

Abstract
Genetic and environmental factors may lead to abnormal growth of the orofacial skeleton, affecting the overall structure of the face. In this study, we investigated the craniofacial abnormalities in a mouse model for Keutel syndrome, a rare genetic disease caused by loss-of-function mutations in the matrix Gla protein (MGP) gene. Keutel syndrome patients show diffuse ectopic calcification of cartilaginous tissues and impaired midface development. Our comparative cephalometric analyses of micro-computed tomography images revealed a severe midface hypoplasia in Mgp−/− mice. In vivo reporter studies demonstrated that the Mgp promoter is highly active at the cranial sutures, cranial base synchondroses, and nasal septum. Interestingly, the cranial sutures of the mutant mice showed normal anatomical features. Although we observed a mild increase in mineralization of the spheno-occipital synchondrosis, it did not reduce the relative length of the cranial base in comparison with total skull length. Contrary to this, we found the nasal septum to be abnormally mineralized and shortened in Mgp−/− mice. Transgenic restoration of Mgp expression in chondrocytes fully corrected the craniofacial anomalies caused by MGP deficiency, suggesting a local role for MGP in the developing nasal septum. Although there was no up-regulation of markers for hypertrophic chondrocytes, a TUNEL assay showed a marked increase in apoptotic chondrocytes in the calcified nasal septum. Transmission electron microscopy confirmed unusual mineral deposits in the septal extracellular matrix of the mutant mice. Of note, the systemic reduction of the inorganic phosphate level was sufficient to prevent abnormal mineralization of the nasal septum in Mgp−/−;Hyp compound mutants. Our work provides evidence that modulation of local and systemic factors regulating extracellular matrix mineralization can be possible therapeutic strategies to prevent ectopic cartilage calcification and some forms of congenital craniofacial anomalies in humans.
Keywords: apoptosis, biomineralization, chondrocyte, craniofacial development, mouse genetics, matrix Gla protein (MGP), midface hypoplasia, nasal septum
Introduction
Congenital anomalies or birth defects are a major cause of perinatal lethality, affecting 2–3% of all newborns (1). A significant number of these infants show abnormal craniofacial development that often disrupts overall body functions and may lead to long-term disabilities (1). Both genetic and epigenetic factors regulating the concerted morphogenesis of two craniofacial tissues, bone and cartilage, may affect craniofacial growth and patterning. Although the primary causes are genetic, maternal exposure to toxic substances and nutritional status during pregnancy may also lead to these inborn deformities (2–6).
Midface hypoplasia is a developmental anomaly in which the nasal, maxillary, and zygomatic bones in the cheek grow slower than other facial structures (7). Genetic mutations, epigenetic factors, or traumatic injuries during the early stage of life may lead to midface hypoplasia (8–11). A mild form of this disorder is usually considered a harmless developmental anomaly of the face, whereas a severe form may seriously affect health and well-being. Clinical complications associated with this disease may include sleep apnea, misalignment of the jaws and eyelids, dental malocclusion, chewing and swallowing difficulties, impaired speech, and overall disfigurement of the face (12–14).
Multiple genetic mutations have been associated with craniofacial malformations with severely impaired midface development. For instance, autosomal dominant mutations affecting fibroblast growth factor signaling cause midface hypoplasia in several diseases, including Pfeiffer syndrome, Crouzon syndrome and Apert syndrome (8, 15, 16). In these diseases, the primary cause of midface abnormalities is early fusion of the cranial sutures (craniosynostosis), which is generally associated with premature closure of the cranial base synchondroses (17, 18).
Although the above developmental anomalies are commonly associated with midface hypoplasia, some rare forms of this disease may have a different etiology that is not yet elucidated. For example, children born to mothers treated with warfarin during pregnancy develop warfarin embryopathy, a disease characterized mainly by brain hemorrhages and severe midface hypoplasia (6, 9, 19). Warfarin is a commonly prescribed anticoagulant that inhibits vitamin K epoxide reductase (VKORC1), which converts the oxidized form of vitamin K1 to its reduced form. The reduced vitamin K1 acts as a co-factor for γ-glutamyl carboxylase (GGCX),3 which is essential for the posttranslational γ carboxylation of specific glutamic acid residues (Gla) in proteins, collectively known as “Gla proteins” (20). Similarly, patients with mutations in matrix Gla protein (MGP) develop Keutel syndrome, a rare genetic disorder primarily characterized by abnormal mineralization (calcification) of all cartilaginous tissues, short stature, stocky distal phalanges, arterial calcification, and severe midface hypoplasia (3, 21).
MGP and a related protein, osteocalcin (a bone Gla protein encoded by Bglap), are known as skeletal Gla proteins (22). Although Bglap is expressed specifically by bone-forming osteoblasts, Mgp is expressed at high levels by chondrocytes in cartilaginous tissues, by vascular smooth muscle cells in the cardiovascular system, and by the endothelial-like cells of the trabecular meshwork of the eye (23, 24). Although initially both of these proteins were proposed to be potent inhibitors of extracellular matrix (ECM) mineralization, our previous work demonstrated that only MGP possesses these anti-mineralization functions (22).
MGP-deficient mice (Mgp−/− mice) recapitulate most of the phenotypic abnormalities of Keutel syndrome patients, including, as reported recently, ectopic arterial calcification (25). However, this latter phenotype is more severe in the mouse model and represents its primary cause of death (26). Although MGP-deficient mice have been well-characterized for their vascular calcification phenotype, their craniofacial anomalies are still understudied.
In this study, we show that midface hypoplasia in MGP-deficient mice is primarily caused by impaired growth of the maxillary and palatine bones, associated with abnormal mineralization and shortening of the nasal septum. For the first time, we show the activity of the Mgp promoter in the craniofacial complex using an in vivo reporter model, and we demonstrate that septal chondrocytes undergo apoptosis in the absence of MGP. Additionally, we show a novel mechanism of cartilage mineralization that, unlike endochondral bones, does not require differentiated hypertrophic chondrocytes. Also of importance, we demonstrate that the extent of ectopic nasal septum mineralization in MGP deficiency is phosphate-dependent, where reduction of systemic phosphate levels impedes nasal septum mineralization, preventing craniofacial malformations. Considering their phenotypic similarities to human patients, MGP-deficient mice may be a useful tool to understand the pathology and underlying cause of Keutel syndrome.
Results
MGP deficiency causes midface hypoplasia
To investigate the effects of MGP and osteocalcin (BGLAP) deficiency in craniofacial development, images of lateral and frontal heads of 5-week-old male WT, Mgp−/−, and Bglap−/− mice were visually examined for gross craniofacial abnormalities. Mgp−/− mice showed severe blunting of the snout and a more rounded and wider face, whereas Bglap−/− mice were indistinguishable from WT mice, as demonstrated by the superimposition of the facial profiles presented in the lateral and frontal images (Fig. 1, A–C).
Figure 1.
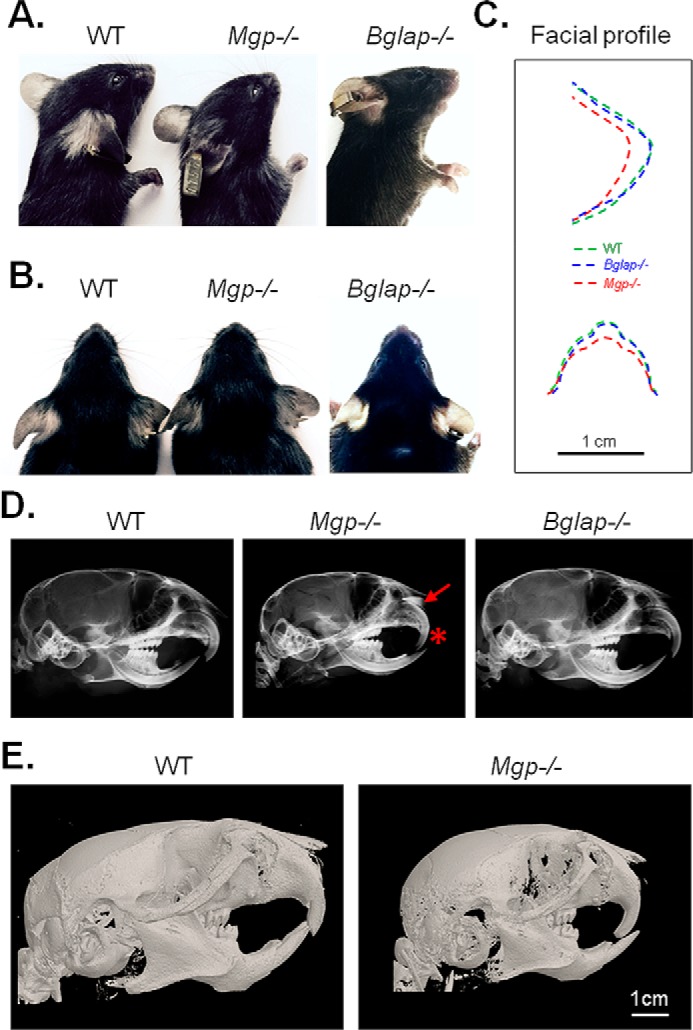
Ablation of Mgp, but not Bglap, causes craniofacial malformations. A, lateral cephalic photographs of WT, Mgp−/−, and Bglap−/− mice showing a blunt and shorter snout in Mgp−/− mice. B, frontal cephalic photographs confirming the abnormal craniofacial phenotype in Mgp−/− mice. C, superimposition of the lateral and frontal cephalic photographs showing a comparable facial profile between WT and Bglap−/− mice and a blunt and wider profile in Mgp−/− mice. D, lateral cephalic X-ray of WT, Mgp−/−, and Bglap−/− mice. Mgp−/− mice show a severe anterior cross-bite with a radiopaque nasal structure (asterisk and arrow, respectively). E, 3D reconstruction of micro-CT scans of WT and Mgp−/− mice confirming the dental malocclusion and craniofacial deformities in the latter genotype.
