

Abstract
It has only recently been appreciated that the human body contains many long-lived proteins. Their gradual degradation over time contributes to human aging and probably also to a range of age-related disorders. Indeed progressive damage of proteins may be implicated in the fact that many neurological diseases do not appear until after middle age. A major factor responsible for the deterioration of old proteins is the spontaneous breakdown of susceptible amino acid residues resulting in racemisation, truncation, deamidation, and cross-linking. When proteins decompose in this way, their structures and functions may be altered and novel epitopes can be formed that can induce an autoimmune response.
Keywords: Protein aging, post-translational modification, racemization, age-related disease
Many of your body’s proteins are present for decades
In the human body, old proteins are everywhere. Indeed the most abundant protein in the body, collagen which represents ~ 30% of total protein is one such old, or long-lived protein (LLP). Its half-life varies, depending on the anatomical site and type of collagen, but has been estimated to be 117 years in articular cartilage [1] and between 95–215 years in intervertebral discs [2].
LLPs are present in the heart [3], arteries [4], lungs [5], skeleton [2], muscle [6], joints [7], skin [8], hair [9], teeth [10], fat cells [11], eye [12–14], epiglottis [15], oocytes [16] and brain [17]. What perhaps is more remarkable is that some proteins in the body do not turn over at all. They are with you for life. Sadly for us, these long-lived macromolecules are not stable; they degrade with age and the consequences for human health, aging and disease are likely to be profound. In this article we propose that age-related protein deterioration plays an important role in each of these conditions.
Why are proteins long-lived?
It is likely that longevity is a consequence of several factors. Firstly it is wasteful energetically, and from a resource standpoint, to continually renew proteins if this is unnecessary. Secondly, it may be difficult or impossible to achieve. For example, it is presumably challenging to rebuild a highly insoluble, crosslinked protein such as elastin, and to regenerate an internal lens cell, considering the architecture of the lens where cells are added continuously throughout life on top of a pre-existing lens. Extracellular renewal/re-synthesis may present particular problems. Many such issues can be considered in terms of reproductive lifespan. We are ‘designed’ to last for approximately 30–40 years, which was sufficient time, when we were evolving, to reproduce and raise children. If a biological structure can achieve this outcome within the timeframe, then it will not be selected against. So a degree of protein decomposition in this period may be tolerated if function is not greatly compromised.
What is an LLP?
For the purposes of this article, LLPs are considered to be those whose half-lives are longer than 48 h. This definition is somewhat arbitrary but is taken in the context of what is known about the typical lifetimes of proteins within cells. This ranges from <1 h to 22 h [18]. In fact, most proteins discussed in this review have lifetimes of months or years, with some showing no turnover at all. It is likely that many more proteins will be discovered to be long-lived.
How do we know that proteins are long-lived?
There are three main ways in which the lifetime of a protein can be determined. In the case of humans, perhaps the most rigorous method is determination of 14C content. This assay takes advantage of the fact that atmospheric levels of 14C reached a maximum in the late 1950s due to above ground testing of nuclear weapons. 14C levels in the atmosphere and food then declined once the 1963 nuclear test ban treaty was implemented. In a sense, all people born after this time are subjects in a pulse chase experiment. If a molecule in the human body does not turnover its content of 14C will reflect that of the atmospheric levels at the time of birth [19].
In animals information about turnover of proteins can be estimated by feeding stable isotopes [17, 20]. Examination of the extent of retention of 15N or 13C in particular proteins by mass spectrometry can reveal if they are subject to degradation or not. Such experiments have revealed some surprising results, but typically can only be performed over a short time period e.g. one or two years. In one recent study [17] components of the nucleus and myelin were found to be LLPs as well as, surprisingly, several cytosolic enzymes. Currently known LLPs are highlighted in Table 1.
Table 1.
Known long-lived proteins.
| Tissue | Long-lived proteins |
|---|---|
| Lens | Crystallins[12] Aquaporin 0 [62] |
| Vitreous | Collagen[63] |
| Retina | Histones[64] Lamins B1 & B2[65] |
| Brain | Myelin basic protein[17] Myelin proteolipid protein [59] Myelin oligodendrocyte glycoprotein[17] 2′,3′-Cyclic-nucleotide 3′-phosphodiesterase[17] Histones [17] Tau[17] Aβ peptide[49] Isoaspartyl peptidase/L-asparaginase[49] NAD-dependent protein deacetylase sirtuin-2[17] Nucleoporins[17] Lamins B1 & B2[17] Neurofilament medium polypeptide [17] Ectonucleotide pyrophosphatase/phosphodiesterase[17] Immunoglobulin superfamily member 8[17] Tubulin[66] Proteoglycans[67] SOD1[20] Versican core protein[17] Immunoglobulin superfamily member 8[17] Collagen[17] |
| Motor Neuron | TPD43[68] |
| Heart, Arteries | Elastin[3] Collagen[69] Histone H4[70] Lamin B1[70] |
| Lung | Elastin[5] Collagen[69] |
| Teeth | Dentine phosphoprotein [71] |
| Liver | Serine hydroxymethyltransferase[72] |
| Muscle | Collagen [6] PKA catalytic subunit[73] Myosin[74] |
| Blood (Erythrocytes) | Membrane protein band 4.1 [75] Ankyrin[76] |
| Skin | Elastin[8] Collagen[69] |
| Hair | Keratin |
| Bone | Collagen[2, 69] Osteocalcin[7] |
| Epiglottis | Elastin[15] |
| Cartilage | Proteoglycans[78] Collagen[42] |
| Oocytes | Rec8[16] Serine hydroxymethyltransferase[9] |
| Fat Cells | Unknown proteins[11] |
| Tendons | Collagen[69] |
| Ligaments | Collagen[69] |
In humans, the most commonly used method to gauge a protein’s lifetime assays the chemical fingerprints of aging which comprise a distinct subset of post-translational modifications (PTMs). Some amino acids within LLPs change in characteristic ways to yield stable products, “signatures” that can be assayed using proteomic techniques [21, 22]. The details of these changes and their functional consequences will be discussed in more detail later in this article.
