Bmal1 and CLOCK genes (23, 26).
Rev-erbs belong to the nuclear receptor superfamily, a group of ligand-responsive transcription factors that include receptors of steroid hormones like estrogen and testosterone. Nuclear receptors have a conserved modular structure consisting of a DNA-binding domain harboring two 4-Cys zinc fingers, a flexible hinge region, and an adjoining globular ligand-binding domain (LBD). In addition to their role as circadian factors, Rev-erbs repress the transcription of genes involved in gluconeogenesis, lipid metabolism, and inflammatory pathways, entraining these processes to the molecular clock (5, 27–33). Rev-erbs do so by interacting with the nuclear receptor co-repressor (NCoR1), which recruits histone deacetylase complexes to the promoters of target genes. Our group and others have shown that Rev-erbs bind heme, which indirectly facilitates the interaction with NCoR1 and has a modest effect on promoting degradation (5, 34, 35). Rev-erbs bind Fe3+-heme in a 6-coordinate complex with conserved His and Cys residues, the latter provided by an HRM (36). Previously, our group reported that under oxidizing conditions, the Rev-erbβ HRM axial thiolate, Cys-384, forms a disulfide with neighboring Cys-374 lowering heme affinity from 23 to 117 nm (37). Moreover, reduction of Fe3+-heme to Fe2+-heme is accompanied by dissociation of the HRM axial thiolate to a mixed 5/6 coordinate system (38); however, based on equilibrium titrations, this iron redox change appears to have little effect on heme affinity (37). These results suggested that the various activities of Rev-erbs, e.g. heme-binding, repression, and degradation, may be modulated by thiol-disulfide conversion or heme redox state.
In this paper, we further investigate the role of the HRM thiolate in regulating Rev-erbβ activity. Utilizing stopped-flow UV-visible spectrophotometry and myoglobin competition assays, we dissected the kinetics of Fe3+- and Fe2+-heme binding to Rev-erbβ. These studies were performed in a unique buffer system containing caffeine, which we and others have found to stabilize monomeric heme and allow for the determination of second-order on-rates for heme binding (39–41). Kinetic analyses provide Kd values for Fe3+-heme in the picomolar range, far lower than those previously reported using equilibrium-based methods. Significantly, loss of the HRM heme axial thiolate by disulfide bond formation or by reduction of Fe3+- to Fe2+-heme has a major effect on lowering heme affinity (by at least 20-fold but by as much as 4 orders of magnitude). Furthermore, substitution of the cysteine thiolate in the HRM with an alanine significantly diminishes NCoR1 binding in a co-immunoprecipitation assay, suggesting the Rev-erbβ HRM is critical for stabilizing the co-repressor complex. These results implicate redox processes, including thiol-disulfide conversion and heme oxidation state, as regulatory mechanisms controlling Rev-erb heme occupancy, repressor activity, and ultimately circadian rhythm and metabolism.
Results
Previous equilibrium titrations yielded Kd values for Fe3+-heme complexes with wild-type and C384A LBDs of 23 ± 3 and 46 ± 14 nm (Table 1), respectively, suggesting the axial thiolate has a minor impact on heme affinity (37). However, recent studies from our laboratory have shown that equilibrium-based methods for determining heme affinities are inaccurate. Standard assays for measuring heme Kd values, such as UV-visible spectrophotometric titrations, use micromolar concentrations of heme and protein. Because heme Kd values are generally low nanomolar and below, during the titration the majority of heme is protein-bound; therefore, the concentration of free heme cannot be assumed to be equal to that of total heme. The expansion of the simple hyperbolic binding curve to include the concentration of free ligand leads to a quadratic form that has been used to fit tight-binding stoichiometric data like those acquired with heme titrations (originally described by Morrison (42)). In our experience, the quadratic fit is also limited in that its accuracy depends on the sharpness of the curve around the point of saturation. When Kd is in the single digit nanomolar range and below, the standard error of the quadratic fit is too high to yield an accurate estimation; therefore, the quadratic equation yields only the upper limit of the Kd value (35, 43). Instead, analysis of heme-binding kinetics provides the most accurate estimation of affinity. Thus, assessing the effect of redox changes on the affinity of Rev-erbβ for heme required the development of a kinetic system to establish accurate Kd values. As described below, by measuring the dissociation (koff) and association (kon) rate constants for interactions of Rev-erbβ with Fe3+-heme, we obtained Kd (koff/kon) values that are significantly lower than reported previously.
Table 1.
Comparison of Rev-erbα/β, HO2(1-248), and myoglobin/hemoglobin dissociation constants obtained with different methods
ND means not determined.
a Heme oxidation state and the presence or absence of caffeine is indicated in parentheses.
b On-rates in parentheses are calculated from kobs2 (see Fig. 3).
c All off-rate data in this study and in Ref. 35 fit best to a double exponential equation yielding two rate constants for the fast and slow phases; Kd is calculated for each off-rate. Off-rates represent the average ± SD for 3–4 replicate experiments except for those rates reported for FLRev-erbβ C384A, which are the average ± range of duplicate experiments.
d Thiol-RED refers to the form of the LBD in which Cys-374 and Cys-384 are in the reduced state.
e Thiol-OX refers to the form of the LBD in which Cys-374 and Cys-384 exist in a disulfide bond.
To perform these kinetic analyses, we overcame two major problems. First, the low yields obtained during purification of full-length Rev-erbβ (FLRev-erbβ) prevented a thorough stopped-flow study to determine the heme kon value. Thus, we generated and purified a truncated construct (the LBD) consisting of an N-terminal MBP solubility tag fused to Rev-erbβ residues 370–579. The LBD exhibits heme Kd values similar to those of FLRev-erbβ (35, 37) and allows vastly (>30-fold) improved protein yields. The fusion protein was proteolytically digested, and the LBD was isolated for transient kinetic experiments.
The second major barrier to overcome in these kinetic studies related to the propensity of heme to form π-π stacked dimers in aqueous solution (44, 45). To ensure that our experiments were performed with monomeric heme solutions, we used methylxanthine (caffeine), which prevents heme dimerization (41, 46).
Caffeine as a reagent to prepare monomeric heme solutions for kinetic analyses
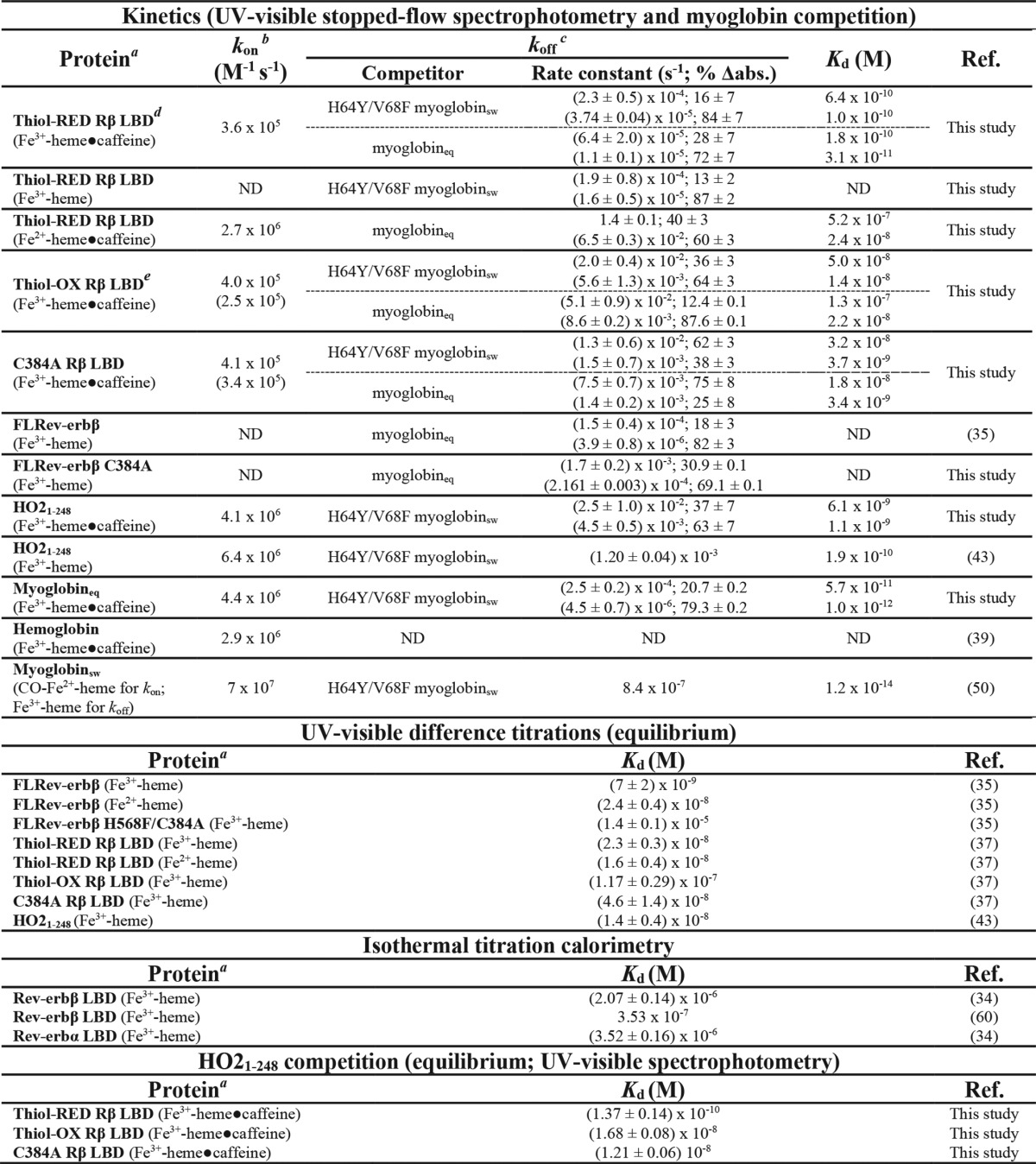
Rapid mixing of Fe3+-heme with varying concentrations of thiol-reduced LBD (thiol-RED; 370–579 contains two Cys residues, Cys-384 and Cys-374, that exist in the dithiol state, see under “Experimental procedures” for details) in 0.5× TNG buffer produced spectral changes consistent with heme binding to Rev-erbβ, namely formation of the Soret band centered at 423 nm (Fig. 1A). The 423-nm kinetic traces fit best to a quadruple exponential equation, but none of the observed rate constants were dependent on [LBD] (Fig. 1, A, inset, and B) suggesting that a rate-limiting first-order process precedes axial ligation of heme, undoubtedly the dissociation of the heme dimer or higher oligomers. Similar observations have been made with hemoglobin binding free heme in aqueous solution, which is also thought to be due to heme oligomer dissociation (47).
Figure 1.
First-order dissociation of heme oligomers precedes Fe3+-heme binding to thiol-RED LBD. A, spectra acquired in PDA mode after rapid mixing of 2.5 μm Fe3+-heme and 12.5 μm thiol-RED LBD in 0.5× TNG buffer lacking caffeine. The initial (0.003 s; red trace), final (500 s; blue trace), and intervening spectra (black traces) depicted are from a representative acquisition. Inset, 2.5 μm Fe3+-heme was rapidly mixed 12.5 μm (maroon circles), 25 μm (orange circles), 50 μm (maize circles), 100 μm (green circles), and 200 μm (teal circles) thiol-RED LBD, and the resulting changes at 423 nm were recorded in single-wavelength mode; there is extensive overlap in kinetic traces due to the rate-limiting first-order dissociation of heme oligomers. The data were normalized to a 100% scale and represent the average of four acquisitions. B, 423-nm traces in A fit best to a quadruple-exponential equation yielding kobs1–4 (kobs1, closed circles; kobs2, open circles; kobs3, closed inverted triangles; kobs4, open triangles) that are plotted as a function of thiol-RED LBD concentration. Data represent the average ± S.D. from four acquisitions, where each replicate acquisition was individually analyzed for rate constants.
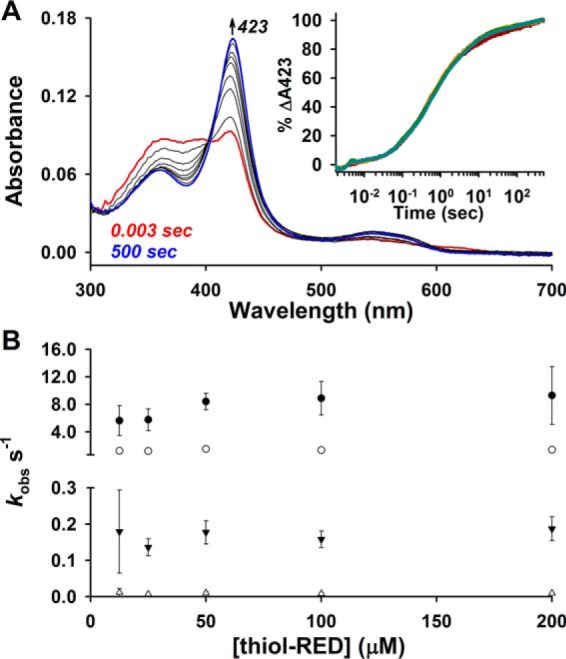
A common method to circumvent heme dimer formation is to perform rapid binding analyses with reduced and CO-treated heme, CO-Fe2+-heme. However, our studies require the use of Fe3+-heme because Cys-384 is dissociated in the CO-Fe2+-heme·LBD complex. High concentrations of caffeine in aqueous solution have been found to dissociate Fe3+- and Fe2+-heme dimers, likely by disrupting π-π interactions (41). Titrating caffeine (>1 mm) into a solution of Fe3+-heme in 0.5× TNG buffer led to spectral changes consistent with those previously observed and ascribed to formation of the monomeric Fe3+-heme·caffeine complex (Fig. 2A) (41). Low concentrations of caffeine (<0.7 mm) led to a uniform decrease in the absorbance of the free Fe3+-heme spectrum, but it did not yield the characteristic Fe3+-heme·caffeine band at 404 nm suggesting low caffeine concentrations may disrupt higher order heme oligomers, whereas high caffeine levels split the heme dimer into monomers (supplemental Fig. S1). Plotting the difference absorbance at 406 nm versus [caffeine] and fitting with a hyperbolic expression yielded a Kd for Fe3+-heme·caffeine of 2.9 ± 0.2 mm (Fig. 2B). Similarly, increasing caffeine concentrations led to spectral changes consistent with formation of the monomeric Fe2+-heme·caffeine complex (Fig. 2C); however, the Kd of 0.29 ± 0.04 mm is 10-fold lower than for Fe3+-heme (Fig. 2D).
