

Editor’s Note:
While drug development has done little to slow the devastating symptoms of amyotrophic lateral sclerosis (ALS), there is some good news in the fact that scientists have identified some 100 related genes and believe that genetic research offers the best hope for treatments. More good news came on the heels of the Ice Bucket Challenge, which raised $220 million globally and has fueled renewed optimism and energy in the ALS community.
“I compare my nervous system to a plaster building. Every day, large crumbly pieces break away. For now, the building remains standing, but the moment of collapse seems inescapable.”
— Frédéric Badré, a French artist and writer, who died in 2016 at 50 years old from ALS
Since Jean-Martin Charcot first described amyotrophic lateral sclerosis (ALS) in 1869, the mechanism underlying the degeneration and death of motor neurons that characterize the disease has remained a mystery. Charcot, considered the Father of Neurology and a professor of pathological anatomy at the University of Paris, was also the first to distinguish Multiple Sclerosis, Parkinson’s, and ALS as separate diseases that involve impaired or lost motor neurons affecting muscle movement. His prognosis for those with ALS was also astute: “Six months to a year after onset, the full complement of symptoms is present and more or less patently manifested. Death occurs on average about two to three years after the appearance of the bulbar symptoms.”1
Beyond Charcot’s core definition, ALS is now known to involve the spinal cord and dysfunction of motor pathways in the cortex. Per the ALS Association, “Once ALS starts, it almost always progresses, eventually taking away the ability to walk, dress, write, speak, swallow, and breathe and shortening the life span. How fast and in what order this occurs is very different from person to person. While the average survival time is three years, about 20 percent of people with ALS live five years, 10 percent will survive ten years, and five percent will live 20 years or more.” ALS is diagnosed and confirmed in about 1 in 500 to 1 in 1,000 adult deaths; 500,000 people in the US will develop this disease in their lifetimes.2
The diagnoses of ALS in the New York Yankees first baseman Lou Gehrig—nicknamed “The Iron Horse” because of his remarkable streak of playing in 2,130 consecutive games—in 1939 brought national and international attention to the disease. After ALS took his life at the age of 37, the illness became known as “Lou Gehrig’s disease.” Other well-known people who are struggling with or have died from ALS include Mao Tse-tung, Stephen Hawking, Charles Mingus, and David Niven. Tuesdays with Morrie, the bestselling memoir about author Mitch Albom’s conversations with his former sociology professor Morrie Schwartz, depicted the disease’s devastating impact.
In 1962, the British neurologist Walter Russell Brain, introduced the concept of “motor neuron disease,” a group of diseases of which ALS is the most common. In the sixth edition of his book Brain, published in 1962, he made it clear that the three syndromes: upper motor neuron degeneration, lower motor neuron degeneration, and mixed upper motor and lower motor neuron degeneration should be regarded as manifestations of the same underlying disorder.3
ALS has been artificially distinct from other motor neuron diseases in the sense that it occurs both sporadically (sALS) and within families (familial ALS, fALS). Although some feel this distinction is practical, others feel it is artificial, since ALS is now believed to be an umbrella term for many different forms of the motor neuron disorder (just as there are various types of flu).
Today, ALS diagnosis remains difficult because these diverse forms involve a spectrum of manifestations whose heterogeneity extends to the site of onset, the degree of upper and lower motor neuron involvement, the rate of motor progression, and the presence and severity of cognitive and behavioral symptoms.
The enormous progress made in both technology and genetics over the past 20 years has led to the discovery of the first genetic mutation that causes ALS and the identification of more than 100 ALS-related genes. These advances have helped us understand the polymorphous nature of the disease, and the ambiguity between sporadic and familial forms of ALS.
