

Abstract
Many Gram-negative bacterial pathogens use a syringe-like apparatus called a type III secretion system to inject virulence factors into host cells. Some of these effectors are enzymes that modify host proteins to subvert their normal functions. NleB is a glycosyltransferase that modifies host proteins with N-acetyl-d-glucosamine to inhibit antibacterial and inflammatory host responses. NleB is conserved among the attaching/effacing pathogens enterohemorrhagic Escherichia coli (EHEC), enteropathogenic E. coli (EPEC), and Citrobacter rodentium. Moreover, Salmonella enterica strains encode up to three NleB orthologs named SseK1, SseK2, and SseK3. However, there are conflicting reports regarding the activities and host protein targets among the NleB/SseK orthologs. Therefore, here we performed in vitro glycosylation assays and cell culture experiments to compare the activities and substrate specificities of these effectors. SseK1, SseK3, EHEC NleB1, EPEC NleB1, and C. rodentium NleB blocked TNF-mediated NF-κB pathway activation, whereas SseK2 and NleB2 did not. C. rodentium NleB, EHEC NleB1, and SseK1 glycosylated host GAPDH. C. rodentium NleB, EHEC NleB1, EPEC NleB1, and SseK2 glycosylated the FADD (Fas-associated death domain protein). SseK3 and NleB2 were not active against either substrate. We also found that EHEC NleB1 glycosylated two GAPDH arginine residues, Arg197 and Arg200, and that these two residues were essential for GAPDH-mediated activation of TNF receptor-associated factor 2 ubiquitination. These results provide evidence that members of this highly conserved family of bacterial virulence effectors target different host protein substrates and exhibit distinct cellular modes of action to suppress host responses.
Keywords: bacterial pathogenesis, glycosyltransferase, innate immunity, signal transduction, virulence factor
Introduction
The type three secretion system is a syringe-like apparatus used by Gram-negative bacteria to deliver virulence proteins (effectors) into infected host cells (1). Enteropathogenic Escherichia coli (EPEC)2 and enterohemorrhagic E. coli (EHEC) inject effectors that target NF-κB pathway components to inhibit host anti-bacterial and inflammatory responses. NleC cleaves the p50 and p65 NF-κB subunits (2). NleD cleaves the host JNK and p38 MAPK (3). NleH1 prevents the nuclear translocation of the NF-κB subunit RPS3 (4).
NleB was previously characterized as a glycosyltransferase that catalyzes the transfer of GlcNAc to protein substrates (5–7). NleB glycosylates arginine residues (6, 7). Several host proteins such as the death domain-containing proteins FADD, TRADD (tumor necrosis factor receptor type 1-associated death domain protein), and RIPK1 (receptor interaction serine/threonine-protein kinase 1) are targets of NleB N-GlcNAcylation activity (6). Glycosylation of TRADD Arg235 abrogates death domain interactions and the assembly of TNFR1, leading to disrupted TNF signaling (6). EPEC NleB1 also antagonizes death domain signaling by GlcNAcylating FADD Arg117 (7). GAPDH is a target of EHEC NleB1, and its glycosylation inhibits GAPDH interaction with TRAF2, which contributes to inhibiting NF-κB pathway activation through an unknown mechanism (5).
Salmonella enterica strains encode up to three NleB orthologs named SseK1, SseK2, and SseK3 (8, 9). Conflicting results of the roles of the SseK proteins in Salmonella virulence have been presented. Deletion of sseK1, sseK2, or sseK3 was reported not to be deleterious to Salmonella persistence (10). Deletion of the sseK genes failed to impact Salmonella replication in RAW264.7 cells but resulted in attenuation in competitive infection models (8). Another study reported that mutation of sseK1 and/or ssek2 did not alter Salmonella virulence during systemic infection of BALB/c mice (9). SseK1 and SseK2 additively inhibit TNF-induced NF-κB activation and cell death in macrophage infection experiments (11). SseK3 binds TRIM32, an E3 ubiquitin ligase involved in TNF signaling and interferon induction, although SseK3 does not GlcNAcylate TRIM32 (12). The contribution of each SseK family member to bacterial virulence remains to be clarified.
Another paradoxical result concerns EHEC and EPEC NleB1 and their activities toward GAPDH. Although EHEC NleB1 glycosylates GAPDH with GlcNAc (5), EPEC NleB1 does not (6), despite 98.6% identity between the two proteins. The present work was undertaken to compare the activities of the different NleB/SseK orthologs and clarify their substrate specificity by combining data from in vitro glycosylation assays and mammalian cell culture assays. Mass spectrometry and site-directed mutagenesis were used to demonstrate that EHEC NleB1 glycosylates GAPDH on Arg197 and Arg200. These residues were essential for GAPDH-mediated activation of TRAF2 ubiquitination.
Results
Differential activities of NleB orthologs against NF-κB activation
NleB is a glycosyltransferase that glycosylates several host proteins with GlcNAcs (5–7). EPEC NleB1 targets TRADD, FADD, RIPK1, and TNFR1 to disrupt TNF signaling (6, 7). EHEC NleB1 and Citrobacter rodentium NleB glycosylate GAPDH to disrupt TRAF2 activation and downstream NF-κB signaling (5). To clarify potential functional differences among the NleB effectors encoded by A/E pathogens, as well as the three NleB orthologs in S. enterica (SseK1/2/3), we transfected HEK293T cells with plasmids expressing each individual effector and then quantified both IκBα degradation and the nuclear translocation of the NF-κB p65 subunit after stimulating the cells with TNF.
Both EHEC and EPEC NleB1, as well as C. rodentium NleB and S. enterica SseK1 and SseK3, prevented IκBα degradation and suppressed p65 nuclear translocation (Fig. 1A). Neither NleB2 (the EHEC and EPEC proteins are identical), SseK2, nor an enzymatically inactive form of EHEC NleB1 in which the NleB1 DAD active site is mutated (NleB1(AAA)) (5) were active in this assay.
Figure 1.
