

Abstract
Diamond-Blackfan anemia (DBA) is a congenital disorder characterized by the failure of erythroid progenitor differentiation, severely curtailing red blood cell production. Because many DBA patients fail to respond to corticosteroid therapy, there is considerable need for therapeutics for this disorder. Identifying therapeutics for DBA requires circumventing the paucity of primary patient blood stem and progenitor cells. To this end, we adopted a reprogramming strategy to generate expandable hematopoietic progenitor cells from induced pluripotent stem cells (iPSCs) from DBA patients. Reprogrammed DBA progenitors recapitulate defects in erythroid differentiation, which were rescued by gene complementation. Unbiased chemical screens identified SMER28, a small-molecule inducer of autophagy, which enhanced erythropoiesis in a range of in vitro and in vivo models of DBA. SMER28 acted through autophagy factor ATG5 to stimulate erythropoiesis and up-regulate expression of globin genes. These findings present an unbiased drug screen for hematological disease using iPSCs and identify autophagy as a therapeutic pathway in DBA.
INTRODUCTION
The blood system is a classical developmental hierarchy in which hematopoietic stem and progenitor cells (HPCs) continuously replenish a pool of short-lived mature cells. The discovery of induced pluripotency has opened new avenues to regenerative medicine, including disease modeling, to gain insights into pathophysiology and drug screening against disease-relevant human cells. A large number of induced pluripotent stem cell (iPSC) models have been established from patients with hematological diseases (1–6). However, inability to derive hematopoietic stem cells (HSCs) and multipotential HPCs has hampered the ability to interrogate disease mechanisms and discover therapeutics using patient-derived iPSCs (1). We previously reported generation of expandable multilineage progenitors from iPSCs using five transcription factors (5F; ERG, HOXA9, RORA, SOX4, and MYB) (7). Doxycycline (Dox)–regulated conditional induction of 5F expanded immature CD34+CD38− blood progenitors (CD34-5F) and removal of Dox initiated differentiation. CD34-5F cells gave rise to short-term engraftment after transplantation in immunodeficient mice, with erythroid progenitors undergoing maturation and hemoglobin switching in vivo. This system has the potential to generate large numbers of engraftable patient-specific cells for modeling hematological diseases.
Diamond-Blackfan anemia (DBA) is a severe macrocytic anemia that usually presents in the first year of life (8). DBA is associated with mutations in ribosomal protein genes, most commonly RPS19 and RPL5 (9). Loss of a single allele of RPS19 perturbs the assembly of 40S ribosomal subunits, and loss of an RPL5 allele perturbs the 60S subunit assembly. These disruptions affect the normal stoichiometry of ribosomal subunits, which leads to ribosomal stress and apoptosis of erythroid progenitors (10, 11). Erythroid differentiation in DBA is arrested at the earliest progenitor stage, the erythroid burst-forming unit (BFU-E) (12, 13). Corticosteroids, such as dexamethasone (DEX), induce proliferation of erythroid progenitors and are a first-line treatment for DBA. Only about half of patients respond to steroids, and some patients lose their response over time and must be managed with lifelong transfusions. Thus, there is a considerable need for new therapeutics for this disorder.
Identifying new therapeutics for DBA is critically dependent on circumventing the paucity of primary patient HPCs. Mouse models of DBA have been reported (14–17) but do not recapitulate all aspects of human disease or enable drug screening. Knockdown of RPS19 by short hairpin RNAs (shRNAs) in human CD34+ progenitors is often used as a model system (10, 18); however, it is difficult to achieve precise haploinsufficient protein dosage or cell expansion owing to reduced proliferative capacity. DBA iPSCs recapitulate aspects of the disease (3), opening the possibility of drug screening against disease-relevant human cells. Here, we use a reprogramming approach to carry out an unbiased drug screen with blood disorder patient iPSCs and identify SMER28, a small-molecule modulator of autophagy, as a candidate therapeutic for DBA.
RESULTS
Generation of reprogrammed progenitors from DBA iPSCs
To establish a model of DBA, we reprogrammed fibroblasts from patients with RPS19 and RPL5 inactivating mutations. We established independent cell lines of normal karyotype (table S1) and confirmed the heterozygous nonsense mutation in iPSCs by Sanger sequencing (Fig. 1A and fig. S1A). Fibroblasts from patient T15 showed the expected decrease in RPS19 protein, but RPS19 expression was not decreased in patient iPSCs (Fig. 1B and fig. S1B), suggesting that the remaining copy of RPS19 is sufficient to maintain normal protein expression in iPSCs. RPS19 protein was decreased in erythroid cells differentiated from patient iPSCs, showing dosage dependence in disease-affected blood cells (Fig. 1C).
Fig. 1. DBA iPSCs phenocopy erythroid defects in vitro.
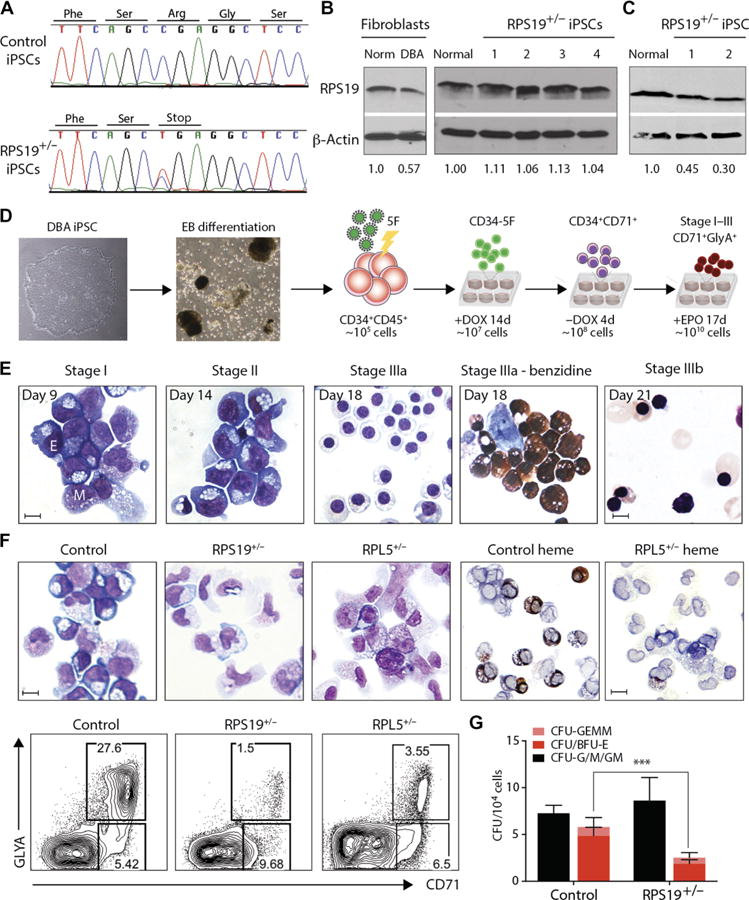
(A) Genomic RPS19 sequence from control and DBA T15 iPSCs to confirm heterozygous C280T nonsense mutation. One representative line is shown. (B) RPS19 expression in normal and DBA patient T15 fibroblasts (left) and iPSCs derived from them (right). RPS19 protein expression normalized to normal control fibroblasts or iPSCs, respectively, is indicated below. (C) RPS19 expression in erythroid cells differentiated from T15 DBA iPSCs. RPS19 protein expression normalized to normal control erythroid cells is indicated below. (D) Experimental outline. Left to right: DBA iPSCs are differentiated into CD34+CD45+ d14 EB-HPCs and transduced with 5F. Progenitors are maintained in an undifferentiated CD34+CD38− state (CD34-5F) with Dox for 14 days. Removal of Dox initiates differentiation into erythroid and myeloid lineages (day 0 to day 4). Erythroid maturation occurs over 17 days in the presence of EPO (stages I to III; days 5 to 21). (E) May-Grunwald-Giemsa staining of normal CD34-5F cells from different stages of erythroid culture. The day of differentiation is marked (Dox withdrawal, day 0). Day 18 includes benzidine staining for hemoglobin (right). E, erythroblast; M, myeloid. Scale bars, 10 μm. (F) Erythroid differentiation of CD34-5F cells derived from normal control, RPS19+/−, and RPL5+/− DBA iPSCs. Top: May-Grunwald-Giemsa and benzidine stain for hemoglobin (day 9). Scale bars, 10 μm. Bottom: Flow cytometry for erythroid markers CD71 and GlyA. Data are representative of four experiments with three normal control iPSC lines, four RPS19+/− DBA iPSC lines, and two RPL5+/− iPSCs. (G) Colony-forming capacity of control (n = 4) and DBA (n = 5) CD34-5F cells showing myeloid (CFU-G/M/GM; black), erythroid (BFU-E and CFU-E; red), and mixed (CFU-GEMM; pink) colonies. Means ± SD of four independent experiments; ***P < 0.001 by unpaired t test.