Lateral cephalic X-rays of age- and gender-matched WT, Mgp−/−, and Bglap−/− mice revealed severe dental malocclusion characterized by an anterior cross-bite only in Mgp−/− mice; this phenotype was consistent in all Mgp-null mice analyzed. Additionally, we detected an intense radiopaque area above the upper incisors in Mgp−/− mice that was not present in Bglap−/− or WT mice (Fig. 1D, asterisk and arrow, respectively). 3D reconstruction of micro-CT scans of the craniofacial complex of WT and Mgp−/− mice confirmed the presence of a severe class III malocclusion (overbite) in the mutant mice (Fig. 1E). Considering the absence of any craniofacial phenotype in Bglap−/− mice, we conclude that, of these two skeletal Gla proteins, only MGP is essential for normal craniofacial development.
We then performed a detailed cephalometric analysis and found that the maxillary and palatine lengths were more severely affected than other craniofacial structures in Mgp−/− mice. In contrast, the skull height, anterior cranial height, and inter-molar maxillary distance were not affected, whereas only modest decreases were observed in the mandibular measurements (Fig. 2, A and B, and supplemental Table S1). Taken together, these data identify midface hypoplasia as the major craniofacial phenotype in MGP-deficient mice.
Figure 2.
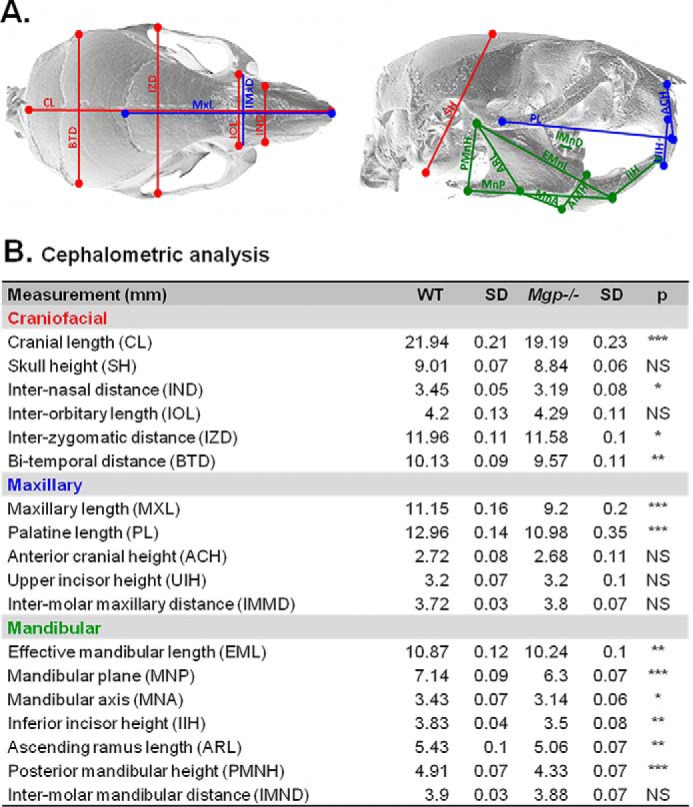
MGP deficiency causes midface hypoplasia. A, depiction of the selected landmarks for cephalometric analyses of WT and Mgp−/− mice. Red, blue, and green lines represent the overall craniofacial, maxillary, and mandibular measurements, respectively. A description of the location of each landmark is presented in supplemental Table S1. B, craniofacial, maxillary, and mandibular measurements in millimeters. Each value represents the mean of six mice analyzed for each genotype at 5 weeks of age. Statistical analysis: Student's t test. SD, standard deviation.
Ectopic calcification of the cartilaginous nasal septum in MGP-deficient mice
The aforementioned phenotype called for an understanding of the mechanism by which MGP could affect craniofacial development. As a first step in addressing this question, we studied Mgp expression in the craniofacial complex. To that end, we used a “knock-in” model (Mgp-Cre) in which a Cre recombinase gene was inserted at the Mgp locus after the stop codon (24). These mice were crossed with a reporter transgenic line, Gtrosa6tm1Sor, carrying a ubiquitous Rosa promoter followed by a “floxed” insulator sequence and the bacterial β-galactosidase gene. In the resultant compound mutants, Mgp-Cre;Gtrosa6tm1Sor (referred to hereafter as Mgp-Cre;LacZ), the β-galactosidase gene is expressed in tissues where the Mgp promoter is active (24). Whole-mount and dissected heads from 2-week-old WT and Mgp-Cre;LacZ mice were stained with X-gal for β-galactosidase activity. Intense blue staining revealed Mgp promoter expression in all cranial sutures and more strongly in the lambdoidal and frontonasal sutures (Fig. 3A). Also, intense β-galactosidase activity was detected in the spheno-occipital synchondrosis (SOS), intersphenoidal synchondrosis (ISS), and cartilaginous nasal septum of the Mgp-Cre;LacZ mice (Fig. 3B).
Figure 3.

Ectopic calcification of the cartilaginous nasal septum in MGP-deficient mice. A and B, whole-mount heads of 2-week-old Mgp-Cre;LacZ and control mice were stained with X-gal for bacterial β-galactosidase detection. Intense blue staining revealed Mgp promoter activity in all craniofacial sutures, more intensely in the lambdoidal (Ld) and frontonasal (FN) sutures. The SOS and ISS in the basicranial region and the NS were also strongly stained (arrows, n = 3 mice for each group). C, 2D images of micro-CT scans of WT and Mgp−/− mice showing the absence of craniosynostosis in the mutant mouse in the lambdoidal, coronal, frontonasal, and palatomaxillary sutures (arrows, n = 6 in each group). D, 2D micro-CT scans (top panels, sagittal view; bottom panels, frontal view) of the basicranium of WT and Mgp−/− mice showing the normal anatomy of the ISS in the mutant mouse. However, the SOS appeared to be disorganized, with an aberrant pattern of mineralization (n = 3 mice in each group). E, 3D reconstruction of WT and Mgp−/− heads in the frontal plane showing severe nasal septum mineralization in Mgp−/− mice (arrows, n = 6 mice in each group). All analyses were done on 5-week-old mice unless indicated otherwise.
The lambdoidal, coronal, frontonasal, and palatomaxillary sutures were analyzed using 2D micro-CT images, which failed to detect any sign of craniosynostosis or other anomalies in the sutures of Mgp−/− mice (Fig. 3C). Similarly, 2D imaging of micro-CT scans of the SOS and ISS (Fig. 3C, top panel, sagittal view; bottom panel, frontal view) did not detect premature closure of the ISS in these mice. However, at 5 weeks of age, the SOS appeared to be disorganized, with an aberrant pattern of mineralization (Fig. 3D).
