

Abstract
CO2 is a physiological gas normally produced in the body during aerobic respiration. Hypercapnia (elevated blood pCO2 >≈50 mm Hg) is a feature of several lung pathologies, e.g. chronic obstructive pulmonary disease. Hypercapnia is associated with increased susceptibility to bacterial infections and suppression of inflammatory signaling. The NF-κB pathway has been implicated in these effects; however, the molecular mechanisms underpinning cellular sensitivity of the NF-κB pathway to CO2 are not fully elucidated. Here, we identify several novel CO2-dependent changes in the NF-κB pathway. NF-κB family members p100 and RelB translocate to the nucleus in response to CO2. A cohort of RelB protein-protein interactions (e.g. with Raf-1 and IκBα) are altered by CO2 exposure, although others are maintained (e.g. with p100). RelB is processed by CO2 in a manner dependent on a key C-terminal domain located in its transactivation domain. Loss of the RelB transactivation domain alters NF-κB-dependent transcriptional activity, and loss of p100 alters sensitivity of RelB to CO2. Thus, we provide molecular insight into the CO2 sensitivity of the NF-κB pathway and implicate altered RelB/p100-dependent signaling in the CO2-dependent regulation of inflammatory signaling.
Keywords: carbon dioxide, chronic obstructive pulmonary disease (COPD), inflammation, innate immunity, NF-κB transcription factor, CO2, hypercapnia, RelB, immunity, p100
Introduction
Oxygen (O2) and carbon dioxide (CO2) are the substrate and product of aerobic respiration, respectively. Atmospheric CO2 levels have recently exceeded 400 ppm (0.04%) with the highest ever daily average CO2 recorded at Mauna Loa Observatory in April, 2016 (409.44 ppm). Although the levels of this greenhouse gas are rising, the levels in the atmosphere are still much lower than that experienced within respiring organisms. CO2 is produced as a consequence of aerobic respiration during the pre-Krebs and Krebs cycle reactions. Thus, in normocapnia the normal pCO2 in the human circulation is ∼40 mm Hg. Because the main mechanism through which CO2 is removed from the body is via exhalation, the circulating pCO2 is closely related to lung function and ventilation. Hyperventilation can result in lower than normal levels of CO2 (hypocapnia), and hypoventilation and/or chronic lung diseases such as chronic obstructive pulmonary disease and cystic fibrosis result in elevated levels of CO2 (hypercapnia) (1). In patients, the degree of hypercapnia can be profound with arterial pCO2 values in excess of 100 mm Hg recorded in exacerbated COPD2 (2). Hypocapnia is associated with a worse outcome in COPD (3) and worsens cerebral ischemia (4). Interestingly, hypercapnia is also associated with a worse outcome in COPD (3) with additional deleterious consequences reported in terms of muscle dysfunction (5) and immunosuppression (6). This evidence indicates that carbon dioxide is not merely a waste product of metabolism and that like oxygen it can elicit a specific repertoire of transcriptional events in a dose-dependent fashion (1, 7). In particular, genes associated with inflammation, immunity, and metabolism appear to be CO2-sensitive and that sensitivity of these cohorts of genes to CO2 is evolutionarily conserved (8–10). The mechanisms, however, are not well characterized. We and others have previously demonstrated sensitivity of the NF-κB pathway to CO2 (11–16), and elevated CO2 is associated with suppression of pro-inflammatory cytokines in a number of different settings (11, 12, 17). The current state of the art is that hypercapnia may be damaging in the context of infection (6, 18) due to immunosuppression. Conversely, hypercapnia may be of benefit in the context of destructive inflammation (19, 20) due to suppression of inflammatory signaling (1, 21). Given the importance of the NF-κB pathway in the regulation of immune and inflammatory signaling, we hypothesize that CO2-dependent alterations in NF-κB are important in determining inflammatory signaling over a range of pCO2 values from hypocapnia to normocapnia to hypercapnia. Our previous work has implicated members of the non-canonical NF-κB pathway (IKK, inhibitor of κB kinase (IKKα) and RelB) as being particularly sensitive to changes in CO2 in the basal (unstimulated) state (11, 12). This study gives molecular insight into CO2-dependent modulation of the NF-κB transcription factor RelB.
NF-κB is a family of five transcription factors: p50, p65, p52, RelB, and c-Rel. Of these, p65 is the transcriptionally active component of the canonical p65-p50 heterodimer that is activated downstream of the heterotrimeric IKKα,β,γ complex (22). Oh the other hand, RelB is the transcriptionally active component of the non-canonical RelB-p52 heterodimer that is activated downstream of an IKKα homodimer complex (23). Canonical signaling is associated with the regulation of classical pro-inflammatory gene activation, e.g. TNFα, IL-6, and IL-1 (mainly via p65/p50 heterodimers), although non-canonical NF-κB signaling is associated with regulation of genes involved in lymphogenesis development (24, 25) (mainly via RelB/p52 heterodimers). Both canonical and non-canonical NF-κB family members demonstrate sensitivity to CO2 in the stimulated state (11); however, non-canonical family members IKKα and RelB appear much more sensitive to CO2 in the basal (non-ligand stimulated) state (12).
RelB is has been dubbed “an outlier in leukocyte biology” (23) and is relatively less well characterized than p65. RelB uniquely possesses an N-terminal leucine zipper domain that affects the ability of RelB to activate transcription of target genes (26); furthermore, it is postulated that RelB can activate a more diverse array of NF-κB consensus binding sequences than other family members (27). The RelB C-terminal contains a transactivation domain, which is conserved among RelA, RelB, and c-Rel (25).
Pathways regulated by RelB include those involved in the following: (i) immune development and signaling (24, 28); (ii) response to xenobiotics (29); (iii) chromatin remodeling, e.g. RelB can associate with the ATP-dependent SWI/SNF nucleosome-remodeling complex (30); (iv) circadian rhythms (31); and (v) cellular metabolism (23). Thus, RelB can modulate an array of cellular responses. The molecular mechanisms underpinning these effects are not yet fully appreciated. Taken together, it is clear that RelB differs significantly from other NF-κB family members in terms of structure, regulation, and target gene expression. Indeed, the relative contribution of both RelA and RelB to lymphotoxin-induced gene expression reveals both overlapping and subunit-specific target genes (32).
RelB is a labile protein subject to significant post-translational modification. Elegant biochemical studies have illustrated that RelB is a target for phosphorylation (33, 34), ubiquitination (35), sumoylation (36), signal-induced degradation, and cleavage (33, 37). These modifications regulate RelB function as well as protein-protein interactions. The protein-protein interaction between RelB and p100 is particularly important for their respective functions (34). p100 acts as an inhibitor of RelB as well as facilitating a “co-operative stabilizing state” between the two proteins (23). p100 in addition to stabilizing and inhibiting RelB has been shown to inhibit canonical NF-κB-dependent transcription via sequestration of RelA-p50 dimers (38). Recent work has highlighted the association of RelB (along with other NF-κB subunits) with p100 as part of a high-molecular-weight repressive “kappaBsome” (39, 40).
Finally, a recent study identified RelB mRNA expression as being associated with acid-base and cardiovascular features in patients with exacerbated COPD (41), suggesting a functional regulation of this NF-κB family member in a serious disease where hypercapnia is prevalent. The detailed mechanisms underpinning the CO2-dependent modulation of p100 and RelB along with the downstream consequences for NF-κB-dependent signaling beyond this have not yet been elucidated and are the focus of this study.