Why are LLPs important?
Unfortunately LLPs decompose over time. From the time that an LLP is formed (Figure 2), it comes under relentless attack by enzymes and reactive small molecules, resulting in degradation. Another source of degradation arises from spontaneous processes, i.e. reactions that take place due to the intrinsic instability of some amino acids and are the inevitable result of heat and time. Spontaneous degradation appears to be the most common cause of LLP decomposition[23].
Figure 2.
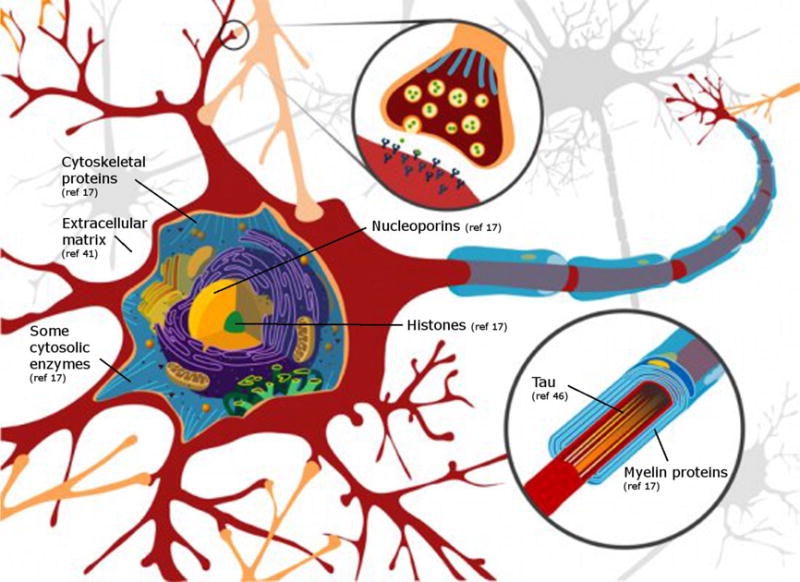
Important structures within cells, and extracellularly, are composed of long-lived proteins. A Neuron is illustrated because the proteins of myelin (derived from oligodendrocytes) are long-lived. Synapsin 1 is a client protein for protein isoaspartate methyl transferase (PIMT) and has been suggested to be long-lived [80].
What happens to long-lived proteins in the body?
The most commonly used clinical method for assessing diabetic compliance (the overall control of blood sugar levels) employs an assay for glycosylated hemoglobin. If glucose levels are raised in the blood then the α-amino group of hemoglobin becomes more highly modified by the aldehyde moiety of glucose. This simple example illustrates that proteins can be significantly modified by molecules that exist throughout the body and that modification can take place over a period of days given that the lifetime of an erythrocyte is about 110 days. In the biochemical soup that is the cell cytosol, there are many reactive small molecules. Each metabolite can attack LLPs and leave an indelible imprint. Exposure to reactive oxygen species can also permanently alter the structure of amino acids present in LLPs [24].
The extent of modification of LLPs due to enzymes is more difficult to assess; however, the adult human lens can be used to investigate this for reasons that are outlined in a later section.
Spontaneous decomposition of amino acids within LLPs is a major source of breakdown
The lens of the eye is composed of proteins that do not turnover [12]. It is an ideal tissue for investigating the sources of LLP breakdown because of its peculiar manner of growth, its simple composition, and the availability of samples across the age range. The center of the lens was present when you were born and the mature fiber cells lack cellular organelles, so the ability to synthesize macromolecules is therefore absent. Over time, the enzymes that were present at birth in the lens centre, become denatured and eventually cease to be active [25]. By following the time course of protein degradation in the lens center i.e. using lenses of different ages, we can therefore indirectly discover which are the most important processes that affect LLPs.
Based on lens data, it has become apparent that spontaneous decomposition is the major reason for the breakdown of LLPs. Three of the twenty amino acids; Asp, Asn and Ser are particularly susceptible to spontaneous processes [26–28].
Surprisingly, given the example of hemoglobin outlined above, modification of lens proteins by metabolites such as glucose was found to occur only at very low levels [29, 30]. Oxidation of lens crystallin proteins, even up until old age, was also barely detectable in normal lenses [31]. Both of these observations can be potentially explained by maintenance of high concentrations of cellular glutathione in the lens, which protects LLPs from covalent modification [32].