Considering that our X-ray analysis showed increased radiopacity in the region corresponding to the nasal septum and the very high expression of Mgp promoter in this tissue, we examined its mineralization status in 5-week-old MGP-deficient mice by micro-CT. As presented in Fig. 3E, frontal sections displayed striking ectopic mineralization of the nasal septum, which normally remains unmineralized throughout life (27).
von Kossa and van Gieson (VKVG) staining of histological sections showed that, although not calcified at birth, by the first week of life, mineral deposition is already detectable in the septal cartilage of MGP-deficient mice. Mineral accumulation then progressively increases, as shown by the analyses of septal cartilage of 2- and 5-week-old mutant mice, whereas it remains unmineralized in WT littermates (Fig. 4).
Figure 4.
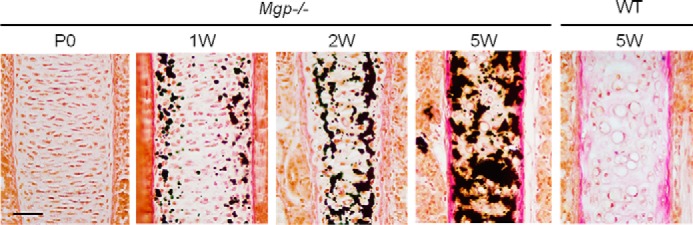
Progressive ectopic mineralization of MGP-deficient nasal septum. Histological analysis of Mgp−/− nasal septa by VKVG staining of 7-μm-thick plastic sections showing the initiation of pathologic cartilage mineralization at 1 week of age and its progression until 5 weeks of age (n = 3 mice for each time point). The last panel represents an unmineralized WT nasal septum at 5 weeks of age stained with VKVG. Scale bar = 40 μm.
Local expression of Mgp in the cartilage corrects midface hypoplasia in MGP-deficient mice
We next investigated whether local expression of Mgp in chondrocytes is sufficient to correct midface hypoplasia in Mgp−/− mice. For this purpose, we generated two transgenic lines that expressed Mgp under the Col2a1 promoter (Fig. 5A, top panel). Semiquantitative PCR analyses showed a higher level of transgene expression in the rib cartilage of one of these founders, although weak expression of the transgene was also detected in the aorta (Fig. 5A, bottom panel). This observation was further confirmed by quantitative real-time PCR (qRT-PCR) (Fig. 5B). We mated Col2a1-Mgp mice with Mgp+/− mice to eventually generate Mgp−/−,Col2a1-Mgp mice in the F2 generation. These mice showed approximately a 6-fold increase of Mgp expression in the nasal septum compared with the WT littermates (Fig. 5C).
Figure 5.

Local expression of Mgp in the cartilage corrects midface hypoplasia in MGP-deficient mice. A, schematic of the genetic construct used to generate Col2a1-Mgp transgenic (Tg) mice expressing Mgp under the cartilage-specific Col2a1 promoter (top panel). The arrows represent the primers used to detect the transgene by PCR. Semiquantitative PCR shows the tissue-specific expression of the transgene in 2-week-old Col2a1-Mgp mice (center panel; 1, cartilage; 2, aorta; 3, calvaria 4, muscle; 5, brain; 6, long bone), strong Mgp expression in cartilage, and minor expression in the aorta. B, qRT-PCR analysis of transgene expression in the same tissues as described in A. RQ, relative quantification. C, qRT-PCR showing ∼6-fold increased Mgp expression in the nasal septum cartilage of Mgp−/−;Col2a1-Mgp (Mgp−/−;Tg) mice compared with WT littermates. Note the absence of Mgp expression in the nasal septum of Mgp−/− mice. D, rib cage skeletal preparations of 5-week-old mice stained with alizarin red and Alcian blue showing the vascular mineralization in Mgp−/− and Mgp−/−;Tg mice. E, VKVG staining showing a complete absence of mineralization in the nasal septum of Mgp−/−;Tg mice. Scale bar = 100 μm. F, lateral cephalic X-ray of WT and Mgp−/−;Tg littermates showing the correction of the craniofacial and dental phenotypes in the latter model. G, craniofacial measurements showing the normalization of the cranial, maxillary, and palatine lengths in Mgp−/−;Tg mice. Statistical analysis: analysis of variance (Tukey's multiple comparisons test). All analyses were done with 5-week-old mice (n = 3 in each group for each experiment).
To examine whether the relatively weak expression of the MGP transgene in the aorta could prevent arterial calcification, we performed alizarin red staining of the thoracic skeleton and vascular tissues of 5-week-old WT, Mgp−/−, and Mgp−/−;Col2a1-Mgp littermates. We confirmed that the weak expression of the transgene in Mgp−/−;Col2a1-Mgp mice was not sufficient to correct the vascular calcification phenotype, suggesting that this is a valid model for our experiments (Fig. 5D).
We next studied the mineralization status of the nasal septum in Mgp−/−;Col2a1-Mgp mice. Histological analyses revealed a complete absence of ectopic mineralization in the septal cartilage. In contrast, the nasal septum of the Mgp−/− littermates was severely mineralized (Fig. 5E). Cephalic X-ray imaging of Mgp−/−;Col2a1-Mgp mice was comparable with that of WT littermates, confirming complete correction of the dental and skeletal malocclusion (Fig. 5F). Similarly, the cranial, maxillary, and palatine lengths that were affected in Mgp−/− mice were normalized in Mgp−/−;Col2a1-Mgp mice (Fig. 5G). Taken together, these data suggest that local expression of MGP by chondrocytes can correct midface hypoplasia in Mgp−/− mice.
MGP-deficient mice have a shorter nasal septum with apoptotic chondrocytes
We next examined the effects of Mgp ablation on the growth of nasal septum. The exposed septal cartilages of WT and Mgp−/− littermates were stained with Safranin O, and the longitudinal lengths were measured. There was a significant reduction in the length of the nasal septum of Mgp−/− mice (Fig. 6A). We then analyzed 2D micro-CT images to measure the rostral, basicranial, and total skull lengths in WT and Mgp−/− mice following landmarks determined previously (28) (Fig. 6B, left panel, and supplemental Table S2). The rostral (point 1-2) and basicranial (points 2-3, 3-4, and 4-5) lengths were measured and individually normalized by total cranial length (point 1-5). We found that the rostral segment, but none of the basicranial lengths measured, were shorter in Mgp−/− mice (Fig. 6B, right panel). This result further suggests that shortening of the nasal septum is the cause of the hypoplastic maxilla in Mgp−/− mice.
Figure 6.

MGP-deficient mice have a shorter nasal septum. A, exposed cartilaginous nasal septa of 5-week-old WT and Mgp−/− mice stained with Safranin O. Measurement of the lengths presented as relative length ratio shows a significantly smaller nasal septum in Mgp−/− mice. B, 2D cephalometric analysis of the viscerocranium and basicranium of 5-week-old WT and Mgp−/− mice. The viscerocranium (1–2) in Mgp−/− mice is significantly smaller compared with WT mice, whereas the total basicranial length (2–5) is not affected. Values represent the ratio of the linear distance between points over the total cranial length (1–5). A description of the anatomical locations of the landmarks is presented in supplemental Table S2 (n = 5 in both groups). Statistical analysis: Student's t test in all cases.
To understand the cause of nasal septum shortening in Mgp−/− mice, we first assessed the cell size by calculating the average chondrocyte area. We did not observe any difference between WT and Mgp−/− septal chondrocytes (Fig. 7A). Then we evaluated chondrocyte proliferation and apoptosis. Anti-Ki67 immunostaining showed a comparable cell proliferation rate between WT and Mgp−/−nasal septum chondrocytes (Fig. 7B). However, we observed an increase in TUNEL-positive immature chondrocytes in the calcified nasal septum of Mgp−/− mice (Fig. 7C).
Figure 7.

MGP-deficient septal chondrocytes undergo apoptosis. A, quantification of cell area on histological sections of WT and Mgp−/− nasal septa stained with H&E shows no difference in chondrocyte size between the genotypes. Three fields were quantified per sample (n = 3 in both groups). Scale bar = 20 μm. B, anti-Ki67 antibody and hematoxylin staining of septal sections showing the proliferating chondrocytes (arrows). Quantification of proliferative cells/total cell count shows no difference between WT and Mgp−/− mice (n = 6 in both groups). Scale bar = 20 μm. C, colorimetric apoptosis detection assay (TUNEL) performed on WT and Mgp−/− septal sections shows the presence of apoptotic cells (arrows) in the MGP-deficient nasal septum but not in the WT nasal septum sections. The sections were counterstained with methyl green (n = 3 in both groups). Scale bars = 20 μm. All analyses were performed on 2-week-old mice. Statistical analysis: Student's t test in all cases.