DBA iPSCs gave rise to comparable numbers of CD34+ hematopoietic progenitors (fig. S2), but these progenitors had reduced potential to differentiate into CD71+ glycophorin A (GlyA)+ erythroid cells (fig. S3A). Expression of p21 was higher in DBA erythroid cells (fig. S3B), as noted in other models of DBA (12). However, we were unable to carry out further characterization because of the limited proliferative capacity of iPSC HPCs. To overcome this limitation, we adopted a transcription factor–based reprogramming system to expand iPSC-derived HPCs (7). HPCs isolated on day 14 of differentiation were transduced with 5F lentiviruses (CD34-5F cells). After 2 weeks of expansion, Dox was removed, and CD34-5F progenitors were differentiated into red blood cells (RBCs) (Fig. 1D) (19). In this protocol, CD34-5F progenitors transitioned through the same morphological stages as cord blood (CB) CD34+ cells (Fig. 1E) (19). CD34-5F RBCs retain some embryonic features in vitro, such as expression of embryonic and fetal globins and low efficiency of enucleation, but undergo globin switching and enucleation after engraftment in vivo (7). Total expansion was >1 × 104–fold, corresponding to >109 RBCs from an average differentiation batch (fig. S3C). This system overcomes inefficient differentiation of iPSCs to generate large numbers of disease-specific HPCs.
DBA iPSCs recapitulate early erythroid defect in vitro
A block in differentiation at the early BFU-E or erythroid colony-forming unit (CFU-E) progenitor stage is a hallmark of DBA, and we observed this phenomenon during differentiation of DBA iPSCs using the CD34-5F system. Normal iPSCs efficiently gave rise to early erythroblasts distinguished by basophilic cytoplasm due to high ribosome content. By contrast, we found virtually no erythroblasts when differentiating the RPS19 and RPL5 mutant patient iPSCs (Fig. 1F). The number and frequency of CD71+GlyA+ cells were also reduced (Fig. 1F, bottom, and fig. S4, A and B). Furthermore, DBA CD34-5F cells generated normal numbers of myeloid but reduced numbers of BFU-E and CFU-E colonies, indicating a specific loss of erythroid progenitors (Fig. 1G). iPSC-derived erythroblasts displayed molecular changes characteristic of DBA patient cells, including increased frequency of apoptotic cells and expression of p21 (fig. S4, C and D). Thus, the CD34-5F in vitro system recapitulates erythroid defects found in DBA.
DBA iPSCs recapitulate erythroid defect after transplant
To further assess the erythroid potential of DBA iPSCs, we transplanted CD34-5F cells into NOD-SCIDγc null (NSG) mice. Four weeks after transplant, CD34-5F cells from normal iPSCs gave rise to both GlyA+CD45− erythroid and GlyA−CD45+ myeloid cells (Fig. 2A). By contrast, DBA CD34-5F cells gave rise to myeloid cells but showed little erythroid engraftment (Fig. 2A, fig. S4E, and table S2). The human erythroid graft consisted predominantly of mature orthochromatic normoblasts and some enucleated reticulocytes (Fig. 2B), consistent with our previous observations that iPSC-derived cells undergo maturation in vivo (7). Because engraftment capacity differed for each iPSC line, we monitored the proportion of erythroid cells relative to total human engraftment to normalize RBC output. Normal iPSC-derived CD34-5F cells yielded 75 ± 6.0% erythroid contribution, whereas CD34-5F cells from DBA patients contributed 13 ± 6.8% erythroid engraftment (Fig. 2C and table S2). These data demonstrate that DBA CD34-5F cells display reduced erythroid potential in vitro and in vivo.
Fig. 2. DBA iPSCs show defective erythropoiesis in vivo and are rescued by gene complementation.
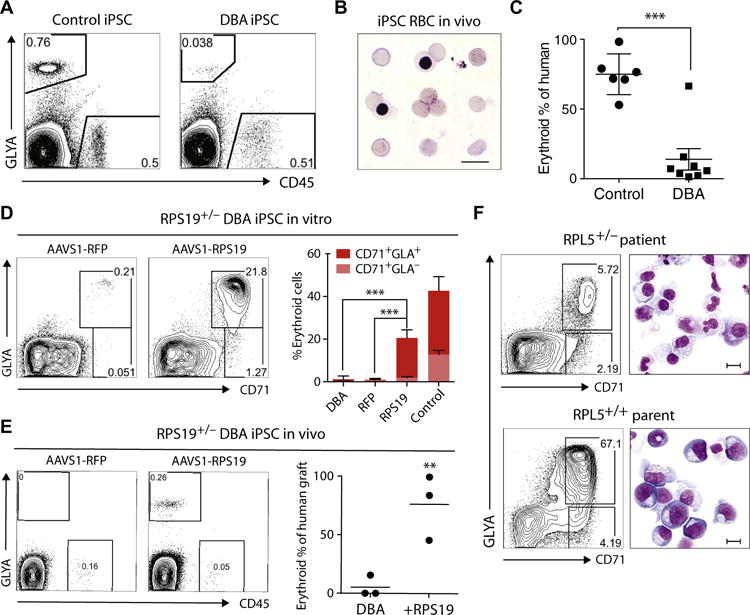
(A) Representative flow cytometry plots of human erythroid (GlyA+) and myeloid (CD45+) engraftment in the bone marrow (BM) of NSG mice transplanted with normal control (n = 6 mice) or DBA (n = 9 mice) CD34-5F cells. Engraftment was analyzed 4 weeks after transplantation using human-specific antibodies. (B) Giemsa stain of sorted GlyA+ RBCs from the BM of mice engrafted with normal control iPSC progenitors. (C) Erythroid cells as proportion of total human engraftment for normal control and DBA iPSCs, plotted as a percentage. Data are shown as means ± SD of two independent experiments with two normal control lines and two RPS19+/− DBA lines. Detailed engraftment data for each mouse are listed in table S2. (D to F) Gene complementation of DBA iPSCs. (D) Erythroid differentiation of RFP- and RPS19-complemented RPS19+/− DBA iPSCs in vitro. Erythroid cells were analyzed on day 9 using flow cytometry for markers CD71 and GlyA. Quantitation on the right is shown as means ± SD for three DBA iPSC, three RFP-corrected (RFP), and four RPS19-corrected (RPS19) iPSC lines independently derived during gene correction. (E) Flow cytometry showing erythroid engraftment of RFP- and RPS19-complemented RPS19+/− DBA iPSCs 4 weeks after transplantation in NSG mice (n = 3 each). Quantitation on the right shows human GlyA+ erythroid cells as a percentage of total human engraftment. (F) Flow cytometry and May-Grunwald-Giemsa staining showing erythroid differentiation of CD34-5F cells derived from the RPL5+/− patient and unaffected parent RPL5+/+ iPSCs. Erythroid cells were analyzed on day 9 using CD71 and GlyA expression. Scale bars, 10 μm. For all panels, **P < 0.01, ***P < 0.001, by unpaired t test.
Gene complementation rescues erythroid defect
To demonstrate that the erythroid defect in RPS19+/− DBA iPSCs is driven by the RPS19 mutation, we inserted a single copy of RPS19 or an irrelevant red fluorescent protein (RFP) gene into the safe harbor adeno-associated virus integration site 1 (AAVS1) locus using CRISPR/Cas9. Multiple independent RPS19-complemented iPSC clones showed significantly improved erythroid potential compared to starting DBA iPSCs and RFP controls in vitro (Fig. 2D; P < 0.001). RPS19 complementation also restored erythroid engraftment of DBA progenitors in vivo (Fig. 2E). To independently validate these findings, we generated iPSC lines from the unaffected parents of an RPL5 patient who did not carry the mutation. Parental lines displayed normal erythroid differentiation (Fig. 2F), indicating that defective erythropoiesis is likely due to the RPL5 mutation rather than to other mutations. These data show that the erythroid defect observed in our DBA model is driven by disease-associated RPS19 and RPL5 mutations.