Racemization, Cleavage and Crosslinking
If indeed spontaneous reactions are a major factor in alterations to LLPs over time, what are they? Four main types of reactions occur, most often involving Asp, Asn and Ser. These are amino acid racemization, deamidation, cleavage and covalent crosslinking. These are briefly summarized in this section.
Deamidation/Racemization
Racemization is the conversion of the normal L- form to the D-form of the amino acid. In the case of Ser [33], this may involve direct removal and re-insertion of the alpha proton of the amino acid; however, for Asn and Asp, racemization in proteins occurs predominantly via a cyclic succinimide intermediate (Figure 3) [34]. In the case of both Asn and Asp, four products result: L-Asp, D-Asp, L-isoAsp and D-isoAsp [34]. A susceptible neutral Asn residue can therefore be converted into four negatively charged products. Thus, in the case of a susceptible Asn residue, racemization and deamidation are intimately linked. Analagous processes occur with Glu and Gln but to a smaller degree. The extent of all of these reactions in LLPs increases with age, and these have been termed “molecular clocks” [35]. To gauge the quantitative importance of this single process, in the human lens every protein chain contains, on average, 2–3 D-amino acids by age 60 [26].
Figure 3.
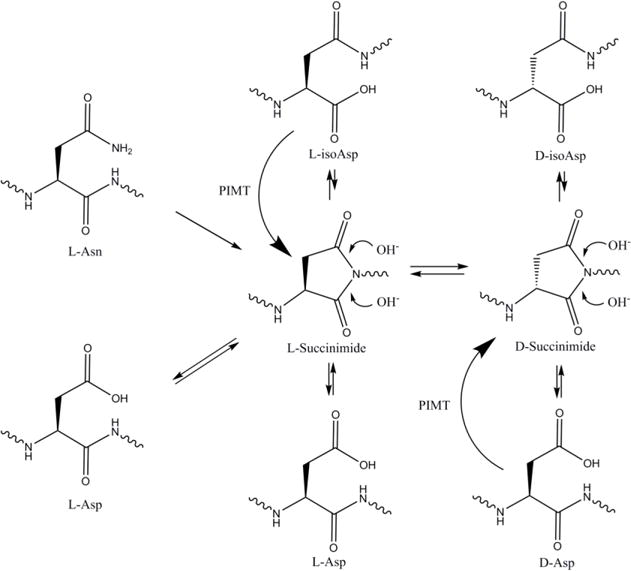
Long-lived proteins (LLPs) decompose in the body. A common site of deterioration is at asparagine and aspartic acid which can undergo racemization via succinimide intermediates. These are spontaneous events that occur primarily in unstructured regions of the protein, leading to four possible Asp isomers: L-Asp, D-Asp, L-isoAsp, and D-isoAsp. Asp residues converted to isoAsp appear to be stable and undergo little interconversion. In most cells protein isoaspartate methyl transferase (PIMT) can ameliorate Asp racemization by methylating L-isoAsp and D-Asp, enabling the conversion back to L-Asp. Currently this is the only known repair enzyme for the repair of racemised Asp in LLPs.
Peptide bond cleavage and covalent crosslinking
Because this is a relatively new area of biochemistry, the reactions underlying cleavage and crosslinking of LLPs are only now being elucidated. Ser is one site where autolytic cleavage of peptide bonds has often been observed. The side chain hydroxyl group is involved and zinc is crucial in catalyzing the hydrolysis [36]. As expected, a similar process occurs with Thr. Cleavage next to Asn/Asp has also been documented [37].
A major pathway of non-disulfide covalent crosslinking of proteins in the lens has been shown to be due to decomposition of phosphoSer and phosphoThr [38]. In the case of phosphoSer the intermediate is reactive dehydroalanine which can also arise from Cys [39].
An important feature observed in the breakdown of all LLPs, is that the spontaneous events are localized within unstructured regions of the proteins [22, 40]. Conformational flexibility is necessary.
Consequences of protein decomposition for aging
While it seems clear that spontaneous processes within LLPs are a major contributor for age-related conditions of the human lens, such as presbyopia and cataract [23], the situation for other tissues is more difficult to determine. Correlations must suffice.
It is well known that the physical properties and functionality of blood vessels, lungs and teeth change over time. For example, forced expiratory volume declines by 1 to 2% per year after the age of 25. The capacity of the heart is diminished over time mainly by a decrease in its elasticity coupled with that of the arterial system [41].
It is likely that racemization of susceptible amino acids is at least, in part, responsible because in these tissues there is a large and remorseless age-related increase in the content of D-Asp. For example, in the aorta and lung, levels increase almost linearly from < 1% in childhood, to ~5% by age 60 years [4], matching closely the profile documented for dentine from teeth. Analysis of D-Asp in teeth is an established method to assess the age of an individual in forensic investigations. If this increase does not seem substantial, it is useful to translate it into residues/polypeptide. For a 40kDa protein with ~40 Asp or Asn residues, this translates into 2 D-Asp residues in every single protein. For any protein, this will almost certainly adversely affect the structure of the protein, its interactions and, if it is an abundant LLP, the tissue where it resides. This is especially true when the most abundant form of D-Asp in aged tissues is D-isoAsp, where an extra methylene group has effectively been inserted into the main protein chain.
It should also be noted that the % calculation of D-Asp content in tissues or individual proteins is an underestimate of LLP change, since it does not measure the content of L-isoAsp.