Still, no known cure exists for ALS. In the last decade alone, 18 clinical and preclinical trials have failed, most recently with a drug called dexpramipexole, which cost the US-based company Biogen $100 million to develop and test. Today, only two drugs are approved by the US Food and Drug Administration (FDA) for treating ALS symptoms. The first, approved in 1995, is Rilutek (riluzole), a costly neuroprotective medication first discovered in the 1940s. It minimally improves bulbar muscle (involved in speech and swallowing) and limb function, but not general muscle strength. Rilutek is relatively safe and probably prolongs median survival by about two to three months.4 Chinese clinicians recently confirmed its modest beneficial effect.5
The second drug, known as either edaravone (Radicava) or MCI-186, was first approved in Japan in 2015§ and then approved by the FDA on May 5, 2017. An anti-oxidant that works in the central nervous system as a potent scavenger of oxygen radicals,7 patients in treatment experienced a 33 percent reduction in the rate of decline in physical function compared with patients on placebo. In Belgium, the country where I work, a patient association called Belgian ALS League found that the intravenous injection necessary to administer the drug presents a “high burden” and renders the drug “less attractive.”
A Cultural Phenomenon
The Ice Bucket Challenge was a campaign to raise money for charity that took the internet by storm in July 2014, and many supporters trace the tipping point and focus on ALS to a video posted by Pete Frates, a former college baseball player who was diagnosed with the disease in 2012. The campaign left scores of notable persons—from Bill Gates and Mark Zuckerberg to George W. Bush and Anna Wintour—shivering and drenched. More importantly, it paid off in the most spectacular way, raising $220 million worldwide for research, education, and patient support for ALS, and more than $115 million of the total for the ALS Association.
Proceeds from the campaign helped the association fund research that identified a new gene, NEK1, that contributes to the disease, the subject of a paper published in Nature Genetics. A year after the challenge went viral, scientists said that the money had a big effect on the gene’s discovery. Says lead researcher Philip Wong, a professor of pathology at Johns Hopkins: “Without it, we wouldn’t have been able to come out with the studies as quickly as we did.”
The Cause of ALS
The molecular era of discovery in ALS began in 1993 with the identification of dominant mutations in a gene that produces a 153-amino acid enzyme, superoxide dismutase (SOD1), that ordinarily protects cells against damaging byproducts of metabolic processes. Specifically, this enzyme8 catalyzes the conversion of the highly reactive and damaging chemical superoxide to hydrogen peroxide or oxygen. More than 170 ALS-causing mutations have now been identified and lie in almost every region of the 153-amino-acid SOD1 polypeptide. Mutations in the SOD1 gene are found both in sporadic and familial ALS cases.9
The consensus is that the disease arises not from the mutant enzymes’ loss of its protective function, but rather their acquisition of toxic properties. But since the discovery of mutations in SOD1, no consensus on the nature of such toxicity has emerged. A prominent finding is that a proportion of each ALS-causing SOD1 mutant fails to fold properly, suggesting that accumulation of misfolded SOD1 may contribute to toxicity in ALS. Misfolded SOD1 forms lead to the inclusion in cells’ cytoplasm of various possibly damaging substances that occur early in ALS and escalate as the disease progresses.10
Human Genome Sequencing
Advances in the genetics of ALS began with the sequencing of the human genome. The 2001 draft sequence by the Human Genome Project (HGP) took some 200 scientists more than a decade and cost almost $3 billion to complete.11 Three years later, the HGP completed mapping the genome, including 99 percent of the active/translatable DNA part sequence12 that comprises its most common form within the cell nucleus. Since then, assembly and refinement of the reference genome has allowed scientists to identify all genes—ca. 25,000—that carry the code for producing all human proteins. The research has also provided a snapshot of genetic variation, most commonly in the form of single nucleotide polymorphisms (SNPs).
The HGP demonstrates the tremendous potential value of coordinated studies to create community resources to propel biomedical research. During the past decade, the price of genome sequencing has dropped as the process has become automated and methods have improved. Now, complete genomes can be sequenced quickly and affordably, enabling the identification of genetic variants that affect heritable phenotypes, including important disease-causing mutations.
“Gene studies [relating to ALS] did not reveal much of use until about six years ago, but they are now rapidly advancing our knowledge,” says Ammar Al-Chalabi, professor of neurology at King’s College in London. “When I started in ALS research in 1994, the only known [genetic] cause accounted for just two percent of cases. Now, we can explain about 20 percent of cases. Through studies of individual lifestyles and the way our DNA changes during our lives, it should become easier to design effective treatments.”13
Excellent recent studies have described the application of genetic methods in discovering new ALS-related genes.14–16 The list (last updated in 2015), with chromosomal location of the ALS related genes, can be seen at http://alsod.iop.kcl.ac.uk/Chromosomes/chromoALL.aspx.
