

Abstract
Background
Chronic post-traumatic headache (PTH) is one of the most common symptoms of mild traumatic brain injury (mTBI) but its underlying mechanisms remain unknown. Inflammatory degranulation of dural mast cells (MCs) is thought to promote headache, and may play a role in PTH. Whether mTBI is associated with persistent degranulation of dural MCs is yet to be determined.
Methods
Histochemistry was used to evaluate time course changes in dural MC density and degranulation level in concussive head trauma and blast mouse models of mTBI. The effects of sumatriptan and the MC stabilizer cromolyn sodium on concussion-evoked dural MC degranulation were also investigated.
Results
Concussive head injury evoked persistent MC degranulation for at least 30 days. Blast trauma gave rise to a delayed MC degranulation response commencing at seven days that also persisted for at least 30 days. Neither sumatriptan nor cromolyn treatment reduced concussion-evoked persistent MC degranulation.
Conclusions
mTBI evoked by closed head injury or blast exposure is associated with persistent dural MC degranulation. Such a response in mTBI patients may contribute to PTH. Amelioration of PTH by sumatriptan may not involve inhibition of dural MC degranulation. If persistent dural MC degranulation contributes to PTH, then cromolyn treatment may not be effective.
Keywords: Minimal traumatic brain injury, concussion, blast, mouse, mast cell, dura, posttraumatic headache
Introduction
It is estimated that every year about 42 million people worldwide suffer a mild traumatic brain injury (mTBI), caused primarily by a direct concussive impact to the head (1). One of the most common symptoms of mTBI is persistent headache, a clinical entity known as post-traumatic headache (PTH) (2,3). For many individuals with persistent or chronic PTH, overall pain management remains unsatisfactory (4), leading to suffering and poor quality of life. One observational study of non-military personnel with chronic PTH reported an overall poor responsiveness to triptans and opiates in patients with a migraine/probable migraine phenotype (5). Another observational study, however, reported that acute triptan treatment is effective for aborting acute headaches in soldiers with chronic PTH related to concussion (6). The notion that the characteristics of PTH in a large number of individuals are similar to those of migraine (7,8) and that some forms of PTH are responsive to triptan treatment suggests a potential shared underlying mechanism for PTH and migraine. Migraine headache is thought to involve the intracranial meninges and the nociceptive primary afferents that innervate them (9–11). One key process that has been suggested to promote the activation of intracranial meningeal nociceptors and the ensuing headache of migraine is sterile meningeal inflammation, induced by the activation and degranulation of meningeal immune cells, in particular dural MCs (12).
We recently demonstrated the degranulation of cephalic, extracranial MCs that populate the periosteal lining of the calvaria in a concussive mouse model of mTBI, evoked by a closed head injury induced by a weight-drop – a model that was also associated with an acute cephalic hyper-nociception (13). The extracranial forces evoked by the traumatic head injury in this model, in addition to affecting the calvaria and brain, may also impact the intracranial meningeal lining (14,15). With this notion in mind, we postulated that closed head trauma leads to persistent activation of intracranial dural MCs. To test this concept, we investigated for the first time temporal changes in the degranulation level and density of dural MCs in a mouse model of mTBI induced by a blunt closed head trauma.
While many cases of mTBI and its ensuing PTH in civilians are the result of blunt head traumas, the most common cause of mTBI and its related PTH in recently deployed military personnel is the exposure to a low-level blast resulting from the detonation of improvised explosive devices (IEDs) (16). Blast related mTBI could also lead to PTH with migrainous features. Blast-related PTH may also respond to acute triptan treatment, albeit with a lower response rate than for concussion-related PTH (6). To compare with the changes seen in dural MCs after a blunt head trauma, we also studied temporal changes in the degranulation level and density of dural MCs in our recently described mouse model of military-related blast evoked mTBI. In this model, the animals are exposed to a low-level blast in a “simulated real-life environment” using a trinitrotoluene (TNT) explosive device (17). Finally, we tested the ability of two pharmacological agents: sumatriptan and cromolyn sodium to inhibit the persistent MC degranulation response following a concussive head injury. The effect of sumatriptan was tested because of its potential efficacy as an abortive treatment in a population of PTH patients, and given its effectiveness in animal models of headache, including inhibition of meningeal sterile neurogenic inflammation (18) and nitroglycerin evoked dural MC degranulation (19). The effect of cromolyn was tested because it is considered a potent inhibitor of MC degranulation and is in clinical use as such (20).
Material and methods
Animals
Adult male ICR mice weighing 25–30 g were kept five per cage under a constant 12-h light/dark cycle at room temperature. Food and water were available ad libitum. For each time point in each experimental group, four different mice were used. Each mouse was used for one time point only. The Institutional Animal Care and Use Committees (IACUCs) of the Sackler Faculty of Medicine and the Beth Israel Deaconess Medical Center approved the experimental protocols, in compliance with the guidelines for animal experimentation of the National Institute of Health.