The increase in D-Asp with age is not always linear
In collagen extracted from cartilage [42] and bone[43] there is also a roughly linear increase in the content of D-Asp with age, although the levels are about half those seen in aorta and lung. Interestingly in the human lens, the age-related increase in modification of LLPs is typically not linear, however the reasons for non-linearity are unclear. The increase in D-Asp content with age in elastin isolated from arteries [4] mirrors more closely the age-dependent profile for total D-Asp from human lenses [26] in that there is more rapid increase up until adult years. The reason for this bi-phasic age response is also unknown.
Racemization and crosslinking not only alter the structures and interactions of LLPs with other molecules but also make the modified LLPs difficult to remove from the cell or tissue is which they are formed. This is because the cellular protease enzymes that recycle proteins are impeded by D-amino acids and covalent crosslinks [44]. This factor, along with LLP modification by reactive lipids, may be responsible for the formation of pigment granules known as lipofuscin, a characteristic feature of old post-mitotic cells.
Consequences of protein decomposition for disease
Once the scale of age-related changes in human LLPs is appreciated, it becomes more apparent how these alterations could translate into the inevitable decline of the body’s parts with age, as well as disease outcomes. This is a new area of medicine and data are, at present scarce, however there are some intriguing observations. It is well known that the incidence of diseases such as cataract, Alzheimer’s disease, Parkinson’s disease, and a host of others, is markedly age-dependent. The LLP changes noted above provide one explanation.
This is a large topic and, for this reason, the following considerations regarding LLPs and disease will be largely confined to the brain.
The brain contains numerous LLPs and many seem to be predominantly unstructured [45, 46]. Thus they are vulnerable to the sort of age-related modifications described here. In the case of motor neuron disease (ALS), transport through the nuclear pore complex may be compromised. Hexapeptide/dipeptide repeat expansions may play a role in this inhibition [47]; however, age-related degradation of the nuclear pore complex (Table 1) may also be a contributing factor.
Both of the protein components of pivotal importance to Alzheimer’s disease (AD); Tau and Aβ, have been shown to contain a large number of racemised amino acids in brain tissue from elderly human donors [48, 49]. Plaque in brain that is characteristic of AD, is composed of highly modified forms of Aβ 1–40 and Aβ 1–42. In a recent paper it was demonstrated that each of the plethora of modifications is due to a spontaneous process [50] implying that plaques are old and also that their toxicity could change with age, as more highly modified forms of Aβ accumulate.
Others have postulated that the reduced flexibility of major blood vessels, such as the aorta, that occurs with age due to LLP degradation (see earlier), could lead to pressure spikes. These, in turn, could promote micro aneurisms in the brain that, in turn, could precipitate AD [51]. In this way, LLP breakdown at one anatomical site can affect distant organs.
Perhaps the most convincing data illustrating the importance of LLPs for normal brain function, has come from animal studies where the repair enzyme, protein isoaspartate methyl transferase (PIMT) was deleted. PIMT knock-out mice suffered seizures and a markedly reduced lifespan [52]. PIMT appears to be the most important enzyme for repairing the ravages outlined in old proteins. PIMT is only a partial solution however, since it is active only on L-isoAsp, and, to a lesser degree, on D-Asp[53]. PIMT is inactive on D-isoAsp and other modified amino acids. It also cannot revert any of the Asp isoforms to Asn. It is currently the only known enzyme that can ameliorate Asp racemization. The existence of one main repair enzyme for aged proteins is in marked contrast with the scores of repair enzymes that exist for DNA [54].
LLPs and autoimmunity
It is apparent that significant changes to an LLP will markedly affect its structure. This has implications for autoimmune diseases because the modified LLP may now be seen by the body as being foreign. This is an area that deserves much greater attention. Mamula and colleagues have shown that changing an L-Asp residue to an isoAsp in a peptide can render it a potent antigen [55, 56]. Supporting this idea, isomerization of Asp25 in histone H2B is implicated in lupus erythematosis [56].
Approximately 80% of people with rheumatoid arthritis have autoantibodies to citrullinated peptides derived from the LLP, collagen [58]. Citrulline can be formed by deiminase action on Arg residues [58] but spontaneous processes could also lead to deimination of Arg, although these have yet to be elucidated. Myelin basic protein isolated from human brain is heavily modified with age, implying that it is an LLP [59]; labeling studies in rodents support this [17]. Myelin basic protein isolated from the brains of multiple sclerosis (MS) patients was also highly modified but appears to have consistent differences in its pattern. Could these MS-specific sites be potent antigens and be responsible for generating the immune response that underpins the etiology of MS?
Concluding remarks and future perspectives
A great deal of research needs to be undertaken before we understand clearly the impact of LLP deterioration on human health. To emphasize this it is not yet widely appreciated that LLPs exist in the human body, or that they probably play an important role in aging, age-related diseases, and even lifespan [60]. The field of old proteins is still in its infancy. To illustrate this point, novel spontaneous PTMs are still being characterized [38, 50, 61]. It will be vital in future research to establish the relationship between the degree of PTM to LLPs within a tissue and age-related changes as well as the onset of disease.