Chemical screens identify compounds that rescue erythroid differentiation
Establishing a robust model that recapitulated clinical features of the disease allowed us to carry out unbiased drug screens to discover candidate therapeutics for DBA. To this end, we developed approaches to screen chemical libraries for enhanced proliferation or differentiation of erythroid cells differentiated from DBA CD34-5F cells. We screened the Sigma LOPAC library of 1280 pharmacologically active small molecules plus 160 selected bioactive compounds (1440 in total) at a concentration of 5 μM and identified compounds that displayed erythroid rescue of DBA lines (Fig. 3A, fig. S5A, and table S3). Hits previously linked with DBA included DEX (18), transforming growth factor–β inhibitor SB431542 (20), and ROCK1 inhibitor Y27632 (21). SMER28, a quinazolinamine derivative previously characterized as an inducer of autophagy (22, 23), displayed the largest dose-dependent effect on erythroid differentiation and was characterized further. SMER28 increased the absolute number of CD71+GlyA+ erythroid cells from RPS19 and RPL5 mutant DBA iPSCs in a dose-dependent manner with a median effective concentration (EC50) of 1.5 μM (95% confidence interval, 0.63 to 2.37 μM) (Fig. 3B and fig. S5, B and C). We did not observe increased cytotoxicity at drug doses up to 20 μM (fig. S5D). SMER28 had a smaller effect on control iPSCs (DBA, 9.8-fold; control, 2.5-fold; EC50 = 1.6 μM) (Fig. 3B). To confirm SMER28 activity in an independent model of DBA, we transduced CB CD34+ progenitors with shRNAs for RPS19 (shRPS19) or control shRNAs targeting an irrelevant luciferase gene (shLUC). As expected, RPS19 knockdown inhibited erythroid differentiation, and treatment withSMER28 stimulated erythroid output, with the highest activity at ~0.5 μM (Fig. 3, C and D). Last, we tested a 1.0 μM dose of the compound on primary CD34+ cells from bone marrow (BM) samples of four DBA patients and observed improved erythroid differentiation (Fig. 3E). These data suggest that SMER28 promotes erythropoiesis in both normal and DBA cells, with a greater proportional response in diseased cells.
Fig. 3. Chemical screens identify compounds that rescue defective erythropoiesis.
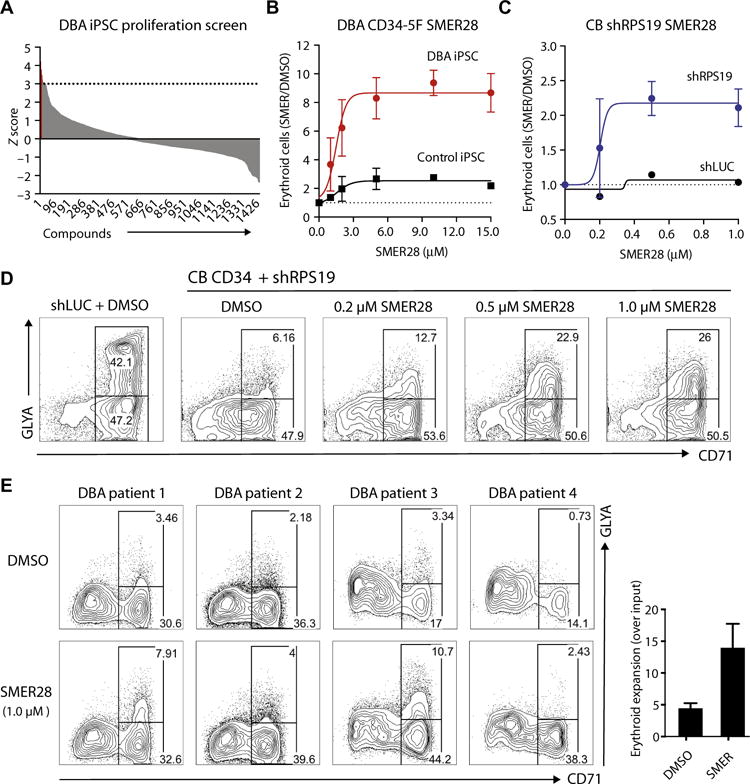
(A) Distribution of 1440 compounds by Z score in a proliferation-based screen. DBA CD34-5F cells were plated under erythroid-promoting conditions in a 384-well format. Erythroid proliferation was measured in the presence of 5 μM of each compound and converted into Z scores. The cutoff for significance was set at Z = 3 (22 compounds, in red; listed in table S3). The screen was carried out with two RPS19+/− DBA and two normal iPSC lines. (B) Dose-dependent effect of SMER28 on erythroid differentiation, shown as the absolute number of erythroid cells in each drug-treated condition normalized to vehicle [dimethyl sulfoxide (DMSO)] control. Erythroid cells were analyzed on day 9 using markers CD71 and GlyA. Dose curves were performed with DBA (n = 5) and normal control (n = 3) iPSCs tested in independent experiments. Nonlinear regression curve was plotted to calculate EC50 values. (C and D) Dose-dependent effect of SMER28 on erythroid differentiation of CB CD34+ shRPS19 cells. (C) Absolute number of erythroid cells (day 9 of differentiation) in each drug-treated condition normalized to vehicle (DMSO) control (four independent experiments). Nonlinear regression curve is plotted. (D) Representative flow cytometry plots of control (shLUC) and shRPS19 cells treated with DMSO or increasing doses of SMER28. (E) Flow cytometry plots illustrating treatment of primary CD34+ cells isolated from the BM of four DBA patients. Progenitors were cultured with vehicle or 1 μM SMER28 and analyzed as in (C). Quantification on the right shows the absolute number of erythroid cells normalized to DMSO. For all panels, data are shown as means ± SEM.
SMER28 enhances erythropoiesis in vivo
To assess the therapeutic potential of SMER28 in vivo, we first tested it in a zebrafish model of DBA. Mutations in ribosomal protein genes, such as rps29, cause DBA-like anemia in zebrafish (24). RPS29 is also mutated in a subset of DBA patients (25). Rps29−/− zebrafish are anemic, as revealed by lack of hemoglobin staining in the yolk sac; treatment with SMER28 for 40 hours postfertilization (hpf) increased hemoglobin staining (Fig. 4A). More than 50% of embryos treated with 1 μM SMER28 showed high expression of hemoglobin, compared to 20% for vehicle controls (P = 0.00002) (Fig. 4B). Thus, SMER28 ameliorates anemia in a zebrafish model of DBA.
Fig. 4. SMER28 promotes erythropoiesis in vivo.
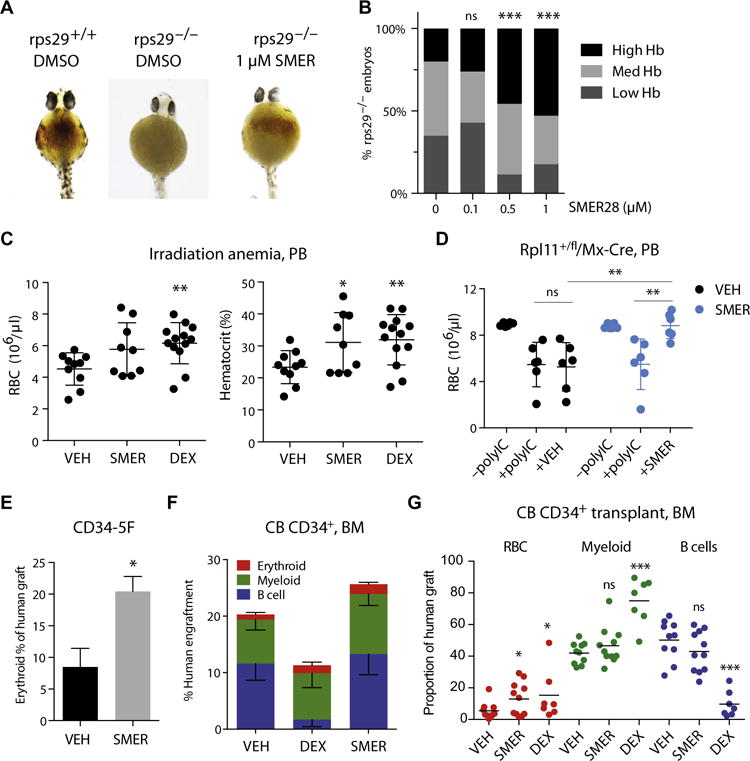
(A) Hemoglobin in the yolk sac was visualized 40 hpf with benzidine staining in zebrafish embryos. Images are representative of wild-type vehicle-treated, rps29−/− poorly hemoglobinized vehicle-treated, and rps29−/− highly hemoglobinized SMER28-treated embryos. (B) Quantitation of SMER28 rescue for a dose range of 0.1 to 1 μM, with embryos grouped by hemoglobin (Hb) staining (high, medium, and low). P = 0.0004 (0.5 μM SMER) and P = 0.00002 (1.0 μM SMER) versus DMSO by binomial test. (C) Peripheral blood (PB) RBC counts and hematocrit in mice with irradiation-induced anemia. Mice were administered DMSO (VEH) (n = 11), SMER28 (10 mg/kg) (n = 9), or DEX (1 mg/kg) (sodium phosphate) (n = 13) in vehicle for 3 weeks. Data combined from three independent experiments. (D) PB RBC counts in Rpl11+/fl/Mx1-Cre DBA mice in which deletion of Rpl11 is under the control of poly(I:C)-inducible Cre. Blood counts were taken before poly(I:C) treatment [−poly(I:C)] and 2 weeks after Cre induction [+poly(I:C)] to verify the onset of anemia. Mice were treated with vehicle containing DMSO (VEH) (n = 6) or SMER28 (20 mg/kg) (n = 6) daily, and RBC counts were taken after 2 weeks. (E) Erythroid cells as proportion of the human graft in mice transplanted with DBA CD34-5F progenitors treated with DMSO (VEH) or SMER28 (10 mg/kg). (F) Human engraftment in NSG mouse BM transplanted with CB CD34+ cells. Mice were treated with DMSO plus vehicle (VEH) (n = 10), DEX (1 mg/kg) (sodium phosphate) (n = 6), or SMER28 (2 mg/kg) (n = 11) for 4 weeks. (G) The proportion of erythroid (RBC), myeloid, and lymphoid (B cells) lineages within the human graft in BM of VEH-, SMER28-, and DEX-treated mice transplanted with CB CD34+ cells. Data in (D) to (G) combined from two independent experiments. Mice were assigned randomly to groups. For all panels, data are shown as means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, unpaired t test or binomial test. ns, not significant.