Results
Elevated CO2 causes a cellular re-organization of the NF-κB family members RelB and p100
We have previously reported that exposure of cells to elevated CO2 induces RelB nuclear localization and cleavage in a number of cell types. The mechanisms underpinning this CO2-dependent modulation of immune signaling are not fully understood and are a focus of this study. Here, we demonstrate in mouse embryonic fibroblasts (MEF) CO2-dependent nuclear localization and cleavage of RelB that are evident over a range of CO2 conditions (0.03, 5, and 10% CO2) (Fig. 1, A and D). We observed a very similar pattern in lung epithelial A549 cells (supplemental Fig. S1, A–C). This CO2 dose-dependent regulation of NF-κB from hypocapnia, to normocapnia, to hypercapnia highlights the importance of considering the impact of microenvironmental CO2 concentrations in a range of conditions.
Figure 1.

Elevated CO2 causes a cellular re-organization of the NF-κB family members RelB and p100. A, MEF were exposed to 0.03, 5, or 10% CO2 in pH-buffered media for 75 min prior to preparation of cytosolic (Cyt) and nuclear (Nuc) protein fractions. Lysates were immunoblotted using specific antibodies against RelB, p100/p52, lamin, and α-tubulin. ns denotes a nonspecific cytosolic band. B, densitometric quantification of cytoplasmic RelB relative to α-tubulin. C, densitometric quantification of cytoplasmic p100 relative to α-tubulin. D, densitometric quantification of nuclear RelB relative to lamin. E, densitometric quantification of nuclear p100 relative to lamin. Data representative of n = 3 experiments.
Given that RelB functionally interacts with p100 within the non-canonical NF-κB pathway, we next investigated whether p100 also demonstrated sensitivity to CO2. Interestingly, we observed sensitivity of the p100 subunit to CO2. To our knowledge, this is the first time that p100 sensitivity to CO2 has been reported. p100 markedly translocates to the nucleus following exposure to 5% CO2 and 10% CO2 (Fig. 1, A and E). We observed a very similar pattern in lung epithelial A549 cells (supplemental Fig. S1, A, D, and E) with a more marked difference observed between 5 and 10% CO2 in these cells. Thus, in the basal (unstimulated) state members of the non-canonical NF-κB family undergo cellular modification and re-organization by CO2 exposure that does not appear to be a consequence of decreased pHe, or pHi, which remained consistent under our experimental conditions (supplemental Fig. S2).
RelB is cleaved at its C terminus in response to elevated CO2
We next developed an overexpression system to test RelB sensitivity in HEK cells. The purpose of this was to allow us to perform mass spectrometric interactome studies in cells exposed to elevated CO2, looking at RelB and its associated proteins (e.g. p100/p52). HEK cells that had been transfected with a full-length N-terminal FLAG-tagged RelB construct (Fig. 2A), re-capitulated the previously observed RelB response to elevated CO2 (Fig. 1, A and D, and supplemental Fig. S1, A–C) (12). Interestingly, the banding pattern for RelB and FLAG from the HEK cells was almost identical (Fig. 2, B and C) suggesting that RelB is cleaved at its C terminus. Thus, we observed CO2-dependent regulation of endogenous NF-κB family members p100 and RelB and also re-capitulated CO2-dependent cleavage of recombinant RelB. Our next experiments were performed to gain insight into the mechanisms governing RelB sensitivity to CO2 as well as the consequences for RelB protein-protein interactions.
Figure 2.
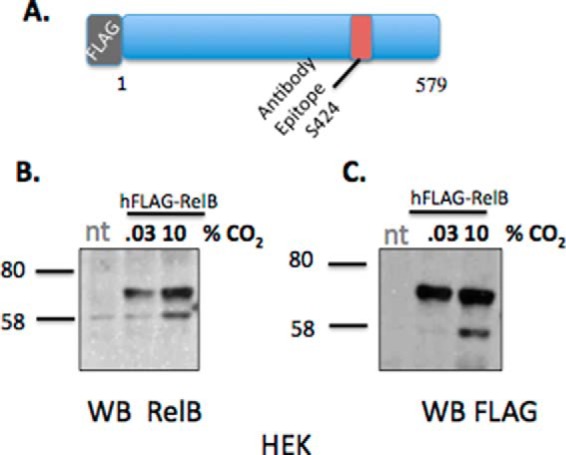
RelB is cleaved at its C terminus in response to elevated CO2. A, schematic indicating the structure of the hFLAG-RelB construct and the region on this protein to which our RelB antibodies are directed (Ser-424). B, HEK cells transiently transfected with hFLAG-RelB were exposed to 0.03 or 10% CO2 in pH-buffered media for 75 min prior to preparation of nuclear protein fractions. Lysates were immunoblotted using specific antibodies against RelB and FLAG (C). Data are representative of n = >3 experiments. nt, non-transfected.
RelB interactome is altered by CO2 exposure
RelB expression and cellular localization are altered in response to CO2 (Figs. 1, A and D, and 2, B and C, and supplemental Fig. S1, A–C) (12). Thus, we hypothesized that these changes were being driven at least in part by altered RelB-protein-protein interactions in the different CO2 environments. We tested this hypothesis in an unbiased and quantitative manner. We first overexpressed FLAG-RelB in HEK cells, exposed the cells to ambient or elevated CO2 under pH-buffered conditions, and performed an immunoprecipitation for FLAG. Precipitated proteins were then analyzed by mass spectrometry (Fig. 3A). Proteins of interest were then determined by meeting specific stringency thresholds for (i) enrichment over control (non FLAG-RelB-expressing cells) and (ii) evidence for CO2 sensitivity in the RelB-specific protein interactions (2-fold difference up or down) (Fig. 3B and supplemental mass spectrometry files). Several known RelB-interacting proteins were identified by the screen, which supports the sensitivity of our experimental approach (Fig. 3C). For example, IκBα, importin α-7, and the ubiquitin C-terminal hydrolase-7 are the most enriched, the 50th most enriched, and the 90th most enriched interaction, respectively (www.ebi.ac.uk/intact). EMBL-EBI IntAct Molecular Interaction Database was searched using the search-term “RelB.” Regarding CO2 sensitivity, 25 proteins met or exceeded the thresholds for being a FLAG-RelB-associated protein that were differentially associated with RelB in a CO2-dependent manner. These proteins are listed in supplemental Fig. S3, with a selection of proteins illustrated in Fig. 4. The seven proteins demonstrated increased association with FLAG-RelB, and 18 proteins demonstrated decreased association with FLAG-RelB at 10% CO2 compared with ambient CO2. Interestingly, when Gene Ontology analysis was performed on the 25 proteins that were found to have differential interactions with RelB in a CO2-dependent manner, there was a strong enrichment of proteins involved in both protein transport and nucleic acid binding (supplemental Fig. S4). These unbiased data support the concept that RelB translocates to the nucleus in a CO2-dependent manner, conceivably facilitated by importin proteins. Selected proteins were chosen for validation of the MS/MS screen by FLAG-RelB overexpression coupled to conventional Western blotting. RelB interactions with p100 were not affected by CO2 exposure (supplemental Fig. S5A). IκBα had reduced association with RelB at 10% CO2 in several experiments (supplemental Fig. S5), whereas Raf-1 had increased association at 10% CO2 (supplemental Fig. S5C). Thus, the data from these IP Western blotting experiments support the IP MS/MS data. Of note, SMARCD2 was markedly enriched with FLAG-IP, but it was not consistently different in 10% CO2 by IP Western blotting (supplemental Fig. S5D). Thus, the IP Western data largely validate the mass spectrometry data, with the MS/MS approach likely more sensitive than IP Western blotting approaches. However, given the SMARCD2 data, we suggest caution in interpreting the data from the less enriched proteins due to a risk of false positives. CO2-dependent changes were more reproducibly validated by Western blotting for highly enriched interactors (e.g. IκBα, p100, and Raf-1).
Figure 3.