There are many issues that need to be addressed (see Outstanding Questions). First, all major age-related PTMs need to be characterized. Each should then be quantified in the LLPs from different human tissues as a function of age. The physical properties of various organs, for example the heart and lungs, also need to be measured and the data correlated with the various PTMs. Do some PTMs contribute to the loss of function more than others? As one specific example, to what degree can the age-related loss of elasticity in the heart/arteries/lungs be attributed to racemization of elastin by comparison with glycosylation, calcification, crosslinking etc? In addition, to what extent can animal models be employed to study changes to LLPs that take place within a short time-frame, and is there significant species variation?
One large impediment to answering these vital questions is that animal models, which have been traditionally used by researchers to investigate other diseases, will be mostly irrelevant. The reason is simply that the sorts of changes to LLPs that are of importance for human health, take years, and often decades, to become evident. This is a new area of science where scientists may be confined to studying humans, or other long-lived primates, and correlations may often have to suffice. Nevertheless, recognition of the ubiquity of LLPs and their inevitable deterioration and its consequences, should be at the forefront of experiments that seek to understand the reasons for human aging and the onset of age-related diseases.
Figure 1.
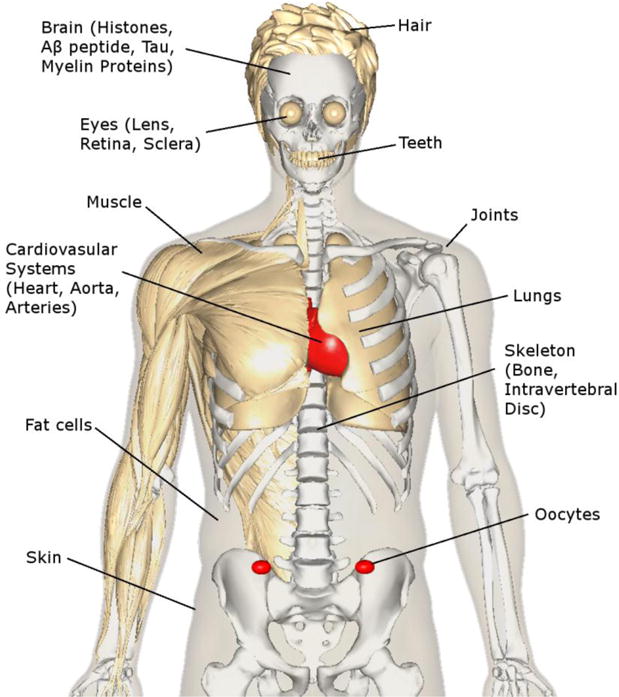
Long-lived proteins are present throughout the body. Their inevitable degeneration over time is associated with the age-related decline of human organs and tissues and may also be implicated in the diseases that accompany old age. This figure was created using BodyParts3D/Anatomography.
References
- 1.Verzijl N, et al. Effect of Collagen Turnover on the Accumulation of Advanced Glycation End Products. J Biol Chem. 2000;275:39027–39031. doi: 10.1074/jbc.M006700200. [DOI] [PubMed] [Google Scholar]
- 2.Sivan SS, et al. Collagen Turnover in Normal and Degenerate Human Intervertebral Discs as Determined by the Racemization of Aspartic Acid. J Biol Chem. 2008;283:8796–8801. doi: 10.1074/jbc.M709885200. [DOI] [PubMed] [Google Scholar]
- 3.Powell JT, et al. On the accumulation of d-aspartate in elastin and other proteins of the ageing aorta. Atherosclerosis. 1992;97:201–208. doi: 10.1016/0021-9150(92)90132-z. [DOI] [PubMed] [Google Scholar]
- 4.Dobberstein RC, et al. Aspartic acid racemisation in purified elastin from arteries as basis for age estimation. Int J Legal Med. 2010;124:269–275. doi: 10.1007/s00414-009-0392-1. [DOI] [PubMed] [Google Scholar]
- 5.Shapiro SD, et al. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J Clin Invest. 1991;87:1828–1834. doi: 10.1172/JCI115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haus JM, et al. Collagen, cross-linking, and advanced glycation end products in aging human skeletal muscle. J Appl Physiol. 2007;103:2068–2076. doi: 10.1152/japplphysiol.00670.2007. [DOI] [PubMed] [Google Scholar]
- 7.Stabler TV, et al. Amino acid racemization reveals differential protein turnover in osteoarthritic articular and meniscal cartilages. Arthritis Res Ther. 2009;11:R34–R34. doi: 10.1186/ar2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ritz-Timme S, et al. Aspartic acid racemization: evidence for marked longevity of elastin in human skin. Br J Dermatol. 2003;149:951–959. doi: 10.1111/j.1365-2133.2003.05618.x. [DOI] [PubMed] [Google Scholar]
- 9.Zhang DS, et al. Multi-isotope imaging mass spectrometry reveals slow protein turnover in hair-cell stereocilia. Nature. 2012;481:520–524. doi: 10.1038/nature10745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Helfman PM, Bada JL. Aspartic acid racemisation in dentine as a measure of ageing. Nature. 1976;262:279–281. doi: 10.1038/262279b0. [DOI] [PubMed] [Google Scholar]
- 11.Spalding KL, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453:783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- 12.Lynnerup N, et al. Radiocarbon Dating of the Human Eye Lens Crystallines Reveal Proteins without Carbon Turnover throughout Life. PLoS ONE. 2008;3:e1529. doi: 10.1371/journal.pone.0001529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klumb K, et al. Age estimation based on aspartic acid racemization in human sclera. Int J Legal Med. 2015;130:207–211. doi: 10.1007/s00414-015-1255-6. [DOI] [PubMed] [Google Scholar]