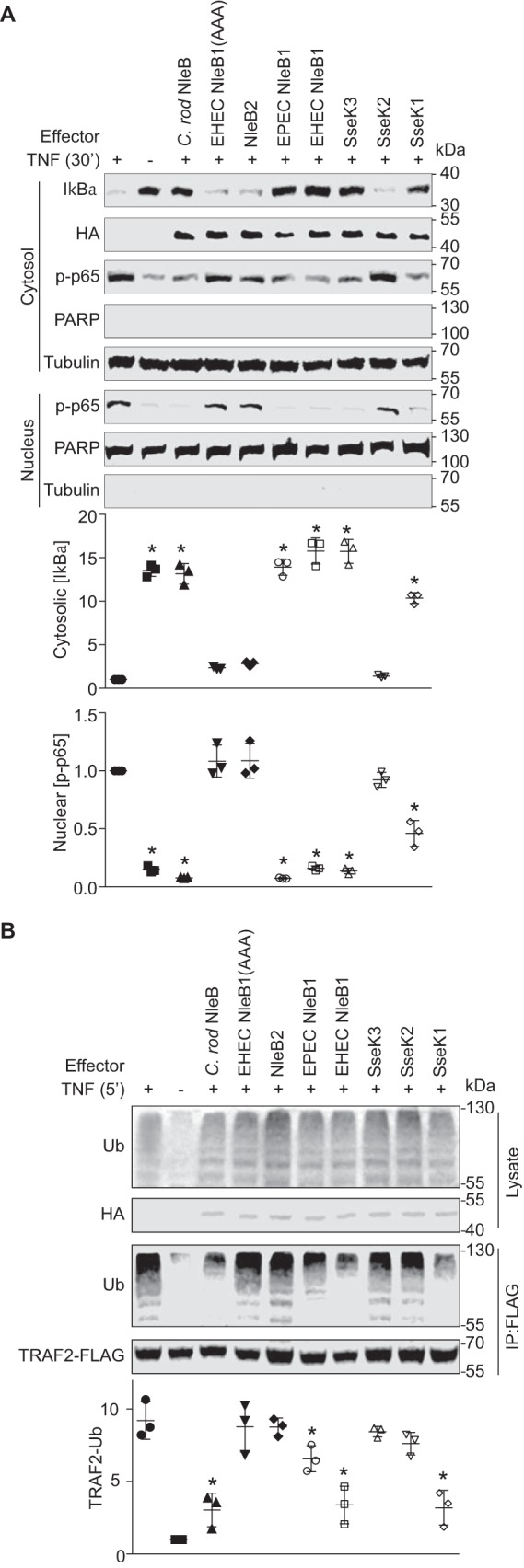
NleB/SseK orthologs have differential activities toward host NF-κB signaling. A, analysis of IκBα degradation and NF-κB p65 subunit nuclear translocation. HEK 293T cells were transfected with the indicated plasmids and treated 24 h later with 50 ng/ml TNF-α for 30 min. The cells were lysed and immunoblotted using the indicated antibodies. Tubulin and poly(ADP-ribose) polymerase (PARP) were used to normalize cytosolic and nuclear protein concentrations, respectively. The asterisks used in quantification panels indicate significantly different protein abundance as compared with the TNF-α control (n = 3, ANOVA). B, analysis of TRAF2 polyubiquitination. HEK 293T cells were transfected with the indicated plasmids and treated 24 h later with 50 ng/ml TNF-α for 5 min. Cell lysates were immunoprecipitated (IP) using FLAG antibody and immunoblotted for ubiquitin (Ub). Asterisks used in quantification panels indicate significantly different TRAF2-Ub signal intensity as compared with the TNF-α control (n = 3, ANOVA). C. rod, C. rodentium.
We also examined the extent to which TRAF2 polyubiquitination was inhibited and found that both EHEC and EPEC NleB1, C. rodentium NleB, and SseK1 inhibited TRAF2 polyubiquitination to a greater extent as compared with the other NleB orthologs (Fig. 1B). However, the extent of inhibition was not as significant for EPEC NleB1, as compared with EHEC NleB1, C. rodentium NleB, and SseK1 (Fig. 1B).
NleB orthologs differentially glycosylate GAPDH and FADD
We then sought to determine whether the NleB/SseK effectors differ in their ability to glycosylate host substrates with GlcNAc. To do this, we incubated either GAPDH or FADD with the NleB/SseK effectors and then monitored substrate glycosylation using Western blotting. C. rodentium NleB and EHEC NleB1 glycosylated both GAPDH and FADD (Fig. 2A). By contrast, EPEC NleB1 and SseK2 only glycosylated FADD, whereas SseK1 activity was limited to GAPDH. No activity was observed for NleB2 or SseK3 toward either GAPDH or FADD. We performed a semiquantitative analysis of these activities by performing glycosylation experiments as a function of effector concentration. Consistent with the qualitative analysis shown in Fig. 2A, we observed that GAPDH and FADD were glycosylated in an effector concentration-dependent manner. (Fig. 2, B–D).
Figure 2.

NleB/SseK orthologs differentially glycosylate GAPDH and FADD. A, in vitro GlcNAcylation assays. NleB/SseK enzymes (200 nm) were incubated for 4 h at room temperature with either with GAPDH or FADD (1 μm) in 50 mm Tris-HCl, pH 7.4, 10 mm UDP-GlcNAc, 10 mm MnCl2, and 1 mm DTT. The samples were then subjected to Western blot detection using an anti-R-GlcNAc monoclonal antibody. B, titration of NleB/SseK orthologs in GAPDH and FADD GlcNAcylation assays. Serial dilutions of NleB/SseK enzymes (6.25–200 nm) were incubated with either GAPDH or FADD (1 μm) and then subjected to Western blot detection using an anti-R-GlcNAc monoclonal antibody. C, quantification of GAPDH glycosylation. The data shown in B were quantified by normalizing GlcNAc-GAPDH signals to total GAPDH signals. The data shown are representative of three independent experiments. D, quantification of FADD glycosylation. The data shown in B were quantified by normalizing GlcNAc-FADD signals to total FADD signals. The data shown are representative of three independent experiments. E and F, pulldown assays to detect binding between the NleB/SseK orthologs and GAPDH (E) or FADD (F). His-GAPDH or His-FADD were individually incubated with each NleB/SseK ortholog then subjected to GST pulldown assays using glutathione-Sepharose beads (GE Healthcare). Protein complexes were eluted with 10 mm reduced glutathione followed by SDS-PAGE analysis. GST was used as negative control. C. rod, C. rodentium.
We also investigated the ability of each ortholog to bind their respective GAPDH and FADD substrates. Only SseK2 and SseK3 were unable to bind GAPDH (Fig. 2E). All effectors that glycosylated FADD, except for SseK2, bound to FADD in the GST pulldown (Fig. 2F). We were unable to detect a stable interaction between SseK2 and FADD, even though SseK2 glycosylated FADD. SseK1 bound to FADD, despite being unable to glycosylate it.