Concussive closed head mTBI model
Closed head injury evoked by blunt trauma was induced using the weight-drop concussive device as described previously (13,21). Briefly, mice were deeply anaesthetized with 3% isoflurane and placed under a weight-drop concussive head trauma instrument. The device consists of a metal tube (inner diameter 13 mm), placed vertically over the mouse head. To induce head injury, a 30 g metal weight was dropped from the top of the tube (80 cm) to strike the head at the temporal right side between the corner of the eye and the ear. A sponge was placed under the animals to support the head while allowing some anterior-posterior motion without any rotational head movement at the moment of the impact. Immediately after the impact, mice were placed in their cages for recovery. Sham-treated animals were anaesthetized but not subjected to the weight-drop. The ability of sumatriptan and cromolyn to inhibit mTBI-related persistent dural MC degranulation was tested in animals subjected to a unilateral concussive head trauma seven and 30 days earlier. Animals were treated acutely with sumatriptan (1 mg/kg i.p., Tocris Bioscience) 2 hours before sacrifice and tissue harvesting. Cromolyn (Sodium cromoglycate, 10 mg/kg i.p. Sigma Aldrich) was administered once a day, starting seven days prior to sacrifice. Saline (0.9%) injections were used as a vehicle control. The doses of sumatriptan and cromolyn were based on previous studies (12,18).
Blast closed head mTBI model
The blast-related mTBI model was conducted as previously described (17). The procedure took place in the experimental site of “Tamar Explosives” and of “Sadwin Consultancy” in central Israel. Mice were anesthetized with an i.p. injection of a mixture of ketamine (100 mg/kg) and xylazine (10 mg/mL). Anesthetized animals were held in a restricted plastic net in a device clamped to the floor with screws, which fixed their position but did not protect them from the blast. A pressure gauge was tied to each device next to the mouse to measure blast levels in PSI. The explosive device was constructed as a cast of 500 g TNT placed four meters from the animals leading to a blast wave pressure ~5.5 PSI. Sham control (i.e., non-blast) mice were placed in the same device, anesthetized but not exposed to the blast.
Tissue preparation, histochemistry and quantitative assessment of changes in dural MCs
Animals subjected to head trauma, blast or sham procedures were deeply anaesthetized and perfused transcardially with 20 ml of phosphate buffered saline (PBS) followed by 20 ml of 4% paraformaldehyde in PBS. The heads were post-fixed overnight in the same fixative solution and then transferred to PBS. Following removal of the calvaria, the dura was removed bilaterally and mounted on a slide. To quantify MC density and level of degranulation, fixed dural whole mount tissues were stained with toluidine blue (TB, 0.1% in 2.5 pH), which binds to glycosaminoglycans in connective tissue MC granules. In different groups of animals, MC density and degranulation levels were examined using an alternative method, the enzyme-histochemical technique of Leder (22), which assesses chloroacetate esterase (CAE) reactivity in MC granules. This method employs naphthol AS-D chloroacetate as the substrate for chymase-like proteinase activity in MCs. Because all mouse MCs that have been analyzed contain large amounts of at least one type of chymotryptic proteinases in their granules, detection of CAE reactivity is another effective way to label mouse MCs (23,24). Fixed dural samples were mounted on a slide and incubated for 15 minutes at 37 C with a solution containing naphthol AS-D chloroacetate (Sigma-Aldrich). Following rinsing, the tissue was counterstained with hematoxylin. For both histological methods, MC density and degranulation level were determined using bright-field illumination, under a 400X magnification (Nikon, Eclipse Ci, Nikon, Tokyo, Japan). Because MC density and degranulation levels were uniform across the dura on each side, MC counts and degranulation levels on each side were averaged based on 20 different randomly chosen visual fields. Dural MCs were considered degranulated if there was an extensive dispersion of more than 15 extruded granules localized near the cell, or when there was an extensive loss of granule staining, giving the cell a ‘ghostly’ appearance (12,24). MC counts and degranulation levels were conducted in a blind fashion.
Data analysis
Data are displayed as the mean±SEM. Statistical analyses were performed using Statview 5.0 (SAS Institute). Group comparisons were analyzed using one-way analysis of variance (ANOVA) with Dunnett’s post-hoc test conducted to examine differences compared to control. Side differences were analyzed using unpaired t-tests. The level of significance was set at p < 0.05.
Results
Time course changes in dural MC degranulation and density levels in the weight-drop concussive mTBI model
We assessed initially, using the TB staining method, the degranulation level and density of MCs from dural samples obtained from sham and mTBI animals. In sham animals, the overall level of dural MC degranulation and MC density did not show any significant side or time (n = 4, for each time point) related differences. Overall, 13.6±2.9% (range 7.7–20.1%) of the dural MCs located over the right hemisphere were considered degranulated compared with 16.6±1.4% (range 10.4–21.2%) of the dural MCs located over the left hemisphere. The overall mean density of dural MCs in the sham groups was 11.4±1.0 cells/visual field (range 9–15) on the right side and 10.8±0.5 cells/visual field (range 7–19) for the contralateral side.