ALS-related Genes
As human beings we are unique; shaped by our environment and life experiences but also by our genetic make-up. Human genetic diversity is associated with phenotypic variation, notably in our physical characteristics but also in determining our susceptibility and response to disease, often as part of a multi-factorial disease process. The most common form of genetic diversity are single nucleotide differences, a spelling change in the DNA code where one nucleotide is replaced by another but other larger structural variants are also being identified.
The challenge amidst so much genetic variation—there are over 10 million single nucleotide differences alone among humans—is to define specific variants responsible for common multi-factorial diseases such as infections, diabetes, or heart disease. There have been major advances in how we define the genetic determinants of common disease over the last few years based on using many hundreds of thousands of genetic markers across the genome to look for association with disease in “genome-wide association studies.” Nevertheless, a major challenge remains once a region of the genome has been implicated in disease to then define the specific functionally important causative variants.
High-throughput DNA sequencing technologies have generated large amounts of sequence data very rapidly and at a substantially lower cost. While these technologies provide unprecedented opportunities to discover new genes and variants underlying human disease, it should be stressed that these discoveries must be rigorously performed and replicated to prevent the proliferation of false-positive findings. A set of guidelines addresses the types of issues to consider in rare variant analysis.17 These guidelines help provide objective, systematic, and quantitative evaluation of the evidence for pathogenicity. Moreover, sharing of these evaluations and data among research and clinical laboratories will maximize the chances that disease-causing genetic variants are correctly differentiated from the many rare non-pathogenic variants seen in all human genomes.
This is particularly true for ALS, in which rare and potentially pathogenic variants in known ALS genes occur in over 25 percent of apparently sporadic and 64 percent of familial patients. The recent work of Janet Cady and others showed that a significant number of subjects carried variants in more than one gene. This finding suggests an explanation for the varied age of symptom onset, and supports oligogenic inheritance (when a few genes cause or modulate a disease) as relevant to ALS pathogenesis.18
Genome-wide association studies, which aim to identify common variants, have been successful in characterizing novel genetic regions associated with complex traits seen in neurodegenerative diseases such as Alzheimer or Parkinson diseases.19 A key discovery related to ALS was the genetic variation observed at the same position (locus) on chromosome 9 that causes sporadic ALS and familial ALS-frontotemporal dementia.20 This indicates that some of the genes that may cause ALS are pleiotropic, meaning that the same genetic mutations may produce different clinical phenotypes. The determinants of this pleiotropy are largely unknown.21
The two sequencing methods used in ALS research are:
-
Exome sequencing, also known as whole exome sequencing (WES or WXS), a technique for sequencing the expressed genes in a genome. This approach identifies genetic variants that alter protein sequences. It costs much less than whole-genome sequencing and is thus a good choice when whole-genome sequencing is not practical or necessary. It may be extended to target functional nonprotein coding elements (e.g., microRNA, long intergenic noncoding RNA, etc.)22 Since these variants can be responsible for both Mendelian and common polygenic diseases, such as Alzheimer’s disease, whole exome sequencing has been applied both in academic research and as a clinical diagnostic. It has rapidly become the primary method for the discovery of causative genes in rare diseases.
With exome sequencing, Elizabeth T. Cirulli and others identified more than 70 distinct pathogenic mutations across several genes; their discovery may guide future efforts to functionally characterize the role of these ALS genes.23
Whole-genome sequencing (WGS) delivers a comprehensive view of the entire genome. It is ideal for discovery applications, such as identifying causative variants. Whole-genome sequencing can detect single nucleotide variants, insertions/deletions, copy number changes, and identify large structural variants. WGS has identified common genetic causes of ALS and frontotemporal dementia (FTD) and transformed our view of these disorders, which share unexpectedly similar signatures, including dysregulation of common molecular players. One player so identified is mutate TBK1, which results in impaired autophagy and contributes to the accumulation of protein aggregates and ALS pathology.24
Oligonucleotides and ALS Therapy
Antisense oligonucleotides (ASOs) are synthetic single stranded strings of nucleic acids, between 8 and 50 nucleotides in length, that target specific species of mRNA. ASOs inhibit gene expression or modify mutant proteins to reduce their toxicity.26 Antisense-mediated gene inhibition was first introduced by Mary Stephenson and Paul Zamecnik in a 1978 article that has been cited more than 700 times.27
For dominantly inherited disorders where data suggest a mutation that confers a toxic activity on a protein (a toxic gain of function), lowering levels of the protein is a potential approach, and antisense oligonucletoides (ASOs) is one means of doing so.