Mass spectrometric analysis of RelB protein-protein interactions. A, schematic illustrating the experimental workflow leading to the identification of proteins associated with FLAG-RelB. B, schematic illustrating the filtering strategy of the LFQ data to identify bona fide RelB interactions and RelB interactions that are altered by exposure to 10% CO2. C, scatter plot of the 135 RelB protein interactions plotted for LFQ FLAG IP intensity (degree of enrichment) versus ratio LFQ FLAG IP 10% CO2, 0.03% CO2 (CO2 sensitivity). CO2 enhanced interactions are shown in red; CO2 diminished interactions are shown in green, and other RelB interactions are shown in blue.
Figure 4.

RelB interactome is altered by CO2 exposure. HEK cells transiently transfected with pcDNA control plasmid or hFLAG-RelB were exposed to 0.03 or 10% CO2 in pH-buffered media for 75 min prior to preparation of whole-cell lysates. Lysates were immunoprecipitated using FLAG-agarose, and precipitated proteins were analyzed by mass spectrometry. Data shown are protein LFQ-intensity values of selected proteins identified in the negative control and the FLAG-RelB IP at 0.03 and 10% CO2. Selected proteins demonstrating increased association with FLAG-RelB at 10% CO2 are highlighted in red, selected proteins demonstrating decreased association with FLAG-RelB at 10% CO2 are highlighted in green. Data are representative of mean peptide intensity values ± S.D. relative to RelB for three biological replicates and two technical replicates per treatment. Statistical analysis comparing FLAG-RelB at 0.03 and 10% CO2 was performed using a Student's t test with a p value ≤ 0.05 deemed significant. ns, not significant. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
Amino acids 484–503 are involved in CO2-dependent processing of RelB
Having identified the CO2-dependent nuclear translocation and processing of RelB (Fig. 1), we next sought to identify the region of RelB that was being cleaved. First, data from Fig. 2, B and C, suggested that RelB was being cleaved in its C-terminal region downstream of Ser-424 (the RelB antibody epitope). Second, the lower molecular weight form of RelB observed in 10% CO2 revealed a relatively small (≈10–15 kDa) increase in electrophoretic mobility. Thus, we performed sequential mutagenesis of the C-terminal region of RelB spanning from before the RelB antibody epitope (Ser-404) (as a control) to beyond where we thought the cleavage site to likely be (Val-524). Thus, to test our hypothesis and gain molecular insight into the effects of CO2 on RelB, we generated six mutants that individually deleted 20 amino acid segments of human RelB spanning from amino acids 404–524 (Fig. 5A). These mutants were screened for CO2 sensitivity alongside a full-length RelB control. As expected, Δ404–423 (which is N-terminal to the Ser-424 RelB antibody epitope and therefore serves as an internal control) demonstrated CO2 sensitivity analogous to that of wild-type RelB. Furthermore, this CO2 sensitivity of Δ404–423 was blocked by pre-treatment with the proteasome inhibitor (as observed for wild-type RelB). Similarly, deletions in the regions 424–443, 444–463, and 464–483 demonstrated the same CO2 sensitivity and MG-132 sensitivity as the wild-type FLAG-RelB construct. Interestingly, the Δ484–503 mutant did not demonstrate CO2-dependent cleavage, whereas CO2 and MG-132 sensitivity was restored in the downstream Δ504–524 mutant (Fig. 5B). Interestingly, these 20 amino acids (LLDDGFAYDPTAPTLFTMLD) reside within a highly conserved region of the protein. Taken together, these data suggest that amino acids 484–503 are within the cleavage site of RelB and/or are involved in transducing the CO2-dependent cleavage of RelB.
Figure 5.

Amino acids 484–503 are involved in CO2-dependent processing of RelB. A, HEK cells transiently transfected with full-length FLAG-RelB or one of six 20-amino acid deletions of RelB (B) were exposed to 0.03 or 10% CO2 in pH-buffered media for 75 min ± Mg-132 (10 μm) prior to preparation of nuclear lysates and immunoblotting using a FLAG antibody and Ponceau S staining of the nitrocellulose membrane. Data are representative of > n = 3 experiments.
RelB Δ484–579 has altered NF-κB-dependent transcriptional activity
Having identified that absence of amino acids 484–503 rendered RelB insensitive to CO2-dependent cleavage, we hypothesized that under conditions of elevated CO2 there is an enriched population of C-terminally truncated RelB in the nucleus. We further hypothesized that the truncated form of RelB has altered transcriptional activity and contributes to CO2-dependent alterations in gene expression. To test the hypothesis that a truncated form of RelB has altered signaling capabilities, we generated a C-terminally truncated form of FLAG-RelB (RelB Δ484–579 also known as RelBshort (Fig. 6A)) and compared it with wild-type FLAG-RelB in an NF-κB-luciferase assay. TNFα-significantly increased NF-κB-luciferase activity at 0.1 and 1 ng/ml.
Figure 6.
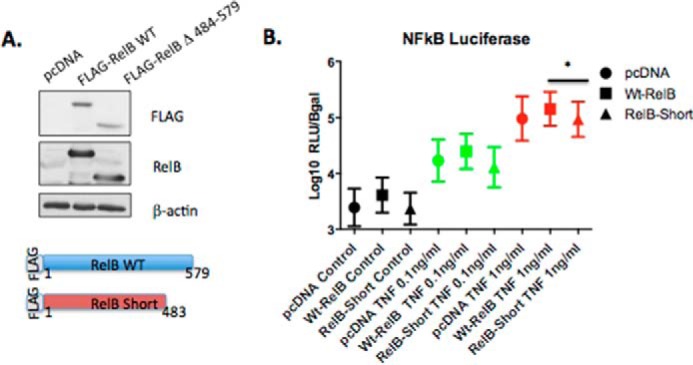
RelB Δ484–579 has altered NF-κB-dependent transcriptional activity. A, HEK cells were transiently co-transfected with pcDNA control, full-length FLAG-RelB (WT), or RelBshort (FLAG-RelB Δ484–579) in addition to a κB-luciferase promoter reporter construct and a β-galactosidase reporter. B, cells were treated with TNFα (0, 0.1, and 1 ng/ml) for 24 h at 5% CO2. Relative light units (RLU) were normalized to β-galactosidase absorbance for each sample. These non-parametric data were normalized by transforming the data by log10. Data presented are mean ± S.E. for n = 4 experiments. Statistical analysis was performed using repeated measures one-way ANOVA, with Tukey's multiple comparisons test. A p value ≤ 0.05 was deemed significant (*, p ≤ 0.05).
Overexpression of RelBshort led to reduced TNFα-stimulated NF-κB-luciferase activity compared with wild-type RelB indicating altered transcriptional activity (Fig. 6B). Thus, overexpression of a truncated form of RelB (that mimics the form of RelB that is enriched in the nucleus at 10% CO2) alters cytokine-stimulated NF-κB-dependent transcriptional activity.
Loss of p100 impairs the CO2-dependent nuclear localization of RelB
To investigate the mechanisms underpinning CO2-dependent nuclear localization of RelB further, we focused on the interaction between p100 and RelB. p100 and RelB regulate each other's stability (34). Furthermore, p100 is known to have an inhibitory role on NF-κB signaling (38) and play a key role in the inhibitory kappaBsome (39, 40). Our earlier data demonstrated that despite a marked nuclear accumulation of RelB in response to CO2, the interaction with p100 remained relatively constant (Figs. 3C and 4 and supplemental Fig. S5A). This suggested that p100 might also become nuclearly localized in response to elevated CO2 and contribute to the CO2-dependent effect on NF-κB signaling. Indeed, we observed clear p100 nuclear localization in response to elevated CO2 (Fig. 1, A and E, and Fig. S1, A and D). These data suggest the possibility of RelB and p100 translocating to the nucleus together as part of a complex. Given the role of p100 in RelB stability (34), we hypothesized that p100 might be required to confer CO2 sensitivity on RelB. To test this hypothesis, we compared the pattern of CO2-dependent RelB nuclear localization in wild-type MEF and in MEF deficient in “canonical” p105 (NFKB1) as well as “non-canonical” p100 (NFKB2). Interestingly, the p100−/− MEF demonstrated an aberrant pattern of nuclear RelB, compared with both the wild-type and p105−/− MEF. In p100−/− MEF, the full-length form of RelB was not enriched in the nucleus in response to elevated CO2; however, the lower molecular weight form of RelB was observed (Fig. 7, B, D, and F). Taken together, this suggests that p100 NF-κB is sensitive to CO2 and that it is required for the normal distribution of RelB in the nucleus under conditions of elevated CO2.