Figure 2.

Caffeine forms a complex with Fe3+/2+-heme leading to monomeric aqueous heme solutions. A, UV-visible spectra acquired during titration of 2.5 μm Fe3+-heme with caffeine in 0.5× TNG. Caffeine (1 m in 0.5× TNG; the stock was kept at 90 °C during the titration to maintain caffeine solubility) was incrementally titrated into both sample (1 ml 0.5× TNG containing 2.5 μm Fe3+-heme) and reference (1 ml buffer) cuvettes in a double-beam spectrophotometer, and the difference spectrum was recorded. Inset, difference spectra generated by subtracting the spectrum of heme in the absence of caffeine from those acquired during the caffeine titration. B, plot of the difference absorbance at 406 nm as a function of caffeine concentration. The data from two independent titrations are plotted with the average fit with a hyperbolic expression (black trace). Based on the fit, 25 mm caffeine in 0.5× TNG yields ∼90% monodisperse heme assuming a direct correlation between those absorbance changes observed and monomeric heme levels. C, same as A, except with Fe2+-heme in 0.5× TNG buffer containing 5 mm dithionite; samples containing heme and various concentrations of caffeine were prepared in an anaerobic chamber, transferred to quartz cuvettes, sealed with rubber septa, and the spectra acquired at the bench. D, plot of the difference absorbance at 416 nm as a function of caffeine concentration. The data represent the average ± S.D. of 3–6 replicates and are fit with a hyperbolic expression (black trace) yielding the depicted Kd. E, UV-visible spectra acquired after rapid mixing of 2.5 μm Fe3+-heme and 1 mm caffeine in 0.5× TNG buffer; inset, kinetic traces at 397 nm were fit with a single-exponential equation, and the observed rate constant (kobs) for caffeine binding plotted against the [caffeine] (data are the average ± S.D. of six acquisitions; the slope of the linear regression (kon) and y intercept (koff) are depicted). F, same as E except with Fe2+-heme; koff was calculated from Kd (obtained by equilibrium titration in D) and the slope of the linear regression of kobs versus [caffeine], i.e. kon. G, effect of varying caffeine concentrations on kobs for the initial Fe3+-heme-binding event to thiol-RED LBD (open circles) or kobs1 (open triangles) and kobs2 (open inverted triangles) for Fe2+-heme binding. 2.5 μm Fe3+- or Fe 2+-heme in 0.5× TNG (5 mm dithionite was present for those experiments with Fe2+-heme) containing various concentrations of caffeine was mixed with 25 μm thiol-RED LBD in the same buffer. Data represent the average ± S.D. from 4 to 7 acquisitions. H, UV-visible spectra of Fe3+-heme complexes with thiol-RED LBD residues 370–579 in 0.5× TNG ± 25 mm caffeine. 3 μm protein was mixed with 1.5 μm Fe3+-heme and the spectrum acquired after reaching equilibrium at 20 °C. Spectra are the result of averaging several replicates acquired on different days.
We also analyzed the transient kinetics of caffeine binding to Fe3+/2+-heme under pseudo first-order conditions. Caffeine binding to Fe3+-heme proceeds through a single kinetic phase with an observed rate constant that is linearly dependent on the concentration of caffeine with a kon value of (8.3 ± 0.3) × 104 m−1 s−1 and a koff value of 109 ± 12 s−1, where koff/kon = Kd = 1.3 mm, similar to the Kd of 2.9 mm determined with an equilibrium titration (Fig. 2, B and E). Although caffeine binding to Fe2+-heme also proceeded through a single kinetic phase, kobs values were >60-fold lower than for Fe3+-heme. Notably, caffeine concentrations less than 8 mm yielded complex Fe2+-heme-binding kinetic traces that did not fit well to a single exponential equation. When caffeine is present at ≥8 mm, kobs is linearly dependent on [caffeine] with a regression yielding a kon value of 56 ± 3 m−1 s−1. The y-intercept of the regression is negative; thus, we cannot estimate koff based on this plot; however, using the Kd of 0.29 mm from the equilibrium titration in Fig. 2D, the koff is calculated to be 0.016 s−1.
Rapid mixing of 2.5 μm Fe3+-heme with 25 μm thiol-RED LBD showed that kobs1 for the initial heme-binding event is dependent on the concentration of caffeine (Fig. 2G). A similar observation was made with Fe2+-heme, although both kinetic phases (kobs1 and kobs2) were dependent on [caffeine]. Counterintuitively, kobs1 for Fe3+-heme binding decreases with caffeine concentrations above 2 mm, whereas those for Fe2+-heme decrease above 5 mm. Thus, it appears as though monomeric heme binds the LBD slower than oligomers, or high concentrations of caffeine compete with the LBD for heme. A similar trend was observed with heme binding to hemopexin in varying concentrations of caffeine indicating the effect is due to the heme oligomeric state and not on the hemoprotein (40). In that study, the authors used 25 mm caffeine to determine the second-order on-rate for heme binding because at that concentration heme is predominantly monomeric and the effect on kobs is nearly saturated. An additional study had successfully utilized 25 mm caffeine to determine the second-order on-rate for Fe3+-heme binding to hemoglobin (39). Thus, we performed our experiments at 25 mm caffeine to compare our results with the aforementioned studies and to ensure, as indicated in Fig. 2, that Fe3+- and Fe2+-heme are ≥90% saturated with caffeine. The presence of 25 mm caffeine also had a negligible effect on the spectrum of the Fe3+-heme·thiol-RED LBD complex suggesting that heme-binding properties are unperturbed by high concentrations of caffeine (Fig. 2H).
Transient kinetic experiments with the LBD: Measurement of kon for Fe3+/2+-heme by stopped-flow UV-visible spectrophotometry
When using a photodiode array (PDA) detector to monitor Fe3+-heme binding to thiol-RED LBD, two major spectral changes were observed as follows: a decrease in absorbance at 404 nm corresponding to the loss of the Fe3+-heme·caffeine complex and an increase at 423 nm representing formation of the Fe3+-heme·thiol-RED LBD complex (Fig. 3A). Kinetic traces for the data obtained at both 423 and 404 nm fit best to a quadruple exponential equation (see supplemental Fig. S2, A–C, H, and I for fits and residuals) indicating the binding process is complex and proceeds through multiple steps. The observed rate constant for the initial rapid phase (kobs1), which makes up >50% of the total Δabsorbance (supplemental Table S1), shows hyperbolic dependence on the concentration of LBD (Fig. 3, B and C, for 423 nm and supplemental Fig. S2, G and J for 404 nm), behavior consistent with a two-step binding mechanism previously observed in other hemoproteins (43, 48, 49). The data fit well to a hyperbolic model (Equation 2) in which heme initially binds weakly to hydrophobic patches on the periphery of the protein (k1) with a first-order fast off-rate (k−1); thus k−1/k1 is the Kd for this initial weak encounter complex. Next, heme associates with the binding pocket, including the axial ligands, with a first-order rate constant, kcoordination; kcoordination/(k−1/k1) represents the second-order rate constant (kon) for heme binding (Fig. 4). The kon, calculated from 404 and 423 nm data, was 2.7 × 105 and 3.6 × 105 m−1 s−1 (Fig. 3C and supplemental Fig. S2J), respectively. Considering the 5–11% error in kcoordination and k−1/k1, these kon values are similar, indicating the process causing initial loss of absorbance at 404 nm is the same one that leads to the increase at 423 nm. The kobs for the remaining three kinetic phases is not dependent on the concentration of LBD, indicating these are first-order reactions (supplemental Fig. S2, D–F, K, and L) and may represent subtle changes in the way heme interacts with the binding pocket or axial ligands that ultimately manifest as a change in extinction coefficient of the Soret.
Figure 3.

Stopped-flow UV-visible spectrophotometry studies to determine the second-order on-rates for Fe3+/2+-heme binding to Rev-erbβ LBD derivatives in 0.5× TNG with 25 mm caffeine. A, spectra acquired in PDA mode after rapid mixing of 2.5 μm Fe3+-heme and 12.5 μm thiol-RED LBD with the initial (0.003 s; red trace), final (200 s; blue trace), and intervening spectra (black traces) depicted, and those wavelengths used for kinetics analyses. B, 2.5 μm Fe3+-heme was rapidly mixed with 12.5 μm (maroon circles), 25 μm (orange circles), 50 μm (maize circles), 100 μm (green circles), 150 μm (teal circles), and 200 μm (blue circles) thiol-RED LBD, and the resulting changes in the absorbance spectrum were recorded in PDA mode. The data at 423 nm were normalized to a 100% scale and fit best to a quadruple-exponential equation (solid black lines). Data represent the average of 4–5 acquisitions. C, plot of kobs1 (representing the dominant initial fast phase that comprises 67% of the total Δabsorbance) versus [thiol-RED LBD]. Data represent the average ± S.D. of 4–5 acquisitions, and are fit with Equation 2. D–F are the same as A–C but with thiol-OX LBD. Data at 414 nm were acquired in single-wavelength mode, and fit best to a double-exponential equation (E; fits are solid black lines). Both kobs1 (blue circles, left y axis) and kobs2 (red circles, right y axis) show hyperbolic dependence on [thiol-OX LBD] and are fit with Equation 2 to yield the depicted second-order on-rates (fits and on-rates are color-coded to match data points and axis labels; data represent the average ± S.D. from five acquisitions). G–I are the same as D–F but with C384A LBD. Data at 414 nm fit best to a triple-exponential equation where kobs1 and kobs2 show hyperbolic dependence on [C384A] and represent the average ± S.D. from 6 to 7 acquisitions; kobs3 is not concentration-dependent (data not shown). J–L are the same as G–I but with Fe2+-heme binding to thiol-RED LBD. The data at 428 nm fit best to a double-exponential equation where the predominant fast phase corresponding to kobs1 demonstrates hyperbolic dependence on [thiol-RED] and is fit with Equation 2 yielding the depicted second-order on-rate (data represent the average ± S.D. of five acquisitions); kobs2 is not dependent on [thiol-RED].
Figure 4.

Reactions of heme with caffeine and Rev-erbβ. A, rate constants of Fe3+/2+-heme·caffeine association (k1) and dissociation (k−1) are depicted (refer to Fig. 2). Based on previous studies, caffeine disrupts π-π interactions between heme dimers (and higher oligomers) to yield a monomeric 1:1 heme·caffeine complex formed largely through hydrophobic interactions (41, 46). B, scheme depicting the two-step binding mechanism of heme and Rev-erbβ. Initially, the heme·caffeine complex binds to the periphery of Rev-erbβ through hydrophobic interactions with concomitant dissociation of caffeine from heme; the association rate constant, k1, is second order, whereas dissociation of the weak complex proceeds via k−1, where k−1/k1 is the Kd value of the weak complex. Heme then associates with axial ligands via the first-order rate constant, kcoordination. The dissociation rate constant for the tight complex, koff, is empirically determined with apomyoglobin competition (see Fig. 6).
Because kcoordination for Fe3+-heme binding to thiol-RED LBD (81 s−1) approaches koff of the Fe3+-heme·caffeine complex (109 s−1), one concern is that the hyperbolic dependence of kobs is due to the rate-limiting dissociation of Fe3+-heme·caffeine at high concentrations of LBD. To address this issue, we followed binding of Fe3+-heme (in the presence of caffeine) to apomyoglobin from equine skeletal muscle (apomyoglobineq) and heme oxygenase-2 (HO2(1–248); a construct encompassing the catalytic core of HO2). If dissociation of the heme·caffeine complex were rate-limiting at high concentrations of protein, one would expect to see identical kcoordination values for different hemoproteins. For apomyoglobineq, we observed two distinct kinetic phases for Fe3+-heme binding: a rapid process constituting 80% of the total amplitude with kobs1 values following a hyperbolic relationship with respect to [apomyoglobineq] and giving a kon = 4.4 × 106 m−1 s−1, followed by a first-order reaction with kobs2 ∼0.4 s−1 (Fig. 5, A–C, Table 1, and supplemental Table S1). These results are similar to those reported for Fe3+-heme binding to apohemoglobin in the presence of 25 mm caffeine, which proceeds via an initial rapid binding event with kon = 2.9 × 106 m−1 s−1 and a slower first-order process (1 s−1) (39). The kcoordination value (357 s−1) from the fit of kobs1 versus [apoglobin] is 4.4-fold greater than that for Fe3+-heme binding to thiol-RED LBD. For HO2(1–248), Fe3+-heme binding proceeds through two kinetic phases, a rapid process and a slower step that appears to be tightly coupled to the initial phase as evidenced by a clear isosbestic point in the kinetic plot (Fig. 5, D and E). The plot of kobs1 versus [HO2(1–248)] for the initial binding phase does not exhibit strong hyperbolic behavior like Rev-erbβ and myoglobineq; however, fitting the data with both Equation 2 and a linear regression give similar kon values of 4.1 × 106 and 2.8 × 106 m−1 s−1, respectively, which agree well with kon for Fe3+-heme in the absence of caffeine of 6.4 × 106 m−1 s−1 (see Table 1) (43). The kcoordination value from the hyperbolic fit in Fig. 5F is 2141 s−1 (supplemental Table S1), 26.4-fold higher than that for thiol-RED LBD; thus, the significant differences in the kcoordination values between myoglobineq, HO2(1–248), and thiol-RED Rev-erbβ indicate that these rate constants indeed relate to the kon, i.e. binding of Fe3+-heme to the LBD and not to dissociation of the heme·caffeine complex. When kobs values approach and exceed the dissociation rate constant of Fe3+-heme·caffeine (109 s−1), it is likely that the heme·caffeine complex initially binds to the protein with concomitant dissociation of caffeine from heme.