As Figure 1(a) depicts, following a concussive closed-head injury elicited by a weight drop there was an overall increase in the level of MC degranulation at all times tested within the ipsilateral dura (F3,16 = 13.49; p < 0.0001) as well as the contralateral dura (F3,16 = 14.39; p < 0.0001). At 72 hours post injury, there was a significant increase in the percentage of degranulated dural MCs ipsilateral to the injury site (mean 47.9±6.2%, range 31.2–66.4%, p < 0.01), constituting a 3.2 fold increase. The percentage of degranulated dural MCs on the contralateral side at 72 hours post injury was also significantly higher than in the control (mean 38.4±3.1%, range 32.1–48.5%, p < 0.01), a 2.3 fold increase (Figure 2). At this time point, there was no difference in the degranulation level between the ipsilateral and contralateral sides. At 1 week post injury, the percentage of degranulated dural MC localized ipsilateral to the injury site remained significantly higher than control (mean 28.7±1.7%, range 25.5–34.4%, p < 0.05), a 1.8 fold increase (Figure 4). On the contralateral side, the mean level of dural MC degranulation was also higher than control (29.8±2.5%, range 24.7–39.6%, p < 0.05), constituting also about 1.8 fold increase. At 30 days post injury, the overall level of dural MCs degranulation on the ipsilateral side was 26.89±3.7% (range 19.8–40%). This represented an overall 1.6 fold increase, which was statistically significant compared to the control group (p < 0.05). On the contralateral side, the level of dural MC degranulation was 22.2±2.5% (range 15.6–29%), a 1.3 fold increase, which was however not statistically different than the control group.
Figure 1.
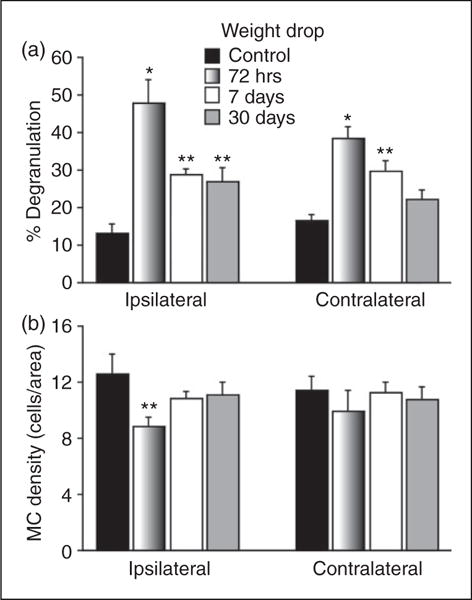
Changes in the mean degranulation (a) and density (b) levels of dural MCs in response to a concussive closed head injury. *p < 0.01, **p < 0.05 Dunnett’s post-hoc test compared to control.
Figure 2.
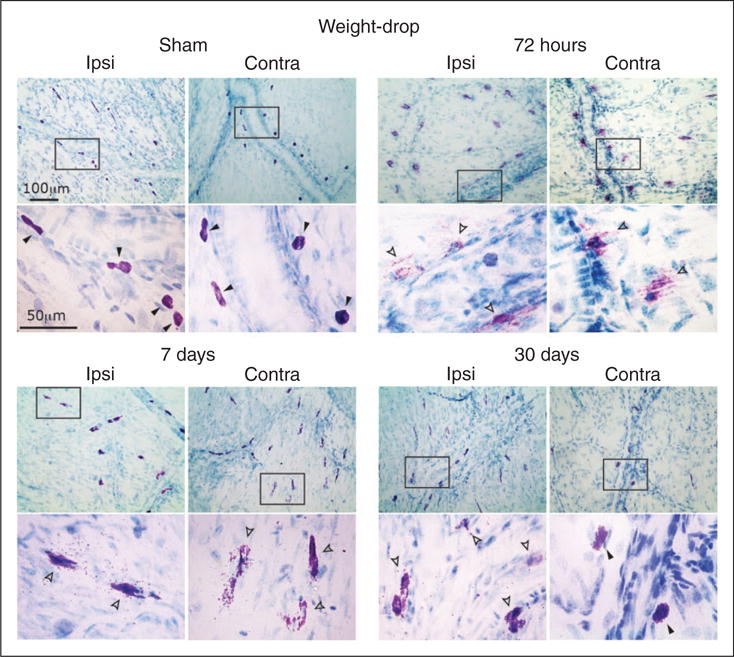
Degranulation of dural MCs following a concussive closed-head trauma evoked by weight-drop apparatus. Representative images of dura mater samples taken from mice subjected to mTBI evoked by weight-drop apparatus or sham injury. The open and closed arrowheads on the higher magnification images mark non-degranulated and degranulated MCs respectively. Note the robust bilateral degranulating effect at 72 hours, as evidenced by the reduced toluidine blue staining, which indicate lost of cytoplasmic MC granules. Also note at the seven and 30 day time-points signs for ongoing MC degranulation as evidenced by the presence of extracellular (i.e. degranulated) MC granules.
Figure 4.