Next, we evaluated the effects of administration of SMER28 to mice with irradiation-induced anemia, a model that enables dose finding and initial testing of drug safety and bioactivity. We measured expression of the autophagic marker LC3B in treated animals to assess drug bioactivity and saw that LC3B-I and LC3B-II were increased in the liver, indicative of drug activity (fig. S6A). Irradiated mice treated with SMER28 (10 mg/kg) had elevated RBC counts (P = 0.07) and hematocrit (P = 0.04) compared to vehicle controls (Fig. 4C). Although Rps19+/− mice do not develop anemia, conditional heterozygous deletion of Rpl11 leads to a DBA-like disease (15). We tested the effect of SMER28 in an Rpl11+/fl/Mx-Cre mouse model in which the excision of exon 2 of Rpl11 is induced by polyinosinic:polycytidylic acid [poly(I:C)] double-stranded RNA. Treatment with poly(I:C) resulted in anemia with decreased RBC counts [Fig. 4D; compare −poly(I:C) to +poly(I:C)]. Anemic mice treated with vehicle alone for 2 weeks displayed stable disease, whereas animals receiving SMER28 (20 mg/kg) showed significant improvement in RBC counts (Fig. 4D; P = 0.004). These data provide preliminary evidence of the efficacy of SMER28 in a mouse model of DBA.
To test the effects of this compound on human cells, we treated mice transplanted with CD34-5F and CB cells. SMER28 increased the output of GlyA+ RBCs from DBA CD34-5F cells (Fig. 4E). Administration of SMER28 (2 mg/kg) for 4 weeks to mice transplanted with CB CD34+ cells increased the erythroid contribution to human graft (13.0% versus 5.5% RBCs as proportion of human graft; P = 0.05) (Fig. 4, F and G). Although DEX also increased RBC output (15.3%; P = 0.04), it nearly abolished human B cells, consistent with its lymphotoxic effects (Fig. 4G). By contrast, SMER28 specifically increased erythroid cells without affecting the distribution of myeloid and lymphoid lineages (Fig. 4G). These data establish preliminary evidence for in vivo efficacy of SMER28 as a therapeutic for DBA in zebrafish, mouse, and human cell models.
SMER28 acts on CD34+ erythroid precursors
Erythropoiesis is a multistep process initiated by erythroid progenitors that culminates with their differentiation into enucleated RBCs. A key advantage of the CD34-5F system is that cells initiate differentiation in a synchronous manner after Dox withdrawal. To identify the cell types responsive to SMER28, we treated CD34-5F cells during sequential stages of erythropoiesis. CD34+ progenitors treated during the initial expansion phase displayed increased output of GlyA+ cells (Fig. 5, A and B). By contrast, treatment of erythroblasts during stage I of differentiation did not affect the output of GlyA+ cells (Fig. 5B; “stage I”). We validated these findings in a CB CD34+ model. SMER28 treatment of CD34+ progenitors transduced with shRPS19 was sufficient to improve erythroid output (Fig. 5C). These data indicate that SMER28 acts on CD34+ progenitors to promote erythroid differentiation. In contrast to SMER28 treatment of CD34+ cells that yielded comparable amounts of RBCs compared to vehicle control, treatment of erythroblasts during stages I to III of differentiation enhanced the output of iPSC- and CB-derived mature RBCs (Fig. 5D) and increased the efficiency of enucleation (2.4 ± 1.2% DMSO versus 5.6 ± 1.6% SMER28) (Fig. 5, E and F). These data indicate that SMER28 acts on immature erythroid precursors and mature erythroblasts to increase generation of RBCs.
Fig. 5. SMER28 promotes erythroid differentiation of CD34+ progenitors.
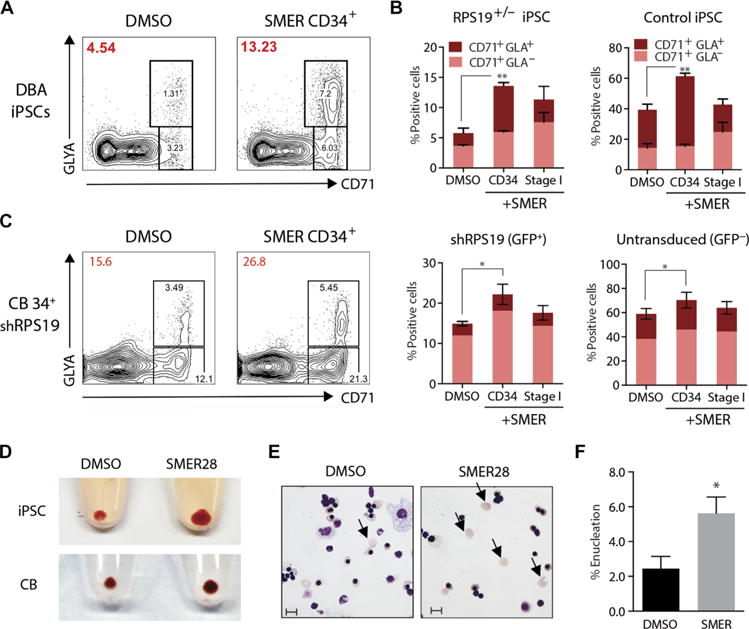
(A) Erythroid differentiation of normal control and DBA CD34-5F cells (flow plots) treated with DMSO vehicle or SMER28 (10 μM) during the initial CD34+ progenitor phase (day 0 to day 4 of differentiation) or stage I of erythroid culture (day 4 to day 9 of differentiation). The percentage of erythroid cells after stage I of differentiation is indicated on the top left (red) for each condition. (B) Quantitation of the percentage of erythroid cells (CD71+/GLA+) from (A). Data from four independent experiments, two normal control, and four DBA iPSCs. (C) Erythroid differentiation of CB CD34+ cells transduced with shRPS19 treated with DMSO or SMER28 (10 μM) as described in (A). shRPS19 [green fluorescent protein–positive (GFP+)] and untransduced (GFP−) cells were quantitated separately; three independent experiments. (D) RBC pellets at the end of stage III initiated with equal numbers of CD34-5F or CB progenitors. SMER28 was added at 10 μM (day 14 to day 21). (E) May-Grunwald-Giemsa stain of erythroid cells from CB CD34+ DMSO- or SMER28-treated cells at the end of stage III. Arrows indicate enucleated RBCs, which were quantitated by flow cytometry. Scale bars, 10 μm. (F) Quantitation of the enucleation efficiency of GlyA+ cells. Data in (E) and (F) represent three independent experiments. For all panels, data are shown as means ± SEM. *P < 0.05, **P < 0.01 by unpaired t test.
SMER28 induces globin expression
To better understand how SMER28 stimulates erythroid differentiation in the context of ribosomal deficiency, we performed expression profiling on CB CD34+ cells 5 days after transduction with shLUC and shRPS19. Genes up-regulated by SMER28 in CB- and iPSC-derived progenitors showed enrichment in the Gene Ontology (GO) categories mitochondrion, hemoglobin, and methyltransferase (Fig. 6A). Ribosomal deficiency causes a delay in the expression of globin proteins and transcripts, which inhibits erythroid differentiation (26), and consistent with this report, we found that globin transcripts were down-regulated in CB shRPS19 cells (19 ± 9% shRPS19 versus shLUC, P = 0.0002) (Fig. 6B). GATA1 targets and genes comprising the erythroid-specific gene-regulatory network (27) remained relatively unperturbed at this stage (Fig. 6B and fig. S6B). SMER28 up-regulated expression of globin genes in shRPS19 compared to shLUC cells (P = 0.0003), but GATA1 targets and erythroid-specific gene expression were relatively unaffected by SMER28 (Fig. 6C and fig. S6C). Early changes in globin expression were indicative of durable changes during erythroid development. CD71+ erythroblasts differentiated from SMER28-treated precursors retained increased expression of globin genes (Fig. 6D), and we found a similar improvement in primary DBA patient cells (fig. S6D). These data suggest that RPS19 deficiency causes a rapid decline in globin transcription, and SMER28 stimulates globin gene expression.
Fig. 6. SMER28 induces globin expression.