Figure 5.

Stopped-flow UV-visible spectrophotometry studies to determine the second-order on-rates for Fe3+-heme binding to HO2(1–248) and apomyoglobineq in 0.5× TNG with 25 mm caffeine. A, spectra acquired in PDA mode after rapid mixing of 2.5 μm Fe3+-heme and 12.5 μm apomyoglobineq. B, data at 408 nm (collected in single-wavelength mode) were normalized to a 100% scale, and fit best to a double-exponential equation (solid black lines; apoglobin concentrations, 12.5 μm (maroon circles); 25 μm (orange circles); 37.5 μm (maize circles); 50 μm (green circles); 62.5 μm (teal circles); 75 μm (blue circles); and 100 μm (purple circles)). Data represent the average of eight acquisitions. C, plot of kobs1 (blue circles, left y axis) and kobs2 (red circles, right y axis) versus [apoglobin]. Data represent the average ± S.D. of eight acquisitions. The data for kobs1 are fit with Equation 2, and the second-order on-rate calculated from the fit parameters is depicted. kobs2 representing the second slow phase is not dependent on [apoglobin]. D–F are the same as A–C but with HO2(1–248). Data at 406 nm were acquired in single-wavelength mode and the data fit best to a double-exponential equation (E; fits are solid black lines; HO2(1–248) concentrations, 12.5 μm (maroon circles); 25 μm (orange circles); 50 μm (maize circles); 100 μm (green circles); 150 μm (teal circles); and 243 μm (blue circles)). kobs1 demonstrates weak hyperbolic dependence on [HO2(1–248)] (blue circles, left y axis; average ± S.D. of 4–5 acquisitions) and is fit with both Equation 2 (solid blue line) and a linear regression (dotted blue line) yielding the depicted second-order on-rates. kobs2 (red circles, right y axis), representing the second slow phase, is inversely dependent on [HO2(1–248)].
To test whether loss of the heme axial thiolate of Rev-erbβ (provided by Cys-384 from the HRM) affects kon, a C384A variant was constructed. Fe3+-heme binding to C384A was monitored in single-wavelength mode at 414 nm (Fig. 3G). At low concentrations of C384A (<100 μm), the kinetic data fit best to a double-exponential equation; however, above 100 μm, an additional kinetic phase (correlating to kobs2) emerged, necessitating the use of a triple-exponential. To maintain consistency in our analysis, a triple-exponential equation was used to analyze all of the data (see under “Experimental procedures” for more details). As observed with wild-type thiol-RED LBD, the initial rapid binding phase makes up the majority of the total Δabsorbance (61%), with kobs1 showing hyperbolic dependence on [C384A]. The data fit well to Equation 2, yielding kon = 4.1 × 105 m−1 s−1, which is very similar to that of the wild-type protein and indicates that Cys-384 has a negligible influence on the initial heme binding kinetics. Curiously, kobs2 for the second kinetic phase, which emerges at LBD concentrations of >100 μm also appears to be hyperbolically dependent on the concentration of C384A yielding (according to Equation 2) a similar kon = 3.4 × 105 m−1 s−1. However, we are skeptical that this phase represents a mixed-order process because only those kobs2 values for C384A concentrations below 100 μm are concentration-dependent, where we fixed the Δabsorbance parameter in the fit. What is more likely is that at low concentrations of C384A the first and second kinetic phases have similar rate constants and overlap; only at high [C384A] is the disparity between kobs1 and kobs2 large enough to observe the two phases independently. In any case, we show the data as a mixed-order process in Fig. 3 and provide kon values in Table 1. kobs3 for the third kinetic phase is not dependent on the concentration of C384A.
In a previous study, we had reported that under oxidizing conditions, HRM Cys-384 forms a disulfide with neighboring Cys-374 (37). To test whether thiol oxidation affects kon, the LBD construct was treated with diamide and desalted, with oxidation confirmed using a DTNB assay. This form of the protein is hereafter referred to as thiol-OX LBD. Interestingly, the stopped-flow results of Fe3+-heme binding to thiol-OX LBD were very similar to those with C384A except the data set was less complex, requiring only a double-exponential fit when [LBD] was >100 μm (the same strategy for fitting C384A data was used here) (Fig. 3, D–F). The kobs1 for the initial binding step is hyperbolically dependent on the concentration of thiol-OX; thus, fitting the data to Equation 2 yielded kon = 4.0 × 105 m−1 s−1, nearly identical to that for C384A and wild type. A second kinetic phase emerged at [thiol-OX] >100 μm as observed with C384A, but it only made up 30% of the total Δabsorbance.
Reduction of Fe3+- to Fe2+-heme causes Cys-384 to dissociate, leading to a mixed 5/6-coordinate complex (38). Based on stopped-flow studies, formation of the Fe2+-heme·thiol-RED LBD complex leads to an increase in absorbance at 428 nm (Fig. 3J). Kinetic plots fit best to a double-exponential equation with the first phase contributing 86% to the total Δabsorbance and kobs1 showing hyperbolic dependence on thiol-RED concentration with kon = 2.7 × 106 m−1 s−1; kobs2 for the second kinetic phase is not dependent on [thiol-RED] and represents a first-order reaction (Fig. 3, K and L). Because kobs1 values (23–133 s−1) far exceed the dissociation rate constant of Fe2+-heme·caffeine (0.016 s−1), we conclude that the Fe2+-heme·caffeine complex initially binds to the LBD. Based on several lines of evidence, we also posit that caffeine concomitantly dissociates from heme upon protein binding as follows: 1) we failed to observe a unique spectrum of a ternary complex between heme, caffeine, and protein, which we would assume would be present in all transient kinetics experiments independent of the protein analyzed; 2) caffeine forms relatively weak complexes with heme through hydrophobic interactions with the porphyrin moiety; thus upon binding of heme to hydrophobic patches of the protein, caffeine would be effectively displaced (41, 46); and 3) if caffeine dissociation from protein-bound heme were slow, we would expect kcoordination values to be similar from protein to protein; however, kcoordination for apomyoglobin and HO2(1–248) are at least 4.4-fold greater than for Fe3+-heme binding to thiol-RED LBD.
Transient kinetic experiments with the LBD: Determination of koff for Fe3+/2+-heme·LBD complexes by apomyoglobin competition and calculation of global dissociation constants
Our stopped-flow studies show that Cys-384 of the HRM has a negligible effect on the initial steps of heme binding with thiol-OX, and C384A LBD derivatives having kon values within 2-fold of the wild-type thiol-RED. A similar observation has been made with myoglobin where site-directed variants of heme axial ligands have a negligible impact on kon for CO-Fe2+-heme binding to sperm whale apomyoglobin (apomyoglobinsw) (50). In contrast, our group and others have shown that the first-order heme dissociation rate constant, koff, is the main determinant of overall affinity and is dramatically affected by mutagenesis of heme axial ligands (35, 43, 51, 52). To establish accurate Kd values for the complexes of Fe2+/3+-heme with LBD derivatives, we utilized apomyoglobin competition assays to measure koff in the same buffer system used to establish kon, i.e. 0.5× TNG with 25 mm caffeine. Traditionally, others have used the H64Y/V68F apomyoglobinsw variant for these studies due to its stability and unique charge transfer band at 600 nm, which allows for easy discrimination between globin and hemoprotein (52). Under the conditions of our assays, apomyoglobineq is sufficiently stable; thus, we measured koff with both competitors (apomyoglobineq and the H64Y/V68F variant of apomyoglobinsw) except for studies with Fe2+-heme when only apomyoglobineq was used due to its high affinity for ferrous heme (Fig. 6 and Table 1).
Figure 6.
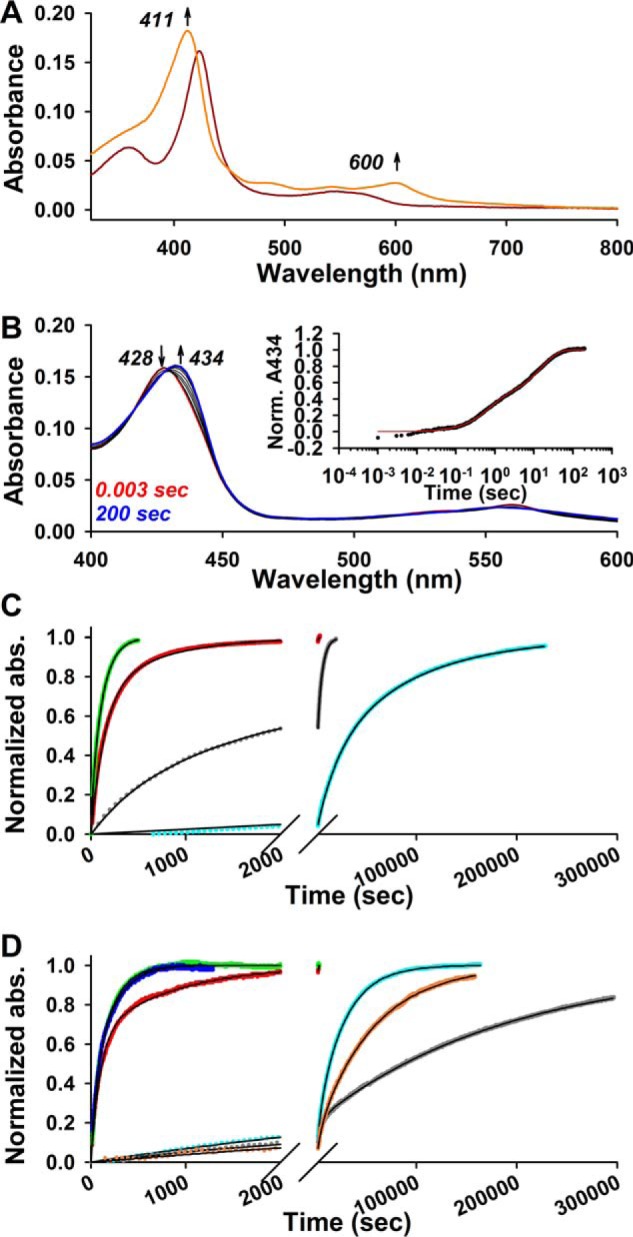
Disparate heme off-rates between wild-type Rev-erbβ and heme axial ligand variants. First-order Fe3+/2+-heme off-rates were measured using apomyoglobin as a heme competitor (see “Experimental procedures” for details). A, UV-visible spectra acquired during a typical off-rate assay. A spectrum of the Fe3+-heme·thiol-RED LBD complex (1.5 μm heme, 3 μm protein) in 0.5× TNG buffer with 25 mm caffeine is acquired prior to the addition of competitor (dark red trace). A 10-fold excess of H64Y/V68F apomyoglobinsw is added, and the absorbance at 411 and 600 nm is recorded over time representing the formation of the Fe3+-heme·H64Y/V68F complex. ∼46 h after the addition of apomyoglobin, heme is completely competed away from thiol-RED LBD and the spectrum acquired (orange trace) revealing the distinct charge transfer band at 600 nm characteristic of the H64Y variant. B, Fe2+-heme off-rate determined with stopped-flow kinetics. Spectra were acquired in PDA mode after mixing Fe2+-heme·thiol-RED LBD complex (1.5 μm heme, 3 μm protein) with 30 μm apomyoglobineq; the increase at 434 nm indicates formation of the Fe2+-heme·myoglobineq complex. Inset, plot of the normalized A434 data (average of four acquisitions; black circles) and the double-exponential fit (red trace). C, Fe3+-heme off-rates measured with apomyoglobineq in 0.5× TNG with 25 mm caffeine (unless otherwise stated). A 10-fold excess of apomyoglobineq was added to the pre-formed complex of 1.5 μm Fe3+-heme·3 μm Rev-erbβ derivative, and the A408 was monitored over time. Kinetic data and fits are normalized for comparison as follows: thiol-OX LBD (green circles), C384A LBD (red circles), FL-C384A (gray circles; no caffeine), and thiol-RED LBD (cyan circles). Data fit best to a double-exponential equation (solid black lines) with the first-order off-rate constants from each phase compiled in Table 1; data are from a representative replicate. D, same as in C, except H64Y/V68F apomyoglobinsw was used as the competitor and the A411 or A600 was monitored and normalized for comparison. Thiol-OX LBD (green circles; A600), C384A LBD (red circles; A600), and thiol-RED LBD in the presence (cyan circles; A411) and absence (salmon circles; A411) of 25 mm caffeine are shown. Off-rate data for HO2(1–248) (blue circles; A600) and myoglobineq (gray circles; A600) using H64Y/V68F as a competitor is also depicted for comparison. All data were fit to a double-exponential equation (solid black lines). Data are from a representative replicate.