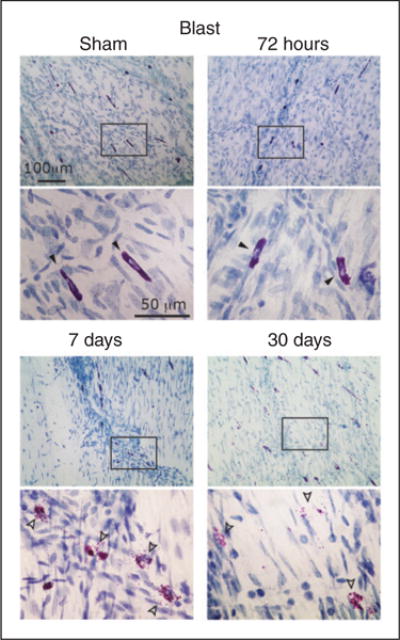
Degranulation of dural MCs following a blast-evoked closed head trauma. Images are taken from samples obtained from the dura outlining the right cortical hemisphere. Note the delayed degranulating effect, first seen at seven days. Also note the robust degranulating effect at 30 days, evidenced by cells with reduced TB staining. Closed arrowheads (non-degranulated MCs) and open arrowheads (degranulated MCs) in the lower magnification point mark the cells shown in the higher magnification images below.
Following the head injury there was an overall change in the density of MCs located within the ipsilateral dura (F3,16 = 4.76, p < 0.05). Post hoc analyses revealed a significant decrease in density at 72 hours post-trauma (mean 8.8±0.6 cells/visual field, range 6–11, p < 0.05, Figure 1(b)). At later time points, however, the density of dural MCs was not different than that seen in sham controls. On the contralateral side, the density of dural MCs was not different than controls at any of the time points tested.
Responses of dural MCs in the blast model of mTBI
All animals exposed to the blast survived and recovered from anesthesia. In sham control animals (n = 4, per group), the overall density and percentage of degranulated dural MCs observed did not show any significant side or time-related differences. Overall, 16.2±1.6% (range 12.5–22.0%) of the MCs located within the dura over the left hemisphere were considered degranulated. On the contralateral side, on average 15.6±1.4% (range 11.3–20.0%) of the dural MCs were considered degranulated. As depicted in Figure 3(a), blast injury led to an overall increase in the level of dural MC degranulation on both the right (F3,16 = 9.75; p < 0.001) and left sides (F3,16 = 4.49; p < 0.01). This increase was however delayed when compared to that seen in the weight-drop concussive model; at 72 hours after the blast exposure, there was no significant change at the level of dural MC degranulation,on both the left and right sides. The mean percentage of degranulated dural MCs located over the left hemisphere was 20.3±1.4% (range 16.6–31%). On the contralateral side, the mean percentage of degranulation, on both the left and degranulation was 18.3±1.8% (range 13–23%). At one week post blast exposure, however, the percentage of dural MCs overlying the left hemisphere that were considered degranulated was significantly higher than control animals (mean 30.4±4.3%, range 20.6–42.0%, p < 0.05; a 1.9 fold increase). The percentage of degranulated dural MCs located above the contralateral hemisphere was also higher than in the control animals (mean 31.2±1.5%, range 27–39.5%, p < 0.01; a two-time fold increase. The higher than control levels of MC degranulation persisted at 30 days bilaterally. On the left side, the mean degranulation level was 34.1±5.9% (range 19.1–51.9%, p < 0.01), a 2.11 fold increase while on the right side the level was 32.4±4.8% (range 21.3–49%, p < 0.01), a 2.1 fold increase.
Figure 3.
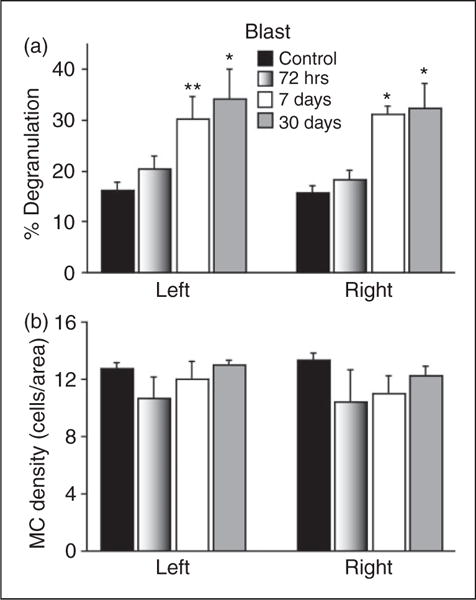
Changes in the mean degranulation (a) and density (b) levels of dural MCs in response to a blast-related closed head injury. Note the delayed increase in degranulation levels. *p < 0.01, **p < 0.05 Dunnett’s post-hoc test compared to control.
The overall density of dural MCs in the control group was 12.7±0.4 cells/area (range 11.8–14.1) for the left side and 13.3±0.5 cells/area (range 12.2–15.1) for the right side. The density of dural MCs was not statistically different than that observed in the sham control group at any of the time points tested following the blast injury.