Chondrocyte hypertrophy is not a prerequisite for nasal septum calcification in MGP-deficient mice
We then interrogated whether ectopic nasal septum calcification in Mgp−/− mice is analogous to growth plate cartilage mineralization, a physiologic event that occurs during endochondral bone development. First we compared the expression of a general chondrogenic marker, Col2a1 (encodes type II collagen), in the nasal septum of 1-week-old WT and Mgp−/− mice; there was no detectable difference in mRNA levels (Fig. 8A). Similarly, there was a comparable deposition of type II collagen, as evident by immunostaining of the histological sections of the septal cartilage from 2-week-old WT and Mgp−/− mice (Fig. 8B). Interestingly, although the cartilaginous ECM was mineralized, there was no up-regulation of the hypertrophic chondrocyte marker Col10a1 by qRT-PCR analysis, nor was there any alteration of the encoded protein type X collagen when analyzed by immunostaining (Fig. 8, C and D). Also, we did not detect any increase in alkaline phosphatase activity, a hallmark of chondrocyte hypertrophy, in tissue extracts or histological sections prepared from Mgp−/− septal cartilage in comparison with that of WT cartilage (Fig. 8, E and F).
Figure 8.

Chondrocyte hypertrophy is not a prerequisite for ectopic mineralization of the nasal septum in MGP-deficient mice. A, gene expression analysis of Col2a1 in 1-week-old WT and Mgp−/− nasal septa showing comparable expression in both groups. RQ, relative quantification. B, immunohistochemistry showing a similar expression pattern of type II collagen in 2-week-old WT and Mgp−/− nasal septa. Embryonic humerus growth plate staining was used as a positive control. C, Col10a1 gene expression in 1-week-old WT and Mgp−/− mice showing no induction in the knockout tissue. D, absence of anti-type X collagen staining in WT and Mgp−/− 2-week-old nasal septum. Embryonic humerus growth plate staining was used as a positive control. E, the alkaline phosphatase (ALPL) basal activity in Mgp−/− nasal septum extracts from 2-week-old mice is comparable with that of WT littermates. F, ALPL staining confirms the absence of ALPL activity in the septal cartilage of Mgp−/− mice. Staining of septal growth plate sections from 2-week-old WT mice was used as a positive control. Scale bars for histological sections = 40 μm. Statistical analysis: Student's t test in all cases.
Amorphous calcium phosphate as main mineral species in MGP-deficient nasal septum
Ultrastructural analysis of Mgp−/− calcified nasal septa by transmission electron microscopy showed globular structures in the ECM in the vicinity of the chondrocytes. We observed incremental growth lines at the peripheral regions of the deposited mineral (Fig. 9A). Detailed characterization of the mineral deposits by energy-dispersive X-ray spectroscopy identified them to contain abundant calcium and phosphorus (Fig. 9B). X-ray diffraction (Fig. 9C) and electron diffraction (Fig. 9C, inset) revealed that the inorganic mineral present in the MGP-deficient nasal septa was primarily an amorphous calcium-phosphate phase with a slight degree of crystallization toward an apatitic mineral phase, as demonstrated by diffraction rings/spectra, labeled 1 and 2 (Fig. 9C). It was also noted that matrix vesicles, which are normally present in the mineralizing growth plates, were generally not observed in the calcified nasal septum.
Figure 9.

Amorphous calcium phosphate as the main mineral species in MGP-deficient nasal septum. A, transmission electron microscopy of 1-week-old Mgp−/− nasal septum showing the presence of chondrocytes (Ch) and mineral deposits (asterisks) having an unusual globular shape. Note the incremental lines (suggesting periodic mineral deposition) within the mineral deposits (arrowheads). B, energy-dispersive X-ray spectroscopy showing the presence of phosphorus and calcium within the mineral deposits. C, X-ray diffraction showing that the mineral phase is largely amorphous calcium phosphate. Minor spectral peaks labeled 1 and 2 indicate tendencies toward crystallization (initiation) of an apatitic phase. a.u., arbitrary units. Inset, electron diffraction pattern confirming the presence of mostly amorphous mineral showing only diffuse (rather than sharp) electron diffraction rings labeled 1 and 2 (n = 3 mice for each group and each experiment).
Prevention of nasal septum mineralization in Mgp−/−;Hyp mice
To understand whether ectopic mineralization in the nasal septum is regulated by global mechanisms that control bone and cartilage mineralization, we generated Mgp−/−;Hyp compound mutants. Hyp mice show an ∼50% reduction of phosphate levels in the serum, with poor bone and tooth mineralization, largely being attributable to this low level of circulating phosphate, otherwise required for appropriate mineralization of the skeleton and dentition (29). As we reported previously, the systemic reduction in phosphate level persists in Mgp−/−;Hyp mice, whereas the serum calcium level was not affected (30) (Fig. 10A). We performed 3D micro-CT on 5-week-old WT, Mgp−/−, and Mgp−/−;Hyp heads, which revealed a complete absence of cartilaginous nasal septum mineralization in the double mutants (Fig. 10B). These findings were further confirmed by histology of nasal septum sections, demonstrating a complete absence of mineralized ECM in Mgp−/−;Hyp mice (Fig. 10C). Interestingly, there were no TUNEL-positive nuclei detected on the septal sections of Mgp−/−;Hyp mice (Fig. 10D). Cephalometric analyses of micro-CT images showed correction of the class III malocclusion together with normal maxillary and palatine lengths (Fig. 10E). Although there was a significant increase in cranial length in Mgp−/−;Hyp mice in comparison with Mgp−/− mice, it remained shorter in comparison with that of WT mice (Fig. 10F).
Figure 10.

Prevention of nasal septum mineralization in Mgp−/−;Hyp mice. A, serum calcium and inorganic phosphate levels in WT, Mgp−/−, and Mgp−/−;Hyp mice show similar calcium levels in all the genotypes and confirm the presence of hypophosphatemia in Mgp−/−;Hyp mice (n = 4). B, 3D reconstruction of micro-CT scans of gender- and age-matched WT, Mgp−/−, and Mgp−/−;Hyp mice showing the correction of nasal septum mineralization in the double mutant. C, histology of nasal septa of Mgp−/− and Mgp−/−;Hyp mice stained with VKVG confirms the absence of apatitic minerals in the latter phenotype. Scale bar = 50 μm. D, fluorometric TUNEL assay shows the presence of apoptotic immature chondrocytes in 3-week-old Mgp−/− (arrows) but not in Mgp−/−;Hyp nasal septa. Images of the same field with fluorescein-12-dUTP (which labels fragmented DNA ends) and H33258 (a nuclear stain) fluorescence overlaid to create the combined images. Scale bar = 25 μm. E, 3D reconstruction of micro-CT scans of Mgp−/− and Mgp−/−;Hyp mice. Note the correction of midface hypoplasia and class III malocclusion in the compound mutant. F, cephalometric analysis of WT, Mgp−/−, and age- and gender-matched Mgp−/−;Hyp mice showing complete correction of the palatine and maxillary lengths in Mgp−/−;Hyp mice. Although the cranial length is corrected in Mgp−/−;Hyp mice compared with Mgp−/− mice, it remains shorter than that of the WT controls. All analyses were performed in 5-week-old mice unless indicated otherwise (n = 3 male mice in each group for each experiment). Statistical analysis: analysis of variance (Tukey's multiple comparisons test).
Discussion
Cephalometric analyses of Mgp−/− heads at 5 weeks of age revealed severe midface abnormalities. Until now, these abnormalities were not fully characterized, and the underlying cause of this phenotype was unknown. Interestingly, the facial morphology of this mouse model closely resembles that of Keutel syndrome and warfarin embryopathy patients, which justifies a thorough mechanistic study of this pathology.
Our micro-CT analyses clearly identified the key features of the facial phenotype in MGP-deficient mice. These measurements suggest that, overall, the craniofacial bones are undersized in the mutant mice, which is expected considering their stunted growth. However, the maxilla and palatine bones are disproportionately smaller, affecting the overall anteroposterior length. Taken together, the cephalometric analyses confirm that midface hypoplasia is the major cause of craniofacial defects in MGP-deficient mice.
As evident in the published literature, the two major causes of midface hypoplasia are premature cranial base synchondroses fusion and craniosynostosis—premature closure of the cranial vault sutures (17). It is now believed that the sutures play a passive role during the growth of the craniofacial complex, whereas the synchondroses act as growth centers, providing sites for rapid bone growth until fusion. Particularly, the spheno-occipital synchondrosis has been shown to play a major role, as its late ossification and the closure timing correlate with the severity of the midface hypoplasia seen in syndromic patients (17, 28, 31, 32).