- 14.Kaji Y, et al. Localization of D-β-Aspartic Acid–Containing Proteins in Human Eyes. Invest Ophthalmol Vis Sci. 2007;48:3923–3927. doi: 10.1167/iovs.06-1284. [DOI] [PubMed] [Google Scholar]
- 15.Matzenauer C, et al. Estimation of age at death based on aspartic acid racemization in elastic cartilage of the epiglottis. Int J Legal Med. 2013;128:995–1000. doi: 10.1007/s00414-013-0940-6. [DOI] [PubMed] [Google Scholar]
- 16.Tachibana-Konwalski K, et al. Rec8-containing cohesin maintains bivalents without turnover during the growing phase of mouse oocytes. Genes Dev. 2010;24:2505–2516. doi: 10.1101/gad.605910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toyama Brandon H, et al. Identification of Long-Lived Proteins Reveals Exceptional Stability of Essential Cellular Structures. Cell. 2013;154:971–982. doi: 10.1016/j.cell.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eden E, et al. Proteome Half-Life Dynamics in Living Human Cells. Science. 2011;331:764–768. doi: 10.1126/science.1199784. [DOI] [PubMed] [Google Scholar]
- 19.Spalding KL, et al. Retrospective Birth Dating of Cells in Humans. Cell. 2005;122:133–143. doi: 10.1016/j.cell.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 20.Crisp MJ, et al. In vivo kinetic approach reveals slow SOD1 turnover in the CNS. The Journal of Clinical Investigation. 2015;125:2772–2780. doi: 10.1172/JCI80705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inoue K, et al. Simultaneous Determination of Post-Translational Racemization and Isomerization of N-Terminal Amyloid-β in Alzheimer’s Brain Tissues by Covalent Chiral Derivatized Ultraperformance Liquid Chromatography Tandem Mass Spectrometry. Anal Chem. 2014;86:797–804. doi: 10.1021/ac403315h. [DOI] [PubMed] [Google Scholar]
- 22.Hooi MYS, et al. Age-dependent deamidation of glutamine residues in human γS crystallin: Deamidation and unstructured regions. Protein Sci. 2012;21:1074–1079. doi: 10.1002/pro.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Truscott RJ, Friedrich MG. The etiology of human age-related cataract. Proteins don’t last forever. Biochimica et Biophysica Acta (BBA)-General Subjects. 2016;1860:192–198. doi: 10.1016/j.bbagen.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu S, et al. The Hydroxyl Radical in Lens Nuclear Cataractogenesis. J Biol Chem. 1998;273:28603–28609. doi: 10.1074/jbc.273.44.28603. [DOI] [PubMed] [Google Scholar]
- 25.Zhu X, et al. Age-Dependent Denaturation of Enzymes in the Human Lens: A Paradigm for Organismic Aging? Rejuvenation Res. 2010;13:553–560. doi: 10.1089/rej.2009.1009. [DOI] [PubMed] [Google Scholar]
- 26.Hooi M, Truscott R. Racemisation and human cataract. d-Ser, d-Asp/Asn and d-Thr are higher in the lifelong proteins of cataract lenses than in age-matched normal lenses. Age. 2011;33:131–141. doi: 10.1007/s11357-010-9171-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilmarth PA, et al. Age-Related Changes in Human Crystallins Determined from Comparative Analysis of Post-translational Modifications in Young and Aged Lens: Does Deamidation Contribute to Crystallin Insolubility? J Proteome Res. 2006;5:2554–2566. doi: 10.1021/pr050473a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su SP, et al. Molecular signatures of long‐lived proteins: autolytic cleavage adjacent to serine residues. Aging Cell. 2012;11:1125–1127. doi: 10.1111/j.1474-9726.2012.00860.x. [DOI] [PubMed] [Google Scholar]
- 29.Ahmed MU, et al. Nε-(Carboxyethyl)lysine, a product of the chemical modification of proteins by methylglyoxal, increases with age in human lens proteins. Biochem J. 1997;324:565–570. doi: 10.1042/bj3240565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunn JA, et al. Oxidation of glycated proteins: age-dependent accumulation of N.epsilon.-(carboxymethyl)lysine in lens proteins. Biochemistry. 1989;28:9464–9468. doi: 10.1021/bi00450a033. [DOI] [PubMed] [Google Scholar]
- 31.Sochaski MA, et al. Isotope Dilution Gas Chromatography/Mass Spectrometry Method for the Determination of Methionine Sulfoxide in Protein. Anal Chem. 2001;73:4662–4667. doi: 10.1021/ac010228k. [DOI] [PubMed] [Google Scholar]
- 32.Bova LM, et al. Major Changes in Human Ocular UV Protection with Age. Invest Ophthalmol Vis Sci. 2001;42:200–205. [PubMed] [Google Scholar]
- 33.Hooi MYS, et al. Age-dependent racemization of serine residues in a human chaperone protein. Protein Sci. 2013;22:93–100. doi: 10.1002/pro.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geiger T, Clarke S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J Biol Chem. 1987;262:785–794. [PubMed] [Google Scholar]