In 2006, Richard Smith and others28 pioneered the idea of ASO infusion into the nervous system as therapy for human neurodegenerative diseases. Seven years later, Timothy Miller and others,29 using the experiments made on transgenic SOD1 mutant mice, succeeded in launching the first clinical study of delivery into spinal fluid of an ASO. The ASO was well tolerated, demonstrating the feasibility of this approach.
The most common inherited cause of ALS and FTD is the expansion of six toxic nucleotides within a gene called C9orf72. Per Genetics Home Reference, the C9orf72 gene contains a segment of DNA made up of a series of six DNA building blocks (nucleotides), four guanines followed by two cytosines (written as GGGGCC). This segment (known as a hexanucleotide repeat) can occur once or be repeated multiple times; estimates suggest repeats of up to 30 times have no negative effect on gene function. Mutations in the C9orf72 gene affect the GGGGCC segment of the gene. When this series of nucleotides is repeated too many times, it can cause ALS and/or FTD. This type of mutation is called a hexanucleotide repeat expansion. The application of ASO-mediated therapy reduced the toxicity of this hexanucleotide, further validating its feasibility.30
The Next Steps
From the remarkable progress already made, there is little doubt that our understanding of the genetic basis of ALS will continue to improve through conventional genetics and enhanced association studies that identify the role of rare genetic variants, including those found in non-coding DNA. However, the success of future initiatives involving the assembly of sequence and clinical data from very large-scale cohorts will require the full translation of human genetic findings into understandable biological and clinical terms. Such efforts will undoubtedly need the cooperation of multiple stakeholders, including clinicians, biochemists, informaticians, statisticians, patient participants or partners,31 and official agencies such as the FDA.
The FDA has, in fact, taken the initiative to prepare a guidance document, “Use of Public Human Genetic Variant Databases to Support Clinical Validity for Next Generation Sequencing (NGS)-Based In Vitro Diagnostics,” that describes the agency’s current thinking on this subject.32 It outlines the FDA’s criteria in determining whether a genetic variant database is a valid source of scientific evidence that supports the clinical validity of an NGS-based test. Upon finalization approval of this document, test developers will be able to follow these recommendations when preparing a premarket submission—just one more step forward in the fight against a disease that must be stopped.?
Figures 1.
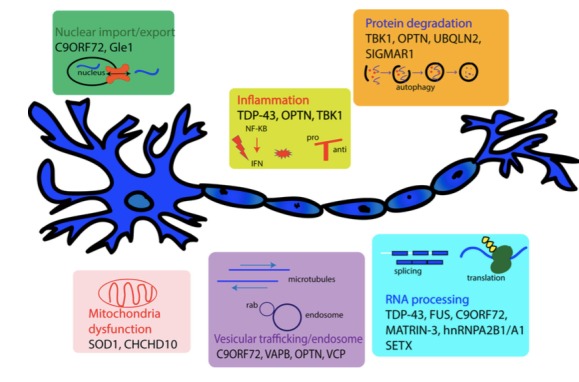
The following diagram illustrates some of the ALS-related genes, clustered per their role in the disease.26 (Reproduced with the permission of Guy Rouleau, M.D., Ph.D.)25
Acknowledgment
I would like to dedicate this article to Mrs. Marie-Henriette Burelle, surnamed Myette, who supported my research and died March 20, 2017. I thank Jacques Haiech for fruitful advice, the European Brain Council and the Belgian Brain Council executive committee for their support, and Alison Turner for having revised the English.