Impaired vitamin K metabolism has also been associated with midface hypoplasia. For instance, inactivating mutations in the gene for VKORC1, which generates the reduced form of vitamin K1, and GGCX, which uses it as a co-factor, both lead to vitamin K-dependent clotting factor deficiency and midface hypoplasia (33, 34). Similarly, babies born to mothers under anticoagulation therapy with warfarin suffer from warfarin embryopathy and also show midface abnormalities (6). Considering that GGCX/VKORC1 mutations or fetal exposure to warfarin both affect γ carboxylation of Gla proteins, it is expected that these conditions would lead to inactivation of the skeletal Gla proteins, e.g. MGP and osteocalcin. However, our experimental data presented here show that only MGP, but not osteocalcin deficiency, causes midface hypoplasia in mice.
The role of the nasal septum in midface development has been a matter of controversy for over half a century. Scott (35), in 1951, proposed that the nasal septum, as all other primary cartilages, acts as a growth center, separating the facial structures, allowing the sagittal growth of the face. This observation was supported by the findings that the nasal septum responds to hormones and growth factors and has an intrinsic ability to grow. Additionally, extirpation of the nasal septum has been shown to impair midface development in rats and rabbits (10, 36). However, such experimental approaches were criticized, as facial development is slower in humans than in the animal models used; thus, the observed midface malformations were attributed to surgical trauma.
In 1968, Moss et al. (37) proposed the nasal septum as a passive structure that serves a supportive role for the other craniofacial structures. This notion was challenged by in vitro and in vivo studies showing that the intrinsic growth capacity of the nasal septum is comparable with that of epiphyseal cartilages (38, 39). Furthermore, the early corrections of nasal septum deformities in infants with cleft lip and palate or other facial defects show a greater effect on the correction of the overall craniofacial irregularities (40). In agreement with these findings, we provide evidence that, during the early phases of facial development, ectopic nasal septum mineralization affects the growth of the midface. We report here that the prevention of nasal septum mineralization by transgenic expression of Mgp or by systemic reduction of inorganic phosphate levels both corrected the midface abnormalities in MGP-deficient mice. These data support the original theory proposed by Scott (35) that the nasal septum acts as a critical growth element during facial development.
We performed an in vivo reporter assay to determine Mgp promoter activity in the developing craniofacial complex. For this purpose, we used a Cre transgenic line in which the gene encoding Cre recombinase was knocked in at the Mgp locus by placing it directly under the control of the endogenous Mgp promoter and its regulatory elements. Crossing this model with the R26R-lacZ reporter line enabled us to faithfully establish endogenous Mgp expression by following β-galactosidase activity in the resultant offspring (24). The knock-in strategy we used is superior to the conventional transgenic approach, as the reporter gene expression is not driven by a truncated promoter at a random location in the genome but by the single copy-endogenous promoter with all its proximal and distal regulatory elements. We found strong Mgp promoter activity in the cranial vault sutures, cranial base synchondroses, and nasal septum. However, our reporter assay only suggests that the promoter is/was active in the β-galactosidase-positive tissues but does not precisely indicate its time of activity during development. Considering that the cartilaginous part of the nasal septum remains unmineralized throughout adulthood (27), it is likely that Mgp is constitutively expressed in this tissue. Collectively, our data from the in vivo reporter assay and qRT-PCR gene expression analyses performed at two different time points (2 and 3 weeks of age, respectively) suggest that this is indeed the case.
It is interesting to note that, despite the high level of Mgp expression in both SOS and ISS, only the SOS growth plates in Mgp−/− mice showed mild mineralization irregularities, whereas the ISS growth plates remained unaffected. In addition, the cranial sutures were also not abnormally mineralized in these mice. The absence of ectopic mineralization at some of these sites could be explained by the action of other mineralization inhibitor(s) (41), which may have differential distributions in various connective tissues. In fact, inorganic pyrophosphate, a potent mineralization inhibitor, may provide complementary anti-mineralization functions in some cartilaginous tissues. This notion is supported by the observation that the severe articular cartilage mineralization caused by impaired pyrophosphate metabolism is absent in MGP-deficient mice (42, 43)
Despite the presence of ectopic mineralization at the SOS, the proportional length of the cranial base was not smaller in Mgp−/− skulls. Considering this observation, it is unlikely that the mild ectopic mineralization of the SOS significantly contributed to the disproportionate shortening of the midface. On the other hand, the relative length of the nasal septum as well as the viscerocranium were significantly smaller, suggesting that ectopic mineralization of the septal cartilage is the major cause of midface hypoplasia in MGP-deficient mice.
To investigate whether MGP acts locally to prevent abnormal nasal septum mineralization, we generated Mgp−/−;Col2a1-Mgp mice. Unexpectedly, the Col2a1-Mgp transgene showed a weak expression in the aorta. This can be explained by the positional effects of the transgene integration site in the chromosome alone or combined effects of both the integration site and copy number of the transgene. However, this weak “leaky” expression of the transgene was not sufficient to prevent ectopic mineralization of the arteries in Mgp−/−;Col2a1-Mgp mice. Our observation that abnormal nasal septum mineralization was fully prevented despite the presence of mineralized arteries has two important implications: first, it rules out the possibility that the calcified blood vessels adjacent to the nasal septum perichondrium serve as nidi for the observed septal cartilage calcification; second, in agreement with our previous findings, it establishes that MGP acts locally to prevent ectopic calcification (22).
During endochondral bone development, growth plate cartilage mineralization is a normal process that requires chondrocyte hypertrophy, hallmarked by type X collagen expression, and the release of matrix vesicles. Interestingly, chondrocyte hypertrophy and matrix vesicles appear not to be a prerequisite for ectopic mineralization of the cartilaginous ECM in the nasal septum of MGP-deficient mice. This observation suggests that mineral accumulation in the septal cartilage is likely not analogous to growth plate cartilage mineralization, where terminally differentiated chondrocytes undergo hypertrophy, and matrix vesicles seem to participate in the initiation of mineralization (44, 45). We reported a similar nonchondrogenic ectopic mineralization event in the arteries of MGP-deficient mice (46). Based on our observation, we conclude that the initiation of ECM mineralization in the MGP-deficient nasal septum does not require cellular differentiation (or release of abundant matrix vesicles) but happens spontaneously by calcium phosphate precipitation. As is the case in blood vessels, whether any particular ECM protein promotes this mineral precipitation in the absence of MGP has yet to be determined.
Our analyses of the deposited mineral in the calcified nasal septum of MGP-deficient mice indicate that the mineral is primarily amorphous (noncrystalline) calcium phosphate. This finding indicates that MGP normally influences the early stages of calcium phosphate precipitation. Although, at this point, it is not known which structural features in MGP are essential for this function, it may not be solely attributable to the posttranslational γ carboxylation of its glutamic acid residues. Our earlier work showed that osteocalcin, a closely related structural Gla protein, does not possess any significant anti-mineralization function (22). In agreement with this, we did not observe nasal septum mineralization or craniofacial abnormalities in osteocalcin-deficient mice. Recent published in vitro data indicate that N-terminal serine residues in MGP undergo phosphorylation and may facilitate the interaction between MGP and hydroxyapatite minerals (47). The in vivo significance of these findings has yet to be determined. Identification of the functional residues in MGP is essential to understand the mechanism underlying the anti-mineralization function of MGP.
Vascular smooth muscle cells produce large amounts of MGP, which prevents mineralization of the elastic lamina in arterial tissues (46). The vascular mineralization phenotype in MGP-deficient mice is fully penetrant and appears at the same time as the initiation of nasal septum mineralization. This raises the question of whether mineral deposition in the nasal septum is actually within the blood vessels present in the tissue. However, this possibility can be ruled out by our observation that, although the blood vessels are extensively mineralized in Mgp−/−;Col2a1-Mgp mice, as is the case in Mgp−/− mice, there was no detectable presence of mineral deposits in the septal cartilage of Mgp−/−;Col2a1-Mgp mice. Furthermore, although vascular calcification is not reported in most Keutel syndrome patients, midface hypoplasia has always been associated with this disease, suggesting that these two phenotypic traits may not be interrelated.