The effect of sumatriptan and cromolyn on concussion-related persistent dural MC degranulation
We used the CAE enzyme histochemistry method to 1) verify the histological finding obtained with TB of persistent dural MC degranulation in animals subjected to a unilateral concussive head injury 7 or 30 days earlier, and 2) determine whether clinically relevant treatment regimens with sumatriptan or cromolyn can inhibit this response. An example of dural MC staining, using the CAE protocol, is shown in Figure 5. At seven days post head injury, vehicle administration (n = 5) resulted in a mean ipsilateral MC degranulation level of 35.3±5.1% (range 26.7–53.5%), which was not significantly different than that observed on the contralateral side (mean 30.1±4.3%, range 21.1–48.9%). MC density in these animals did not differ between the ipsilateral (mean 10.9±0.6 cells/visual field, range 8.1–12.1) and contralateral sides (10.5±1.1 cells/visual field, range 8.4–11.9). At 30 days post-concussion, vehicle treatment (n = 5), resulted in a mean ipsilateral degranulation rate of 34.8±1.8% (range 30.0–39.7%), which was significantly higher than that observed on the contralateral side (22.8±2.1%, range 17.8–28.1%, p < 0.01). Dural MC density, at this time point, did not differ between the ipsilateral (mean 10.2±1.1 cells/visual field, range 7.2–12.4) and contralateral sides (mean 10.0±0.6 cells/visual field, range 8.1–11.7). Importantly, the values obtained in these vehicle-treated animals, as assessed using the CAE method, were similar to those observed using the TB histological method in the untreated animals, at both the seven and 30 day time points (data not shown).
Figure 5.
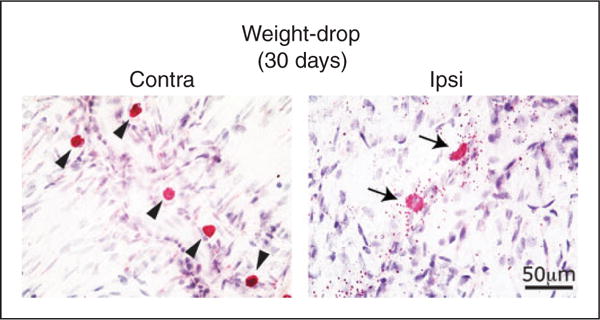
MC labeling using enzyme histochemistry with naphthol AS-D chloroacetate of the dura of animals subjected to a unilateral concussive head trauma 30 days earlier and treated with vehicle. Note the compact shape and lack of extracellular granules (i.e. degranulation) in non-activated perivascular MCs localized to the contralateral side (arrowheads in a) and the robust degranulating effect, as demonstrated by the numerous extracellular CAE labeled granule, seen in MCs localized to the dural ipsilateral to the trauma side (arrows in b).
As Figure 6 depicts, when compared to vehicle, sumatriptan treatment at seven days post head injury (n = 5) did not reduce the level of MC degranulation neither on the ipsilateral side (mean 36.7±3.5%, range 16.7–56%) nor on the contralateral side (mean 31.5±6.3, range 27.5–46%). When compared to vehicle, treatment with cromolyn (n = 5) was also ineffective in reducing the degranulation level of dural MCs on the injury side (mean 37.1±3.4%, range 30.6–49%) as well as on the contralateral side (mean 27.7±3.6%, range 18.4–42.4%). At 30 days post injury sumatriptan treatment (n = 5), when compared to vehicle did not reduce the elevated level of dural MC degranulation ipsilateral to the head injury side (mean 38.3±1.3%, range 35– 41%). Treatment with cromolyn (n = 5) was also ineffective in reducing the increased MC degranulation level ipsilateral to the injury side at this time point (mean 30.7±1.7%, range 27.1–34.5%). Neither sumatriptan nor cromolyn had significant effects on the density of dural MCs, either ipsilateral or contralateral to the head injury at both time points tested (data not shown).
Figure 6.
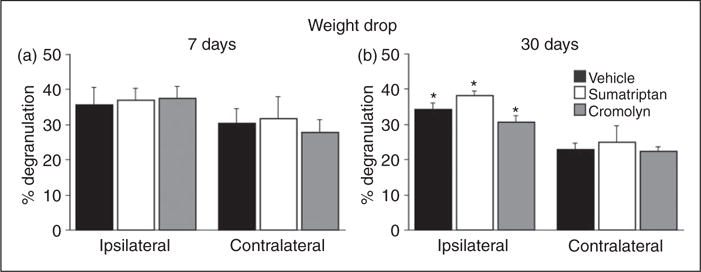
Effect of sumatriptan and cromolyn on concussive head injury-evoked dural MC degranulation. Neither sumatriptan nor cromolyn treatment reduced the persistent increase in MC degranulation level seen at seven and 30 days post-trauma when compared to the vehicle treated group. p < 0.05, unpaired t-test, ipsilateral vs. contralateral sides.