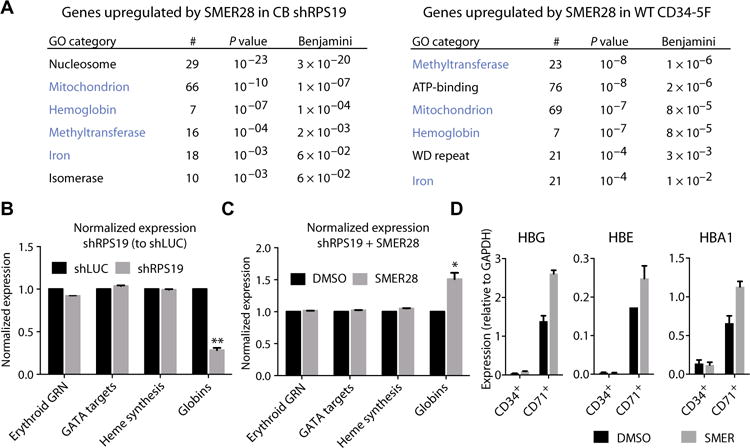
CB CD34+ progenitors transduced with shLUC or shRPS19 were treated with DMSO or SMER28 (10 μM) for 72 hours under expansion conditions. RNA was isolated from sorted GFP+ cells (positive for shRNA) after a total of 5 days in culture. (A) GO annotation of transcripts up-regulated by SMER28 (P < 0.1, >1.5-fold) in CB shRPS19 (left) or CD34-5F progenitors (right). Number of genes annotated (#), P value, and Benjamini-corrected P value for each GO category are shown. Common GO terms in blue. ATP, adenosine 5′-triphosphate. (B) Normalized expression of genes specific to the erythroid lineage after knockdown of RPS19 in CB CD34+ cells. Erythroid gene regulatory network (GRN) is composed of 235 erythroid-specific genes as predicted by CellNet (28). Predicted GATA1 targets (TRANSFAC database), heme synthesis (GO: 0006783), and expressed globin genes are included. Expression in CB shRPS19 is shown relative to shLUC control. (C) Normalized expression of genes comprising the erythroid GRN, predicted GATA1 targets, heme synthesis, and globin genes after SMER28 treatment of CB shRPS19 cells. Expression is shown relative to DMSO-treated CB shRPS19 cells. For (B) and (C), data are shown as means ± SD of two CB shRPS19 and shLUC samples. *P < 0.05, **P < 0.001 by paired t test with Bonferroni multiple testing correction. (D) Expression of transcripts for globin genes HBG, HBE, and HBA1 during erythroid differentiation in CD34+ precursors and CD71+ erythroblasts. CD34-5F cells were treated with SMER28 for the last 48 hours of expansion. Compound was removed, and cells were switched to stage I conditions. CD71+ erythroblasts were purified after 48 hours, and quantitative polymerase chain reaction (qPCR) was performed on CD34+ precursors (end of expansion) and purified CD71+ cells. Shown as means ± SD of two independent experiments.
Autophagy factor ATG5 is required for SMER28 activity
SMER28 was first identified as a mechanistic target of rapamycin (mTOR)–independent inducer of autophagy in models of neurodegenerative disease (22, 23, 28). Autophagy is a prosurvival pathway induced by stresses, such as nutrient limitation or protein aggregation, and is required for clearance of mitochondria in terminal erythroid maturation (29), but it has not been linked with earlier stages of erythropoiesis. To confirm that SMER28 modulates autophagy in erythroid cells, we interrogated the expression of autophagy substrate p62 and autophagosome marker LC3B in K562 erythroleukemia cells. SMER28 increased the expression of lipidated LC3B-II isoform associated with autophagosomes and enhanced clearance of p62 (Fig. 7A). Conversion of cytosolic LC3B-I into LC3B-II was also induced by cytotoxic 5-fluorouracil and occurred in the presence of actinomycin D, indicating that it was independent of transcription (Fig. 7A). p62 expression was increased after knockdown of RPS19 or RPL5, indicating that ribosomal deficiency interferes with autophagic clearance (fig. S7A). We next used an LC3-GFP reporter to monitor autophagosomal LC3 during erythroid differentiation of DBA CD34-5F cells and saw that LC3 expression was increased in CD71+ erythroblasts treated with SMER28 (fig. S7B). These data show that SMER28 induces autophagy during erythroid differentiation.
Fig. 7. SMER28 acts through autophagy factor ATG5.
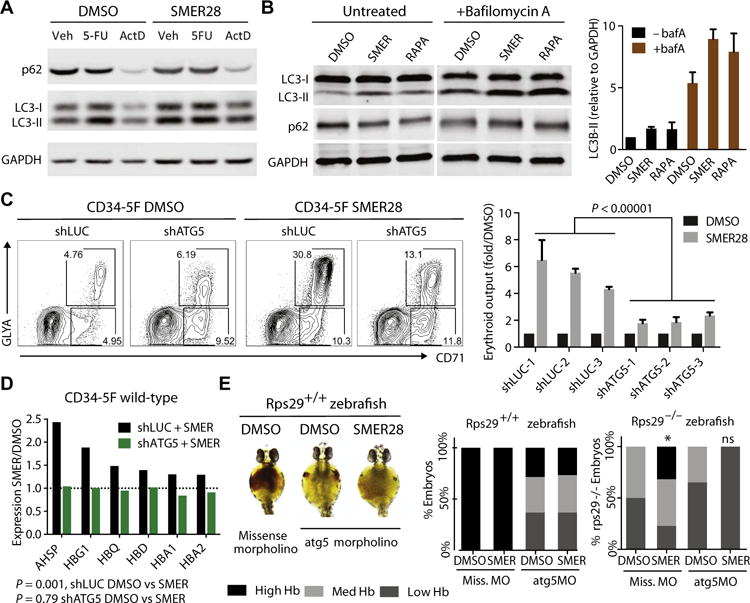
(A) Protein expression of p62, LC3-I, and LC3-II after treatment with SMER28 for 24 hours in K562 cells. Cells were also treated with 5-fluorouracil (5-FU) (100 nM) or actinomycin D (ActD; 100 nM). (B) Induction of autophagy by SMER28 (40 μM) monitored by protein expression of p62, LC3-I, and LC3-II after 24 hours of treatment with or without lysosomal blockade with bafA (200 nM for 4 hours). Untreated samples are shown at higher exposure to capture dynamic changes in the LC3-II expression. Rapamycin (200 nM) is a positive control for induction of autophagy. LC3-II expression relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is quantitated on the right as means ± SD of three experiments. p62 expression is quantitated in fig. S7 (C and D). (C) Erythroid differentiation of CD34-5F cells transduced with shRNAs for luciferase (LUC) or ATG5 treated with SMER28 or DMSO control. Representative flow cytometry plots and quantitation showing the number of erythroid cells induced by SMER28 (10 μM) normalized to DMSO. Three control LUC shRNAs and three ATG5 shRNAs are included (knockdown efficiency, 57, 48, and 40% respectively); means ± SEM of two independent lines, three independent experiments. P = 0.000001, unpaired t test. (D) Induction of globin transcripts by SMER28 in CD34-5F cells transduced with shLUC or shATG5. Cells stably expressing shRNAs were treated with DMSO or SMER28 for 72 hours. Expression in SMER28-treated samples is shown relative to DMSO. P = 0.001 shLUC DMSO versus SMER28; P = 0.76 shATG5 DMSO versus SMER28, paired t test. qPCR validation is in fig. S7G. (E) Representative images of hemoglobin visualized with benzidine staining at 40 hpf in wild-type (rps29+/+) zebrafish embryos injected with 1.6 ng of atg5 or missense morpholino (MO) and treated with SMER28 (1 μM) or DMSO. Embryos are grouped by high, medium, and low Hb staining. Quantitation (right) of the percentage of wild-type (rps29+/+) or rps29−/− embryos showing high, medium, and low Hb staining. n = 20 to 26 embryos per condition.
To measure autophagic flux, we monitored LC3B and p62 expression after blocking lysosomal fusion with bafilomycin A (bafA). As expected, LC3B-II expression increased after lysosomal blockade, indicative of an accumulation of autophagosomes (Fig. 7B; compare DMSO −bafA to +bafA). Similar to the mTOR inhibitor rapamycin, SMER28 enhanced this accumulation, suggesting that it promotes autophagosome formation (Fig. 7B; compare +bafA DMSO to SMER). p62 expression was not affected by the lysosomal blockade (Fig. 7B and fig. S7C); however, clearance of p62 by SMER28 and rapamycin was blocked in the presence of bafA, whereas its transcript expression was unchanged (fig. S7, C and D). These data suggest that the effect of SMER28 in erythroid cells is consistent with enhanced autophagic flux.