Our experimental data revealed an increase in apoptosis in MGP-deficient septal chondrocytes, which may explain, at least in part, the observed shortening of the nasal septum in these mice. Hayano et al. (48) found that augmented BMP signaling leads to apoptosis in the developing nasal cartilage through p53 up-regulation. Additionally, other laboratories have reported that MGP inhibits BMP signaling in vascular smooth muscle cells (49). Therefore, it is possible that MGP deficiency in the nasal septum may induce BMP signaling and cause chondrocyte apoptosis. However, these data needs to be confirmed experimentally. A second hypothesis that can explain the increased apoptosis in the nasal septum of Mgp−/− mice is the local increase in inorganic phosphate in the cartilaginous ECM. Inorganic phosphate has been shown to be a major regulator of apoptosis of hypertrophic chondrocytes in developing endochondral bones (50). As reported here, we found the presence of amorphous calcium phosphate precipitates in the calcified nasal septum. The unstable nature of this transient phase may allow the minerals to be readily dissolved, increasing local inorganic phosphate levels and inducing apoptosis of the neighboring cells. In addition to chondrocyte apoptosis, another possible mechanism that may affect the anteroposterior growth of the nasal septum is the stiffening of its ECM caused by ectopic mineral deposition. Future studies of the mechanical properties of the mineralized septal cartilage will reveal whether this is indeed the case.
Last, we found that systemic regulation of inorganic phosphate levels in the serum is sufficient to prevent nasal septum mineralization in Mgp−/−;Hyp double mutant mice. Mutations in the Phex gene lead to X-linked hypophosphatemia (Hyp mice), characterized by phosphaturia and increased osteoid volume. Interestingly, lowering serum phosphate levels in Mgp−/− mice also completely prevents the vascular calcification phenotype, as we reported previously (30). This finding has a substantial clinical implication in that it demonstrates that nasal septum mineralization and craniofacial malformations can be modulated by systemic factors. Future preventive interventions can be developed based on this finding.
Experimental procedures
Mice
The generation of Mgp−/−, Bglap−/− (Ocn−/−), and Mgp-Cre;Gtrosa6tm1Sor mice has already been described (24, 26, 51). All experiments were performed on mice with a C57BL/6 background. Col2a1-Mgp transgenic mice were generated by pronuclear injection at the Transgenic Core Facility at the Goodman Cancer Center of McGill University following standard techniques. Hyp mice were purchased from The Jackson Laboratory. Mgp−/−;Hyp double mutants were generated through breeding, and only male mice were used for our analyses. Mice were maintained in a pathogen-free standard animal facility.
Genotypes were determined by PCR on genomic DNAs isolated from tail biopsies. The sequences of the primers used for genotyping can be provided upon request.
Skeletal preparation
Skeletal tissues from adult mice were fixed overnight in 95% ethanol, stained in 0.015% Alcian blue dye (Sigma-Aldrich) in a 1:4 solution of glacial acetic acid and absolute ethanol for 24 h. Tissues were then treated with 2% potassium hydroxide for another 24 h (or until the soft tissues were dissolved) and stained with 0.005% Alizarin Red (Sigma-Aldrich) in a 1% potassium hydroxide solution. Finally, the stained skeletal tissues were clarified in 1% potassium hydroxide/20% glycerol for 2 days.
Radiography and X-ray micro-computed tomography (micro-CT)
Radiographic analyses of the mouse heads were performed at the Centre for Bone and Periodontal Research core facility at McGill University, with an XPERT X-ray imaging system (Kubtec). Micro-CT scanning of mouse skulls was performed with a SkyScan model 1072 instrument (SkyScan) set at a resolution of 8.0 μm and 0.5-mm Al filter. Micro-CT image processing and analysis was performed with version 2.2f of the SkyScan software. Cephalometric measurements, sutures, and cranial base analyses were done using Data Viewer software (SkyScan). The 3D reconstructions of head scans were done using CtAn and CtVol software (SkyScan).
Cephalometric analysis
Cephalometric analyses of micro-CT scans of whole heads were done following the method reported by Eimar et al. (52). Cephalometric analysis of the basicranium was done according to the modified method reported by Laurita et al. (28). Only male mice were used for the cephalometric and basicranial studies.
Histology and tissue imaging
Mouse skulls were fixed overnight in 10% formalin, embedded in methyl methacrylate, sectioned (7 μm), and stained with VKVG or hematoxylin and eosin for cell size assessment. For type II (ab21291, Abcam Inc.) and type X collagen (a generous gift from Dr. Pierre Moffatt, Shriners Hospital for Children, Montreal (53)) and Ki-67 (ab66155, Abcam Inc.) immunohistochemistry, 7-μm-thick paraffin sections of decalcified tissues were prepared. Hematoxylin (Fisher Diagnostics) was used for visualization of cell nuclei. Images were taken using a light microscope (DM200, Leica Microsystems) with ×20 (numerical aperture of 0.40) and ×40 (numerical aperture of 0.65) objectives. Exposed cartilaginous nasal septa were stained with Safranin-O (BioShop) and scanned using an Epson V700 PHOTO scanner. Cell area (square micrometers) was measured using PhotoShop (Adobe). All histological images were captured using a digital camera (DP72, Olympus Canada Inc.), acquired with DP2-BSW software (XV3.0, Olympus Canada Inc.) and processed using PhotoShop software (Adobe).
TUNEL assay
A TUNEL assay was performed using a colorimetric (Trevigen) or fluorometric (Promega) kit following the instructions of the manufacturer. Samples from 2- and 3-week-old mice were decalcified in 25% EDTA (Sigma-Aldrich) in PBS and embedded in paraffin to cut 7-μm sections. Upon labeling, sections were counterstained with 1% methyl green or H33258 (Sigma-Aldrich). TUNEL-positive cells were visualized using light (DM200, Leica Microsystems Inc.) and fluorescence (EVOS FL cell imaging system, Thermo Fisher Scientific) microscopes and quantified using ImageJ software (National Institutes of Health).
Alkaline phosphatase activity assay
Tissue extracts were prepared with 1× passive lysis buffer (Promega), and total proteins were measured with the Micro BCA protein assay (Thermo Scientific). Alkaline phosphatase activity was measured using p-nitrophenyl phosphate substrate (Sigma-Aldrich) and then normalized by the respective protein concentration in the extracts.
X-gal staining
For X-gal (BioShop) staining, mouse heads were dissected and fixed for 5 min in 2% formalin and 0.2% glutaraldehyde in PBS containing 5 mm EGTA and 2 mm MgCl2. Fixed tissues were rinsed with PBS containing 2 mm MgCl2 and 0.2% IGEPAL® CA 630 and left for 4 h in the same buffer supplemented with 5 mm K3Fe(CN)6 and 5 mm K4Fe(CN)6.3H2O. Later, the whole-mount heads were stained for 4 h at 37 °C in 25 mg/ml X-gal diluted in the same buffer.
Gene expression analysis
Gene expression analyses were performed using a qRT-PCR system (model 7500, Applied Biosystems, Mississauga, ON, Canada). Total RNA was extracted from different tissues with TRIzol reagent (Invitrogen) and subjected to DNase I (Invitrogen) treatment. The first-strand cDNA synthesis and qRT-PCR were performed using a high-capacity cDNA reverse transcription kit (Applied Biosystems) and Maxima SYBR Green quantitative PCR master mix (Fermentas), respectively. Relative gene expression was analyzed by SDS software (Applied Biosystems) using comparative threshold cycles (CT) and hypoxanthine guanine phosphoribosyl transferase (Hprt, a housekeeping gene) expression as an endogenous control. To calculate the Δ threshold cycle value, the mean CT value of the expression of a gene in a sample was first normalized to the mean CT value of Hprt expression in that sample. The Δ threshold cycle value of the calibrator sample was subtracted from that of the sample of interest to obtain the ΔΔCT value. The relative expression was reported as 2−ΔΔCT.
Electron microscopy
Ultrastructural characterization was performed by transmission electron microscopy (TEM). Tissues were fixed with 2% glutaraldehyde (Electron Microscopy Sciences) in 0.1 m sodium cacodylate buffer (pH 7.2), followed by dehydration through a series of graded ethanol dilutions. Samples were embedded in LR White acrylic resin (Electron Microscopy Sciences). Ultrathin sections (80-nm-thick) were cut using a Leica EM UC6 ultramicrotome (Leica Microsystems Inc.), placed on formvar-coated nickel grids (Electron Microscopy Sciences), and stained conventionally with uranyl acetate and lead citrate (Electron Microscopy Sciences) for viewing by TEM. A field-emission FEI Tecnai 12 BioTwin TEM (FEI) was used to image the stained sections at 120 kV.