All off-rate data fit best to a double-exponential equation suggesting either the dissociation mechanism is a multistep process or our LBD preparations are heterogeneous with subpopulations having different Kd values for heme. The former is implicit in the two-step binding mechanism offered by Equation 2 and in Fig. 4B. Equation 2 assumes that dissociation of the weak, nonspecific complex between heme and apolar regions of the protein (k−1) far exceeds the rate of kcoordination and that koff is infinitesimally small (48, 53). Under such conditions, dissociation of heme from Rev-erbβ is rate-limited by koff, and k−1 is rapid (especially in the presence of a heme scavenger like apomyoglobin) and would presumably manifest as a single kinetic phase. The latter scenario might arise if the LBD is in equilibrium between multiple quaternary structures, e.g. monomer, dimer, etc. However, analytical gel filtration analysis of thiol-RED and C384A LBDs indicates the preparations are monomers in both apo- and holo-forms (Fig. 7), except that apo- and holo-thiol-OX LBD contain populations of dimers and higher oligomers (≤32% of the total based on peak integration). Biphasic heme off-rates have been observed with many heme proteins (51, 52, 54–57), including HO2(1–248) and apomyoglobineq described herein (Table 1 and Fig. 6). Without sufficient evidence to describe the molecular basis of the two kinetic phases, we provide Kd values for Fe2+/3+-heme·LBD complexes using both off-rates and the second-order kon from the initial rapid binding step. The global dissociation constant for the two-step binding mechanism is shown in Equation 1 (58),
| (Eq. 1) |
Figure 7.
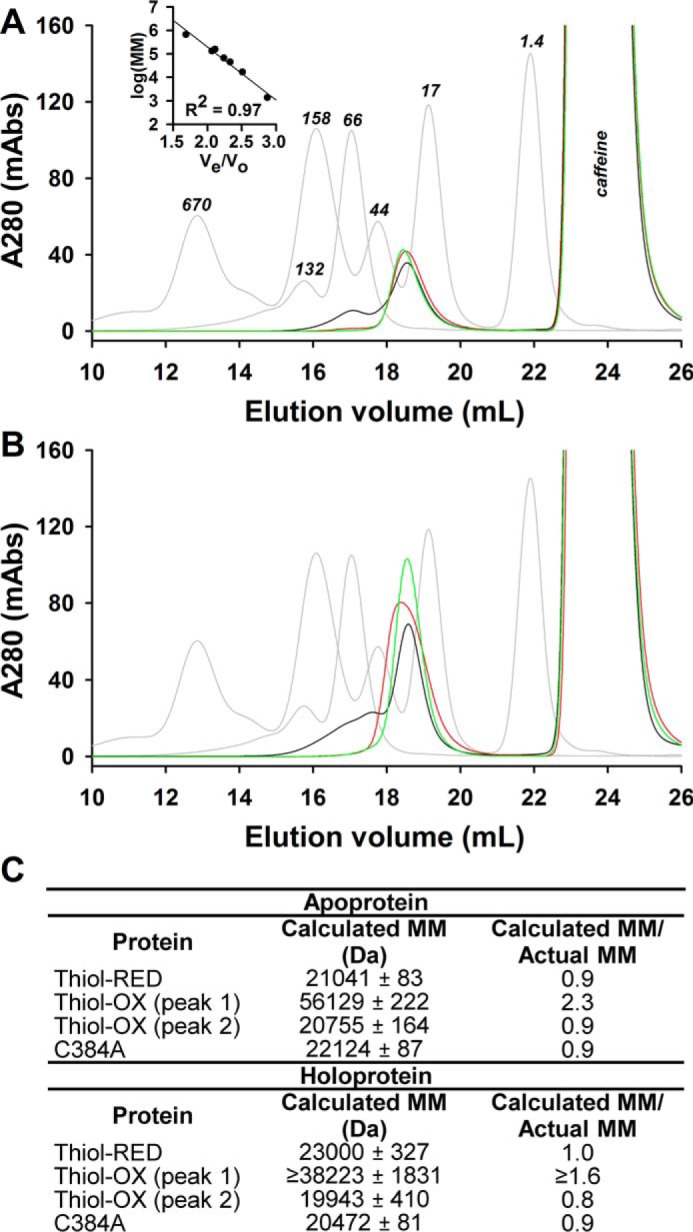
Quaternary structure determination of thiol-RED, thiol-OX, and C384A LBDs using size-exclusion chromatography. A, Superose 6 10/300 GL elution profiles of molecular mass standards (Bio-Rad gel filtration standards and bovine serum albumin (monomer and dimer), gray traces) versus as purified LBD samples (thiol-OX, black trace; thiol-RED, red trace; C384A, green trace). Inset, the plot of Ve/Vo versus the logarithm of the standards molecular masses. Caffeine in the protein samples elutes at ∼24 ml as indicated. B, elution profiles of holoproteins. C, summary of the calculated molecular masses for LBD derivatives using the linear regression equation for the standards in A. Thiol-OX LBD is a mixture of monomers, dimers, and higher oligomers, and thiol-RED and C384A proteins are monomers. Heme has a negligible effect on quaternary structure under these conditions. Calculated molecular masses represent the average ± S.D. of three FPLC runs except for holo-thiol-OX protein, which is the average ± range of duplicate samples.
The expression simplifies to Kd = koff/(kcoordination/(k−1/k1)) or koff/kon when koff ≪ kcoordination and is eliminated from the denominator.
Off-rates for Fe3+-heme·thiol-RED LBD are between 2.3 × 10−4 s−1 and 1.1 × 10−5 s−1 with the slow kinetic phase providing the majority of the total amplitude (>70%) both in the presence and absence of 25 mm caffeine (Fig. 6, A, C, and D, for spectra and traces and Table 1 for rate constants). Depending on the competitor, koff-slow for the LBD is ∼3–10 times faster than that for FLRev-erbβ indicating the dimeric quaternary structure of the full-length protein or the presence of additional domains has a modest effect on stabilizing heme binding. Using those off-rates obtained with apomyoglobineq and kon = 3.6 × 105 m−1 s−1, we calculate a Kd range of 31–178 pm. Similar values are obtained in the studies using apomyoglobinsw H64Y/V68F (104–639 pm). Because the amplitude of the phase related to koff-slow predominates, the majority of the LBD preparation interacts with Fe3+-heme with a Kd ≤∼100 pm. Significantly, reduction of Fe3+-heme to Fe2+-heme with dithionite accelerates heme dissociation by several orders of magnitude with koff-fast = 1.4 s−1 and koff-slow = 0.065 s−1, which yield a Kd range of 24–519 nm. Based on the Kd value for Fe3+-heme of ∼100 pm, the affinity of Rev-erbβ for Fe2+-heme is at least 240-fold lower indicating that the heme redox state could act as a regulatory switch governing Rev-erbβ heme occupancy and thus its interaction with NCoR1.
Apomyoglobin competition assays with C384A LBD yield Fe3+-heme off-rates between 1.3 × 10−2 and 1.4 × 10−3 s−1 (Table 1), providing a Kd range of 3–32 nm using kon = 4.1 × 105 m−1 s−1. Sequestration of the axial thiolate as a disulfide in thiol-OX LBD provides Fe3+-heme off-rates from 5.1 × 10−2 s−1 to 5.6 × 10−3 s−1 and a Kd range of 14–128 nm using kon = 4.0 × 105 m−1 s−1. Thus, the loss of axial thiolate ligation to Fe3+-heme leads to at least a 30-fold decrease in affinity based on the thiol-RED Kd of ∼100 pm. Because Rev-erbβ has a high propensity to form the Cys-384–Cys-374 disulfide under oxidizing conditions, thiol-disulfide interconversion is also a potentially relevant redox switch controlling Rev-erbβ heme occupancy.
As a point of reference to compare those off-rates and Kd values described for Rev-erbβ, we also determined koff of Fe3+-heme from myoglobineq using H64Y/V68F apomyoglobinsw as a competitor due to its unique Tyr-Fe3+-heme charge transfer band at 600 nm (Table 1 and Fig. 6D). Similar to Rev-erbβ, kinetic traces for Fe3+-heme dissociation from myoglobineq were biphasic yielding koff-fast = (2.5 ± 0.2) × 10−4 s−1 and koff-slow = (4.5 ± 0.7) × 10−6 s−1, with the latter phase contributing ∼79% of the total amplitude. Using kon = 4.4 × 106 m−1 s−1 (Fig. 5C) and koff-slow, the Kd for the Fe3+-heme·myoglobineq complex is 1 pm; earlier estimates for heme·myoglobinsw Kd of 12 fm were based on kon values obtained with CO-Fe2+-heme, which binds >10-fold faster than Fe3+-heme in caffeine, at least partially accounting for the discrepancy (50). The role of heme as a scaffold for oxygen binding and transport in globins requires that it bind with high affinity; a Kd of 1 pm is well below estimated intracellular heme concentrations in the low nanomolar range (10), providing confidence that our kinetic measurements yield values that correlate with function.
Transient kinetic experiments with full-length Rev-erbβ: Determination of koff for Fe3+-heme·C384A FLRev-erbβ by apomyoglobin competition experiments
Previous biophysical and crystallographic analyses as well as the transient kinetics studies described herein using truncated Rev-erbβ LBD constructs have demonstrated Cys-384 is a Fe3+-heme axial ligand that dissociates upon heme reduction (36, 38). To ensure these properties are maintained in the context of the full-length protein, UV-visible spectra were acquired of wild-type and C384A FLRev-erbβ (Fig. 8). The spectra of wild-type Fe2+-heme, CO-Fe2+-heme, and NO-Fe2+-heme complexes are nearly identical to those of C384A in terms of wavelength maxima and extinction coefficients (<13% difference in ϵ for the Soret), indicating that the absence of Cys-384 has a negligible influence on the electronic properties of ferrous heme and gas complexes. In contrast, the Soret band of the C384A·Fe3+-heme complex of FLRev-erbβ is blue-shifted 6 nm in comparison with that of wild-type protein. We had previously observed a similar blue shift when comparing truncated C384A and wild-type LBD complexes with Fe3+-heme, and we confirmed by electron paramagnetic resonance experiments the loss of the heme axial thiolate in C384A (37). Thus, the current data confirm those studies done with truncated LBDs showing that in the full-length protein Cys-384 is a ligand to Fe3+-heme that dissociates upon heme reduction and gas binding.
Figure 8.
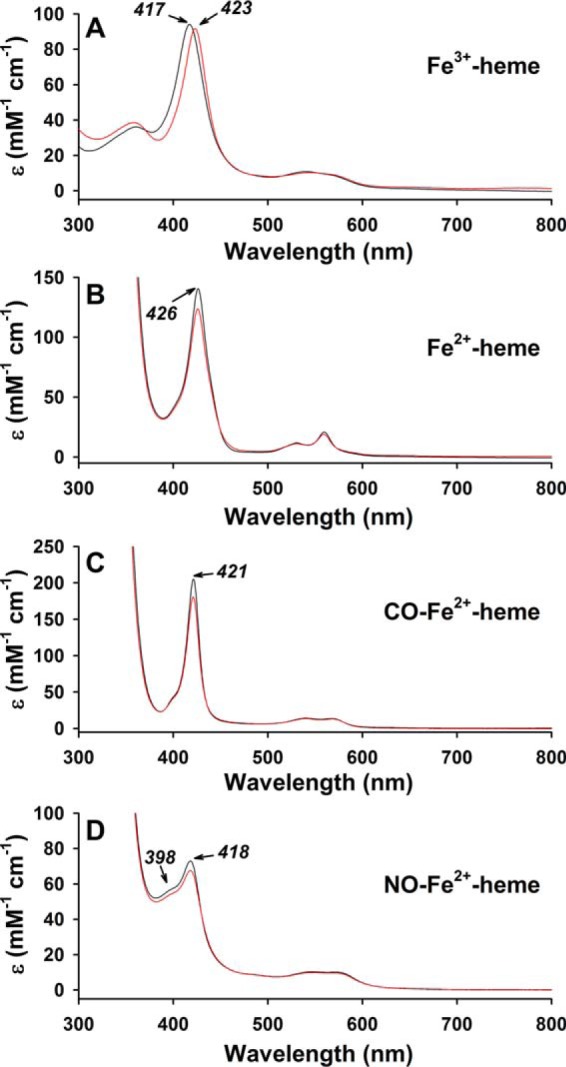
UV-visible spectra of wild-type (red traces) and C384A (black traces) FLRev-erbβ·heme complexes. A, 3 μm Fe3+-heme was mixed with 6 μm thiol-reduced wild-type or C384A FLRev-erbβ in 0.5× TNG buffer under anaerobic conditions, and the spectrum was recorded at 20 °C. B, 1 mm dithionite was added to prepare Fe2+-heme complexes (the reduction process is slow requiring 10–15 min to achieve spectral stability subsequent to dithionite addition), and the cuvette headspace was purged with CO gas, and the sample was mixed to form the CO-Fe2+-heme adduct (C). D, NO-Fe2+-heme adduct was prepared by first adding 0.5 mm dithionite to the Fe3+-heme·protein complex followed by 100 μm proli-NONOate. The NO-Fe2+-heme complex formed rapidly with a stable spectrum achieved within minutes. The addition of 100 μm proline (parent compound) had no effect on the spectrum of the Fe2+-heme·protein complex indicating the spectral changes were due to the formation of the NO adduct (data not shown). Spectra are plotted as ϵ (mm−1 cm−1) based on the concentration of heme.
Although we were unable to generate sufficient amounts of FLRev-erbβ for the stopped-flow studies required for measuring kon, we were able to determine koff, which is known to be the primary determinant of Kd for hemoproteins, most of which exhibit similar kon values for CO-Fe2+-heme (50, 51). Mixing excess apomyoglobineq with the FL-C384A·Fe3+-heme complex leads to an increase in absorbance at 408 nm, indicative of heme competition and binding to globin (Fig. 6C). The data fit best to a double-exponential equation yielding koff-fast = (1.7 ± 0.2) × 10−3 s−1 and koff-slow = (2.161 ± 0.003) × 10−4 s−1; the latter is the predominant kinetic phase contributing 69% of the total absorbance change. Comparison of that value with the koff for the predominant kinetic phase of wild-type FLRev-erbβ (see Table 1) (35), koff-slow = (3.9 ± 0.8) × 10−6 s−1 (82 ± 3% of the total Δabsorbance) indicates that loss of the axial thiolate leads to a 55-fold decrease in heme affinity, assuming a direct correlation between off-rate and Kd. Thus, Cys-384 does in fact provide a strong axial ligand to Fe3+-heme, suggesting that any processes, e.g. redox or gas-binding, that cause Cys-384 dissociation significantly decreases the affinity of Rev-erbβ for heme.