Discussion
The current study investigated time course changes in the degranulation and density levels of intracranial dural MCs in two distinct, clinically relevant mouse models of non-penetrating head injury, which lead to numerous cognitive deficits reminiscent of mTBI in humans (17,21,25). Our data indicates that following a unilateral concussive mild head trauma the dural MC population demonstrates a robust acute degranulating effect at 72 hours post trauma. The acute increase in MC degranulation documented in our concussive mTBI model agrees with the earlier report of Stokely and Orr (14), who observed an immediate ipsilateral increase in dural MC degranulation levels following a unilateral mild grazing injury of the skull in mice. The extensive ipsilateral degranulation response observed at 72 hours post trauma was associated with an ipsilateral decrease in the density of dural MCs, which recovered at seven days. A similar temporary ipsilateral decrease in dural MC density was also noted in another, yet more invasive mTBI model, involving a controlled cortical impact made through an open skull (26). The acute decrease in dural MC density ipsilateral to the injury side suggests that the MC degranulating response at this time point may have been even larger given that fully degranulated MCs are not visible using the histological techniques employed in this study, which only stain the cytoplasmic granules of MCs. Alternatively, this decrease in dural MC density could have been the result of MC migration outside the dura or MC apoptosis. At later time points, a re-granulation effect and/or enhanced recruitment of MCs to the dura may have led to the observed increase in dural MC density to baseline levels.
The data obtained using two distinct histochemical methods - TB staining and CAE reactivity - suggest that the elevated MC degranulation level, ipsilateral to the trauma side, persists for at least 30 days following the concussion. Our findings also point to a sizable, short-term increase in dural MC degranulation levels contralateral to the trauma side that return to baseline level before the 30 days post-trauma time point. The mechanisms underlying the ipsilateral MC degranulating response may involve the intracranial transmission of the mechanical trauma through the calvaria. The factors that mediate the contralateral degranulating affect may be different, however. One possible factor that may contribute to the contralateral degranulating effect (and which may also mediate the ipsilateral one) is a bilateral cortical response; for example cell death (27) with ensuing neuroinflammation. Parenchymal inflammatory response after cortical injury, for example as a result of cortical spreading depression, has been shown to promote dural MC degranulation (28). While it is possible that a bilateral cortical neuroinflammation may contribute, at least in part to the degranulation of dural MCs at the earlier time points, the notion that activation of meningeal MCs after a brain trauma may initially suppress post-traumatic brain inflammation (29) argues nevertheless against such a mechanism.
Our data suggests that the responses of dural MCs following a blast injury are somewhat distinct from those seen after a direct unilateral concussive trauma: the degranulating effect is delayed, first observed later than 72 hours post-trauma and then persisting at a similar level bilaterally for at least 30 days. The exact mechanism underlying this delayed effect is not known but may be related to a delayed cortical response; we have shown in a previous study that in this model of blast-evoked mTBI, there is also a delayed cortical inflammatory vascular response, with only a marginal trend towards an alteration of blood–brain barrier integrity at seven days but a pronounced cortical vascular response, including increased blood–brain barrier permeability at 30 days (17).
The long-term increase in the degranulation level of dural MCs ipsilateral to the trauma site in the concussive model and bilaterally in the blast model may play a role in PTH. In particular, the persistent release of pro-nociceptive MC mediators could lead to persistent activation and sensitization of dural nociceptors (12,30), a nociceptive response that could potentially contribute to the emergence and perhaps maintenance of PTH. The activation of dural nociceptors with subsequent peripheral release of pro-inflammatory neuropeptides, such as calcitonin gene-related peptide (CGRP) and substance P, has been suggested to promote dural MC degranulation, through an axonal reflex mechanism known as neurogenic inflammation (31). This mechanism could theoretically mediate the persisted concussion-related dural MC degranulating response, as observed in our study. We found however that acute treatment with the anti-migraine drug sumatriptan, which has been shown to inhibit neurogenic inflammation and dural MC degranulation in animal studies (18,32), did not reduce the level of dural MC degranulation at seven and 30 days following the head injury. Our data thus does not support dural neurogenic inflammation as a major contributing mechanism that sustains the persistent MC degranulation response following a concussion. The ability of acute sumatriptan treatment to ameliorate the headache in a subpopulation of PTH patients thus may not be related to its proposed meningeal anti-inflammatory action but possibly to its antinociceptive effect within the trigeminal nucleus caudalis (33). Surprisingly, we also found that treatment with cromolyn sodium, an agent commonly used to inhibit MC degranulation (20), was also ineffective in reducing the concussion-related persistent dural MC degranulation response. Although a similar treatment regimen with cromolyn can inhibit MC degranulation in numerous MC-related inflammatory conditions, it is not always effective in patients (20). If dural MC degranulation plays a role in mediating PTH, then cromolyn treatment may not be therapeutically effective.
In conclusion, we show that mouse models of mTBI evoked by a concussive head trauma and blast injury are associated with persistent degranulation of dural MCs, a response mediated through an, as yet, undefined mechanism. While it is conceivable that concussion-related damage to brain areas that mediate nociception could also play a role in mediating PTH (34), mTBI-related persistent activation of dural MCs could lead to ongoing activation and sensitization of the headache pain pathway (12), thus potentially promoting the headache as well as the associated cranial hyperalgesia seen in PTH patients (34,35).