Biography
Roland Pochet, Ph.D., Honorary Professor of Cell Biology at the Faculty of Medicine of Université Libre de Bruxelles, is a neuroscientist who studies stem cell transplantation for amyotrophic lateral sclerosis. Pochet studied for two years in Israel under a European Molecular Biology Organization (EMBO) fellowship and in labs at the University of Texas and Vanderbilt University. From 2006 to 2013, he was chair of the Biomedicine Domain of the European Cooperation in Science and Technology (COST), an international organization consisting of 36 countries. He founded the European Calcium Society, is general secretary of the Belgian Brain Council, and evaluates projects for the European Commission. Pochet is a member of the Dana Alliance for Brain Initiatives, the European Dana Alliance for the Brain, and the International Scientific Committee of AriSLA.
References
- 1.Goetz CG. Amyotrophic Lateral Sclerosis: Early Contributions of Jean-Martin Charcot. Muscle Nerve. 2000;23:336–343. doi: 10.1002/(sici)1097-4598(200003)23:3<336::aid-mus4>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 2.Peters OM, Ghasemi M, Brown RH., Jr Emerging mechanisms of molecular pathology in ALS. J Clin Invest. 2015 May;125(5):1767–79. doi: 10.1172/JCI71601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brain WR. Motor Neuron Disease. Oxford University Press; Oxford: 1962. Diseases of the nervous system; pp. 531–43. [Google Scholar]
- 4.Miller RG1, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND) Cochrane Database Syst Rev. 2012 Mar;14(3):CD001447. doi: 10.1002/14651858.CD001447.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L, Liu X, Tang L, Zhang N, Fan D. Long-Term Use of Riluzole Could Improve the Prognosis of Sporadic Amyotrophic Lateral Sclerosis Patients: A Real-World Cohort Study in China. Front Aging Neurosci. 2016 Oct 24;8:246. doi: 10.3389/fnagi.2016.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abe K, Itoyama Y, Sobue G, Tsuji S, Aoki M, Doyu M, Hamada C, Kondo K, Yoneoka T, Akimoto M, Yoshino H Edaravone ALS Study Group. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014 Dec;15(7–8):610–7. doi: 10.3109/21678421.2014.959024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watanabe T, Yuki S, Egawa M, Nishi H. Protective effects of MCI-186 on cerebral ischemia: possible involvement of free radical scavenging and antioxidant actions. J Pharmacol Exp Ther. 1994 Mar;268(3):1597–604. [PubMed] [Google Scholar]
- 8.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 9.Andersen PM, Sims KB, Xin WW, Kiely R, O’Neill G, Ravits J, Pioro E, Harati Y, Brower RD, Levine JS, Heinicke HU, Seltzer W, Boss M, Brown RH., Jr Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003 Jun;4(2):62–73. doi: 10.1080/14660820310011700. [DOI] [PubMed] [Google Scholar]
- 10.Bruijn LI, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 11.International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 12.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 13.https://www.theguardian.com/society/2015/may/30/als-after-the-ice-bucket-challenge
- 14.Paul Taylor J, Brown Robert H, Jr, Cleveland Don W. Decoding ALS: from genes to mechanism. Nature. 2016 Nov 10;539(7628):197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014 Jan;17(1):17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol. 2016 Dec 16; doi: 10.1038/nrneurol.2016.182. [DOI] [PubMed] [Google Scholar]
- 17.MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE, Ashley EA, Barrett JC, Biesecker LG, Conrad DF, Cooper GM, Cox NJ, Daly MJ, Gerstein MB, Goldstein DB, Hirschhorn JN, Leal SM, Pennacchio LA, Stamatoyannopoulos JA, Sunyaev SR, Valle D, Voight BF, Winckler W, Gunter C. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014 Apr 24;508(7497):469–476. doi: 10.1038/nature13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cady J, Allred P, Bali T, Pestronk A, Goate A, Miller TM, Mitra RD, Ravits J, Harms MB, Baloh RH. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol. 2015 Jan;77(1):100–13. doi: 10.1002/ana.24306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harari O, Cruchaga C. Paving the road for the study of epigenetics in neurodegenerative diseases. Acta Neuropathol. 2016 Oct;132(4):483–5. doi: 10.1007/s00401-016-1614-5. [DOI] [PubMed] [Google Scholar]