Electron diffraction
Electron diffraction in the selected area configuration and energy-dispersive X-ray spectroscopy were performed at 200 kV with an FEI Tecnai G2 F20 200 cryoscanning transmission electron microscope equipped with a Gatan Ultrascan 4000 × 4000 charged coupled device (CCD) camera system (model 895) and an EDAX Octane T Ultra W/Apollo XLT2 Silicon Drift Detector (SSD) and TEAMTM energy-dispersive X-ray spectroscopy analysis system (FEI). Samples were as those used for electron microscopy imaging, but sections were left unstained.
X-ray diffraction
X-ray diffraction analysis was performed using a D8 Discover diffractometer (Bruker-AXS Inc.) equipped with a copper X-ray tube (wavelength, 1.54056 Å) and a HI-STAR general area detector diffraction system mounted on a vertical θ–θ goniometer (Bruker-AXS Inc). Measurements were run in coupled θ–θ scan in microbeam analysis mode (50-μm X-ray beam spot size). The samples examined were the same as those used for electron microscopy, but here the microtomed LR White plastic block face (from which TEM sections were obtained) was analyzed directly in the X-ray beam spot mode.
Statistical analysis
All results are shown as mean ± SD. Statistical analyses were performed by Student's t test or analysis of variance (Tukey's multiple-comparisons test) using GraphPad Prism software (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Study approval
All animal experiments were performed according to Animal Use Protocol 7132, approved by the Animal Care Committee of McGill University.
Author contributions
J. M. contributed to the research experimental design, conducted experiments, analyzed data, and prepared the manuscript. H. E. performed the initial micro-CT scans and cephalometric analysis. M. D. M. conducted EM and mineral characterization experiments and contributed to manuscript preparation. M. B. contributed to the analysis of the phenotype of Mgp−/−;Col2a1-Mgp mice. V. N. conducted mineral characterization experiments and proofread the manuscript. H. R. generated the Col2a1-Mgp mice. T. B. generated the Mgp-Cre;Gtrosa6tm1Sor mice. F. T. contributed to the micro-CT analyses and proofread the manuscript. M. F. provided the Bglap−/− mice and contributed to the experimental design. M. M. contributed to the research experimental design, conducted experiments, analyzed data, and prepared the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Gerard Karsenty for critical reading of the manuscript. We also thank Mia Esser, Louise Marineau, and Maude Danis Ladouceur for animal husbandry. The core facility for skeletal phenotyping was supported by Le Réseau de Recherche en Santé Buccodentaire et Osseuse.
This work was supported by operating grant 123310 from the Canadian Institutes of Health Research Fund (to M. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1 and S2.
- GGCX
- γ-glutamyl carboxylase
- MGP
- matrix Gla protein
- ECM
- extracellular matrix
- micro-CT
- micro-computed tomography
- SOS
- spheno-occipital synchondrosis
- ISS
- intersphenoidal synchondrosis
- VKVG
- von Kossa and van Gieson
- qRT-PCR
- quantitative real-time PCR
- TEM
- transmission electron microscopy
- ALPL
- alkaline phosphatase
- BMP
- bone morphogenetic protein.
References
- 1. WHO, RMo, C. A. (2000) Craniofacial anomalies and associated birth defects. Global registry and database on craniofacial anomalies.
- 2. Wang Y., Liu G., Canfield M. A., Mai C. T., Gilboa S. M., Meyer R. E., Anderka M., Copeland G. E., Kucik J. E., Nembhard W. N., Kirby R. S., and National Birth Defects Prevention Network (2015) Racial/ethnic differences in survival of United States children with birth defects: a population-based study. J. Pediatr. 166, 819–826.e1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Munroe P. B., Olgunturk R. O., Fryns J. P., Van Maldergem L., Ziereisen F., Yuksel B., Gardiner R. M., and Chung E. (1999) Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat. Genet. 21, 142–144 [DOI] [PubMed] [Google Scholar]
- 4. Hur D. J., Raymond G. V., Kahler S. G., Riegert-Johnson D. L., Cohen B. A., and Boyadjiev S. A. (2005) A novel MGP mutation in a consanguineous family: review of the clinical and molecular characteristics of Keutel syndrome. Am. J. Med. Genet. A 135, 36–40 [DOI] [PubMed] [Google Scholar]
- 5. Lefebvre M., Dufernez F., Bruel A. L., Gonzales M., Aral B., Saint-Onge J., Gigot N., Desir J., Daelemans C., Jossic F., Schmitt S., Mangione R., Pelluard F., Vincent-Delorme C., Labaune J. M., et al. (2015) Severe X-linked chondrodysplasia punctata in nine new female fetuses. Prenat. Diagn. 35, 675–684 [DOI] [PubMed] [Google Scholar]
- 6. Starling L. D., Sinha A., Boyd D., and Furck A. (2012) Fetal warfarin syndrome. BMJ Case Rep. 2012, bcr2012007344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weaver K. N., El Hallek M., Hopkin R. J., Sund K. L., Henrickson M., Del Gaudio D., Yuksel A., Acar G. O., Bober M. B., Kim J., and Boyadjiev S. A. (2014) Keutel syndrome: report of two novel MGP mutations and discussion of clinical overlap with arylsulfatase E deficiency and relapsing polychondritis. Am. J. Med. Genet. A 164A, 1062–1068 [DOI] [PubMed] [Google Scholar]
- 8. Martelli H. Jr, Paranaíba L. M., de Miranda R. T., Orsi J. Jr., and Coletta R. D. (2008) Apert syndrome: report of a case with emphasis on craniofacial and genetic features. Pediatr. Dent. 30, 464–468 [PubMed] [Google Scholar]
- 9. Mehndiratta S., Suneja A., Gupta B., and Bhatt S. (2010) Fetotoxicity of warfarin anticoagulation. Arch. Gynecol. Obstet. 282, 335–337 [DOI] [PubMed] [Google Scholar]
- 10. Wong K. K., Filatov S., and Kibblewhite D. J. (2010) Septoplasty retards midfacial growth in a rabbit model. Laryngoscope 120, 450–453 [DOI] [PubMed] [Google Scholar]
- 11. Sarnat B. G., and Wexler M. R. (1967) Rabbit snout growth after resection of central linear segments of nasal septal cartilage. Acta Otolaryngol. 63, 467–478 [DOI] [PubMed] [Google Scholar]
- 12. Patel N., and Fearon J. A. (2015) Treatment of the syndromic midface: a long-term assessment at skeletal maturity. Plast. Reconstr. Surg. 135, 731e–742e [DOI] [PubMed] [Google Scholar]
- 13. Cielo C. M., and Marcus C. L. (2015) Obstructive sleep apnoea in children with craniofacial syndromes. Paediatr. Respir. Rev. 16, 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hall B. K., and Precious D. S. (2013) Cleft lip, nose, and palate: the nasal septum as the pacemaker for midfacial growth. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 115, 442–447 [DOI] [PubMed] [Google Scholar]
- 15. Forte A. J., Alonso N., Persing J. A., Pfaff M. J., Brooks E. D., and Steinbacher D. M. (2014) Analysis of midface retrusion in Crouzon and Apert syndromes. Plast. Reconstr. Surg. 134, 285–293 [DOI] [PubMed] [Google Scholar]
- 16. Chokdeemboon C., Mahatumarat C., Rojvachiranonda N., Tongkobpetch S., Suphapeetiporn K., and Shotelersuk V. (2013) FGFR1 and FGFR2 mutations in Pfeiffer syndrome. J. Craniofac. Surg. 24, 150–152 [DOI] [PubMed] [Google Scholar]
- 17. Goldstein J. A., Paliga J. T., Wink J. D., Bartlett S. P., Nah H. D., and Taylor J. A. (2014) Earlier evidence of spheno-occipital synchondrosis fusion correlates with severity of midface hypoplasia in patients with syndromic craniosynostosis. Plast. Reconstr. Surg. 134, 504–510 [DOI] [PubMed] [Google Scholar]
- 18. Paliga J. T., Goldstein J. A., Vossough A., Bartlett S. P., and Taylor J. A. (2014) Premature closure of the spheno-occipital synchondrosis in Pfeiffer syndrome: a link to midface hypoplasia. J. Craniofac. Surg. 25, 202–205 [DOI] [PubMed] [Google Scholar]