Key findings.
A unilateral concussive, closed head injury model of mTBI is associated with a persistent ipsilateral activation of dural mast cells.
A blast model of mTBI, using exposure to TNT is associated with a delayed but long lasting activation of dural mast cells.
The persistent activation of dural mast cells after concussion and exposure to blast suggests persistent intracranial meningeal inflammation.
Concussion-related persistent mast cell degranulation is resistant to clinically-relevant treatment with sumatriptan or cromolyn.
mTBI-related persistent mast cell degranulation with ensuing meningeal inflammation may play a role in the genesis and maintenance of post-traumatic headache.
Acknowledgments
Funding/Acknowledgments
The study was supported by grants from the NIH/NINDS (NS077882, NS086830, NS078263 to DL).
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Gardner RC, Yaffe K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol Cell Neurosci. 2015;66(Pt B):75–80. doi: 10.1016/j.mcn.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keidel M, Ramadan N. Acute post traumatic headache. In: Olesen J, Goadsby PJ, Ramadan NM, Tfelt-Hansen P, Welch KMA, editors. The Headaches. 3rd. Philadelphia: Lippincott: Willimas & Wilkins; 2006. pp. 863–872. [Google Scholar]
- 3.Nampiaparampil DE. Prevalence of chronic pain after traumatic brain injury: a systematic review. JAMA. 2008;300(6):711–719. doi: 10.1001/jama.300.6.711. [DOI] [PubMed] [Google Scholar]
- 4.Lucas S. Posttraumatic Headache: Clinical Characterization and Management. Curr Pain Headache Rep. 2015;19(10):520. doi: 10.1007/s11916-015-0520-1. [DOI] [PubMed] [Google Scholar]
- 5.DiTommaso C, Hoffman JM, Lucas S, et al. Medication usage patterns for headache treatment after mild traumatic brain injury. Headache. 2014;54(3):511–519. doi: 10.1111/head.12254. [DOI] [PubMed] [Google Scholar]
- 6.Ericsson A, Arias C, Sawchenko PE. Evidence for an intramedullary prostaglandin-dependent mechanism in the activation of stress-related neuroendocrine circuitry by intravenous interleukin-1. J Neurosci. 1997;17(18):7166–7179. doi: 10.1523/JNEUROSCI.17-18-07166.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lucas S, Hoffman JM, Bell KR, et al. Characterization of headache after traumatic brain injury. Cephalalgia. 2012;32(8):600–606. doi: 10.1177/0333102412445224. [DOI] [PubMed] [Google Scholar]
- 8.Walker WC, Marwitz JH, Wilk AR, et al. Prediction of headache severity (density and functional impact) after traumatic brain injury: A longitudinal multicenter study. Cephalalgia. 2013;33(12):998–1008. doi: 10.1177/0333102413482197. [DOI] [PubMed] [Google Scholar]
- 9.Olesen J, Burstein R, Ashina M, et al. Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol. 2009;8(7):679–90. doi: 10.1016/S1474-4422(09)70090-0. [DOI] [PubMed] [Google Scholar]
- 10.Levy D, Strassman AM, Burstein R. A critical view on the role of migraine triggers in the genesis of migraine pain. Headache. 2009;49(6):953–957. doi: 10.1111/j.1526-4610.2009.01444.x. [DOI] [PubMed] [Google Scholar]
- 11.Messlinger K. Migraine: where and how does the pain originate? Exp Brain Res. 2009;196(1):179–193. doi: 10.1007/s00221-009-1756-y. [DOI] [PubMed] [Google Scholar]
- 12.Levy D, Burstein R, Kainz V, et al. Mast cell degranulation activates a pain pathway underlying migraine headache. Pain. 2007;130(1–2):166–176. doi: 10.1016/j.pain.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benromano T, Defrin R, Ahn AH, et al. Mild closed head injury promotes a selective trigeminal hypernociception: Implications for the acute emergence of post-traumatic headache. Eur J Pain. 2014;19(5):621–628. doi: 10.1002/ejp.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stokely ME, Orr EL. Acute effects of calvarial damage on dural mast cells, pial vascular permeability, and cerebral cortical histamine levels in rats and mice. J Neurotrauma. 2008;25(1):52–61. doi: 10.1089/neu.2007.0397. [DOI] [PubMed] [Google Scholar]
- 15.Roth TL, Nayak D, Atanasijevic T, et al. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 2014;505(7482):223–228. doi: 10.1038/nature12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Theeler BJ, Flynn FG, Erickson JC. Headaches after concussion in US soldiers returning from Iraq or Afghanistan. Headache. 2010;50(8):1262–1272. doi: 10.1111/j.1526-4610.2010.01700.x. [DOI] [PubMed] [Google Scholar]