- 35.Robinson NE, Robinson AB. Molecular clocks. Proceedings of the National Academy of Sciences. 2001;98:944–949. doi: 10.1073/pnas.98.3.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyons B, et al. Spontaneous cleavage of proteins at serine and threonine is facilitated by zinc. Aging Cell. 2016;15:237–244. doi: 10.1111/acel.12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schey KL, et al. Characterization of Human Lens Major Intrinsic Protein Structure. Invest Ophthalmol Vis Sci. 2000;41:175–182. [PubMed] [Google Scholar]
- 38.Wang Z, et al. Human protein aging: modification and crosslinking through dehydroalanine and dehydrobutyrine intermediates. Aging Cell. 2014;13:226–234. doi: 10.1111/acel.12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bar-Or R, et al. Dehydroalanine derived from cysteine is a common post-translational modification in human serum albumin. Rapid Commun Mass Spectrom. 2008;22:711–716. doi: 10.1002/rcm.3421. [DOI] [PubMed] [Google Scholar]
- 40.Wearne SJ, Creighton TE. Effect of protein conformation on rate of deamidation: ribonuclease A. Proteins. 1989;5:8–12. doi: 10.1002/prot.340050103. [DOI] [PubMed] [Google Scholar]
- 41.Fernandes VRS, et al. Arterial Stiffness Is Associated With Regional Ventricular Systolic and Diastolic Dysfunction: The Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:194–201. doi: 10.1161/ATVBAHA.107.156950. [DOI] [PubMed] [Google Scholar]
- 42.Ritz-Timme S, Collins MJ. Racemization of aspartic acid in human proteins. Ageing Res Rev. 2002;1:43–59. doi: 10.1016/s0047-6374(01)00363-3. [DOI] [PubMed] [Google Scholar]
- 43.Ohtani S, et al. Comparison of age estimated from degree of racemization of aspartic acid, glutamic acid and alanine in the femur. J Forensic Sci. 2004;49:441–445. [PubMed] [Google Scholar]
- 44.Böhme L, et al. Isoaspartate residues dramatically influence substrate recognition and turnover by proteases. Biol Chem. 2008:1043. doi: 10.1515/BC.2008.123. [DOI] [PubMed] [Google Scholar]
- 45.Harauz G, et al. Myelin basic protein—diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron. 2004;35:503–542. doi: 10.1016/j.micron.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Luo Y, et al. Structural Insight into Tau Protein’s Paradox of Intrinsically Disordered Behavior, Self-Acetylation Activity, and Aggregation. The Journal of Physical Chemistry Letters. 2014;5:3026–3031. doi: 10.1021/jz501457f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang K, et al. The C9orf72 repeat expansion disrupts necleocytoplasmic transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saido TC, et al. Dominant and differential deposition of distinct β-amyloid peptide species, AβN3(pE), in senile plaques. Neuron. 1995;14:457–466. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- 49.Shimizu T, et al. Isoaspartate Formation and Neurodegeneration in Alzheimer’s Disease. Arch Biochem Biophys. 2000;381:225–234. doi: 10.1006/abbi.2000.1955. [DOI] [PubMed] [Google Scholar]
- 50.Lyons B, et al. Amyloid Plaque in the Human Brain Can Decompose from Aβ(1–40/1–42) by Spontaneous Nonenzymatic Processes. Anal Chem. 2016;88:2675–2684. doi: 10.1021/acs.analchem.5b03891. [DOI] [PubMed] [Google Scholar]
- 51.Stone J, et al. The Mechanical Cause of Age-Related Dementia (Alzheimer’s Disease): The Brain is Destroyed by the Pulse. Journal of Alzheimer’s Disease. 2015;44:355–373. doi: 10.3233/JAD-141884. [DOI] [PubMed] [Google Scholar]
- 52.Yamamoto A, et al. Deficiency in Protein l-Isoaspartyl Methyltransferase Results in a Fatal Progressive Epilepsy. The Journal of Neuroscience. 1998;18:2063–2074. doi: 10.1523/JNEUROSCI.18-06-02063.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lowenson JD, Clarke S. Recognition of D-aspartyl residues in polypeptides by the erythrocyte L-isoaspartyl/D-aspartyl protein methyltransferase. Implications for the repair hypothesis. J Biol Chem. 1992;267:5985–5995. [PubMed] [Google Scholar]
- 54.Sancar Aziz, et al. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 55.Doyle HA, et al. Altered immunogenicity of isoaspartate containing proteins. Autoimmunity. 2007;40:131–137. doi: 10.1080/08916930601165180. [DOI] [PubMed] [Google Scholar]
- 56.Mamula MJ, et al. Isoaspartyl Post-translational Modification Triggers Autoimmune Responses to Self-proteins. J Biol Chem. 1999;274:22321–22327. doi: 10.1074/jbc.274.32.22321. [DOI] [PubMed] [Google Scholar]
- 57.Doyle HA, et al. Autoimmunity to isomerized histone H2B in systemic lupus erythematosus. Autoimmunity. 2012;46:6–13. doi: 10.3109/08916934.2012.710859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uysal H, et al. Antibodies to citrullinated proteins: molecular interactions and arthritogenicity. Immunol Rev. 2010;233:9–33. doi: 10.1111/j.0105-2896.2009.00853.x. [DOI] [PubMed] [Google Scholar]