EHEC NleB1 GlcNAcylates GAPDH on Arg197 and Arg200
EPEC NleB1 glycosylates the arginine residues Arg117 of FADD and Arg235 of TRADD (6, 7). To determine whether EHEC NleB1 targets GAPDH arginines, we mutated all GAPDH arginines to alanines (Fig. 3A, GAPDH R-free) and incubated the recombinant mutant protein with EHEC GST-NleB1 and UDP-GlcNAc. EHEC NleB1 glycosylated WT GAPDH but did not glycosylate Arg-free GAPDH (Fig. 3A).
Figure 3.
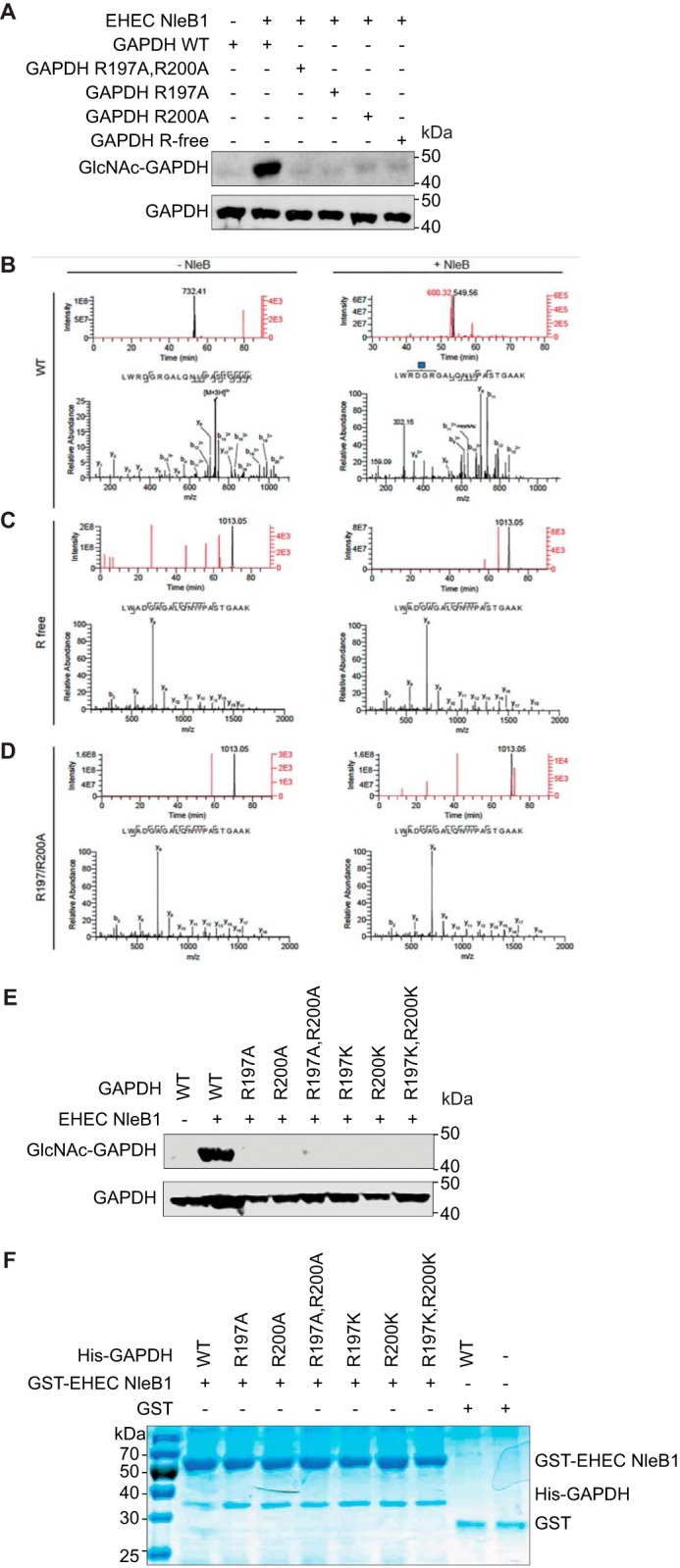
EHEC NleB1 glycosylates GAPDH on Arg197/Arg200. A, in vitro GlcNAcylation of WT and GAPDH mutants by EHEC NleB1. The indicated GAPDH proteins (1 μm) were incubated for 4 h at room temperature with EHEC NleB1 (200 nm) in 50 mm Tris-HCl, pH 7.4, 10 mm UDP-GlcNAc, 10 mm MnCl2, and 1 mm DTT. Samples were then subjected to Western blot detection using an anti-R-GlcNAc monoclonal antibody. B, mass spectrometric analysis of WT GAPDH tryptic digests. An Arg-GlcNAc modified peptide LWRDGRGALQNIIPASTGAAK was identified only when WT GAPDH was incubated with UDP-GlcNAc and NleB (right panels). The top right panel shows extracted ion chromatograms for the GlcNAc-modified LWRDGRGALQNIIPASTGAAK peptide (red trace, m/z = 600.314+) and for the unmodified LWRDGRGALQNIIPASTGAAK peptide (black trace, m/z = 549.564+). Unlabeled ion chromatogram peaks are not related to the LWRDGRGALQNIIPASTGAAK peptide. The corresponding HCD-MS2 spectrum of the Arg-GlcNAc-modified LWRDGRGALQNIIPASTGAAK is shown in the bottom right panel. The blue square denotes the GlcNAc residue. The top left panel shows extracted ion chromatograms for the GlcNAc-modified (red trace, m/z = 800.103+) and unmodified (black trace, m/z = 731.413+) LWRDGRGALQNIIPASTGAAK peptides, demonstrating that only the unmodified peptide is detected when UDP-GlcNAc and NleB are omitted. The corresponding HCD-MS2 spectrum of the unmodified LWRDGRGALQNIIPASTGAAK peptide is shown on the bottom left. C, mass spectrometric analysis of arginine-free (R free) GAPDH tryptic digests. The top panels show extracted ion chromatograms for unmodified (black trace, m/z = 1013.052+) and GlcNAc-modified LWADGAGALQNIIPASTGAAK (red trace, m/z = 1114.582+) peptides, demonstrating that only the unmodified peptide is detected when GDP-GlcNAc and NleB are included (top right panel) or excluded (top left panel) from the reaction. The corresponding HCD-MS2 spectra for the unmodified LWADGAGALQNIIPASTGAAK peptides are shown in the bottom panels. D, mass spectrometric analysis of the R197A/R200A GAPDH tryptic digests. The top panels show extracted ion chromatograms for unmodified (black trace, m/z = 1013.052+) and GlcNAc-modified LWADGAGALQNIIPASTGAAK (red trace, m/z = 1114.582+) peptides, demonstrating that only the unmodified peptide is detected when GDP-GlcNAc and NleB are included (top right panel) or excluded (top left panel) from the reaction. The corresponding HCD-MS2 spectra for the unmodified LWADGAGALQNIIPASTGAAK peptides are shown in the bottom panels. E, in vitro GlcNAcylation of GAPDH alanine and lysine mutants. The indicated GAPDH proteins (1 μm) were incubated for 4 h at room temperature with EHEC NleB1 (200 nm) in 50 mm Tris-HCl, pH 7.4, 10 mm UDP-GlcNAc, 10 mm MnCl2, and 1 mm DTT. The samples were then subjected to Western blot detection using an anti-R-GlcNAc monoclonal antibody. F, mutating GAPDH Arg197/Arg200 does not affect NleB binding. Pulldown assays to detect binding between the GAPDH mutants and EHEC NleB1. His-GAPDH mutants were individually incubated with each EHEC NleB1 and then subjected to GST pulldown assays using glutathione-Sepharose beads (GE Healthcare). Protein complexes were eluted with 10 mm reduced glutathione followed by SDS-PAGE analysis. GST was used as negative control.