Fe3+-heme competition assays with HO2(1–248) corroborate Kd values determined by kinetic analyses
Although we are confident that the initial rapid binding phase in stopped-flow studies accurately represents kon for the heme·LBD complex, the biphasic kinetics of heme dissociation only allows us to provide a range for Kd. Thus, we established an equilibrium-based competition assay in which we varied the concentration of a competitor protein, whose Kd for heme is well established and is similar to that of the unknown against a constant concentration of heme and LBD. In this case, the distribution of heme bound to LBD or competitor depends on the relative Kd values of both proteins. Mechanistically, this type of assay is akin to competitive enzyme inhibition where both the substrate and inhibitor compete for binding to the enzyme, except here the LBD and competitor protein compete for heme. A complicating factor is that heme binds with high affinity to both LBD and competitor, similar to “tight-binding inhibitors” that do not follow the typical scenario where substrate and inhibitor concentrations far exceed their Km value for the enzyme. The Morrison equation (42) was developed to account for such behavior and is used herein (see “Experimental procedures” for more detail).
We chose HO2(1–248) as the competitor protein because its Soret band at ∼404 nm has a higher extinction and is blue-shifted in comparison with those of thiol-RED, thiol-OX, and C384A LBDs. The Kd value for the Fe3+-heme·HO2(1–248) complex was previously determined to be ∼0.2 nm in Tris-HCl buffer lacking caffeine (43). However, for consistency we measured kon (discussed above) and koff for Fe3+-heme·HO2(1–248) in 0.5× TNG buffer with 25 mm caffeine. The Fe3+-heme off-rate from HO2(1–248) is also biphasic with koff-fast = (2.5 ± 1.0) × 10−2 s−1 and koff-slow = (4.5 ± 0.5) × 10−3 s−1 (Table 1 and Fig. 6D), leading to a Kd range of 1.1–6.1 nm (using kon = 4.1 × 106 m−1 s−1, Fig. 5F). The amplitudes of the fast and slow phases are relatively similar, 37 and 63%, respectively; thus, we used the median value of the range, 3.6 nm, as the Kd for the Fe3+-heme·HO2(1–248) complex. Although HO2(1–248) has a lower affinity for Fe3+-heme in caffeine, it still serves as a good competitor because its Kd of 3.6 nm lies between those Kd values for thiol-RED, thiol-OX, and C384A LBDs.
Increasing concentrations of apoHO2(1–248) result in a blue shift of the Soret band of thiol-RED, thiol-OX, and C384A LBDs to ∼404 nm (Fig. 9, A–C), indicative of forming the Fe3+-heme·HO2(1–248) complex. The spectrum of Fe3+-heme-bound HO2(1–248) (Fig. 9A, red trace) was used to normalize the data in terms of the ratio of HO2(1–248) occupancy versus the concentration of HO2(1–248). In fitting the data to the Morrison equation (Equation 3) to determine the Kd value of the Fe3+-heme·Rev-erbβ complex, the Ki value for Fe3+-heme and HO2(1–248) was held constant at 3.6 nm and the concentration of Fe3+-heme at 2.5 μm for C384A or 2.76 μm for thiol-RED/OX (to account for the residual heme in the preparation of wild-type LBD). Using this method, we find the Kd of Fe3+-heme for thiol-RED LBD is 137 pm (Fig. 9D), in excellent agreement with the value of ≤100 pm determined by transient kinetics. Strikingly, the concentration of HO2(1–248) required to compete for 50% of the Fe3+-heme bound to thiol-OX and C384A is much lower than wildtype, ∼2 μm versus 60 μm, respectively (Fig. 9, D–F). Furthermore, Equation 3 yields Kd for Fe3+-heme and thiol-OX or C384A of 16.8 and 12.1 nm, respectively, both values lying within the range determined with kinetics (see Table 1). These data demonstrate that the loss of Cys-384 as a heme axial ligand decreases affinity by >80-fold.
Figure 9.

Determination of Kd for the Fe3+-heme·LBD complex using HO2(1–248) as a competitor. Increasing concentrations of apo-HO2(1–248) were incubated with the pre-formed complex of 2.5 μm heme and 3 μm wild-type thiol-RED (A and D), C384A (B and E), or thiol-OX LBD (C and F). In the case of thiol-RED and -OX proteins, the final concentration of heme in the assay is 2.76 μm due to residual heme in the protein preparation. After reaching equilibrium, the UV-visible spectra were recorded (A–C) to measure the extent of heme competition between LBD and HO2(1–248); blue traces, Rev-erbβ spectra in the absence of HO2(1–248); red traces, spectrum of fully heme-bound HO2(1–248) used for normalization acquired on the same day as the experimental spectra. Difference spectra were generated, and the absorbance maximum for the HO2(1–248)·heme complex (∼404 nm) was plotted as a function of [HO2(1–248)] (D–F). The data were normalized to the ratio of heme bound to HO2(1–248) based on the spectrum of 2.5–2.76 μm heme·HO2(1–248) (fully bound), and the data were fit with Equation 3. Data represent the average ± S.D. of three replicate experiments; thiol-RED data represent the average of two replicates ± range. Kd values are generated from the fit with the standard error reported.
HRM C384 regulates Rev-erbβ's capacity to bind NCoR1 and susceptibility to proteasomal degradation
Several studies have shown that heme binding to Rev-erbα and Rev-erbβ facilitates their interaction with NCoR1 (5, 31, 34, 35). Recently, we provided evidence that this process occurs through an indirect mechanism, i.e. heme is not required for the interaction of purified FLRev-erbβ and NCoR1 but is necessary in the presence of cell extracts. Thus, an unidentified cellular component indirectly regulates Rev-erbβ-binding to NCoR1 in a heme-dependent manner (35). Here, we show with EMSA that FLRev-erbβ C384A binds MBP-NCoR1(1–540) (a purified fusion of MBP and the C-terminal 540 amino acids of NCoR1 encompassing all three nuclear receptor interaction domains) with the same capacity as wild-type protein, in a heme-independent manner (Fig. 10A). On the contrary, co-immunoprecipitation of endogenous NCoR1 with recombinant FLRev-erbβ C384A in HEK293 extracts is compromised (Fig. 10B). Densitometric analyses indicate that the variant binds 74% less co-repressor compared with wild type. The H568F/C384A variant, which has a Kd for heme in the micromolar range, is nearly devoid of NCoR1 binding. Thus, the affinity of Rev-erbβ for heme is strongly modulated by the ligation state of His-568 and Cys-384, with thiol-RED (picomolar Kd) > C384A (nanomolar) > H568F/C384A (micromolar) and directly correlates with its capacity to bind NCoR1 (thiol-RED > C384A > H568F/C384A).
Figure 10.

Heme binding to the Rev-erbβ HRM axial thiolate indirectly facilitates its interaction with NCoR1. A, electrophoretic mobility shift assay demonstrates heme is not necessary for the interaction of recombinant wild-type and C384A FLRev-erbβ with DNA and NCoR1 in a purified system. Lane 1, free probe; lane 2, wild-type FLRev-erbβ binds the FAM-Bmal1 promoter fragment yielding three complexes with differing mobility; lanes 3 and 4, MBP-NCoR1(1–540) binds to and supershifts the FLRev-erbβ·DNA complexes although the addition of heme has a negligible effect on mobility or band density. Lanes 5–7 are the same as 2–4 except with FLRev-erbβ C384A demonstrating the same heme-independent DNA and co-repressor binding as for wild type. FAM-Bmal1-promoter is present at 1 μm in all lanes, as is 25 μg ml−1 nonspecific poly(dI-dC) competitor. B, Rev-erbβ heme axial ligand variants are compromised in NCoR1 binding in a co-immunoprecipitation assay. Recombinant wild-type, H568F/C384A, and C384A FLRev-erbβ (5 μg) are added to HEK293 cells extracts (1 mg) and immunoprecipitated, and the levels of co-precipitating NCoR1 are measured with Western blotting. Input, 40 μg of IP prior to the addition of primary antibody demonstrating equal levels of NCoR1 and recombinant Rev-erbβ, except in the no Rβ lane in which FLRev-erbβ was omitted as a control showing specificity of NCoR1 binding for the recombinant protein. Nonspecific chicken IgY was used as a primary antibody in an additional specificity control. IP:Rβ, IP elutions; the density of the NCoR1 bands from three independent experiments were normalized to the density of Rev-erbβ from the same co-IP, and the average ± S.D. of the replicates was plotted.
Although the heme dependence of NCoR1 binding is robust, we have also observed a more modest effect of heme on Rev-erbβ stability where H568F variants are stabilized against proteasomal degradation (35). Interestingly, we find that the steady-state levels of C384A and H568F/C384A are ∼2-fold higher than wild-type protein in transiently transfected HEK293 cells (Fig. 11). Thus, it appears that loss of the heme axial thiolate promotes stability, which we assume relates to reduced heme occupancy of the C384A variant. There also is precedent for Rev-erbα stabilization in heme-depleted cells, indicating that heme-binding plays a role in regulating cellular stability of both Rev-erb isoforms (59).
Figure 11.

Substitution of Rev-erbβ heme axial ligands promotes stability. A, Western blotting comparing steady-state levels of wild-type, H568F/C384A, and C384A Rev-erbβ in transiently transfected HEK293 cells. Recomb., 10 ng of recombinant FLRev-erbβ as a positive control; Empty, denotes an empty lane on the blot; pcDNA3.1(+) indicates cells transfected with the parent vector as a negative control. B, densitometry analysis of three independent experiments; bars represent the average ± S.D.
Discussion
HRMs are emerging as regulators of key cellular processes. Although their involvement in maintaining homeostatic heme levels through proteins like heme oxygenase 2, ALAS-1, and BACH1 is well documented, more recent studies, including these herein, have implicated HRMs in the regulation of the cell cycle and DNA-damage repair via p53, and circadian rhythm maintenance/metabolism via Period 2 and Rev-erbs. From the perspective of evolution, the HRM·heme signaling axis provides a simple feedback mechanism for regulating heme/globin biosynthesis and degradation, but why did the cell select for and maintain HRMs for the regulation of circadian rhythms and metabolism? One thought is that heme is a form of energy “currency” that reflects the metabolic state of the cell. Heme biosynthesis is linked to the tricarboxylic acid cycle through the common intermediate succinyl-CoA, and heme is a critical component of the respiratory chain and metabolic enzymes. Because metabolism is tightly entrained to the diurnal cycle by the molecular clock, the HRM·heme circuit, specifically via Period 2 and Rev-erbs, seemingly provides a mechanism to couple these processes. In this paper, we explored the function of the Rev-erbβ HRM and found that it not only provides a strong axial thiolate ligand to Fe3+-heme but also mediates the effects of Fe3+/2+-heme and thiol-disulfide redox regulation on Rev-erbβ heme occupancy and ultimately on its interaction with NCoR1.
Kinetic complexity of interactions between heme and Rev-erb
One of the major goals of the work described in this study was to provide accurate rate constants for binding and dissociation of heme and Kd values for the Fe3+/2+-heme·Rev-erbβ complex. This is important because heme occupancy regulates affinity for NCoR1 and proteolytic degradation (34, 35, 59). Furthermore, if Rev-erbβ plays a role within the cell as a heme sensor, it must have thermodynamic and kinetic properties that allow it to equilibrate quickly with changes in the regulatory heme pool. As summarized in Table 1, there is a vast disparity in Kd for the Rev-erbβ·heme complex depending on the technique employed. For instance, isothermal titration calorimetry (ITC) measurements provided Kd values ranging from 0.35 to 2.07 μm (34, 60), whereas the UV-visible monitored heme titrations yielded values from 6 to 24 nm (35). Because the concentration of Rev-erbβ in the aforementioned ITC studies (∼50 μm) far exceeded those in the equilibrium titrations (< 1 μm), it has been suggested that aggregation of the LBD at high concentrations may lead to the disparity in Kd (37, 60). However, we did not observe higher order oligomers in the current gel filtration study with LBD concentrations of >200 μm (Fig. 7); thus, it remains unclear why ITC measurements are not congruent with spectrophotometry. Ultimately, systems with sub-micromolar Kd values require the use of specialized techniques like rapid kinetics to establish accurate heme-binding constants.
Traditionally, monomeric CO-Fe2+-heme has been used to determine second-order on-rates for heme binding to various hemoproteins (50, 51, 61). The dicyano adduct of Fe3+-heme has also been used; however, the binding process is complicated in that CN− is a strong ligand to heme iron that must dissociate prior to protein axial ligation (50, 62). Because Rev-erbβ Cys-384 is dissociated in the CO-Fe2+-heme state, we required a system with more flexibility in which both monomeric Fe3+- or Fe2+-heme could be used. Caffeine is an ideal reagent in that it splits heme dimers, but it does so by forming relatively weak hydrophobic interactions with the porphyrin, and it appears to dissociate rapidly upon binding of the heme·caffeine complex to protein; it also has a negligible influence on Rev-erbβ stability. Use of this system also allowed us to establish rate constants that were determined with the same redox form of heme, and in the same buffer system as opposed to studies that provide heme Kd values using kon from stopped-flow experiments with CO-Fe2+-heme, and koff of Fe3+-heme using apoglobin competition (50, 51, 61).