- 17.Rubovitch V, Ten-Bosch M, Zohar O, et al. A mouse model of blast-induced mild traumatic brain injury. Exp Neurol. 2011;232(2):280–289. doi: 10.1016/j.expneurol.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buzzi MG, Moskowitz MA. The antimigraine drug, sumatriptan (GR43175), selectively blocks neurogenic plasma extravasation from blood vessels in dura mater. Br J Pharmacol. 1990;99(1):202–206. doi: 10.1111/j.1476-5381.1990.tb14679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pedersen SH, Ramachandran R, Amrutkar DV, et al. Mechanisms of glyceryl trinitrate provoked mast cell degranulation. Cephalalgia. 2015;35(14):1287–1297. doi: 10.1177/0333102415574846. [DOI] [PubMed] [Google Scholar]
- 20.Zhang T, Finn DF, Barlow JW, et al. Mast cell stabilisers. Eur J Pharmacol. 2015 Jun 27; doi: 10.1016/j.ejphar.2015.05.071. [DOI] [PubMed] [Google Scholar]
- 21.Zohar O, Schreiber S, Getslev V, et al. Closed-head minimal traumatic brain injury produces long-term cognitive deficits in mice. Neuroscience. 2003;118(4):949–955. doi: 10.1016/s0306-4522(03)00048-4. [DOI] [PubMed] [Google Scholar]
- 22.Leder LD. The chloroacetate esterase reaction. A useful means of histological diagnosis of hematological disorders from paraffin sections of skin. Am J Dermatopathol. 1979;1(1):39–42. [PubMed] [Google Scholar]
- 23.Humphries DE, Wong GW, Friend DS, et al. Heparin is essential for the storage of specific granule proteases in mast cells. Nature. 1999;400(6746):769–772. doi: 10.1038/23481. [DOI] [PubMed] [Google Scholar]
- 24.Bankova LG, Lezcano C, Pejler G, et al. Mouse mast cell proteases 4 and 5 mediate epidermal injury through disruption of tight junctions. J Immunol. 2014;192(6):2812–2820. doi: 10.4049/jimmunol.1301794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milman A, Rosenberg A, Weizman R, et al. Mild traumatic brain injury induces persistent cognitive deficits and behavioral disturbances in mice. J Neurotrauma. 2005;22(9):1003–1010. doi: 10.1089/neu.2005.22.1003. [DOI] [PubMed] [Google Scholar]
- 26.Shimada R, Nakao K, Furutani R, et al. A rat model of changes in dural mast cells and brain histamine receptor H3 expression following traumatic brain injury. J Clin Neurosci. 2012;19(3):447–451. doi: 10.1016/j.jocn.2011.06.033. [DOI] [PubMed] [Google Scholar]
- 27.Tashlykov V, Katz Y, Volkov A, et al. Minimal traumatic brain injury induce apoptotic cell death in mice. J Mol Neurosci. 2009;37(1):16–24. doi: 10.1007/s12031-008-9094-2. [DOI] [PubMed] [Google Scholar]
- 28.Karatas H, Erdener SE, Gursoy-Ozdemir Y, et al. Spreading depression triggers headache by activating neuronal Panx1 channels. Science. 2013;339(6123):1092–1095. doi: 10.1126/science.1231897. [DOI] [PubMed] [Google Scholar]
- 29.Hendrix S, Kramer P, Pehl D, et al. Mast cells protect from post-traumatic brain inflammation by the mast cell-specific chymase mouse mast cell protease-4. FASEB J. 2013;27(3):920–929. doi: 10.1096/fj.12-204800. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Strassman AM, Burstein R, et al. Sensitization and activation of intracranial meningeal nociceptors by mast cell mediators. J Pharmacol Exp Ther. 2007;322(2):806–812. doi: 10.1124/jpet.107.123745. [DOI] [PubMed] [Google Scholar]
- 31.Russo AF. Calcitonin gene-related peptide (CGRP): a new target for migraine. Annu Rev Pharmacol Toxicol. 2015;55:533–552. doi: 10.1146/annurev-pharmtox-010814-124701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buzzi MG, Dimitriadou V, Theoharides TC, et al. 5-Hydroxytryptamine receptor agonists for the abortive treatment of vascular headaches block mast cell, endothelial and platelet activation within the rat dura mater after trigeminal stimulation. Brain Res. 1992;583(1–2):137–149. doi: 10.1016/s0006-8993(10)80017-4. [DOI] [PubMed] [Google Scholar]
- 33.Levy D, Jakubowski M, Burstein R. Disruption of communication between peripheral and central trigeminovascular neurons mediates the antimigraine action of 5HT 1B/1D receptor agonists. Proc Natl Acad Sci USA. 2004;101(12):4274–4279. doi: 10.1073/pnas.0306147101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Defrin R, Riabinin M, Feingold Y, et al. Deficient pain modulatory systems in patients with mild traumatic brain and chronic post-traumatic headache: implications for its mechanism. J Neurotrauma. 2015;32(1):28–37. doi: 10.1089/neu.2014.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Defrin R, Gruener H, Schreiber S, et al. Quantitative somatosensory testing of subjects with chronic post-traumatic headache: implications on its mechanisms. Eur J Pain. 2010;14(9):924–931. doi: 10.1016/j.ejpain.2010.03.004. [DOI] [PubMed] [Google Scholar]