We subjected these protein samples to mass spectrometry analysis and observed mass increments corresponding to one and two single GlcNAc modifications on the LWRDGRGALQNIPASTGAAK peptide of GAPDH. These GlcNAc modifications were mapped to Arg197 and Arg200 by using MS/MS fragmentation (Fig. 3B). Neither the Arg-free GAPDH nor R197A/R200A or R197K/R200K GAPDH mutants were glycosylated, as measured by mass spectrometry or Western blotting (Fig. 3, C–E). Mutating either Arg197 or Arg200 affected the glycosylation of the other arginine, indicating a potential cooperativity between the amino acids. The GAPDH mutants still bound to EHEC NleB1, as determined using in vitro pulldown assays (Fig. 3F).
GAPDH Arg197 and Arg200 are required for GAPDH stimulation of TRAF2 ubiquitination
We previously observed that GAPDH interacts with TRAF2 and that this interaction enhances TRAF2 ubiquitination (5). To determine the functional importance of the GAPDH Arg197 and Arg200 residues in the context of TRAF2 activity, we mutated Arg197 and/or Arg200 to either alanine or lysine for use in transfection experiments in a stable GAPDH knockdown cell line (Fig. 4A). We transfected these cells with TRAF2-FLAG and with different Myc-GAPDH mutants. By contrast to WT GAPDH, the GAPDH mutants neither interacted with nor stimulated TRAF2 polyubiquitination (Fig. 4B). Thus, GAPDH Arg197 and Arg200 are essential both for the GAPDH-TRAF2 interaction and for activating TRAF2 polyubiquitination. Although we had predicted that the GAPDH Arg197/Arg200 mutants would thus be resistant to EHEC NleB1-mediated inhibition in these cell culture experiments, our finding that the GAPDH mutants failed to interact with TRAF2 precluded us from testing this hypothesis directly.
Figure 4.
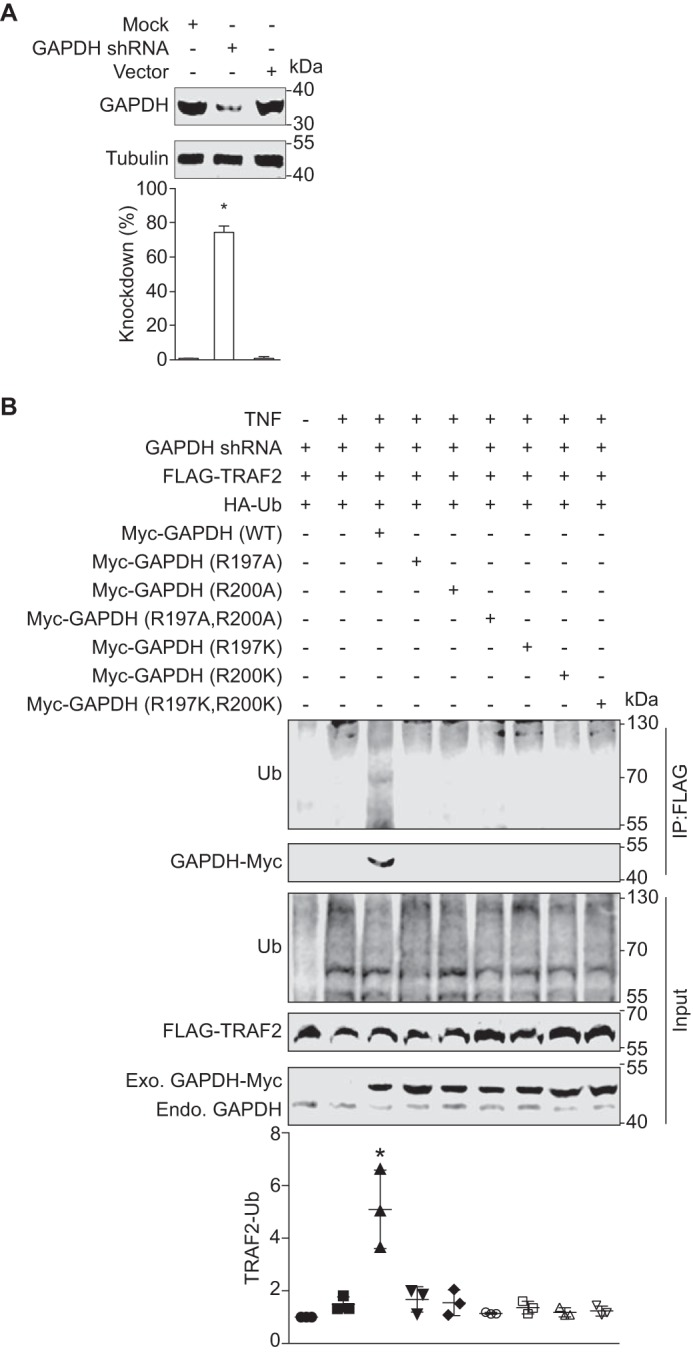
Function of GAPDH arginines 197 and 200 in TRAF2 polyubiquitination. A, GAPDH knock down efficiency. HEK293T cells were transfected with the shRNA plasmid psi-LVRU6GP targeting the GAPDH 3′-UTR mRNA. Stable cell lines were created using puromycin selection. Tubulin was used to normalize GAPDH abundance. Asterisks indicate protein abundance significantly different from that of the control (n = 3, ANOVA). B, GAPDH R197/Arg200 mutants neither interact with nor activate TRAF2 polyubiquitination in HEK293T cells. GAPDH stable cell line was co-transfected with FLAG-TRAF2, HA-ubiquitin together with either Myc-GAPDH WT or the indicated GAPDH mutants. After 48 h, cell lysates were immunoprecipitated (IP) with anti-FLAG antibody, followed by immunoblotting with anti-ubiquitin or anti-Myc antibody. The abundance of endogenous GAPDH and transfected forms of GADPH-Myc is also shown. Asterisks used in quantification panels indicate significantly different TRAF2-Ub signal intensity as compared with the TNF-α control (n = 3, ANOVA). Endo., endogenous; Exo., exogenous; Ub, ubiquitin.