An interesting observation from our stopped-flow studies is the presence of multiple kinetic phases during heme binding. All proteins analyzed yield an initial rapid binding phase with an observed rate constant that is dependent on [protein] and makes up >60% of the total Δabsorbance, followed by one or more first-order reactions. A similar observation was made with CO-Fe2+-heme binding to apomyoglobin where the slow first-order phase (k2 ∼9 s−1; 5–15% of the total amplitude change) is attributed to nonspecific interactions of heme with fully bound hologlobin (50). We also observed first-order Fe3+-heme binding to apomyoglobineq (20% of the total amplitude, Fig. 5, B and C); however, the rate constant (kobs2), ∼0.4 s−1, is much slower than observed with carboxy-heme, suggesting that heme redox state, carbonylation, or the presence of caffeine influences the binding mechanism. In any case, the initial rapid binding step reflects the mixed-order mechanism depicted in Fig. 4B and includes the interaction of heme (or the heme·caffeine complex) with Rev-erbβ, HO2(1–248), or apomyoglobin, whereas those slower first-order reactions can be attributed to either nonspecific heme binding or subtle changes in the conformation of axial ligands that manifest as a change in extinction of the Soret band.
The hyperbolic nature of kobs for the initial binding phase can be described by Equation 2 and the scheme illustrated in Fig. 4B for a two step-binding mechanism. However, it behooves us to discuss this mechanism in the context of the two widely accepted models of ligand binding, conformational selection and induced-fit (63). The former occurs when a ligand preferentially binds to a specific protein conformation among multiple structures present in equilibrium. The induced fit model describes the scenario when the ligand binds indiscriminately to any protein conformation, but then causes the protein to adopt a structure that best accommodates the ligand. Although the two-step heme-binding mechanism is akin to the induced-fit model, the transient kinetic results collected with HO2(1–248) are more suggestive of a conformational selective scenario. Specifically, the decrease in kobs2 as a function of [HO2(1–248)] is diagnostic of conformational selection (63). Furthermore, the near linear dependence of kobs1 on [HO2(1–248)] is not observed in Rev-erbβ or apomyoglobin, suggesting some dichotomy in the mechanism of heme binding to these different proteins. Perhaps the function of the protein dictates the mechanism by which it binds heme, e.g. heme is a substrate for HO2, while acting as a gas-binding scaffold in myoglobin, and an apparent structural element in Rev-erbβ.
The ratio k−1/k1 (i.e. Kd) for the weak, nonspecific interaction of heme with the periphery of the protein ranges from ∼20 to 500 μm depending on the protein analyzed (supplemental Table S1). Considering that intracellular regulatory heme levels are estimated to be in the low nanomolar range (10), these values appear irrelevant in the context of the cell, unless the heme pool exists not as a disperse and evenly distributed population but as a gradient, with localized “pockets” of high heme concentrations. For the sake of this study, however, Equation 2 provides the framework for fitting data and estimating kon under second-order conditions (e.g. stoichiometric concentrations of heme and LBD), which are more likely to exist in the cell than pseudo first-order conditions as we employed in this work.
In the cell, regulatory heme is highly unlikely to exist in a “free” state but as a transient monomeric complex with free thiols (glutathione, cysteine, etc.) or amino acid side chains on the surface of proteins. Thus, we would argue the caffeine·heme complex is a more representative form of heme for binding studies than free Fe3+-heme, CO-Fe2+-heme, or dicyanoheme.
Implications of redox-dependent heme binding and the role of HRM Cys-384 in regulating Rev-erbβ repressor activity
We have shown that redox processes that cause HRM Cys-384 dissociation from heme, specifically thiol-disulfide interconversion, and heme reduction are important regulators of Rev-erbβ heme occupancy; moreover, Cys-384 is critical for facilitating the heme-dependent interaction of Rev-erbβ with NCoR1. The question is whether these potential redox switches interface and respond to changes in cellular redox buffers. Significantly, several couples, including FAD/FADH2, NADP+/NADPH, glutathione/glutathione disulfide, and ascorbic acid/dehydroascorbic acid, show diurnal changes in the ratio of reduced to oxidized species (64). Thus, it is tempting to speculate that, as circadian factors, Rev-erbs might sense changes in cellular redox state through HRM Cys-384, which ultimately regulates heme occupancy, NCoR1 interaction, and degradation. However, further studies are required to confirm that these redox rheostats are occurring in the cell.
Of great significance to this work is a recent study by Reddi and co-workers (10) describing the use of fluorescent heme sensors in determining the concentration of the regulatory heme pool in yeast. Their results indicate that the pool is 20–40 nm in the cytosol but <2.5 nm in the mitochondria and nucleus. Previous estimates of Rev-erbα/β affinity for Fe3+-heme in the micromolar to low nanomolar range suggest that Rev-erbs would exist exclusively as apoproteins in the nucleus negating a function for Rev-erbs as heme-binding proteins. The Kd reported herein of ∼100 pm for Fe3+-heme lies well below 2.5 nm, whereas those for thiol-OX or Fe2+-heme are above that concentration. Thus, heme reduction or conversion to the disulfide form of the protein would cause dissociation of heme and NCoR1 under the limiting heme concentrations in the nucleus. These data support the hypothesis we had posed in a recent study that Rev-erbβ binds Fe3+-heme too tightly to be a direct heme sensor (35) but also expand on that idea in that Rev-erbβ possesses qualities of a redox sensor, where heme binding is coupled to the redox-sensing function and acts to poise the receptor for NCoR1 binding or degradation.
Experimental procedures
Production and purification of Rev-erbβ and NCoR1 derivatives
DNA encoding Rev-erbβ residues 370–579 encompassing the LBD was amplified with PCR using pcDNA3.1(+)FLRev-erbβ (35) as a template, a forward primer (5′-TACTTCCAATCCAATGCAAATAGTTACCTGTGCAACACTGGAGGAAGA-3′) and a reverse primer (5′-TTATCCACTTCCAATGTTAAGGGTGAACTTTAAAGGCCAAGAGCTCCTC-3′), and PfuUltra HS II polymerase (Agilent Technologies, Santa Clara, CA). Ligation-independent cloning methods were used to insert the PCR fragment into pMCSG9 (65) resulting in pMBP-LBD(370–579). The vector encodes a translational fusion between maltose-binding protein (MBP) and the Rev-erbβ LBD with an internal tobacco etch virus (TEV) protease cleavage site for downstream removal of MBP. The C384A variant was generated as described previously (35). Escherichia coli BL21(DE3) cells transformed with pMBP-LBD(370–579) (or site-directed variants) were grown with shaking at 37 °C in Terrific Broth (TB) (Thermo Fisher Scientific) supplemented with 0.4% glycerol and 200 μg ml−1 ampicillin. Once the cells reached an absorbance of 600 nm (A600) of 3–4, the temperature was reduced to 15 °C for 2 h prior to inducing protein production with 0.5 mm isopropyl β-d-1-thiogalactopyranoside (IPTG) and continuing to grow the cells at 15 °C for 16 h. Alternatively, when experiments required LBD preparations with little residual heme, cells harboring the expression plasmid were grown in M9 minimal medium containing 0.4% glucose and 133 μg ml−1 ampicillin at 37 °C with shaking until they reached an A600 of 1.0. Expression was induced with 1 mm IPTG and the temperature reduced to 16 °C, and the cells were grown for an additional 16 h. Cells were harvested by centrifugation, and the pellets (a typical purification was performed on 2 liters of TB culture or 12–18 liters of M9) suspended in TNG buffer (50 mm Tris-HCl, pH 8.0, 300 mm NaCl, and 10% glycerol) containing 1 mm dithiothreitol (DTT), 6 mm benzamidine, 1 mm EDTA, 0.5 mm phenylmethanesulfonyl fluoride, and 1× protease inhibitor mixture (Roche Applied Science) on ice. The slurry was sonicated and centrifuged at 100,000 × g to pellet insoluble material, and the soluble cell-free extract was applied to a 2.5 × 17-cm amylose resin column (New England Biolabs) equilibrated in TNG with 1 mm DTT at 4 °C. The column was washed with 3–4 volumes of the same buffer prior to eluting bound protein with 20 mm maltose. Pure MBP-LBD in the elution fraction was verified with denaturing SDS-PAGE, and the concentration of the fusion was determined with the Bradford assay (66). TEV protease was added at a (w/w) ratio of 0.5:20, and the proteolysis reaction was dialyzed against 50 mm Tris-HCl, pH 8.0, 10 mm NaCl, 10% glycerol, and 1 mm DTT for 16 h at 4 °C. The dialysis bag was transferred to 20 mm sodium phosphate, pH 7.2, with 0.2 m NaCl at ambient temperature for 4 h prior to applying the pool to a 2.5 × 42-cm Bio-Gel HT hydroxyapatite column equilibrated in the same buffer, also at ambient temperature. Pure LBD slowly elutes from the column in ∼10 volumes of equilibration buffer, whereas free MBP, undigested fusion, and TEV protease remain bound to the resin. The elution pool was concentrated, and buffer was exchanged into 0.5× TNG buffer (25 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 5% glycerol) containing 5 mm DTT with an Amicon ultrafiltration device (Millipore) equipped with a 10-kDa cutoff membrane at 4 °C. The protein (∼50 mg ml−1) was aliquoted, snap-frozen in liquid nitrogen, and stored at −80 °C until use. Typical yields were 60–100 mg of LBD liter−1 of TB culture, and ∼2 mg of LBD liter−1 of M9 culture. Wild-type LBD purified from cells grown in TB and M9 contains 8.7 and 1.1% residual heme, respectively, based on the pyridine hemochrome assay (67) and UV-visible spectra. Wild type, C384A, H568F/C384A FLRev-erbβ, and MBP-NCoR1(1–540) were purified as described previously (35), as was HO2(1–248) (43).
Production and purification of myoglobin derivatives
Myoglobineq was obtained from Sigma. For the preparation of myoglobinsw variant H64Y/V68F, E. coli BL21(DE3) was transformed with an expression vector encoding the aforementioned variant kindly provided by Dr. John Olson (Rice University). Cells were grown at 37 °C with shaking in LB broth (Lennox formulation, Thermo Fisher Scientific) supplemented with 0.1 mm IPTG and 200 μg ml−1 ampicillin to stationary phase (at least 16 h) prior to harvesting by centrifugation. Purification was performed as described with modifications (68). The cell pellet was suspended in 50 mm Tris-HCl, pH 8.0, 1 mm EDTA, 0.5 mm DTT, 1 mm phenylmethanesulfonyl fluoride, 40 units ml−1 DNase I (Roche Applied Science), 3 units ml−1 RNase A (Roche Applied Science), and 2 mg ml−1 lysozyme and stirred at 4 °C for 15 min prior to sonication. Insoluble material was pelleted at 100,000 × g for 1 h, and the soluble cell-free extract was collected. Ammonium sulfate crystals were added to 50% saturation and stirred on ice for 15 min before pelleting precipitated proteins at 17,000 × g for 15 min. We collected the supernatant and added ammonium sulfate to 95% saturation, stirred, and pelleted precipitated proteins as before. Pellets were dissolved in 20 mm Tris-HCl, pH 8.0, and 1 mm EDTA and extensively dialyzed against the same buffer before loading onto a 2.5 × 23-cm DEAE-Sepharose (Sigma) column equilibrated in the dialysate. The flow-through containing H64Y/V68F was collected, titrated to pH 6 with 50% acetic acid, and loaded onto a 2.5 × 26-cm CM-Sepharose (Sigma) column equilibrated in 20 mm sodium phosphate pH 6. Protein was eluted with a linear gradient from equilibration buffer to 50 mm sodium phosphate dibasic (∼pH 9.5); fractions containing pure H64Y/V68F were verified with SDS-PAGE. Apomyoglobineq and apo-H64Y/V68F were prepared using methyl ethyl ketone extraction as described (35).
Preparation of thiol-reduced and oxidized LBD
In an anaerobic chamber, the as-purified LBD was mixed with ≥11-fold excess of tris(2-carboxyethyl)phosphine (TCEP; a stock solution was prepared by dissolving TCEP in water and titrating to pH 8.0 with NaOH) to [protein], incubated at ambient temperature for 1 h, and exchanged into the desired buffer using either PD-10 columns (GE Healthcare) or Microbiospin 6 columns (Bio-Rad); the C384A variant was also thiol-reduced prior to use. Thiol-oxidation was performed using either thiol-reduced protein or as-purified LBD that had been buffer exchanged to eliminate reductant. Diamide (Sigma) was added at a 30-fold excess to [protein], incubated for 2 h at ambient temperature, and buffer exchanged as above. The extent of thiol reduction was confirmed using a modified 5,5′-dithiobis-(2-nitrobenzoic acid), DTNB assay (69). Briefly, 50 μl of 5 mm DTNB in 0.1 m potassium phosphate, pH 7.2, was mixed with 950 μl of 6 m guanidine-HCl in 0.1 m Tris-HCl, pH 8.0, and 20 μl of sample was added, mixed, and incubated at ambient temperature for 15 min prior to recording the UV-visible spectrum. The assay was standardized with DTT and N-acetylcysteine, both of which generated ϵ = 11,800 m−1 cm−1 at 420 nm. 20.4 μm thiol-RED LBD yielded 41.9 μm free thiols or 2.1 thiols per LBD protomer accounting for the two thiols, Cys-374 and Cys-384, in the LBD construct. The same concentration of thiol-OX protein yielded no absorbance at 420 nm indicating complete oxidation of Cys-374 and Cys-384 to a disulfide (37). Thiol reduction of FLRev-erbβ was carried out as described (35).
Protein assays, PAGE, Western blotting, and electrophoretic mobility shift assays (EMSA)
Concentrations of the LBD, FLRev-erbβ, MBP-NCoR1(1–540), and HO2(1–248) were determined with the Bradford assay (66), and the following molecular masses: wild-type LBD, 23,935 Da; C384A LBD, 23,903 Da; wild-type FLRev-erbβ (mass is for the TEV-digested purified protein related to the human genome sequence, see Ref. 35), 64,784 Da; C384A FLRev-erbβ, 64,752 Da; H568F/C384A FLRev-erbβ, 64,762 Da; MBP-NCoR1(1–540), 102,306 Da; HO2(1–248), 28,698 Da. The concentrations of H64Y/V68F apomyoglobinsw and apomyoglobineq were determined by their absorbance at 280 nm in water, according to ϵ values of 15.2 mm−1 cm−1 (52) and 14 mm−1 cm−1 (35), respectively. PAGE, Western blotting, and EMSA were performed as described (35).