Figure 7.
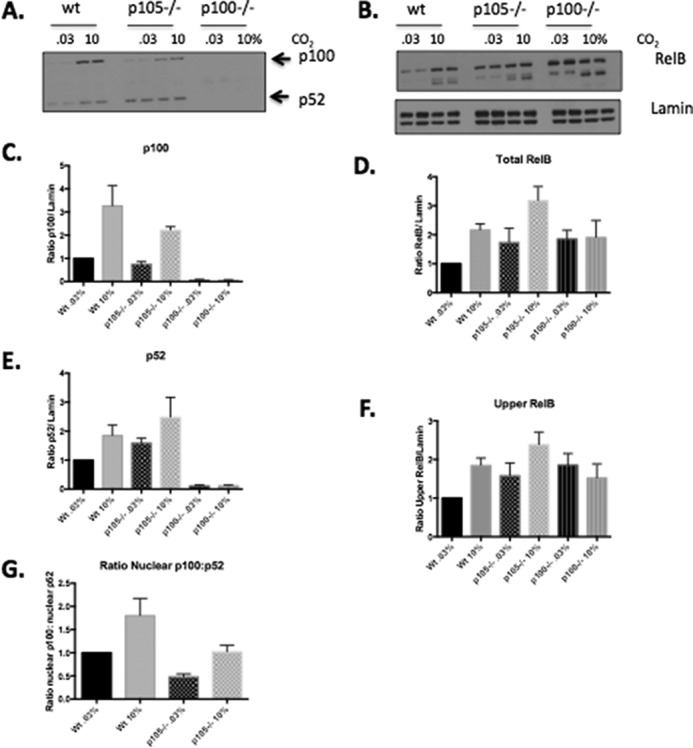
Loss of p100 NF-κB impairs the CO2-dependent nuclear localization of RelB. MEF (WT NFkB1−/− (p105−/−) and NFkB2−/− (p100−/−)) were exposed to 0.03 or 10% CO2 in pH-buffered media for 75 min prior to preparation of nuclear lysates. Lysates were immunoblotted using (A) p100/p52, (B) RelB and lamin-specific antibodies. Densitometric analysis was performed to determine the ratio of (C) p100, (D), total RelB, (E) p52, and (F) upper band of RelB relative to lamin. Data shown are the mean protein expression ± S.E. relative to control (WT MEF exposed to 0.03% CO2). G, densitometric analysis of the ratio of nuclear p100 relative to nuclear p52 in response to elevated CO2. Densitometric data are presented as mean ± S.E. for n = 4 experiments.
Discussion
Alterations in CO2 levels are increasingly being associated with human pathologies, e.g. COPD, where either hypo- or hypercapnia increases the hazard ratio for death (3). This observation is supported by in vivo experimentation illustrating the deleterious consequences of elevated CO2 in the context of infection (6) and in a clinical trial where reducing hypercapnia in COPD patients was beneficial (43). Against this background, it is perhaps counterintuitive that therapeutic hypercapnia is being investigated in the context of single lung ventilation (44). There is, however, emerging evidence for elevated CO2 being associated in a better outcome in models of inflammation (19), ventilator-induced lung injury (45, 46), skin graft survival (47), and stretch-induced epithelial injury (48). The mechanisms reconciling these seemingly opposing outcomes in hypercapnia are not fully elucidated and are a basis for this study.
Studies in both model organisms as well as human cells and tissues have implicated a role for altered NF-κB-dependent signaling in response to different CO2 concentrations (9, 11, 12, 14, 15, 46, 49, 50). Members of the non-canonical or “alternative” NF-κB family (IKKα and RelB) have been reported to be CO2-sensitive (11, 12), as have genes downstream of the Drosophila orthologue Relish in flies (9). Thus, we focused our attention on the transcriptionally active component of the non-canonical NF-κB pathway to gain insight into CO2 sensing and how CO2 affects NF-κB signaling.
Our data suggest a significant re-arrangement of RelB and p100 within the cell under conditions of elevated CO2. Mass spectrometry analysis of immunoprecipitated RelB reveals 135 proteins that are 3-fold-enriched compared with control. Of these, several bona fide interactors (p100, IκBα, and Raf-1) were found to be significantly enriched by mass spectrometry and separately validated by conventional immunoprecipitation coupled to Western blots. Approximately 19% of these proteins were differentially associated with RelB in a CO2-dependent manner. Thus, most RelB interactions were not significantly changed by CO2. This is an interesting observation given that we can clearly observe marked nuclear translocation of RelB in response to elevated CO2. This suggests that a subpopulation of RelB is actually moving into the nucleus in response to CO2 and/or that when RelB moves into the nucleus, it does so as a complex with a number of other proteins, including p100, for example. This proposed scenario might also explain why the ratio of RelB/p100 is unchanged by CO2. What is clear from the mass spectrometry experiment is that there is a difference in the degree of RelB interaction with proteins associated with the nucleus regarding nuclear shuttling (importins), nuclear pore, and DNA binding, for example (SMARCD2 and RB1), upon exposure to CO2. RelB has previously been shown to have a physical association with members of the KPNA family (51). Our observed decrease in association between RelB and α-importin proteins suggests the possibility that RelB is associated with the importin α complex in advance of a stimulus, and following CO2 exposure this interaction is reduced, as a subpopulation of RelB translocates to the nucleus. Taken together, these data point to a selective re-arrangement of RelB with its interacting partners in response to CO2, which facilitates localization to the nucleus and may interfere with existing RelB-DNA-binding complexes.
RelB is subject to multiple post-translational modifications as well as cleavage by a variety of enzymes (33, 52, 53). Our data indicate that RelB is cleaved at its C terminus (Fig. 2, B and C). Scanning mutagenesis was employed to determine the precise site involved, to gain insight into how CO2 modulates NF-κB-dependent signaling. Here, we report that a Δ484–503 deletion mutant of RelB demonstrates aberrant CO2-dependent processing (Fig. 5B). The 484–503 region is C-terminal to the RelB nuclear localization motif, which explains why both full-length and truncated forms of RelB can accumulate in the nucleus in a CO2-dependent manner. Furthermore, this site is distinct from other sites that have previously been reported to control RelB processing, for example Asp-205 (54), Thr-84/Ser-552 (53), and Arg-85 (52). Thus, we propose that this 20-amino acid CO2-responsive domain of RelB is involved in CO2-dependent cleavage of RelB. The crystal structure for this region of the RelB protein has not yet been solved, and in silico structural predictions of this region are of low confidence. However, region 484–503 does lie within the C-terminal transactivation domain of RelB (55). This suggests that RelB proteins deficient in the region C-terminal to the cleavage site may have impaired transcriptional activity. Previous studies have demonstrated the requirement of both N- and C-terminal regions of RelB in the presence of p50-NF-κB for full transactivation (26).
Thus, our data suggest that under conditions of elevated CO2 RelB is cleaved and that both full-length and truncated forms of RelB can then translocate to the nucleus. It is unlikely that both of these forms of RelB have identical transcriptional activity, and consequently, we generated a RelBshort construct to test this. This truncated form of RelB (which mimics a form of RelB generated in hypercapnia) has impaired NF-κB-dependent transcriptional activity compared with full-length RelB (Fig. 6B).