Rapamycin induces autophagy but suppresses protein synthesis, which is deleterious to DBA cells. However, rapamycin enhanced the erythroid potential of wild-type progenitors and synergized with SMER28 (fig. S7E). Because chemical inhibition or induction of autophagy typically affects multiple pathways, we transduced CD34-5F progenitors with shRNAs for ATG5, which is required for LC3B lipidation and autophagosome formation, to directly test the role of autophagy factors in SMER28 function. Knockdown of ATG5 using three independent shRNAs reduced the effect of SMER28 on erythroid differentiation (5.4-fold shLUC versus 1.8-fold shATG5, P < 0.00001) (Fig. 7C). Erythroid differentiation was not affected by the ~50% knockdown of ATG5 (fig. S7F). We next examined the effect of ATG5 on globin gene expression. Similar to results seen in CB CD34+ cells, globin transcripts were up-regulated by SMER28 in CD34-5F cells (P = 0.001) (Fig. 7D). Knockdown of ATG5 did not affect the expression of globin transcripts (P = 0.79) but completely blocked their induction by SMER28 (Fig. 7D and fig. S7G). These data suggest that reduction of ATG5 to haploinsufficiency does not perturb erythropoiesis in our iPSC model but reveals its critical role in mediating SMER28 function.
To test the role of ATG5 in erythropoiesis in vivo, we depleted atg5 in zebrafish using a previously characterized morpholino (30), which did not cause morphological or developmental defects (fig. S7H). Injection of atg5, but not missense, morpholino robustly depleted ATG5 protein expression (fig. S7I). Depletion of atg5 in wild-type zebrafish caused anemia, and SMER28 failed to stimulate erythropoiesis after atg5 depletion (Fig. 7E; rps29+/+). By contrast, loss of atg5 did not affect the remaining erythropoiesis in rps29−/− zebrafish, suggesting that ribosomal protein loss is epistatic to autophagy (Fig. 7E and fig. S7J; rps29−/− missense versus atg5 morpholino). Although SMER28 stimulated erythropoiesis in rps29−/− zebrafish injected with missense morpholino, it had no effect in the context of atg5 depletion (Fig. 7E). Together, these findings indicate that autophagy factor ATG5 plays an important role in erythropoiesis by mediating canonical autophagy or alternative pathways, and SMER28 stimulates erythropoiesis by acting through ATG5.
DISCUSSION
Reprogramming cells to an induced pluripotent state is a powerful tool to create models of human disease. Patient-specific iPSCs have been characterized for many hematological disorders, including anemias, neutropenia, myeloproliferative disease, chronic myeloid leukemia, and immunodeficiencies (1–6). iPSCs overcome the limitations of accessing primary patient cells to enable drug screening against pathologic human cellular phenotypes. Blood progenitors can be readily generated by direct differentiation from iPSCs; however, they are largely myeloid lineage–restricted and lack proliferative and engraftment potential. These shortcomings have curtailed the power of iPSC models, and drug screens for blood disorders have not been reported to date. We combined directed differentiation with reprogramming to endow iPSC-derived HPCs with self-renewal and multilineage differentiation potential (7). As a proof of principle, we used this platform to carry out drug screens for DBA and identified SMER28, a small-molecule inducer of autophagy, as a candidate therapeutic.
Our findings suggest that SMER28 stimulates erythropoiesis by promoting autophagy in erythroid progenitors. Autophagy plays important roles in development, homeostasis, and cancer, and is induced to cope with cellular stresses in response to nutrient deprivation and protein aggregation (31, 32). The role of autophagy in hematopoiesis is only beginning to be elucidated. Autophagy serves a cytoprotective function in HSCs (33, 34) and is required for clearance of mitochondria in terminal erythroid maturation (29). Expression of autophagic genes is directly activated by the erythroid master regulator GATA1 (35); however, the role of autophagy in early erythroid development has not been established. SMER28 increases autophagic flux in erythroid cells, and loss of ATG5, which is required for autophagosome assembly, blocks the effects of SMER28 on erythropoiesis. It remains possible that ATG5 mediates these effects independent of autophagic cargo degradation because ATG5 has been shown to regulate innate immunity and pathogen resistance through LC3B-assisted phagocytosis (36–38). Although these findings suggest that autophagy may be dysregulated in DBA, it may also be a generally favorable mechanism in early erythropoiesis. A previous study has found increased autophagic activity in DBA (39), although our findings point to a defect in autophagic turnover. Detailed genetic and biochemical studies are required to elucidate the status of autophagy and its contribution to DBA pathophysiology.
Delayed expression of globin genes in primary DBA cells has been reported to lead to accumulation of free heme and heme toxicity, which perturbs erythroid differentiation (26). We show that knockdown of RPS19 in CD34+ progenitors rapidly inhibited expression of globin transcripts. SMER28 up-regulated globins in RPS19-deficient cells, providing an explanation for its rescue of erythroid differentiation in DBA. This globin induction was dependent on ATG5, suggesting that modulation of autophagy or closely related pathways alters erythroid gene expression. Atg5-deficient zebrafish are anemic, indicating that ATG5 plays a role in erythroid development. Autophagy has been recently shown to affect erythropoiesis through degradation of the iron storage protein ferritin (40, 41). Future studies will need to establish how autophagic signals induce erythroid gene expression and stimulate erythroid differentiation.
The current standard of care for DBA involves treatment with corticosteroids. However, steroids have potent immunosuppressive effects, as seen by the ablation of human lymphocytes in DEX-treated mice in our studies. Because SMER28 will undergo extensive derivatization to improve pharmacokinetics, it is too early to gauge its efficacy relative to steroids. SMER28 did not perturb other hematopoietic lineages and was well tolerated in our dose escalation studies. For patients who do not respond to steroids, SMER28 or its derivatives may be viable alternatives to lifelong transfusions. In summary, we report an unbiased drug screen for hematological disorders using patient iPSCs leading to the discovery of a candidate therapeutic for DBA. The reprogrammed progenitor cell model developed here will enable large-scale genetic and chemical screens in a variety of disease contexts and will serve as a paradigm for continued efforts to reprogram readily accessible patient cells into HSCs.
MATERIALS AND METHODS
Study design
The goal of the study was to identify therapeutics for DBA. The study used iPSCs as a drug discovery model. A candidate therapeutic, SMER28, was tested in independent in vitro and in vivo DBA models including primary patient samples, CB, zebrafish, and mouse. The drug’s mechanism of action during erythroid differentiation was studied in erythroid cell lines, iPSCs, and zebrafish using genetic tools and biochemical assays. Cells from two DBA patients were reprogrammed, and two independent iPSC lines were derived from each patient. Three control iPSC lines were obtained from unaffected individuals. Experiments were performed in triplicate, unless otherwise stated, with at least three biological replicates (independent DBA and control iPSC lines). Drug effects were measured relative to vehicle control. Mice were assigned randomly to groups. Zebrafish hemoglobin staining was scored blindly as low (little/no staining), medium (patched staining), and high (consistent staining across yolk sac). Data points were combined from all independent biological replicates and experiments, and outliers were not excluded.
Patient samples and reprogramming
Skin fibroblasts were obtained by biopsy from DBA patient T15 (UPN NCI 131-1) enrolled in an open cohort study (NCI 02-C-0052, ClinicalTrials.gov ID: NCT00027274) approved by the National Cancer Institute (NCI) Institutional Review Board (IRB). Skin fibroblasts were obtained from patient T5 according to the institutional guidelines approved by the Boston Children’s Hospital (BCH) IRB. Patient fibroblasts were independently reprogrammed with episomal and Sendai methods. Control iPSC lines used in this study, CD45-IPS and CD34-IPS, were reprogrammed from normal donors and MSC-IPS1 (42). The episomal protocol was used as previously described with modifications (43). The Amaxa nucleofector was used according to the manufacturer’s instructions with 1 μg of each plasmid (Addgene). After plating, medium was changed daily for 6 days. At day 6, cells were split, and after 24 hours, medium was changed to KnockOut serum replacement (KOSR)–based human embryonic stem cell (ESC) medium. Integration analysis was performed using the EBNA TaqMan assay. For Sendai reprogramming, fibroblasts were reprogrammed using the CytoTune-iPS Sendai Reprogramming Kit (Thermo). All lines were characterized by karyotyping. The summary of iPSC lines is shown in table S1.
DBA patient BM samples were obtained from the NCI BM failure cohort. Patient 1 (ID: NCI UPN 50-1) had an RPS19 R62Q mutation, patient 2 (ID: NCI UPN 193-1) had an RPS29 I31F mutation, patient 3 (NCI UPN 131-1) had an RPS19 R94X mutation, and patient 4 (NCI UPN 282-1) had an RPS19 c.43G>T (p.Val15Phe) mutation. BM samples were separated with Ficoll (GE), and CD34+ cells were isolated by flow sorting as detailed below.
iPSC culture
Human iPSC lines were maintained on Matrigel (BD) in mTeSR1 (STEMCELL Technologies; subsequently Stemcell) medium. Media were changed daily, and cells were passaged at a ratio of 1:8 to 1:10 every 5 to 7 days using standard passaging techniques with Dispase (Stemcell). Before initiating differentiation, colonies were passaged once onto mouse embryonic fibroblasts (GlobalStem) in human ESC medium [Dulbecco’s modified Eagle’s medium (DMEM)/F12 + 20% KOSR, 1 mM L-glutamine, 1 mM nonessential amino acids (NEAA), 0.1 mM β-mercaptoethanol (β-Me) (all purchased from Life Technologies), and basic fibroblast growth factor (10 ng/ml) (Gemini)].