Discussion
Here we have unequivocally demonstrated that the NleB/SseK orthologs have different acceptor substrate specificities using both in vitro glycosylation assays and cell-based assays. We found that EHEC NleB1 and C. rodentium NleB glycosylate both GAPDH and FADD, whereas SseK1 only glycosylates GAPDH, and SseK2 and EPEC NleB1 are only active with FADD. Moreover, NleB2 and SseK3 were inactive with these substrates (Fig. 2). The observed glycosylation activities among the NleB/SseK effectors were not tightly linked to their substrate binding, because EPEC NleB1 and NleB2 both bound to GAPDH and SseK1 bound to FADD, despite having no apparent ability to glycosylate these substrates.
Similarly, catalytically inactive mutants such as EPEC NleB1 (active site DAD mutated to AAA) still bind to FADD but do not label it (13). By contrast, we failed to detect stable binding between SseK2 and FADD in pulldown assays, even though SseK2 glycosylated FADD in vitro. A similar result was obtained in a previous mutagenesis study of EPEC NleB1 in which several EPEC NleB1 mutants glycosylated FADD yet exhibited weak or no binding in pulldown assays (13). Thus, it is clear that catalytic activity and substrate recognition and binding are distinct aspects of the NleB/SseK effectors.
EHEC NleB1 glycosylated GAPDH, whereas EPEC NleB1 did not, even though the two proteins are almost identical (98% identity) with only 7 amino acid differences (P35S, K42R, S106N, Y111H, M193I, D302N, and C326S) among the 329 amino acid proteins. Unexpectedly, EPEC NleB1, similar to EHEC NleB1, showed binding affinity for GAPDH despite lacking catalytic activity with this substrate (Fig. 2E), providing evidence that enzyme activity and substrate recognition are distinct aspects that appear not to be coupled in some particular cases. Future site-directed mutagenesis studies may be informative in determining the NleB1 amino acids that govern and dictate substrate specificity and catalysis.
NleB2 did not exhibit detectable glycosylation activity in our assays. Nevertheless, NleB2 bound to GAPDH, but it did not inhibit NF-κB in transfection experiments. NleB2 may glycosylate unknown targets with GlcNAc or another sugar or may lack glycosylation activity. The role of NleB2 is currently unclear, as is why both NleB1 and NleB2 are maintained in EPEC and EHEC genomes.
We found that SseK3 has a similar phenotype as compared with EHEC NleB1, EPEC NleB1, and SseK1 in our transfection assays that examined NF-κB pathway inhibition (Fig. 1), despite our in vitro assays lacking activity toward FADD or GAPDH (Fig. 2). SseK3 binds but does not glycosylate TRIM32, and this interaction is required for inhibiting NF-κB (12), suggesting that SseK3 may not be a highly active enzyme. SseK1 and SseK3 have additive effects on inhibiting NF-κB, whereas SseK2 has a moderate impact on this pathway (11). Our in vitro data show that SseK2 glycosylates FADD, whereas data from transfection experiments (11) indicate that SseK1, rather than SseK2, glycosylates FADD. It is conceivable that effector activities may differ between in vitro glycosylation assays from their in vivo context as studied in transfection experiments.
NleB2 and SseK3 both contain the conserved DXD motif found in glycosyltransferases, suggesting that they may be enzymatically active, although host substrates for these proteins have not been identified. Additional glycosyltransferase motifs are also found in these two orthologs. NleB2 is 62% identical to EHEC NleB1, whereas SseK3 is 60% identical to SseK1. The absence of their detectable activities toward GAPDH and FADD in vitro does not exclude their potential activities against other substrates that may affect host NF-κB pathway activity. NleB2 and SseK3 might also require different in vitro conditions for optimal enzyme activity.
We showed that EHEC NleB1 glycosylates GAPDH with GlcNAc on two different arginines, Arg197 and Arg200 (Fig. 3), in contrast to the single arginine residue GlcNAcylated in FADD or TRADD (6, 7). Glycosylation of both residues are critical for GAPDH-mediated activation of TRAF2 ubiquitination (Fig. 4B). Our data also suggest that the two Arg197 and Arg200 residues may be essential to maintain the complex in a proper conformation that may be necessary to recruit other proteins to the TRAF2 complex.
The amino acid region surrounding the GAPDH arginine residue targeted by EHEC NleB1 is partly conserved within the other EPEC NleB1 targets TRADD and FADD (WRDGRGAL for GAPDH, WRKVGRSL for TRADD, and WRRLARQL for FADD) (6, 7), suggesting some commonalities among the recognition sequences. The identification of additional NleB targets, together with site-directed mutagenesis and structural studies, may be helpful in establishing a consensus NleB recognition motif. In summary, we present compelling evidence for distinct substrate specificities of virulence effector enzymes and further demonstrate that substrate binding and catalytic activity of these effectors may be distinct.
Experimental procedures
Cloning
The primers used in this study are listed in Table 1. The plasmids used in this study are listed in Table 2.
Table 1.