Finally, given the known reciprocal role of p100 in stabilizing RelB, we demonstrated for the first time a profound nuclear localization of p100 in response to elevated CO2 (Figs. 1 and 7, A and C, and supplemental Fig. S1, A and D). This is consistent with our mass spectrometry data indicating that the ratio of RelB/p100 is unchanged at 10% CO2 and that the two proteins may translocate to the nucleus as a complex. Thus, using MEF deficient in p100, we investigated the requirement of p100 on the RelB response to elevated CO2. Interestingly, loss of p100 significantly altered the CO2-dependent nuclear localization profile of RelB in the nucleus (Fig. 7, B and D). Notably, a lower molecular weight form of RelB was still evident in the nucleus in response to CO2 in p100−/− MEF. However, the full-length form of RelB that normally accumulates under those conditions did not (Fig. 7, B and F). Together, this suggests that p100 is important in the regulation of the RelB response to elevated CO2 but is dispensable for the cleavage. Thus, nuclear p100 localization appears to be a key event in coordinating the NF-κB-dependent response to elevated CO2. Interestingly, although we observed both p100 and p52 translocation to the nucleus in response to CO2, they did not accumulate to the same extent, with p100 relatively more enriched in the nucleus at 10% CO2 compared with p52 (Fig. 7G and supplemental Fig. S1F). This observation first suggests that elevated CO2 is not driving the non-canonical processing of p100 to p52 and second that a repressive p100- and RelB- containing complex is enriched in response to CO2.
In summary, hypercapnia is a feature of a number of pathologies and is known to modulate innate and immune signaling. A role for the NF-κB pathway downstream of hypercapnia has been proposed; however, the mechanisms are not fully elucidated. Targeting CO2-dependent signaling may represent a new anti-inflammatory strategy in the treatment of human disease; however, the molecular mechanisms downstream of CO2 need to be more fully described. Recently, RelB has been proposed as a potential novel marker of health outcomes in exacerbated COPD (57), a condition linked to hypercapnia. Here, we show that RelB is a CO2-sensitive transcription factor that undergoes a complex cellular rearrangement under conditions of elevated CO2. RelB demonstrates decreased association with importin proteins, CO2-dependent cleavage at its C-terminal that requires amino acids 484–503, and translocates to the nucleus both as a full-length protein and as a cleaved short form. RelBshort (a truncated form of RelB) demonstrates decreased NF-κB-dependent transcriptional activity compared with wild type, which may be due to impairment of its transactivation domain and consequent ability to bind nuclear proteins. The RelB interactome is altered in response to CO2, but several RelB-protein interactions are maintained at 10% CO2, e.g. interactions with p100. p100, like RelB, also translocates to the nucleus under conditions of elevated CO2, and loss of p100 impairs RelB nuclear localization when CO2 levels are elevated. Interestingly, in a recent cohort study of patients requiring acute mechanical ventilation, PaCO2 was an independent predictor of survival to hospital discharge over a linear range of PaCO2 pressures from hypocapnia (<35 mm Hg) to hypercapnia (66–75 mm Hg) (42). Thus, a better understanding of the molecular mechanisms underpinning CO2-dependent NF-κB regulation will enhance our understanding of human pathologies where hypercapnia is a feature and help to develop CO2-dependent therapeutic strategies.
Materials and methods
Cell culture and exposure to different CO2 environments
Human embryonic kidney, mouse embryonic fibroblast, and A549 cells were cultured at ambient O2 and 5% CO2 and maintained in a humidified tissue culture incubator prior to exposure to the conditions indicated in the individual experiments.
Temperature was maintained at 37 °C in a humidified environment. CO2 incubation was achieved by exposure of cells to preconditioned medium in an environmental chamber (COY Laboratories) set at 5 or 10% CO2 with a balance of air. Ambient CO2 experiments were carried out in a 37 °C humidified incubator with room air.
For experiments involving exposure to 0.03, 5, and 10% CO2, pH buffering was achieved by supplementing high glucose DMEM powder (D1152 Sigma) with different amounts of NaHCO3 as described previously (12). Media were then reconstituted, filter-sterilized, and supplemented with FCS (10%) and penicillin/streptomycin. NaCl was supplemented to correct for osmolality differences. Taken together, this approach can maintain pHe over a range of CO2 concentrations (0.03–10%).
Western blot analysis
Nuclear, cytosolic, whole-cell, or immunoprecipitated lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted as described previously (11). Primary antibodies against RelB (4954) (Figs. 1, 6, and 7), RelB (4922) (supplemental Fig. S1), p100 (4882), and lamin (4777) (Cell Signaling Technology), α-tubulin (sc-8035) (Santa Cruz Biotechnology), and FLAG (F7425) and β-actin (A5316) (Sigma) were used, as well as species-specific HRP-conjugated secondary antibodies.
Molecular cloning/mutagenesis
hFLAG-RelB in a pCR3 (Invitrogen) backbone underwent site-directed mutagenesis of its C-terminal region using QuikChange XL mutagenesis kit according to the manufacturer's instructions. The QuikChange primer design tool was used to generate a series of deletion mutants using the following specific primers: mutant Δ404–423 F, 5′-ctcgcgaccatgacagctctgaccccca-3′, and Δ404–423 R, 5′-tgggggtcagagctgtcatggtcgcgag-3′; B mutant Δ424–443 F, 5′-cttggggagctgaaccacttcctgcccaac-3′, and Δ424–443 R, 5′-gttgggcaggaagtggttcagctccccaag-3′; C mutant Δ444–463 F, 5′-ccggccatcctggaccctgacttcttctct-3′, and Δ444–463 R, 5′-agagaagaagtcagggtccaggatggccgg-3′; mutant Δ464–483 F, 5′-ccctgctgccagacctcctggacgatgg-3′, and Δ464–483 R, 5′-ccatcgtccaggaggtctggcagcaggg-3′; mutant Δ484–503 F, 5′-gcgggcctgacctgctgccccc-3′, and Δ484–503 R, 5′-gggggcagcaggtcaggcccgc-3′; F mutant Δ504–523 F, 5′-ttcaccatgctggacgtggttggggagacc-3′, and Δ504–523 R, 5′-ggtctccccaaccacgtccagcatggtgaa-3′; and RelBshort Δ484–579 F, 5′-ccctggcgggcctgactagaagcttgaattctg-3′, and Δ484–579 R, 5′-cagaattcaagcttctagtcaggcccgccaggg-3′. Mutations were confirmed by sequencing using forward T7 and/or pCR3.1-BGHrev primers.
Transfection
Cells were transfected using plasmid DNA, OptiMEM1 serum-free media (Gibco), and Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions in antibiotic-free media. Plasmids for RelB and RelB mutants are described above.
Immunoprecipitation
HEK cells overexpressing recombinant FLAG-tagged proteins were lysed in whole-cell lysis buffer (1% Triton X-100, 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm MgCl2) and incubated with anti-FLAG M2 affinity gel (7.5–15 μl/Eppendorf) end-over-end with rotation at 4 °C for 1–2 h. Samples were centrifuged at 500 rpm for 1 min to pellet the beads, which were then washed two times in lysis buffer and two times in wash buffer (lysis buffer without Triton X-100) with centrifugation in between each wash step. Beads were then incubated with NuPAGE sample buffer (≈30–60 μl) and boiled for 5 min. The supernatant was collected, supplemented with 100 mm DTT, and boiled for a further 5 min. Samples were then frozen at −20 °C or immediately run on a Western blot.