Embryoid body differentiation
Embryoid body (EB) differentiation was performed as previously described (44). Briefly, iPSC colonies were scraped into nonadherent rotating 10-cm plates. EB medium was KO-DMEM (Thermo) + 20% fetal bovine serum (FBS) (Stemcell), 1 mM L-glutamine, 1 mM NEAA, penicillin/streptomycin, 0.1 mM β-Me, holo-transferrin (200 μg/ml) (Sigma), and ascorbic acid (50 μg/ml) (Sigma). After 24 hours, medium was changed and supplemented with the following growth factors: bone morphogenic protein 4 (BMP4) (50 ng/ml) (R&D), stem cell factor (SCF) (300 ng/ml), Fms-related tyrosine kinase 3 ligand (FLT3) (300 ng/ml), granulocyte colony-stimulating factor (50 ng/ml), interleukin-6 (IL-6) (20 ng/ml), and interleukin-3 (IL-3) (10 ng/ml) (all purchased from PeproTech). Medium was changed on days 5 and 10. EBs were dissociated on day 14 with collagenase B (Roche), followed by treatment with enzyme-free dissociation buffer (Gibco). Dissociated EBs were frozen in 10% DMSO (Sigma) and 40% FBS.
Lentivirus and shRNA plasmids
5F lentiviral plasmids HOXA9, ERG, RORA, SOX4, and MYB were in pInducer-21 lentiviral vector (Addgene). shRNAs for RPS19 (TRCN0000074915 and TRCN0000074914), RPL5 (TRCN0000291973 and TRCN0000291975), and ATG5 (TRCN0000330392, TRCN0000330394, and TRCN0000330395) were from Sigma in pLKO.1 lentiviral vector. Lentiviral particles were produced by transfecting 293T-17 cells (American Type Culture Collection) with the lentiviral plasmids and third-generation packaging plasmids. Virus was harvested 24 hours after transfection and concentrated by ultracentrifugation. All viruses were titered by serial dilution on 293T cells.
EB progenitor sorting
Dissociated EB cells were thawed using Lonza Poietics protocol and resuspended at 1 × 106/100-μl staining buffer (PBS + 2% FBS). Cells were stained with a 1:50 dilution of CD45 PE-Cy5 (Immu19.2; Clontech), CD34 PE-Cy7 (8G12; BD), and 4′,6-diamidino-2-phenylindole (Molecular Probes) for 20 min at room temperature. All sorting was performed on a BD FACS Aria II cell sorter using a 70-μm nozzle.
CD34-5F culture
Sorted CD34+CD45+ EB progenitors were seeded on RetroNectincoated (Takara; 10 μg/cm2) 96-well plates at a density of 2 × 104 to 5 × 104 cells per well in Serum-Free Expansion Medium (SFEM) (Stemcell) with SCF (50 ng/ml), FLT3 (50 ng/ml), thrombopoietin (TPO) (50 ng/ml), IL-6 (50 ng/ml), and IL-3 (10 ng/ml) (R&D). Lentiviral infections were carried out in a total volume of 150 μl. The multiplicity of infection (MOI) for ERG and HOXA9 is 5, whereas the MOI for RORA, SOX4, and MYB is 3. Virus was concentrated onto cells by centrifuging at 2500 rpm for 30 min. Infections were carried out for 24 hours. After gene transfer, CD34-5F cells were cultured in SFEM + SCF (50 ng/ml), FLT3 (50 ng/ml), TPO (50 ng/ml), IL-6 (50 ng/ml), and IL-3 (10 ng/ml) (R&D). Dox was added at 2 μg/ml (Sigma). Cultures were maintained at a density of <1 × 106 cells/ml, and medium was changed every 3 to 4 days. After 14 days, CD34-5F cells were plated according to the erythroid protocol or transplanted in vivo.
Erythroid differentiation
Differentiation was performed as previously described (19), with the following modifications. CD34 expansion phase (4 days): To initiate differentiation, we cultured CD34-5F cells in SFEM progenitor medium (see CD34-5F culture) without Dox. Stage I (5 days): Medium was changed to IMDM + 1% bovine serum albumin (BSA) (Gibco), 20% FBS, 1 mM L-glutamine, penicillin/streptomycin, holo-transferrin (500 μg/ml) (Sigma), human insulin (10 μg/ml) (Cell Sciences), 1 μM β-estradiol, 1 μM DEX, 6 U erythropoietin (EPO) (Cell Sciences), SCF (100 ng/ml), and IL-3 (5 ng/ml) (PeproTech). Cells were seeded at a density of <1 × 105 cells/ml in 24-well plates. Stage II (4 days): Medium was changed to IMDM + 1% BSA, 20% FBS, 1 mM L-glutamine, penicillin/streptomycin, h-transferrin (500 μg/ml), human insulin (10 μg/ml), 6 U EPO, and SCF (50 ng/ml). Cells were seeded at a density of <2 × 105 cells/ml in 24-well plates. Stage III (10 days): Medium was changed to IMDM + 1% BSA, 20% FBS, 1 mM L-glutamine, penicillin/streptomycin, holo-transferrin (500 μg/ml), human insulin (10 μg/ml), and 2 U EPO. Cells were seeded at a density of <5 × 105 cells/ml in 24-well plates.
Statistics
All statistical calculations were performed using GraphPad Prism. Tests between two groups used two-tailed unpaired or paired Student’s t test. Significance between high, medium, and low levels of hemoglobin staining was defined by binomial test. Data are presented as means ± SEM or SD as indicated.
Inducing autophagy to improve anemia
Diamond-Blackfan anemia (DBA) is a rare blood disorder characterized by insufficient red blood cell production that is treated with corticosteroids and transfusion therapy. To identify additional therapeutics for DBA, Doulatov et al. performed a chemical screen with hematopoietic progenitor cells derived from iPSCs from two DBA patients with RPS19 and RPL5 genetic mutations. The autophagy inducing small-molecule SMER28 rescued erythroid differentiation in an autophagy factor ATG5-dependent manner in iPSC-derived patient cells, in zebrafish models of DBA, and in several mouse models. These results demonstrate the utility of iPSC-based screens for drug discovery for rare blood disorders and identify SMER28 and the autophagy pathway as promising targets for DBA therapy.
Supplementary Material
Fig. S1. Reprogramming of patient iPSCs.
Fig. S2. Hematopoietic differentiation efficiency of iPSC lines used in this study.
Fig. S3. In vitro characterization of DBA iPSCs.
Fig. S4. Characterization of CD34-5F cells derived from DBA iPSC lines.
Fig. S5. Chemical screening for compounds that rescue defective DBA erythropoiesis.
Fig. S6. Additional characterization of SMER28 activity.
Fig. S7. SMER28 promotes erythropoiesis through autophagy factor Atg5.
Table S1. Summary of normal and DBA iPSC lines used in this study.
Table S2. Detailed engraftment information for control and DBA CD34-5F cells.
Table S3. List of significant hits from the chemical library screen.
Acknowledgments
We thank BCH ESC core for assistance with reprogramming and R. Mathieu from BCH flow cytometry core. DBA patient samples from NCI were provided by L. Mirabello. M. Weiss provided the RPS19 gene correction plasmid.
Funding:
This work was supported by grants from the NIH National Institute of Diabetes and Digestive and Kidney Diseases (R24-DK092760, R24-DK49216, and U54DK110805) and National Heart, Lung, and Blood Institute (NHLBI) Progenitor Cell Biology Consortium (UO1-HL100001 and U01HL134812) (NHLBI R01HL04880 and NIH R24OD017870-01); Alex’s Lemonade Stand Foundation; The Taub Foundation Grants Program for MDS Research; and the Doris Duke Medical Foundation. This study was funded in part by the intramural research program of the Division of Cancer Epidemiology and Genetics, NCI, NIH (B.P.A.). G.Q.D. is an associate member of the Broad Institute and an investigator of the Howard Hughes Medical Institute and the Manton Center for Orphan Disease Research. S.D. was supported by NHLBI 1K99HL123484, R00HL123484-03, and the Helen Hay Whitney Foundation. H.T.G. was supported by NIH K02HL111156 and NHLBI R01HL107558. L.T.V. was supported by an NSF Graduate Research Fellowship. E.R.M. was supported by NIH NHLBI 1F32HL124948-01. L.W. was supported by a Career Development Award from the University of Heidelberg Medical School. M.A.K. was supported by a T32 hematology training grant from Brigham and Women’s Hospital. H.-Y.L. was supported by a postdoctoral fellowship from the Charles H. Hood Foundation.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/9/376/eaah5645/DC1
Materials and Methods
Author contributions: G.Q.D., L.I.Z., A.N., B.L.E., S.D., and L.T.V. conceived the project. B.P.A., H.T.G., C.A.S., S.A., and A.H.B. provided essential clinical samples. H.F.L. and H.-Y.L. developed protocols. S.D., L.T.V., E.R.M., L.W., A.M.T., T.M.S., L.I.Z., and G.Q.D. designed the experiments; S.D., L.T.V., E.R.M., L.W., A.M.T., J.B., M.G., A.D., K.M., and J.M.H. generated the data. S.D., L.T.V., E.R.M., M.A.K., and L.W. analyzed the data. S.D., L.T.V., E.R.M., L.I.Z., and G.Q.D. prepared the manuscript.