Primers used in this study
| Description | Sequence |
|---|---|
| C. rodentium nleB BamHI forward | G4ATC2ATGT2ATCTC2AT2A3TGT2C |
| C. rodentium nleB XhoI reverse | C3TCGAGC2ATGA2CTGT2G2TATAC |
| C. rodentium nleB NleB forward | ATATCTAGAGC2AC2ATGTAC3ATACGACGTC3G2ACTACGCGT2ATCTC2AT2A3T |
| C. rodentium nleB NleB reverse | ATATGCG2C2GCT2AC2ATGA2CTGT2G2TATACAT |
| EHEC nleB1 NcoI forward | ATCGCATATGC3ATG2CAT2ATCT2CAT2A3TGTC2T2CA2T |
| EHEC nleB1 SalI reverse | ATCG2TCGAC3ATGA2CTGCAG2TATACATACTG2T |
| EHEC nleB2 XbaI forward | ATATCTAGAGC2AC2ATGTAC3ATACGACGTC3G2ACTACGCGC3ATG2CACT3CA |
| EHEC nleB2 NotI reverse | ATATGCG2C2GCT2AC2ATGA2CTGCATGTATACTGACT2 |
| EHEC nleB2 BamHI forward | CGCG3ATC2ATGCT3CAC2GATA2G2ACA2CT3C |
| EHEC nleB2 XhoI reverse | CGCGCTCGAGC2ATGA2CTGCATGTATACTGACT |
| EHEC nleB1 XbaI forward | ATATCTAGAGC2AC2ATGTAC3ATACGACGTC3G2ACTACGCGT2ATCT2CAT2A3T |
| EHEC nleB1 NotI reverse | ATATGCG2C2GCT2AC2ATGA2CTGCAG2TATACATACTG2T |
| EPEC nleB1 NcoI forward | ATCGCATATGC3ATG2CAT2ATCT2CAT2A3TGTC2T2CA2T |
| EPEC nleB1 SalI reverse | ATCG2TCGAC3ATGA2CTGCTG2TATACATACT |
| EPEC nleB1 NotI reverse | ATATGCG2C2GCT2AC2ATGA2CTGCTG2TATACATACT |
| FADD NdeI forward | CG2A2T2CTATATG3TGA2GA2GATCTGTGCGC2 |
| FADD BamHI reverse | CG2A2T2CG2ATC2TCA2GCGC2GCTACGAT4GCAG |
| GAPDH BamHI forward | G4ATC2ATG4A2G2TG |
| GAPDH XhoI reverse | C3TCGAGCTC2T2G2AG2 |
| GAPDH R200A forward | G2A3CTGTG2CGTGATG2CGC2G4CTCTC2 |
| GAPDH R200A reverse | G2AGAGC3G2CGC2ATCACGC2ACAGT3C2 |
| GAPDH R197A forward | G2A3CTGTG3CTGATG2C2GCG3CTCTC2AGA2CATCATC2 |
| GAPDH R197A reverse | G2ATGATGT2CTG2AGAGC4GCG2C2ATCAGC3ACAGT3C2 |
| GAPDH R197A,R200A forward | G3A3CTGTG3CTGATG2CGCTG4CT |
| GAPDH R197A,R200A reverse | AGC4AGCGC2ATCAGC3ACAGT3C3 |
| GAPDH R197K forward | G2ATG2C4TC2G3A3CTGTG2A2G2ATG2C2GCG4CTCT |
| GAPDH R197K reverse | AGAGC4GCG2C2ATC2T2C2ACAGT3C3G2AG4C2ATC2 |
| GAPDH R200K forward | G2ATG2C4TC2G3A3CTGTG2CGTGATG2CA2G5CTCTC2AGA2CATC |
| GAPDH R200K reverse | GATGT2CTG2AGAGC4CT2GC2ATCACGC2ACAGT3C3G2AG4C2ATC2 |
| GAPDH R197K,R200K forward | G2ATG2C3TC2G3A2CTGTG2A2G2ATG2CA2G5CTCTC2AGA2CATC |
| GAPDH R197K,R200K reverse | GATGT2CTG2AGAGC4CT2GC2ATC2T2C2ACAGT3C3G2AG4C2ATC2 |
| GAPDH R197A,R200A forward | G3A3CTGTG3CTGATG2CGCTG4CT |
| GAPDH R197A,R200A reverse | AGC4AGCGC2ATCAGC3ACAGT3C3 |
| GAPDH shRNA UTR-1 forward | GATC2G2AGC2GCAC2T2GTCATGTACTCA2GAG2TACATGACA2G2TGCG2CTCT6G2 |
| GAPDH shRNA UTR-1 reverse | A2T2C2A6GAGC2GCAC2T2GTCATGTAC2TCT2GAGTACATGACA2G2TGCG2CTC2G |
| Salmonella sseK1 XbaI forward | ATATCTAGAGC2AC2ATGTAC3ATACGACGTC3G2ACTACGCGATC3AC2AT2A3T |
| Salmonella sseK1 NotI reverse | ATATGCG2C2GC2TACTGCACATGC2TCGC3ATGA2CT |
| Salmonella sseK2 XbaI forward | ATATCTAGAGC2AC2ATGTAC3ATACGACGTC3G2ACTACGCG2CACGT4A2TGC2 |
| Salmonella sseK2 NotI reverse | ATATGCG2C2GCT2AC2TC2A2GA2CTG2CAGT2A3CT |
| Salmonella sseK3 XbaI forward | ATATCTAGAGC2AC2ATGTAC3ATACGACGTC3G2ACTACGCGT4CTCGAGTCAGA |
| Salmonella sseK3 NotI reverse | ATATGCG2C2GCT2ATCTC2AG2AGCTGATAGTCA3 |
| Salmonella sseK1 BamH1 forward | CGCG3ATC2ATGATC3AC2AT2A3TAGATATGT2C |
| Salmonella sseK1 XhoI reverse | CGCGCTCGAGCTGCACATGC2TCGC3ATGA2 |
| Salmonella sseK2 BamHI forward | CGCG3ATC2ATG2CACGT4A2TGC2GCT4A |
| Salmonella sseK2 XhoI reverse | CGCGCTCGAGC2TC2A2GA2CTG2CAGT2A3CT |
| Salmonella sseK3 NcoI forward | CGCGC2ATG2T3CTCGAGTCAGAG2T4CT3C |
| Salmonella sseK3 SalI reverse | CGCG2TCGACATC2TCTC2AG2AGCTGATAGTC |
| TRAF2 BamHI forward | GCG2ATC2ATG2CTGCAGCTAGCGTGAC2 |
| TRAF2 HindIII reverse | GCA2GCT2G2AGC3TGTCAG2TC2ACA2TG2 |
Table 2.