Analytical gel filtration
Thiol-RED, thiol-OX, and C384A LBDs (apoproteins) were buffer exchanged into 0.5× TNG with 25 mm caffeine and diluted to 5 mg ml−1. Alternatively, samples were reconstituted with heme at a ratio of 0.9:1 heme/protein in the same buffer. 0.1 ml was injected onto a 10/300 GL Superose 6 column (GE Healthcare) equilibrated in 0.5× TNG (caffeine was omitted from the running buffer to avoid saturation of the UV detector) and connected to an Akta Pure FPLC system housed in a Coy chamber maintained at 3–5% H2 in N2. The samples were chromatographed at 0.5 ml min−1, and the eluent monitored at 280 nm. The column was standardized using blue dextran to determine the void volume, Bio-Rad gel filtration standards (thyroglobulin, 670 kDa; γ-globulin, 158 kDa; ovalbumin, 44 kDa; myoglobin, 17 kDa; and vitamin B12, 1.35 kDa), and bovine serum albumin (66-kDa monomer and 132-kDa dimer).
UV-visible stopped-flow spectrophotometry
A heme stock was prepared by dissolving hemin in 0.1 m NaOH and 10% DMSO (20 mg ml−1), filtering, and diluting to ∼1 mm in 0.5× TNG. The concentration of the dilution was confirmed in 0.1 m NaOH and ϵ = 58.44 mm−1 cm−1 at 385 nm (70) and further diluted to 5 μm in anaerobic 0.5× TNG with 25 mm caffeine. LBD preparations were thiol-reduced or oxidized as described above and buffer exchanged into 0.5× TNG with 25 mm caffeine. For the experiment with Fe2+-heme, 5 mm dithionite was present in the heme and protein solutions; a 1 m dithionite stock was prepared in 1 m Tris-HCl, pH 8.0, under anaerobic conditions to avoid pH effects, thereby leading to a final concentration of 30 mm Tris-HCl in the assay buffer after dilution. Protein solutions of varying concentration were rapidly mixed with heme (final concentration 2.5 μm in all experiments) using an Applied Photophysics stopped-flow spectrophotometer with the sample handling unit (drive syringes, mixing chamber, and optical cell) housed in a Vacuum Atmospheres anaerobic chamber held at <1 ppm O2. All measurements were carried out at 20 °C using the 1-cm pathlength configuration. An initial measurement was carried out in photodiode array mode to observe overall spectral changes associated with heme binding to LBDs. However, most kinetic measurements (unless otherwise stated) used for analyses of rate constants were collected in single-wavelength mode to maximize the data acquired at early time points (e.g. <5 ms).
Apomyoglobin competition assays to determine Fe2+/3+-heme off-rates
Most off-rate experiments were performed as described previously (35) with minor modifications. Under anaerobic conditions in stoppered quartz cuvettes (0.8–1 ml volumes), apomyoglobineq or H64Y/V68F apomyoglobinsw (30 μm) was mixed with the Rev-erbβ·heme complex (3 μm Rev-erbβ, 1.5 μm heme) at 20 °C in 0.5× TNG ± 25 mm caffeine. The rate of heme transfer from Rev-erbβ to globin was monitored with a Shimadzu UV-2600 or UV-2501PC double beam spectrophotometer equipped with a TCC temperature controller at the specified wavelength. HO2(1–248)·Fe3+-heme off-rates were determined as described but under aerobic conditions. The fast off-rate of Fe2+-heme necessitated use of the stopped-flow spectrophotometer; the complex of 3 μm thiol-RED wild-type LBD and 1.5 μm Fe2+-heme was mixed with 30 μm apomyoglobineq in 0.5× TNG with 25 mm caffeine and 5 mm dithionite (final [Tris] = 30 mm), and the transfer of heme was monitored in PDA mode. To determine the Fe3+-heme off-rate from myoglobineq, 80 μm H64Y/V68F apomyoglobinsw was mixed with the complex of 8 μm apomyoglobineq and 7 μm Fe3+-heme in 0.5× TNG with 25 mm caffeine under aerobic conditions. High concentrations of protein were used to better resolve the slow changes at 600 nm arising from the formation of the H64Y/V68F·heme complex, which yields a Tyr-Fe3+-heme charge transfer band (52).
HO2(1–248) competition assays
In 1 ml of 0.5× TNG with 25 mm caffeine, varying concentrations of apoHO2(1–248) were mixed with the pre-formed complex of 3 μm LBD (thiol-RED or -OX wild-type, and C384A) and 2.5 μm Fe3+-heme. The assays were incubated at ambient temperature until equilibrium was achieved (calculated based on off-rates determined with apomyoglobin competition), and the UV-visible spectra were acquired. All assays were carried out in an anaerobic glove box, and the samples were sealed in stoppered quartz cuvettes to maintain O2-free conditions when spectra were acquired at the bench. Residual absorbance in the HO2(1–248) preparation (likely arising from remaining minor levels of heme or biliverdin due to the high concentrations of HO2(1–248) used in that study) was manually subtracted from the assay spectra acquired with thiol-RED LBD.
Data analysis
Stopped-flow traces were fit with exponential equations using Pro-data Viewer software provided by Applied Photophysics. The instrument dead time of 1.8 ms used in curve fitting was determined using the reduction of ferricyanide by ascorbate in 20 mm CHES, pH 9.0, and 200 mm NaCl at 20 °C, 100 p.s.i. drive pressure (71, 72). For thiol-OX and C384A LBDs, an additional kinetic phase corresponding to kobs2 was observed only at high concentrations of protein >100 μm. For example, when analyzing the data for Fe3+-heme binding to C384A (see Fig. 3, G–I), a double-exponential equation was sufficient to fit the data for C384A concentrations below 100 μm; however, at >100 μm, another phase emerged that necessitated the use of a triple-exponential equation. The fractional contribution of the amplitudes of the additional phase to the total amplitude change for acquired kinetic traces at [C384A] >100 μm was averaged and that value was fixed when analyzing the traces using a triple exponential equation. There is little (<20%) effect on the rate constants for the initial rapid binding phase when comparing double and triple-exponential fits at low [LBD], i.e. the method does not introduce significant bias. Interestingly, Fe2+/3+-heme binding to thiol-RED wild-type LBD and Fe3+-heme binding to apomyoglobineq and HO2(1–248) did not yield data with concentration-dependent emergence of kinetic phases. Plots of kobs versus [apoprotein] that demonstrate hyperbolic dependence were fit with Equation 2,
| (Eq. 2) |
which is used to describe the observed rate constant under pseudo first-order conditions in the context of the two-step binding mechanism (Fig. 4B) when k1 and k-1 ≫ kcoordination, and koff is ≪ kcoordination. Under such circumstances, koff is omitted from the equation resulting in a hyperbolic expression with a y-intercept equal to zero (48, 53).
Off-rate data obtained from myoglobin competition experiments were fit with double-exponential equations using SigmaPlot statistical software. Heme competition experiments between Rev-erbβ and HO2(1–248) were fit with Equation 3,
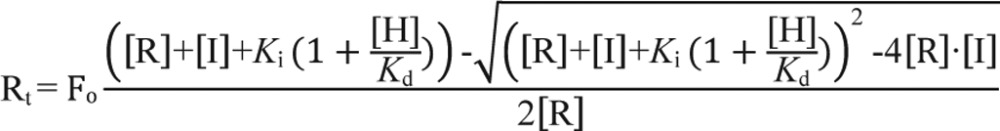 |
also known as the Morrison equation (42) for tight-binding competitive inhibitors, where Rt is the ratio of heme transferred to HO2(1–248) at any given [HO2(1–248)]; Fo is the fractional occupancy of HO2(1–248) as [HO2(1–248)] approaches ∞; [R] is the fixed concentration LBD; [I] is the concentration of HO2(1–248); Ki is tantamount to the Kd for the Fe3+-heme·HO2(1–248) complex; [H] is the fixed concentration of Fe3+-heme present in the assay, and Kd is the dissociation constant for the LBD·Fe3+-heme complex.
Author contributions
E. L. C. conducted most of the experiments, analyzed the data, and wrote the paper. Y. R. performed the cloning and initial purification of the LBD construct, early heme-binding studies, and the EMSA. S. W. R. wrote the paper and analyzed data with E. L. C. All authors reviewed and approved the manuscript prior to submission.
Supplementary Material
Acknowledgments
We thank members of the Ragsdale laboratory, namely Dr. Angela Fleischhacker and Dr. Mehmet Can, for helpful discussions pertaining to this manuscript and Dr. Frederick Stull for providing valuable feedback on stopped-flow kinetics studies.
This work was supported by National Institutes of Health Grants F32HL114150 (to E. L. C.) and R01-GM123513 (to S. W. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Table S1 and Figs. S1 and S2.
- HRM
- heme-regulatory motif
- apomyoglobineq
- apomyoglobin from equine skeletal muscle myoglobin
- apomyoglobinsw
- apomyoglobin from sperm whale
- HO2(1–248)
- purified heme-oxygenase 2 residues 1–248
- LBD
- Rev-erbβ ligand-binding domain
- MBP
- maltose-binding protein
- NCoR1
- nuclear receptor co-repressor 1
- TB
- Terrific Broth
- TEV
- tobacco etch virus
- TCEP
- tris(2-carboxyethyl)phosphine
- DTNB
- 5,5′-dithiobis(nitrobenzoic acid)
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- PDA
- photodiode array
- IP
- immunoprecipitation
- ITC
- isothermal titration calorimetry.