- 19. Howe A. M., Hawkins J. K., and Webster W. S. (2004) The growth of the nasal septum in the 6–9 week period of foetal development: warfarin embryopathy offers a new insight into prenatal facial development. Aust. Dent. J. 49, 171–176 [DOI] [PubMed] [Google Scholar]
- 20. Tie J. K., and Stafford D. W. (2016) Structural and functional insights into enzymes of the vitamin K cycle. J. Thromb. Haemost. 14, 236–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meier M., Weng L. P., Alexandrakis E., Rüschoff J., and Goeckenjan G. (2001) Tracheobronchial stenosis in Keutel syndrome. Eur. Respir. J. 17, 566–569 [DOI] [PubMed] [Google Scholar]
- 22. Murshed M., Schinke T., McKee M. D., and Karsenty G. (2004) Extracellular matrix mineralization is regulated locally; different roles of two gla-containing proteins. J. Cell Biol. 165, 625–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luo G., D'Souza R., Hogue D., and Karsenty G. (1995) The matrix Gla protein gene is a marker of the chondrogenesis cell lineage during mouse development. J. Bone Miner. Res. 10, 325–334 [DOI] [PubMed] [Google Scholar]
- 24. Borrás T., Smith M. H., and Buie L. K. (2015) A novel Mgp-Cre knock-in mouse reveals an anticalcification/antistiffness candidate gene in the trabecular meshwork and peripapillary scleral region. Invest. Ophthalmol. Vis. Sci. 56, 2203–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ayyildiz P., Ceyhan Bilgici M., Ozyilmaz B., Sungur M., Baysal K., and Ogur G. (2012) Keutel syndrome: a case report with aortic calcification. Firat Tip Dergisi 17, 167–169 [Google Scholar]
- 26. Luo G., Ducy P., McKee M. D., Pinero G. J., Loyer E., Behringer R. R., and Karsenty G. (1997) Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 386, 78–81 [DOI] [PubMed] [Google Scholar]
- 27. Palhazi P., Daniel R. K., and Kosins A. M. (2015) The osseocartilaginous vault of the nose: anatomy and surgical observations. Aesthet. Surg. J. 35, 242–251 [DOI] [PubMed] [Google Scholar]
- 28. Laurita J., Koyama E., Chin B., Taylor J. A., Lakin G. E., Hankenson K. D., Bartlett S. P., and Nah H. D. (2011) The Muenke syndrome mutation (FgfR3P244R) causes cranial base shortening associated with growth plate dysfunction and premature perichondrial ossification in murine basicranial synchondroses. Dev. Dyn. 240, 2584–2596 [DOI] [PubMed] [Google Scholar]
- 29. Eicher E. M., Southard J. L., Scriver C. R., and Glorieux F. H. (1976) Hypophosphatemia: mouse model for human familial hypophosphatemic (vitamin D-resistant) rickets. Proc. Natl. Acad. Sci. U.S.A. 73, 4667–4671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murshed M., Harmey D., Millán J. L., McKee M. D., and Karsenty G. (2005) Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 19, 1093–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rosenberg P., Arlis H. R., Haworth R. D., Heier L., Hoffman L., and LaTrenta G. (1997) The role of the cranial base in facial growth: experimental craniofacial synostosis in the rabbit. Plast. Reconstr. Surg. 99, 1396–1407 [DOI] [PubMed] [Google Scholar]
- 32. Nah H. D., Koyama E., Agochukwu N. B., Bartlett S. P., and Muenke M. (2012) Phenotype profile of a genetic mouse model for Muenke syndrome. Childs Nerv. Syst. 28, 1483–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tie J. K., Carneiro J. D., Jin D. Y., Martinhago C. D., Vermeer C., and Stafford D. W. (2016) Characterization of vitamin K-dependent carboxylase mutations that cause bleeding and nonbleeding disorders. Blood 127, 1847–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Watzka M., Geisen C., Scheer M., Wieland R., Wiegering V., Dörner T., Laws H. J., Gümrük F., Hanalioglu S., Unal S., Albayrak D., and Oldenburg J. (2014) Bleeding and non-bleeding phenotypes in patients with GGCX gene mutations. Thromb. Res. 134, 856–865 [DOI] [PubMed] [Google Scholar]
- 35. Scott J. (1956) Growth at facial sutures. Am. J. Orthod. Dentofacial Orthop. 42, 381–387 [Google Scholar]
- 36. Kvinnsland S. (1974) Partial resection of the cartilaginous nasal septum in rats: its influence on growth. Angle Orthod. 44, 135–140 [DOI] [PubMed] [Google Scholar]
- 37. Moss M. L., Bromberg B. E., Song I. C., and Eisenman G. (1968) The passive role of nasal septal cartilage in mid-facial growth. Plast. Reconstr. Surg. 41, 536–542 [DOI] [PubMed] [Google Scholar]
- 38. Copray J. C. (1986) Growth of the nasal septal cartilage of the rat in vitro. J. Anat. 144, 99–111 [PMC free article] [PubMed] [Google Scholar]
- 39. Al Dayeh A. A., Rafferty K. L., Egbert M., and Herring S. W. (2013) Real-time monitoring of the growth of the nasal septal cartilage and the nasofrontal suture. Am. J. Orthod. Dentofacial Orthop. 143, 773–783 [DOI] [PubMed] [Google Scholar]
- 40. Delaire J., and Precious D. (1986) Influence of the nasal septum on maxillonasal growth in patients with congenital labiomaxillary cleft. Cleft Palate J. 23, 270–277 [PubMed] [Google Scholar]
- 41. Marulanda J., Alqarni S., and Murshed M. (2014) Mechanisms of vascular calcification and associated diseases. Curr. Pharm. Des. 20, 5801–5810 [DOI] [PubMed] [Google Scholar]
- 42. Ho A. M., Johnson M. D., and Kingsley D. M. (2000) Role of the mouse ank gene in control of tissue calcification and arthritis. Science 289, 265–270 [DOI] [PubMed] [Google Scholar]
- 43. Okawa A., Nakamura I., Goto S., Moriya H., Nakamura Y., and Ikegawa S. (1998) Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat. Genet. 19, 271–273 [DOI] [PubMed] [Google Scholar]
- 44. Anderson H. C. (2003) Matrix vesicles and calcification. Curr. Rheumatol. Rep. 5, 222–226 [DOI] [PubMed] [Google Scholar]
- 45. Golub E. E. (2009) Role of matrix vesicles in biomineralization. Biochim. Biophys. Acta 1790, 1592–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khavandgar Z., Roman H., Li J., Lee S., Vali H., Brinckmann J., Davis E. C., and Murshed M. (2014) Elastin haploinsufficiency impedes the progression of arterial calcification in MGP-deficient mice. J. Bone Miner. Res. 29, 327–337 [DOI] [PubMed] [Google Scholar]
- 47. O'Young J., Liao Y., Xiao Y., Jalkanen J., Lajoie G., Karttunen M., Goldberg H. A., and Hunter G. K. (2011) Matrix Gla protein inhibits ectopic calcification by a direct interaction with hydroxyapatite crystals. J. Am. Chem. Soc. 133, 18406–18412 [DOI] [PubMed] [Google Scholar]
- 48. Hayano S., Komatsu Y., Pan H., and Mishina Y. (2015) Augmented BMP signaling in the neural crest inhibits nasal cartilage morphogenesis by inducing p53-mediated apoptosis. Development 142, 1357–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zebboudj A. F., Imura M., and Boström K. (2002) Matrix GLA protein, a regulatory protein for bone morphogenetic protein-2. J. Biol. Chem. 277, 4388–4394 [DOI] [PubMed] [Google Scholar]
- 50. Mansfield K., Rajpurohit R., and Shapiro I. M. (1999) Extracellular phosphate ions cause apoptosis of terminally differentiated epiphyseal chondrocytes. J. Cell. Physiol. 179, 276–286 [DOI] [PubMed] [Google Scholar]
- 51. Ducy P., Desbois C., Boyce B., Pinero G., Story B., Dunstan C., Smith E., Bonadio J., Goldstein S., Gundberg C., Bradley A., and Karsenty G. (1996) Increased bone formation in osteocalcin-deficient mice. Nature 382, 448–452 [DOI] [PubMed] [Google Scholar]
- 52. Eimar H., Tamimi F., Retrouvey J. M., Rauch F., Aubin J. E., and McKee M. D. (2016) Craniofacial and dental defects in the Col1a1Jrt/+ mouse model of osteogenesis imperfecta. J. Dent. Res. 95, 761–768 [DOI] [PubMed] [Google Scholar]
- 53. Lee E. R., Lamplugh L., Kluczyk B., Leblond C. P., and Mort J. S. (2009) Neoepitopes reveal the features of type II collagen cleavage and the identity of a collagenase involved in the transformation of the epiphyses anlagen in development. Dev. Dyn. 238, 1547–1563 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.