Plasmids used in this study
| Description | Source |
|---|---|
| HA-C. rodentium_NleB | This study |
| HA-EHEC_NleB1 | Ref. 5 |
| HA-EHEC_NleB1(AAA) | Ref. 5 |
| HA-EPEC_NleB1 | This study |
| HA-NleB2 | This study |
| HA-SseK1 | This study |
| HA-SseK2 | This study |
| HA-SseK3 | This study |
| GST-C. rodentium_NleB | This study |
| GST-EHEC_NleB1 | This study |
| GST-EPEC_NleB1 | This study |
| GST-NleB2 | This study |
| GST-SseK1 | This study |
| GST-SseK2 | This study |
| GST-SseK3 | This study |
| His-GAPDH | Ref. 5 |
| His-GAPDH (R197A) | This study |
| His-GAPDH (R200A) | This study |
| His-GAPDH (R197A,R200A) | This study |
| His-GAPDH (R197K) | This study |
| His-GAPDH (R200K) | This study |
| His-GAPDH (R197K,R200K) | This study |
| GAPDH shRNA in psi-LVRU6GP | This study |
| CSHCTR001-LVRU6GP, scrambled shRNA control | GeneCopoeia |
| Myc-GAPDH | Ref. 15 |
| Myc-GAPDH (R197A) | This study |
| Myc-GAPDH (R200A) | This study |
| Myc-GAPDH (R197A,R200A) | This study |
| Myc-GAPDH (R197K) | This study |
| Myc-GAPDH (R200K) | This study |
| Myc-GAPDH (R197K,R200K) | This study |
| Ubiquitin-HA | Ref. 16 |
| FLAG-TRAF2 | Ref. 5 |
| GST-TRAF2 | This study |
| His-FADD | This study |
Protein purification
E. coli BL21 (DE3) was transformed with pET42a-nleB, pET42a-traf2, pET28a-gapdh, or pET15b-fadd and grown in LB to an A600 of 0.4. Isopropyl β-d-thiogalactopyranoside (0.5 mm) was added for 4 h, bacteria were pelleted using centrifugation, and the pellet was suspended in 50 mm sodium phosphate, pH 8.0, 0.5 mg/ml lysozyme). The suspension was incubated on ice for 30 min with occasional shaking. An equal volume of 50 mm sodium phosphate, pH 8.0, 2 m NaCl, 8 mm imidazole, 20% glycerol, 1% Triton X-100 was added, and after 30 min on ice, the bacterial lysate was sonicated and then clarified by centrifugation. The supernatant was incubated with nickel-nitrilotriacetic acid beads (Qiagen) with gentle rotation for 2 h at 4 °C, then loaded on a Poly-Prep Chromatography Column (Bio-Rad), and washed with 5 bead volumes of 50 mm sodium phosphate, pH 8.0, 600 mm NaCl, 10% glycerol, 60 mm imidazole. Proteins were eluted in 50 mm sodium phosphate, pH 8.0, 600 mm NaCl, 10% glycerol, 250 mm imidazole.
GlcNAcylation assays
NleB/SseK effector proteins (200 nm) were incubated with either GAPDH or FADD (1 μm) for 4 h at room temperature in 50 mm Tris, pH 7.4, 10 mm MnCl2, 1 mm UDP-GlcNAc, 1 mm DTT. The reactions were terminated by adding 250 mm Tris-HCl, pH 6.8, 10% sodium dodecyl sulfate, 30% glycerol, 5% β-mercaptoethanol, 0.02% bromphenol blue and then immunoblotted using an anti-R-GlcNAc monoclonal antibody (Abcam).
Pulldown assays
Pulldown experiments were performed as described (14). GST-tagged proteins (10 μm) were immobilized on glutathione-Sepharose beads (GE Healthcare) and then mixed with His-tagged prey proteins (10 μm) in 20 mm Tris-HCl, pH 7.9, 20% glycerol, 1 mm EDTA, 5 mm MgCl2, 0.1% Nonidet P-40, 1 mm DTT, 0.2 mm PMSF, 100 mm NaCl, supplemented with 0.33 unit/μl of RNase A and DNase I. After overnight incubation at 4 °C, the beads were washed four times with 20 mm Tris-HCl, pH 7.9, 20% glycerol, 1 mm EDTA, 5 mm MgCl2, 0.1% Nonidet P-40, 1 mm DTT, 0.2 mm PMSF, 1 m NaCl. Proteins were eluted with 10 mm reduced glutathione and analyzed using SDS-PAGE.
Mass spectrometry
Proteins (10 μg) were precipitated and washed in 90% ice-cold acetone, air-dried briefly, and then resolubilized in 50 mm ammonium bicarbonate containing 0.1% Rapigest (Waters). The samples were reduced in 5 mm DTT at 56 °C for 30 min and subsequently alkylated at room temperature for 30 min by the addition of 10 mm iodoacetamide. Each sample was digested overnight with 0.1 μg of LysC protease (Roche) at 37 °C. Peptides were purified and concentrated using reversed-phased C18 chromatography with in-house packed Stagetips (Empore disk-C18, 3M).
LysC digests were separately analyzed using a setup composed of an EASY-nLC 1000 UHPLC (Thermo Scientific) interfaced via a nanoSpray Flex ion source to an LTQ-Orbitrap Velos Pro hybrid mass spectrometer. The EASY-nLC 1000 was operated using a single analytical column setup (PicoFrit Emitters, New Objectives, 75-μm inner diameter) packed in-house with Reprosil-Pure-AQ C18 phase (Dr. Maisch, 1.9-μm particle size). Peptides were separated using a 90-min LC gradient operated at 200 nl/min. The mass spectrometer was operated in data-dependent mode. MS1 precursor ion acquisition was performed in the Orbitrap (nominal resolution of 30,000), followed by HCD and electron-transfer dissociation (ETD) fragmentations of the top five multiply charged ions. MS2 scans were acquired in the Orbitrap mass analyzer using a resolution setting of 15,000.
Data processing and analysis were performed using Proteome Discoverer 1.4 (Thermo Fisher Scientific). Enzyme specificity was set to LysC allowing for one missed cleavage site. Full specificity and semispecific cleavages were considered. Peptide mass tolerance was 10 ppm; fragment ion mass tolerance was 0.05 atomic mass unit; carbamidomethyl was set as a fixed modification for cysteine residues. Methionine oxidation and HexNAc modification of arginine were used as variable modifications. The data were filtered for only high confidence (p < 0.01) identifications. Peptide identifications with HexNAc modifications were inspected manually to verify the accuracy of the assignments.
Cell fractionation
Cytosolic and nuclear protein extracts were prepared from HEK293T cells using the NE-PER nuclear and cytoplasmic extraction reagents (Thermo). TNF-α stimulation (30 min, 50 ng/ml) was used to monitor IκBα degradation and phosphorylated p65 nuclear translocation. Poly(ADP-ribose) polymerase and β-tubulin were used to normalize the protein concentrations of nuclear and cytoplasmic fractions, respectively.