Mass spectrometry
Following immunoprecipitation, samples were treated as followed for MS analysis. After washing twice with 300 μl of ice-cold PBS, beads with bound proteins were eluted in two steps. First, 60 μl of eluting buffer I (50 mm Tris-HCl (pH 7.5), 2 m urea, and 50 μg/ml trypsin (modified sequencing grade trypsin); Promega) were incubated while shaking at 27 °C for 30 min, and second, 25 μl of elution buffer II (50 mm Tris-HCl (pH 7.5), 2 m urea, and 1 mm DTT) was added twice. Both supernatants were combined and incubated overnight at room temperature. Samples were alkylated (20 μl of iodoacetamide, 5 mg/ml, 30 min in the dark). Then, the reaction was stopped with 1 μl of 100% trifluoroacetic acid (TFA), and 100 μl of the sample was immediately loaded into equilibrated hand-made C18 StageTips containing Octadecyl C18 disks (Supelco). Samples were desalted by using two times 50 μl of 0.1% TFA and eluted with two times 25 μl of 50% acetonitrile and 0.1% TFA solution. Final eluates were combined and concentrated until the volume was reduced to 5 μl, using a CentriVap concentrator (Labconco). Samples were diluted to obtain a final volume of 12 μl by adding 0.1% TFA. The samples were run on a Q-Exactive mass spectrometer (Thermo Fisher Scientific) connected to a Dionex Ultimate 3000 (RSLCnano) chromatography system (Thermo Fisher Scientific). Each sample was loaded onto Biobasic Picotip Emitter (120 mm length, 75 μm internal diameter) packed with Reprocil Pur C18 (1.9 μm) reverse phase media column and was separated by an increasing acetonitrile gradient, using a 53-min reverse phase gradient at a flow rate of 250 nl/min. The mass spectrometer was operated in positive ion mode with a capillary temperature of 220 °C and a capillary voltage of 1,900 V applied to the capillary. All data were acquired with the mass spectrometer operating in automatic data-dependent switching mode. A high-resolution-MS scan (350–2,000 Da) was performed using the Orbitrap to select the 12 most intense ions before MS/MS analysis using the ion trap. Raw files were analyzed, and relative protein concentration and identifications were determined by label-free quantification using the MaxQuant software suite (9). MS/MS spectra were searched against the human UniProt database. Triplicate biological samples for each treatment were performed in each case. Each individual biological sample was then run in duplicate on the mass spectrometer. Each of the triplicate biological samples was considered as an individual n-number for the purposes of this experiment and to determine statistical significance. We have used a similar approach previously (56).
Filtering of mass spectrometry/mass spectrometry data
Protein mass spectrometry label-free quantification (LFQ) intensity values were normalized in each replicate for the respective experimental treatments. To discriminate specific FLAG-RelB-associated interactions from nonspecific FLAG-agarose interactions, we defined several inclusion criteria.
Enriched interactions
Proteins that were enriched >3-fold in the 0.03% CO2 and 10% CO2 FLAG-RelB sample were compared with their respective pcDNA control sample at 0.03 and 10% CO2 with a p value of ≤0.05 in each case. 135 proteins were enriched with FLAG-RelB using this analysis.
CO2-sensitive interactions
Proteins that were enriched as under “Enriched interactions” above were additionally filtered for CO2 sensitivity (ratio FLAG-RelB 10% CO2/FLAG-RelB 0.03% CO2, >2 or <0.05) (2-fold difference up or down). 25 proteins had a CO2-sensitive interaction with FLAG-RelB using this analysis.
Pathway analysis of mass spectrometry/mass spectrometry data
25 CO2-sensitive protein interactions (see under “CO2-sensitive interactions” above for filtering of mass spectrometry data and supplemental Fig. S1) were analyzed for protein class using Panther bioinformatic software (www.pantherdb.org)3 (58).
Densitometry
Densitometric analysis was carried out using ImageJ software to determine band size/intensity of target proteins on Western blottings and normalized to respective controls, e.g. α-tubulin, lamin A/C, or FLAG.
Luciferase assay
HEK cells were seeded on 24-well plates (50,000 cells/well). 24 h later, cells were co-transfected with NFκB-Luc PEST (Promega) and β-galactosidase control plasmid along with pcDNA or WT hFLAG-RelB or FLAG-RelBshort mutant RelB. 24 h later cells were treated ±TNFα (0.1–1 ng/ml) (Sigma) for 24 h prior to lysis. Lysate was incubated with luciferase substrate (Promega), and chemiluminescence was detected on a BIO-TEK Synergy-HT plate reader.
pHi assay
This experiment was performed as described previously (11). Briefly, cells were washed in OptiMEM1 (Gibco) serum-free media and loaded with 5 μm 2′,7′-bis-(2-carboxyethyl)-5-(and -6)-carboxyfluorescein, acetoxymethyl ester (BCECF-AM) (Molecular Probes Code B1170) in OptiMEM1 for 30 min at 37 °C, 21% O2, 5% CO2. Dye was removed, and cells were incubated in full DMEM for 30 min at 37 °C, 21% O2, 5% CO2. Cells were then exposed to pre-equilibrated buffered media at 0.03% CO2, 5 or 10% CO2 for 75 min. Following exposure, cells were removed and immediately assayed in a fluorescent plate reader at room temperature at 21% O2, ambient CO2. The fluorophore was excited at 485 nm (λ1) and 444 nm (λ2), and emission was recorded at 538 nm in each case. The ratio λ1/λ2 is directly proportional to intracellular pH, which was confirmed using a standard curve of nigericin (Sigma)-permeabilized cells exposed to a high potassium buffer (KCl (140 mm), MgCl2 (1 mm), CaCl2 (2 mm), d-glucose (5 mm)) adjusted to a range of pH values (pH 5–8) using MES (20 mm)-acidifying solution or Tris base (20 mm) alkaline solution.
Author contributions
C. E. K., C. C. S., J. R., A. C. S., A. von K., and E. P. C. designed, performed, and analyzed experiments. E. P. C. conceived and coordinated the study and wrote the paper. All authors reviewed the results and approved the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Margot Thome (University of Lausanne) for the generous gift of hFLAG-RelB and Prof. Alex Hoffmann (UCLA) for generously providing the wild-type, RelB−/−, p100−/−, and p105−/− MEF.
This work was supported by Science Foundation Ireland Grant 15/CDA/3490, University College Dublin School of Medicine, and University College Dublin Research Grant SF1146. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S5 and Mass spectrometry raw data file.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- COPD
- chronic obstructive pulmonary disease
- MEF
- in mouse embryonic fibroblast
- IP
- immunoprecipitation
- F
- forward
- R
- reverse
- h
- human
- LFQ
- label-free quantification
- IKK
- inhibitor of κB kinase
- EMBL-EBI
- European Molecular Biology Laboratory-European Bioinformatics Institute.