- 59.Shapira R, et al. Racemization of individual aspartate residues in human myelin basic protein. J Neurochem. 1988;50:649–654. doi: 10.1111/j.1471-4159.1988.tb02960.x. [DOI] [PubMed] [Google Scholar]
- 60.Truscott RJW. Macromolecular deterioration as the ultimate constraint on human lifespan. Ageing Res Rev. 2011;10:397–403. doi: 10.1016/j.arr.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 61.Lyons B, et al. Spontaneous cyclization of polypeptides with a penultimate Asp, Asn or isoAsp at the N-terminus and implications for cleavage by aminopeptidase. FEBS J. 2014;281:2945–2955. doi: 10.1111/febs.12833. [DOI] [PubMed] [Google Scholar]
- 62.Korlimbinis A, et al. Protein aging: Truncation of aquaporin 0 in human lens regions is a continuous age-dependent process. Exp Eye Res. 2009;88:966–973. doi: 10.1016/j.exer.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Halfter W, et al. Origin and turnover of ECM proteins from the inner limiting membrane and vitreous body. Eye. 2008;22:1207–1213. doi: 10.1038/eye.2008.19. [DOI] [PubMed] [Google Scholar]
- 64.Amed A, et al. Protein Turnover in Retina. J Neurochem. 1980;35:131–142. doi: 10.1111/j.1471-4159.1980.tb12498.x. [DOI] [PubMed] [Google Scholar]
- 65.Razafsky D, et al. Lamin B1 and lamin B2 are long-lived proteins with distinct functions in retinal development. Mol Biol Cell. 2016 doi: 10.1091/mbc.E16-03-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Najbauer J, et al. Molecular Aging of Tubulin: Accumulation of Isoaspartyl Sites in Vitro and in Vivo. Biochemistry. 1996;35:5183–5190. doi: 10.1021/bi953063g. [DOI] [PubMed] [Google Scholar]
- 67.David CL, et al. Isoaspartate in Chrondroitin Sulfate Proteoglycans of Mammalian Brain. J Biol Chem. 1998;273:32063–32070. doi: 10.1074/jbc.273.48.32063. [DOI] [PubMed] [Google Scholar]
- 68.Lee EB, et al. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2012;13:38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gineyts E, et al. Racemization and isomerization of type I collagen C-telopeptides in human bone and soft tissues: assessment of tissue turnover. Biochem J. 2000;345:481–485. [PMC free article] [PubMed] [Google Scholar]
- 70.Lau E, et al. A large dataset of protein dynamics in the mammalian heart proteome. Scientific Data. 2016;3:160015. doi: 10.1038/sdata.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Masuda W, et al. d-Aspartic acid in bovine dentine non-collagenous phosphoprotein. Arch Oral Biol. 2002;47:757–762. doi: 10.1016/s0003-9969(02)00064-x. [DOI] [PubMed] [Google Scholar]
- 72.Artigues A, et al. Evidence for the in vivo deamidation and isomerization of an asparaginyl residue in cytosolic serine hydroxymethyltransferase. J Biol Chem. 1990;265:4853–4858. [PubMed] [Google Scholar]
- 73.Kinzel V, et al. The amino terminus of PKA catalytic subunit—A site for introduction of posttranslation heterogeneities by deamidation: D-Asp2 and D-isoAsp2 containing isozymes. Protein Sci. 2000;9:2269–2277. doi: 10.1110/ps.9.11.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dreyfus JC, et al. Metabolism of myosin and life time of myofibrils. Biochem J. 1960;75:574–578. doi: 10.1042/bj0750574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brunauer LS, Clarke S. Age-dependent accumulation of protein residues which can be hydrolyzed to D-aspartic acid in human erythrocytes. J Biol Chem. 1986;261:12538–12543. [PubMed] [Google Scholar]
- 76.D’Angelo S, et al. Accumulation of altered aspartyl residues in erythrocyte membrane proteins from patients with sporadic amyotrophic lateral sclerosis. Neurochem Int. 2013;63:626–634. doi: 10.1016/j.neuint.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 77.Ritz S, et al. Identification of osteocalcin as a permanent aging constituent of the bone matrix: basis for an accurate age at death determination. Forensic Sci Int. 1996;77:13–26. doi: 10.1016/0379-0738(95)01834-4. [DOI] [PubMed] [Google Scholar]
- 78.Maroudas A, et al. Racemization of aspartic acid in human articular cartilage. Connect Tissue Res. 1992;28:161–169. doi: 10.3109/03008209209015033. [DOI] [PubMed] [Google Scholar]
- 79.Artigues A, et al. Cytosolic serine hydroxymethyltransferase. Deamidation of asparaginyl residues and degradation in Xenopus laevis oocytes. J Biol Chem. 1993;268:13784–13790. [PubMed] [Google Scholar]
- 80.Paranandi MV, Aswad DW. Spontaneous alternations in the covalent structure of synapsin I during in vitro aging. Biochem Biophys Res Commun. 1995;212:442–448. doi: 10.1006/bbrc.1995.1989. [DOI] [PubMed] [Google Scholar]