- 20.Shatunov A, Mok K, Newhouse S, Weale ME, Smith B, Vance C, Johnson L, Veldink JH, van Es MA, van den Berg LH, Robberecht W, Van Damme P, Hardiman O, Farmer AE, Lewis CM, Butler AW, Abel O, Andersen PM, Fogh I, Silani V, Chiò A, Traynor BJ, Melki J, Meininger V, Landers JE, McGuffin P, Glass JD, Pall H, Leigh PN, Hardy J, Brown RH, Jr, Powell JF, Orrell RW, Morrison KE, Shaw PJ, Shaw CE, Al-Chalabi A. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 2010 Oct;9(10):986–94. doi: 10.1016/S1474-4422(10)70197-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benatar M, Stanislaw C, Reyes E, Hussain S, Cooley A, Fernandez MC, Dauphin DD, Michon SC, Andersen PM, Wuu J. Presymptomatic ALS genetic counseling and testing: Experience and recommendations. Neurology. 2016 Jun 14;86(24):2295–302. doi: 10.1212/WNL.0000000000002773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warr A, Robert C, Hume D, Archibald A, Deeb N, Watson M. Exome Sequencing: Current and Future Perspectives. G3 (Bethesda) 2015 Jul 2;5(8):1543–50. doi: 10.1534/g3.115.018564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cirulli Elizabeth T, Lasseigne Brittany N. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015 Mar 27;347(6229):1436–1441. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, Brown P, Ravenscroft T, van Blitterswijk M, Nicholson AM, DeTure M, Knopman DS, Josephs KA, Parisi JE, Petersen RC, Boylan KB, Boeve BF, Graff-Radford NR, Veltman JA, Gilissen C, Murray ME, Dickson DW, Rademakers R. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015 Jul;130(1):77–92. doi: 10.1007/s00401-015-1436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Therrien M, Dion PA, Rouleau GA. ALS: Recent Developments from Genetics Studies. Curr Neurol Neurosci Rep. 2016 Jun;16(6):59. doi: 10.1007/s11910-016-0658-1. [DOI] [PubMed] [Google Scholar]
- 26.Evers MM, Toonen LJ, van Roon-Mom WM. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv Drug Deliv Rev. 2015 Jun 29;87:90–103. doi: 10.1016/j.addr.2015.03.008. Review. [DOI] [PubMed] [Google Scholar]
- 27.Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A. 1978 Jan;75(1):285–8. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G, Lobsiger CS, Ward CM, McAlonis-Downes M, Wei H, Wancewicz EV, Bennett CF, Cleveland DW. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116:2290–2296. doi: 10.1172/JCI25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, Andres PL, Mahoney K, Allred P, Alexander K, Ostrow LW, Schoenfeld D, Macklin EA, Norris DA, Manousakis G, Crisp M, Smith R, Bennett CF, Bishop KM, Cudkowicz ME. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013 May;12(5):435–42. doi: 10.1016/S1474-4422(13)70061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, Stauffer JE, Jafar-Nejad P, Drenner K, Schulte D, Chun S, Sun S, Ling SC, Myers B, Engelhardt J, Katz M, Baughn M, Platoshyn O, Marsala M, Watt A, Heyser CJ, Ard MC, De Muynck L, Daughrity LM, Swing DA, Tessarollo L, Jung CJ, Delpoux A, Utzschneider DT, Hedrick SM, de Jong PJ, Edbauer D, Van Damme P, Petrucelli L, Shaw CE, Bennett CF, Da Cruz S, Ravits J, Rigo F, Cleveland DW, Lagier-Tourenne C. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron. 2016 May 4;90(3):535–50. doi: 10.1016/j.neuron.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terry SF. The study is open: Participants are now recruiting investigators. Sci Transl Med. 2017 Jan 4;9(371) doi: 10.1126/scitranslmed.aaf1001. [DOI] [PubMed] [Google Scholar]
- 32.http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM509837.pdf