References
- 1. Cummins E. P., Selfridge A. C., Sporn P. H., Sznajder J. I., and Taylor C. T. (2014) Carbon dioxide-sensing in organisms and its implications for human disease. Cell. Mol. Life Sci. 71, 831–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crummy F., Buchan C., Miller B., Toghill J., and Naughton M. T. (2007) The use of noninvasive mechanical ventilation in COPD with severe hypercapnic acidosis. Respir. Med. 101, 53–61 [DOI] [PubMed] [Google Scholar]
- 3. Ahmadi Z., Bornefalk-Hermansson A., Franklin K. A., Midgren B., and Ekström M. P. (2014) Hypo- and hypercapnia predict mortality in oxygen-dependent chronic obstructive pulmonary disease: a population-based prospective study. Respir. Res. 15, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roberts B. W., Karagiannis P., Coletta M., Kilgannon J. H., Chansky M. E., and Trzeciak S. (2015) Effects of PaCO2 derangements on clinical outcomes after cerebral injury: a systematic review. Resuscitation 91, 32–41 [DOI] [PubMed] [Google Scholar]
- 5. Jaitovich A., Angulo M., Lecuona E., Dada L. A., Welch L. C., Cheng Y., Gusarova G., Ceco E., Liu C., Shigemura M., Barreiro E., Patterson C., Nader G. A., and Sznajder J. I. (2015) High CO2 levels cause skeletal muscle atrophy via AMPK, FoxO3a and muscle-specific ring finger protein1 (MuRF1). J. Biol. Chem. 290, 9183–9194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gates K. L., Howell H. A., Nair A., Vohwinkel C. U., Welch L. C., Beitel G. J., Hauser A. R., Sznajder J. I., and Sporn P. H. (2013) Hypercapnia impairs lung neutrophil function and increases mortality in murine pseudomonas pneumonia. Am. J. Respir. Cell Mol. Biol. 49, 821–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cummins E. P., and Keogh C. E. (2016) Respiratory gases and the regulation of transcription. Exp. Physiol. 101, 986–1002 [DOI] [PubMed] [Google Scholar]
- 8. Sharabi K., Hurwitz A., Simon A. J., Beitel G. J., Morimoto R. I., Rechavi G., Sznajder J. I., and Gruenbaum Y. (2009) Elevated CO2 levels affect development, motility, and fertility and extend life span in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 106, 4024–4029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Helenius I. T., Krupinski T., Turnbull D. W., Gruenbaum Y., Silverman N., Johnson E. A., Sporn P. H., Sznajder J. I., and Beitel G. J. (2009) Elevated CO2 suppresses specific Drosophila innate immune responses and resistance to bacterial infection. Proc. Natl. Acad. Sci. U.S.A. 106, 18710–18715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li G., Zhou D., Vicencio A. G., Ryu J., Xue J., Kanaan A., Gavrialov O., and Haddad G. G. (2006) Effect of carbon dioxide on neonatal mouse lung: a genomic approach. J. Appl. Physiol. 101, 1556–1564 [DOI] [PubMed] [Google Scholar]
- 11. Cummins E. P., Oliver K. M., Lenihan C. R., Fitzpatrick S. F., Bruning U., Scholz C. C., Slattery C., Leonard M. O., McLoughlin P., and Taylor C. T. (2010) NF-κB links CO2 sensing to innate immunity and inflammation in mammalian cells. J. Immunol. 185, 4439–4445 [DOI] [PubMed] [Google Scholar]
- 12. Oliver K. M., Lenihan C. R., Bruning U., Cheong A., Laffey J. G., McLoughlin P., Taylor C. T., and Cummins E. P. (2012) Hypercapnia induces cleavage and nuclear localization of RelB protein, giving insight into CO2 sensing and signaling. J. Biol. Chem. 287, 14004–14011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Taylor C. T., and Cummins E. P. (2011) Regulation of gene expression by carbon dioxide. J. Physiol. 589, 797–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takeshita K., Suzuki Y., Nishio K., Takeuchi O., Toda K., Kudo H., Miyao N., Ishii M., Sato N., Naoki K., Aoki T., Suzuki K., Hiraoka R., and Yamaguchi K. (2003) Hypercapnic acidosis attenuates endotoxin-induced nuclear factor-κB activation. Am. J. Respir. Cell Mol. Biol. 29, 124–132 [DOI] [PubMed] [Google Scholar]
- 15. O'Toole D., Hassett P., Contreras M., Higgins B. D., McKeown S. T., McAuley D. F., O'Brien T., and Laffey J. G. (2009) Hypercapnic acidosis attenuates pulmonary epithelial wound repair by an NF-κB dependent mechanism. Thorax 64, 976–982 [DOI] [PubMed] [Google Scholar]
- 16. Abolhassani M., Guais A., Chaumet-Riffaud P., Sasco A. J., and Schwartz L. (2009) Carbon dioxide inhalation causes pulmonary inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 296, L657–L665 [DOI] [PubMed] [Google Scholar]
- 17. Wang N., Gates K. L., Trejo H., Favoreto S. Jr., Schleimer R. P., Sznajder J. I., Beitel G. J., and Sporn P. H. (2010) Elevated CO2 selectively inhibits interleukin-6 and tumor necrosis factor expression and decreases phagocytosis in the macrophage. FASEB J. 24, 2178–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. O'Croinin D. F., Nichol A. D., Hopkins N., Boylan J., O'Brien S., O'Connor C., Laffey J. G., and McLoughlin P. (2008) Sustained hypercapnic acidosis during pulmonary infection increases bacterial load and worsens lung injury. Crit. Care Med. 36, 2128–2135 [DOI] [PubMed] [Google Scholar]
- 19. Laffey J. G., Honan D., Hopkins N., Hyvelin J. M., Boylan J. F., and McLoughlin P. (2004) Hypercapnic acidosis attenuates endotoxin-induced acute lung injury. Am. J. Respir. Crit. Care Med. 169, 46–56 [DOI] [PubMed] [Google Scholar]
- 20. Costello J., Higgins B., Contreras M., Chonghaile M. N., Hassett P., O'Toole D., and Laffey J. G. (2009) Hypercapnic acidosis attenuates shock and lung injury in early and prolonged systemic sepsis. Crit. Care Med. 37, 2412–2420 [DOI] [PubMed] [Google Scholar]
- 21. Otulakowski G., and Kavanagh B. P. (2011) Hypercapnia in acute illness: sometimes good, sometimes not. Crit. Care Med. 39, 1581–1582 [DOI] [PubMed] [Google Scholar]
- 22. Ghosh S., and Hayden M. S. (2012) Celebrating 25 years of NF-κB research. Immunol. Rev. 246, 5–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Millet P., McCall C., and Yoza B. (2013) RelB: an outlier in leukocyte biology. J. Leukocyte Biol. 94, 941–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yilmaz Z. B., Weih D. S., Sivakumar V., and Weih F. (2003) RelB is required for Peyer's patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J. 22, 121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weih F., Carrasco D., Durham S. K., Barton D. S., Rizzo C. A., Ryseck R. P., Lira S. A., and Bravo R. (1995) Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/Rel family. Cell 80, 331–340 [DOI] [PubMed] [Google Scholar]
- 26. Dobrzanski P., Ryseck R. P., and Bravo R. (1993) Both N- and C-terminal domains of RelB are required for full transactivation: role of the N-terminal leucine zipper-like motif. Mol. Cell. Biol. 13, 1572–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moorthy A. K., Huang D.-B., Wang V. Y., Vu D., and Ghosh G. (2007) X-ray structure of a NF-κB p50/RelB/DNA complex reveals assembly of multiple dimers on tandem κB sites. J. Mol. Biol. 373, 723–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Caamaño J., Alexander J., Craig L., Bravo R., and Hunter C. A. (1999) The NF-κB family member RelB is required for innate and adaptive immunity to Toxoplasma gondii. J. Immunol. 163, 4453–4461 [PubMed] [Google Scholar]