References
- 1. Chapman S. K., Daff S., and Munro A. W. (1997) in Metal Sites in Proteins and Models: Iron Centres (Hill H. A. O., Sadler P. J., and Thomson A. J., eds) pp. 39–70, Springer Berlin Heidelberg, Berlin [Google Scholar]
- 2. Tsiftsoglou A. S., Tsamadou A. I., and Papadopoulou L. C. (2006) Heme as key regulator of major mammalian cellular functions: molecular, cellular, and pharmacological aspects. Pharmacol. Ther. 111, 327–345 [DOI] [PubMed] [Google Scholar]
- 3. Mense S. M., and Zhang L. (2006) Heme: a versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 16, 681–692 [DOI] [PubMed] [Google Scholar]
- 4. Kubota Y., Nomura K., Katoh Y., Yamashita R., Kaneko K., and Furuyama K. (2016) Novel mechanisms for heme-dependent degradation of ALAS1 protein as a component of negative feedback regulation of heme biosynthesis. J. Biol. Chem. 291, 20516–20529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yin L., Wu N., Curtin J. C., Qatanani M., Szwergold N. R., Reid R. A., Waitt G. M., Parks D. J., Pearce K. H., Wisely G. B., and Lazar M. A. (2007) Rev-erbα, a heme sensor that coordinates metabolic and circadian pathways. Science 318, 1786–1789 [DOI] [PubMed] [Google Scholar]
- 6. Shimizu T., Huang D., Yan F., Stranava M., Bartosova M., Fojtíková V., and Martínková M. (2015) Gaseous O2, NO, and CO in signal transduction: structure and function relationships of heme-based gas sensors and heme-redox sensors. Chem. Rev. 115, 6491–6533 [DOI] [PubMed] [Google Scholar]
- 7. Muraki N., Kitatsuji C., and Aono S. (2015) A new biological function of heme as a signaling molecule. J. Porphyrins Phthalocyanines 19, 9–20 [Google Scholar]
- 8. Hou S., Reynolds M. F., Horrigan F. T., Heinemann S. H., and Hoshi T. (2006) Reversible binding of heme to proteins in cellular signal transduction. Acc. Chem. Res. 39, 918–924 [DOI] [PubMed] [Google Scholar]
- 9. Atamna H., Brahmbhatt M., Atamna W., Shanower G. A., and Dhahbi J. M. (2015) ApoHRP-based assay to measure intracellular regulatory heme. Metallomics 7, 309–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hanna D. A., Harvey R. M., Martinez-Guzman O., Yuan X., Chandrasekharan B., Raju G., Outten F. W., Hamza I., and Reddi A. R. (2016) Heme dynamics and trafficking factors revealed by genetically encoded fluorescent heme sensors. Proc. Natl. Acad. Sci. U.S.A. 113, 7539–7544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang L., and Guarente L. (1995) Heme binds to a short sequence that serves a regulatory function in diverse proteins. EMBO J. 14, 313–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Munakata H., Sun J.-Y., Yoshida K., Nakatani T., Honda E., Hayakawa S., Furuyama K., and Hayashi N. (2004) Role of the heme-regulatory motif in the heme-mediated inhibition of mitochondrial import of 5-aminolevulinate synthase. J. Biochem. 136, 233–238 [DOI] [PubMed] [Google Scholar]
- 13. Zenke-Kawasaki Y., Dohi Y., Katoh Y., Ikura T., Ikura M., Asahara T., Tokunaga F., Iwai K., and Igarashi K. (2007) Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol. Cell. Biol. 27, 6962–6971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ogawa K., Sun J., Taketani S., Nakajima O., Nishitani C., Sassa S., Hayashi N., Yamamoto M., Shibahara S., Fujita H., and Igarashi K. (2001) Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 20, 2835–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shen J., Sheng X., Chang Z., Wu Q., Wang S., Xuan Z., Li D., Wu Y., Shang Y., Kong X., Yu L., Li L., Ruan K., Hu H., Huang Y., Hui L., Xie D., Wang F., and Hu R. (2014) Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 7, 180–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qi Z., Hamza I., and O'Brian M. R. (1999) Heme is an effector molecule for iron-dependent degradation of the bacterial iron response regulator (Irr) protein. Proc. Natl. Acad. Sci. U.S.A. 96, 13056–13061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ishikawa H., Kato M., Hori H., Ishimori K., Kirisako T., Tokunaga F., and Iwai K. (2005) Involvement of heme-regulatory motif in heme-mediated ubiquitination and degradation of IRP2. Mol. Cell 19, 171–181 [DOI] [PubMed] [Google Scholar]
- 18. Simcox J. A., Mitchell T. C., Gao Y., Just S. F., Cooksey R., Cox J., Ajioka R., Jones D., Lee S.-H., King D., Huang J., and McClain D. A. (2015) Dietary iron controls circadian hepatic glucose metabolism through heme synthesis. Diabetes 64, 1108–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qian J., and Scheer F. A. (2016) Circadian system and glucose metabolism: Implications for physiology and disease. Trends Endocrinol. Metab. 27, 282–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gerhart-Hines Z., and Lazar M. A. (2015) Rev-erbα and the circadian transcriptional regulation of metabolism. Diabetes Obes. Metab. 17, 12–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Milev N. B., and Reddy A. B. (2015) Circadian redox oscillations and metabolism. Trends Endocrinol. Metab. 26, 430–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Asher G., and Sassone-Corsi P. (2015) Time for food: the intimate interplay between nutrition, metabolism, and the circadian clock. Cell 161, 84–92 [DOI] [PubMed] [Google Scholar]
- 23. Takahashi J. S. (2015) Molecular components of the circadian clock in mammals. Diabetes Obes. Metab. 17, 6–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dioum E. M., Rutter J., Tuckerman J. R., Gonzalez G., Gilles-Gonzalez M.-A., and McKnight S. L. (2002) NPAS2: a gas-responsive transcription factor. Science 298, 2385–2387 [DOI] [PubMed] [Google Scholar]
- 25. Rutter J., Reick M., Wu L. C., and McKnight S. L. (2001) Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 293, 510–514 [DOI] [PubMed] [Google Scholar]
- 26. Crumbley C., and Burris T. P. (2011) Direct regulation of CLOCK expression by REV-ERB. PLoS ONE 6, e17290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bugge A., Feng D., Everett L. J., Briggs E. R., Mullican S. E., Wang F., Jager J., and Lazar M. A. (2012) Rev-erbα and Rev-erbβ coordinately protect the circadian clock and normal metabolic function. Genes Dev. 26, 657–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Delezie J., Dumont S., Dardente H., Oudart H., Gréchez-Cassiau A., Klosen P., Teboul M., Delaunay F., Pévet P., and Challet E. (2012) The nuclear receptor REV-ERBα is required for the daily balance of carbohydrate and lipid metabolism. FASEB J. 26, 3321–3335 [DOI] [PubMed] [Google Scholar]
- 29. Raspé E., Duez H., Mansén A., Fontaine C., Fiévet C., Fruchart J.-C., Vennström B., and Staels B. (2002) Identification of Rev-erbα as a physiological repressor of apoC-III gene transcription. J. Lipid Res. 43, 2172–2179 [DOI] [PubMed] [Google Scholar]
- 30. Wang J., Yin L., and Lazar M. A. (2006) The orphan nuclear receptor Rev-erbα regulates circadian expression of plasminogen activator inhibitor type 1. J. Biol. Chem. 281, 33842–33848 [DOI] [PubMed] [Google Scholar]
- 31. Chandra V., Mahajan S., Saini A., Dkhar H. K., Nanduri R., Raj E. B., Kumar A., and Gupta P. (2013) Human IL10 gene repression by Rev-erbα ameliorates Mycobacterium tuberculosis clearance. J. Biol. Chem. 288, 10692–10702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramakrishnan S. N., Lau P., Burke L. J., and Muscat G. E. (2005) Rev-erbβ regulates the expression of genes involved in lipid absorption in skeletal muscle cells: evidence for cross-talk between orphan nuclear receptors and myokines. J. Biol. Chem. 280, 8651–8659 [DOI] [PubMed] [Google Scholar]
- 33. Gibbs J. E., Blaikley J., Beesley S., Matthews L., Simpson K. D., Boyce S. H., Farrow S. N., Else K. J., Singh D., Ray D. W., and Loudon A. S. (2012) The nuclear receptor REV-ERBα mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc. Natl. Acad. Sci. U.S.A. 109, 582–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Raghuram S., Stayrook K. R., Huang P., Rogers P. M., Nosie A. K., McClure D. B., Burris L. L., Khorasanizadeh S., Burris T. P., and Rastinejad F. (2007) Identification of heme as the ligand for the orphan nuclear receptors REV-ERBα and REV-ERBβ. Nat. Struct. Mol. Biol. 14, 1207–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Carter E. L., Gupta N., and Ragsdale S. W. (2016) High affinity heme binding to a heme-regulatory motif on the nuclear receptor Rev-erbβ leads to its degradation and indirectly regulates its interaction with nuclear receptor co-repressor. J. Biol. Chem. 291, 2196–2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pardee K. I., Xu X., Reinking J., Schuetz A., Dong A., Liu S., Zhang R., Tiefenbach J., Lajoie G., Plotnikov A. N., Botchkarev A., Krause H. M., and Edwards A. (2009) The structural basis of gas-responsive transcription by the human nuclear hormone receptor REV-ERBβ. PLoS Biol. 7, e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gupta N., and Ragsdale S. W. (2011) Thiol-disulfide redox dependence of heme binding and heme ligand switching in nuclear hormone receptor Rev-erbβ. J. Biol. Chem. 286, 4392–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marvin K. A., Reinking J. L., Lee A. J., Pardee K., Krause H. M., and Burstyn J. N. (2009) Nuclear receptors Homo sapiens Rev-erbβ and Drosophila melanogaster E75 are thiolate-ligated heme proteins which undergo redox-mediated ligand switching and bind CO and NO. Biochemistry 48, 7056–7071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kawamura-Konishi Y., and Suzuki H. (1985) Binding reaction of hemin to globin. J. Biochem. 98, 1181–1190 [DOI] [PubMed] [Google Scholar]
- 40. Pasternack R. F., Gibbs E. J., Hoeflin E., Kosar W. P., Kubera G., Skowronek C. A., Wong N. M., and Muller-Eberhard U. (1983) Hemin binding to serum proteins and the catalysis of interprotein transfer. Biochemistry 22, 1753–1758 [DOI] [PubMed] [Google Scholar]
- 41. Gallagher W. A., and Elliott W. B. (1967) Caffeine derivatives of haematin compounds. Biochem. J. 105, 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morrison J. F. (1969) Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 185, 269–286 [DOI] [PubMed] [Google Scholar]
- 43. Fleischhacker A. S., Sharma A., Choi M., Spencer A. M., Bagai I., Hoffman B. M., and Ragsdale S. W. (2015) The C-terminal heme-regulatory motifs of heme oxygenase-2 are redox-regulated heme binding sites. Biochemistry 54, 2709–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. de Villiers K. A., Kaschula C. H., Egan T. J., and Marques H. M. (2007) Speciation and structure of ferriprotoporphyrin IX in aqueous solution: spectroscopic and diffusion measurements demonstrate dimerization, but not μ-oxo dimer formation. J. Biol. Inorg. Chem. 12, 101–117 [DOI] [PubMed] [Google Scholar]
- 45. Asher C., de Villiers K. A., and Egan T. J. (2009) Speciation of ferriprotoporphyrin IX in aqueous and mixed aqueous solution is controlled by solvent identity, pH, and salt concentration. Inorg. Chem. 48, 7994–8003 [DOI] [PubMed] [Google Scholar]
- 46. Keilin J. (1943) Effect of caffeine and other iminazole compounds on haematins and their derivatives. Biochem. J. 37, 281–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gibson Q. H., and Antonini E. (1960) Kinetic studies on the reaction between native globin and haem derivatives. Biochem. J. 77, 328–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nygaard T. K., Blouin G. C., Liu M., Fukumura M., Olson J. S., Fabian M., Dooley D. M., and Lei B. (2006) The mechanism of direct heme transfer from the streptococcal cell surface protein Shp to HtsA of the HtsABC transporter. J. Biol. Chem. 281, 20761–20771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu M., Tanaka W. N., Zhu H., Xie G., Dooley D. M., and Lei B. (2008) Direct hemin transfer from IsdA to IsdC in the iron-regulated surface determinant (Isd) heme acquisition system of Staphylococcus aureus. J. Biol. Chem. 283, 6668–6676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hargrove M. S., Barrick D., and Olson J. S. (1996) The association rate constant for heme binding to globin is independent of protein structure. Biochemistry 35, 11293–11299 [DOI] [PubMed] [Google Scholar]
- 51. Owens C. P., Du J., Dawson J. H., and Goulding C. W. (2012) Characterization of heme ligation properties of Rv0203, a secreted heme-binding protein involved in Mycobacterium tuberculosis heme uptake. Biochemistry 51, 1518–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hargrove M. S., Singleton E. W., Quillin M. L., Ortiz L. A., Phillips G. N. Jr., Olson J. S., and Mathews A. J. (1994) His64(E7) → Tyr apomyoglobin as a reagent for measuring rates of hemin dissociation. J. Biol. Chem. 269, 4207–4214 [DOI] [PubMed] [Google Scholar]
- 53. Strickland S., Palmer G., and Massey V. (1975) Determination of dissociation constants and specific rate constants of enzyme-substrate (or protein-ligand) interactions from rapid reaction kinetic data. J. Biol. Chem. 250, 4048–4052 [PubMed] [Google Scholar]
- 54. Honsa E. S., Fabian M., Cardenas A. M., Olson J. S., and Maresso A. W. (2011) The five near-iron transporter (NEAT) domain anthrax hemophore, IsdX2, scavenges heme from hemoglobin and transfers heme to the surface protein IsdC. J. Biol. Chem. 286, 33652–33660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Malmirchegini G. R., Sjodt M., Shnitkind S., Sawaya M. R., Rosinski J., Newton S. M., Klebba P. E., and Clubb R. T. (2014) Novel mechanism of hemin capture by Hbp2, the hemoglobin-binding hemophore from Listeria monocytogenes. J. Biol. Chem. 289, 34886–34899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Le P., Zhao J., and Franzen S. (2014) Correlation of heme binding affinity and enzyme kinetics of dehaloperoxidase. Biochemistry 53, 6863–6877 [DOI] [PubMed] [Google Scholar]
- 57. Hayasaka K., Kitanishi K., Igarashi J., and Shimizu T. (2011) Heme-binding characteristics of the isolated PAS-B domain of mouse Per2, a transcriptional regulatory factor associated with circadian rhythms. Biochim. Biophys. Acta 1814, 326–333 [DOI] [PubMed] [Google Scholar]
- 58. Lin S. X., and Neet K. E. (1990) Demonstration of a slow conformational change in liver glucokinase by fluorescence spectroscopy. J. Biol. Chem. 265, 9670–9675 [PubMed] [Google Scholar]
- 59. Kumar N., Solt L. A., Wang Y., Rogers P. M., Bhattacharyya G., Kamenecka T. M., Stayrook K. R., Crumbley C., Floyd Z. E., Gimble J. M., Griffin P. R., and Burris T. P. (2010) Regulation of adipogenesis by natural and synthetic REV-ERB ligands. Endocrinology 151, 3015–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Matta-Camacho E., Banerjee S., Hughes T. S., Solt L. A., Wang Y., Burris T. P., and Kojetin D. J. (2014) Structure of REV-ERBβ ligand-binding domain bound to a porphyrin antagonist. J. Biol. Chem. 289, 20054–20066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Miksanova M., Igarashi J., Minami M., Sagami I., Yamauchi S., Kurokawa H., and Shimizu T. (2006) Characterization of heme-regulated eIF2α kinase: roles of the N-terminal domain in the oligomeric state, heme binding, catalysis, and inhibition. Biochemistry 45, 9894–9905 [DOI] [PubMed] [Google Scholar]
- 62. Kawamura-Konishi Y., Kihara H., and Suzuki H. (1988) Reconstitution of myoglobin from apoprotein and heme, monitored by stopped-flow absorption, fluorescence and circular dichroism. Eur. J. Biochem. 170, 589–595 [DOI] [PubMed] [Google Scholar]
- 63. Vogt A. D., and Di Cera E. (2013) Conformational selection is a dominant mechanism of ligand binding. Biochemistry 52, 5723–5729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang T. A., Yu Y. V., Govindaiah G., Ye X., Artinian L., Coleman T. P., Sweedler J. V., Cox C. L., and Gillette M. U. (2012) Circadian rhythm of redox state regulates excitability in suprachiasmatic nucleus neurons. Science 337, 839–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Eschenfeldt W. H., Lucy S., Millard C. S., Joachimiak A., and Mark I. D. (2009) A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol. Biol. 498, 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 67. Berry E. A., and Trumpower B. L. (1987) Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal. Biochem. 161, 1–15 [DOI] [PubMed] [Google Scholar]
- 68. Springer B. A., and Sligar S. G. (1987) High-level expression of sperm whale myoglobin in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 84, 8961–8965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ellman G. L. (1959) Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82, 70–77 [DOI] [PubMed] [Google Scholar]
- 70. Dawson R. M., Elliott D. C., Elliott W. H., and Jones K. M. (eds) (1969) Data for Biochemical Research, 2nd Ed., pp. 316–317, Oxford University Press, Oxford, UK [Google Scholar]
- 71. Matsumura K., Enoki Y., Kohzuki H., and Sakata S. (1990) A simple procedure for determination of the dead time of a stopped-flow instrument. Jpn. J. Physiol. 40, 567–571 [DOI] [PubMed] [Google Scholar]
- 72. Hammes G. G., and Haslam J. L. (1968) A kinetic investigation of the interaction of α-methylaspartic acid with aspartate aminotransferase. Biochemistry 7, 1519–1525 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.