Ubiquitination assays
HEK293T cells were transfected and treated with TNF-α (50 ng/ml, 5 min). The cells were washed with cold PBS, and cell pellets were lysed in 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100 on ice for 30 min and then mixed with anti-FLAG M2 affinity resin and rotated at 4 °C overnight. The resins were centrifuged at 8,000 × g for 30 s at 4 °C and then washed three times with cold TBS. The resins were resuspended in SDS sample loading buffer, boiled for 10 min, and immunoblotted with appropriate antibodies.
GAPDH stable cell line construction
shRNAs targeting the GAPDH 3′-UTR mRNA were synthesized and cloned into the psi-LVRU6GP shRNA mammalian expression vector (GeneCopoeia). CSHCTR001-LVRU6GP (GeneCopoeia) was used as a scrambled sequence control. HEK 293T cells were seeded onto 100-mm dishes 24 h before transfection and then transfected with 5 μg of plasmids using PolyJet transfection reagent (SignaGen Laboratories). After 48 h of incubation at 37 °C, the cells were passaged in medium containing puromycin (2 μg/ml; InvivoGen) for selection. Puromycin-resistant colonies were isolated by serial dilution in 96-well plates, after which individual clones were expanded in selection medium.
Statistical analyses
Protein abundance was quantified using Li-COR Image Studio software. IκBα degradation, p65 nuclear translocation, and TRAF2 polyubiquitination were analyzed statistically using one-way analysis of variance (ANOVA). p values < 0.05 were considered significant.
Author contributions
P. R. H. conceived and coordinated the study and wrote the paper. S. E. Q., K. C., A. H., and L. S. designed, performed, and analyzed the experiments. C. R., R. H.-G., and H. C. assisted with analysis and interpretation of data. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by Grants AI093913 and AI127973 from the NIAID, National Institutes of Health (to P. R. H.) and Danish National Research Foundation Grant DNRF107 (to A. H. and H. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- EPEC
- enteropathogenic E. coli
- EHEC
- enterohemorrhagic E. coli
- TRAF
- TNF receptor-associated factor
- ANOVA
- analysis of variance
- HCD
- higher-energy collisional dissociation.
References
- 1. Galán J. E., Lara-Tejero M., Marlovits T. C., and Wagner S. (2014) Bacterial type III secretion systems: specialized nanomachines for protein delivery into target cells. Annu. Rev. Microbiol. 68, 415–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yen H., Ooka T., Iguchi A., Hayashi T., Sugimoto N., and Tobe T. (2010) NleC, a type III secretion protease, compromises NF-κB activation by targeting p65/RelA. PLoS Pathog. 6, e1001231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baruch K., Gur-Arie L., Nadler C., Koby S., Yerushalmi G., Ben-Neriah Y., Yogev O., Shaulian E., Guttman C., Zarivach R., and Rosenshine I. (2011) Metalloprotease type III effectors that specifically cleave JNK and NF-κB. EMBO J. 30, 221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gao X., Wan F., Mateo K., Callegari E., Wang D., Deng W., Puente J., Li F., Chaussee M. S., Finlay B. B., Lenardo M. J., and Hardwidge P. R. (2009) Bacterial effector binding to ribosomal protein s3 subverts NF-κB function. PLoS Pathog. 5, e1000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gao X., Wang X., Pham T. H., Feuerbacher L. A., Lubos M. L., Huang M., Olsen R., Mushegian A., Slawson C., and Hardwidge P. R. (2013) NleB, a bacterial effector with glycosyltransferase activity, targets GAPDH function to inhibit NF-κB activation. Cell Host Microbe 13, 87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li S., Zhang L., Yao Q., Li L., Dong N., Rong J., Gao W., Ding X., Sun L., Chen X., Chen S., and Shao F. (2013) Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature 501, 242–246 [DOI] [PubMed] [Google Scholar]
- 7. Pearson J. S., Giogha C., Ong S. Y., Kennedy C. L., Kelly M., Robinson K. S., Lung T. W., Mansell A., Riedmaier P., Oates C. V., Zaid A., Mühlen S., Crepin V. F., Marches O., Ang C. S., et al. (2013) A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature 501, 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brown N. F., Coombes B. K., Bishop J. L., Wickham M. E., Lowden M. J., Gal-Mor O., Goode D. L., Boyle E. C., Sanderson K. L., and Finlay B. B. (2011) Salmonella phage ST64B encodes a member of the SseK/NleB effector family. PLoS One 6, e17824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kujat Choy S. L., Boyle E. C., Gal-Mor O., Goode D. L., Valdez Y., Vallance B. A., and Finlay B. B. (2004) SseK1 and SseK2 are novel translocated proteins of Salmonella enterica serovar typhimurium. Infect. Immun. 72, 5115–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kidwai A. S., Mushamiri I., Niemann G. S., Brown R. N., Adkins J. N., and Heffron F. (2013) Diverse secreted effectors are required for Salmonella persistence in a mouse infection model. PLoS One 8, e70753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Günster R. A., Matthews S. A., Holden D. W., and Thurston T. L. (2017) SseK1 and SseK3 type III secretion system effectors inhibit NF-κB signaling and necroptotic cell death in Salmonella-infected macrophages. Infect. Immun. 85, e00010–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang Z., Soderholm A., Lung T. W., Giogha C., Hill M. M., Brown N. F., Hartland E., and Teasdale R. D. (2015) SseK3 is a Salmonella effector that binds TRIM32 and modulates the host's NF-κB signalling activity. PLoS One 10, e0138529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wong Fok Lung T., Giogha C., Creuzburg K., Ong S. Y., Pollock G. L., Zhang Y., Fung K. Y., Pearson J. S., and Hartland E. L. (2016) Mutagenesis and functional analysis of the bacterial arginine glycosyltransferase effector NleB1 from enteropathogenic Escherichia coli. Infect. Immun. 84, 1346–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nguyen T. N., and Goodrich J. A. (2006) Protein-protein interaction assays: eliminating false positive interactions. Nat. Methods 3, 135–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sen N., Hara M. R., Ahmad A. S., Cascio M. B., Kamiya A., Ehmsen J. T., Agrawal N., Hester L., Doré S., Snyder S. H., and Sawa A. (2009) GOSPEL: a neuroprotective protein that binds to GAPDH upon S-nitrosylation. Neuron 63, 81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lim K. L., Chew K. C., Tan J. M., Wang C., Chung K. K., Zhang Y., Tanaka Y., Smith W., Engelender S., Ross C. A., Dawson V. L., and Dawson T. M. (2005) Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci. 25, 2002–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]