- 29. Baglole C. J., Maggirwar S. B., Gasiewicz T. A., Thatcher T. H., Phipps R. P., and Sime P. J. (2008) The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-κB. J. Biol. Chem. 283, 28944–28957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tando T., Ishizaka A., Watanabe H., Ito T., Iida S., Haraguchi T., Mizutani T., Izumi T., Isobe T., Akiyama T., Inoue J., and Iba H. (2010) Requiem protein links RelB/p52 and the Brm-type SWI/SNF complex in a noncanonical NF-κB pathway. J. Biol. Chem. 285, 21951–21960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bellet M. M., Zocchi L., and Sassone-Corsi P. (2012) The RelB subunit of NFκB acts as a negative regulator of circadian gene expression. Cell Cycle 11, 3304–3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lovas A., Radke D., Albrecht D., Yilmaz Z. B., Möller U., Habenicht A. J., and Weih F. (2008) Differential RelA- and RelB-dependent gene transcription in LTβR-stimulated mouse embryonic fibroblasts. BMC Genomics 9, 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Neumann M., Klar S., Wilisch-Neumann A., Hollenbach E., Kavuri S., Leverkus M., Kandolf R., Brunner-Weinzierl M. C., and Klingel K. (2011) Glycogen synthase kinase-3β is a crucial mediator of signal-induced RelB degradation. Oncogene 30, 2485–2492 [DOI] [PubMed] [Google Scholar]
- 34. Maier H. J., Marienfeld R., Wirth T., and Baumann B. (2003) Critical role of RelB serine 368 for dimerization and p100 stabilization. J. Biol. Chem. 278, 39242–39250 [DOI] [PubMed] [Google Scholar]
- 35. Leidner J., Palkowitsch L., Marienfeld U., Fischer D., and Marienfeld R. (2008) Identification of lysine residues critical for the transcriptional activity and polyubiquitination of the NF-κB family member RelB. Biochem. J. 416, 117–127 [DOI] [PubMed] [Google Scholar]
- 36. Leidner J., Voogdt C., Niedenthal R., Möller P., Marienfeld U., and Marienfeld R. B. (2014) SUMOylation attenuates the transcriptional activity of the NF-κB subunit RelB. J. Cell. Biochem. 115, 1430–1440 [DOI] [PubMed] [Google Scholar]
- 37. Hailfinger S., Nogai H., Pelzer C., Jaworski M., Cabalzar K., Charton J.-E., Guzzardi M., Décaillet C., Grau M., Dörken B., Lenz P., Lenz G., and Thome M. (2011) Malt1-dependent RelB cleavage promotes canonical NF-κB activation in lymphocytes and lymphoma cell lines. Proc. Natl. Acad. Sci. U.S.A. 108, 14596–14601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Basak S., Kim H., Kearns J. D., Tergaonkar V., O'Dea E., Werner S. L., Benedict C. A., Ware C. F., Ghosh G., Verma I. M., and Hoffmann A. (2007) A fourth IκB protein within the NF-κB signaling module. Cell 128, 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fusco A. J., Mazumder A., Wang V. Y., Tao Z., Ware C., and Ghosh G. (2016) The NF-κB subunit RelB controls p100 processing by competing with the kinases NIK and IKK1 for binding to p100. Sci. Signal. 9, ra96. [DOI] [PubMed] [Google Scholar]
- 40. Tao Z., Fusco A., Huang D. B., Gupta K., Young Kim D., Ware C. F., Van Duyne G. D., and Ghosh G. (2014) p100/IκBδ sequesters and inhibits NF-κB through kappaBsome formation. Proc. Natl. Acad. Sci. U.S.A. 111, 15946–15951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Labonté L., C. P., Zago M., Bourbeau J., and Baglole C. J. (2014) Alterations in the expression of the NF-κB family member RelB as a novel marker of cardiovascular outcomes during acute exacerbations of chronic obstructive pulmonary disease. PLoS ONE 10.1371/journal.pone.0112965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fuller B. M., Mohr N. M., Drewry A. M., Ferguson I. T., Trzeciak S., Kollef M. H., and Roberts B. W. (2017) Partial pressure of arterial carbon dioxide and survival to hospital discharge among patients requiring acute mechanical ventilation: A cohort study. J. Crit. Care 41, 29–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Köhnlein T., Windisch W., Köhler D., Drabik A., Geiseler J., Hartl S., Karg O., Laier-Groeneveld G., Nava S., Schönhofer B., Schucher B., Wegscheider K., Criée C. P., and Welte T. (2014) Non-invasive positive pressure ventilation for the treatment of severe stable chronic obstructive pulmonary disease: a prospective, multicentre, randomised, controlled clinical trial. Lancet Respir. Med. 2, 698–705 [DOI] [PubMed] [Google Scholar]
- 44. Gao W., Liu D. D., Li D., and Cui G. X. (2015) Effect of therapeutic hypercapnia on inflammatory responses to one-lung ventilation in lobectomy patients. Anesthesiology 122, 1235–1252 [DOI] [PubMed] [Google Scholar]
- 45. Otulakowski G., Engelberts D., Gusarova G. A., Bhattacharya J., Post M., and Kavanagh B. P. (2014) Hypercapnia attenuates ventilator-induced lung injury via a disintegrin and metalloprotease-17. J. Physiol. 592, 4507–4521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Contreras M., Ansari B., Curley G., Higgins B. D., Hassett P., O'Toole D., and Laffey J. G. (2012) Hypercapnic acidosis attenuates ventilation-induced lung injury by a nuclear factor-κB-dependent mechanism. Crit. Care Med. 40, 2622–2630 [DOI] [PubMed] [Google Scholar]
- 47. Tzeng Y. S., Wu S. Y., Peng Y. J., Cheng C. P., Tang S. E., Huang K. L., and Chu S. J. (2015) Hypercapnic acidosis prolongs survival of skin allografts. J. Surg. Res. 195, 351–359 [DOI] [PubMed] [Google Scholar]
- 48. Horie S., Ansari B., Masterson C., Devaney J., Scully M., O'Toole D., and Laffey J. G. (2016) Hypercapnic acidosis attenuates pulmonary epithelial stretch-induced injury via inhibition of the canonical NF-κB pathway. Intensive Care Med. Exp. 4, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li A. M., Quan Y., Guo Y. P., Li W. Z., and Cui X. G. (2010) Effects of therapeutic hypercapnia on inflammation and apoptosis after hepatic ischemia-reperfusion injury in rats. Chin. Med. J. 123, 2254–2258 [PubMed] [Google Scholar]
- 50. Masterson C., O'Toole D., Leo A., McHale P., Horie S., Devaney J., and Laffey J. G. (2016) Effects and mechanisms by which hypercapnic acidosis inhibits sepsis-induced canonical nuclear factor-κB signaling in the lung. Crit. Care Med. 44, e207–e217 [DOI] [PubMed] [Google Scholar]
- 51. Bouwmeester T., Bauch A., Ruffner H., Angrand P.-O., Bergamini G., Croughton K., Cruciat C., Eberhard D., Gagneur J., Ghidelli S., Hopf C., Huhse B., Mangano R., Michon A.-M., Schirle M., et al. (2004) A physical and functional map of the human TNF-α/NF-κB signal transduction pathway. Nat. Cell Biol. 6, 97–105 [DOI] [PubMed] [Google Scholar]
- 52. Hailfinger S., Lenz G., Ngo V., Posvitz-Fejfar A., Rebeaud F., Guzzardi M., Penas E. M., Dierlamm J., Chan W. C., Staudt L. M., and Thome M. (2009) Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 106, 19946–19951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Marienfeld R., Berberich-Siebelt F., Berberich I., Denk A., Serfling E., and Neumann M. (2001) Signal-specific and phosphorylation-dependent RelB degradation: a potential mechanism of NF-κB control. Oncogene 20, 8142–8147 [DOI] [PubMed] [Google Scholar]
- 54. Kuboki M., Ito A., Simizu S., and Umezawa K. (2015) Activation of apoptosis by caspase-3-dependent specific RelB cleavage in anticancer agent-treated cancer cells: involvement of positive feedback mechanism. Biochem. Biophys. Res. Commun. 456, 810–814 [DOI] [PubMed] [Google Scholar]
- 55. Perkins N. D. (2007) Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 8, 49–62 [DOI] [PubMed] [Google Scholar]
- 56. Rodriguez J., Pilkington R., Garcia Munoz A., Nguyen L. K., Rauch N., Kennedy S., Monsefi N., Herrero A., Taylor C. T., and von Kriegsheim A. (2016) Substrate-trapped interactors of PHD3 and FIH cluster in distinct signaling pathways. Cell Rep. 14, 2745–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Labonté L., Coulombe P., Zago M., Bourbeau J., and Baglole C. J. (2014) Alterations in the expression of the NF-κB family member RelB as a novel marker of cardiovascular outcomes during acute exacerbations of chronic obstructive pulmonary disease. PLoS ONE 9, e112965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mi H., Muruganujan A., and Thomas P. D. (2013) PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acid Res. 41, D377–D386 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.