Competing interests: H.F.L. is a founder and shareholder of Rubius. H.-Y.L. and H.F.L. are inventors on patent application PCT/US2014/037554 “In vitro production of red blood cells with sortaggable proteins” held by the Whitehead Institute for Biomedical Research that covers the human erythroid culture system that was used in this manuscript. All other authors declare that they have no competing interests.
Data and materials availability: Microarray data have been deposited in the Gene Expression Omnibus (GSE90940).
REFERENCES AND NOTES
- 1.Vo LT, Daley GQ. De novo generation of HSCs from somatic and pluripotent stem cell sources. Blood. 2015;125:2641–2648. doi: 10.1182/blood-2014-10-570234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tulpule A, Kelley JM, Lensch MW, McPherson J, Park IH, Hartung O, Nakamura T, Schlaeger TM, Shimamura A, Daley GQ. Pluripotent stem cell models of Shwachman-Diamond syndrome reveal a common mechanism for pancreatic and hematopoietic dysfunction. Cell Stem Cell. 2013;12:727–736. doi: 10.1016/j.stem.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garçon L, Ge J, Manjunath SH, Mills JA, Apicella M, Parikh S, Sullivan LM, Podsakoff GM, Gadue P, French DL, Mason PJ, Bessler M, Weiss MJ. Ribosomal and hematopoietic defects in induced pluripotent stem cells derived from Diamond Blackfan anemia patients. Blood. 2013;122:912–921. doi: 10.1182/blood-2013-01-478321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saliba J, Hamidi S, Lenglet G, Langlois T, Yin J, Cabagnols X, Secardin L, Legrand C, Galy A, Opolon P, Benyahia B, Solary E, Bernard OA, Chen L, Debili N, Raslova H, Norol F, Vainchenker W, Plo I, Di Stefano A. Heterozygous and homozygous JAK2V617F states modeled by induced pluripotent stem cells from myeloproliferative neoplasm patients. PLOS ONE. 2013;8:e74257. doi: 10.1371/journal.pone.0074257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumano K, Arai S, Hosoi M, Taoka K, Takayama N, Otsu M, Nagae G, Ueda K, Nakazaki K, Kamikubo Y, Eto K, Aburatani H, Nakauchi H, Kurokawa M. Generation of induced pluripotent stem cells from primary chronic myelogenous leukemia patient samples. Blood. 2012;119:6234–6242. doi: 10.1182/blood-2011-07-367441. [DOI] [PubMed] [Google Scholar]
- 6.Nayak RC, Trump LR, Aronow BJ, Myers K, Mehta P, Kalfa T, Wellendorf AM, Valencia CA, Paddison PJ, Horwitz MS, Grimes HL, Lutzko C, Cancelas JA. Pathogenesis of ELANE-mutant severe neutropenia revealed by induced pluripotent stem cells. J Clin Invest. 2015;125:3103–3116. doi: 10.1172/JCI80924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doulatov S, Vo LT, Chou SS, Kim PG, Arora N, Li H, Hadland BK, Bernstein ID, Collins JJ, Zon LI, Daley GQ. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell. 2013;13:459–470. doi: 10.1016/j.stem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narla A, Ebert BL. Ribosomopathies: Human disorders of ribosome dysfunction. Blood. 2010;115:3196–3205. doi: 10.1182/blood-2009-10-178129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig TN, Dianzani I, Ball S, Tchernia G, Klar J, Matsson H, Tentler D, Mohandas N, Carlsson B, Dahl N. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet. 1999;21:169–175. doi: 10.1038/5951. [DOI] [PubMed] [Google Scholar]
- 10.Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, Wilson FH, Currie T, Khanna-Gupta A, Berliner N, Kutok JL, Ebert BL. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood. 2011;117:2567–2576. doi: 10.1182/blood-2010-07-295238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choesmel V, Bacqueville D, Rouquette J, Noaillac-Depeyre J, Fribourg S, Crétien A, Leblanc T, Tchernia G, Da Costa L, Gleizes P-E. Impaired ribosome biogenesis in Diamond-Blackfan anemia. Blood. 2007;109:1275–1283. doi: 10.1182/blood-2006-07-038372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moniz H, Gastou M, Leblanc T, Hurtaud C, Crétien A, Lécluse Y, Raslova H, Larghero J, Croisille L, Faubladier M, Bluteau O, Lordier L, Tchernia G, Vainchenker W, Mohandas N, Da Costa L, DBA Group of Société d’Hématologie et d’Immunologie Pédiatrique-SHIP Primary hematopoietic cells from DBA patients with mutations in RPL11 and RPS19 genes exhibit distinct erythroid phenotype in vitro. Cell Death Dis. 2012;3:e356. doi: 10.1038/cddis.2012.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nathan DG, Clarke BJ, Hillman DG, Alter BP, Housman DE. Erythroid precursors in congenital hypoplastic (Diamond-Blackfan) anemia. J Clin Invest. 1978;61:489–498. doi: 10.1172/JCI108960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsson H, Davey EJ, Draptchinskaia N, Hamaguchi I, Ooka A, Levéen P, Forsberg E, Karlsson S, Dahl N. Targeted disruption of the ribosomal protein S19 gene is lethal prior to implantation. Mol Cell Biol. 2004;24:4032–4037. doi: 10.1128/MCB.24.9.4032-4037.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morgado-Palacin L, Varetti G, Llanos S, Gómez-López G, Martinez D, Serrano M. Partial loss of Rpl11 in adult mice recapitulates Diamond-Blackfan anemia and promotes lymphomagenesis. Cell Rep. 2015;13:712–722. doi: 10.1016/j.celrep.2015.09.038. [DOI] [PubMed] [Google Scholar]
- 16.McGowan KA, Li JZ, Park CY, Beaudry V, Tabor HK, Sabnis AJ, Zhang W, Fuchs H, de Angelis MH, Myers RM, Attardi LD, Barsh GS. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat Genet. 2008;40:963–970. doi: 10.1038/ng.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devlin EE, DaCosta L, Mohandas N, Elliott G, Bodine DM. A transgenic mouse model demonstrates a dominant negative effect of a point mutation in the RPS19 gene associated with Diamond-Blackfan anemia. Blood. 2010;116:2826–2835. doi: 10.1182/blood-2010-03-275776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narla A, Dutt S, McAuley JR, Al-Shahrour F, Hurst S, McConkey M, Neuberg D, Ebert BL. Dexamethasone and lenalidomide have distinct functional effects on erythropoiesis. Blood. 2011;118:2296–2304. doi: 10.1182/blood-2010-11-318543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee HY, Gao X, Barrasa MI, Li H, Elmes RR, Peters LL, Lodish HF. PPAR-α and glucocorticoid receptor synergize to promote erythroid progenitor self-renewal. Nature. 2015;522:474–477. doi: 10.1038/nature14326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge J, Apicella M, Mills JA, Garçon L, French DL, Weiss MJ, Bessler M, Mason PJ. Dysregulation of the transforming growth factor β pathway in induced pluripotent stem cells generated from patients with Diamond Blackfan anemia. PLOS ONE. 2015;10:e0134878. doi: 10.1371/journal.pone.0134878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vemula S, Shi J, Mali RS, Ma P, Liu Y, Hanneman P, Koehler KR, Hashino E, Wei L, Kapur R. ROCK1 functions as a critical regulator of stress erythropoiesis and survival by regulating p53. Blood. 2012;120:2868–2878. doi: 10.1182/blood-2011-10-384172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O’Kane CJ, Schreiber SL, Rubinsztein DC. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tian Y, Bustos V, Flajolet M, Greengard P. A small-molecule enhancer of autophagy decreases levels of Aβ and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 2011;25:1934–1942. doi: 10.1096/fj.10-175158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor AM, Humphries JM, White RM, Murphey RD, Burns CE, Zon LI. Hematopoietic defects in rps29 mutant zebrafish depend upon p53 activation. Exp Hematol. 2012;40:228–237.e5. doi: 10.1016/j.exphem.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mirabello L, Macari ER, Jessop L, Ellis SR, Myers T, Giri N, Taylor AM, McGrath KE, Humphries JM, Ballew BJ, Yeager M, Boland JF, He J, Hicks BD, Burdett L, Alter BP, Zon L, Savage SA. Whole-exome sequencing and functional studies identify RPS29 as a novel gene mutated in multicase Diamond-Blackfan anemia families. Blood. 2014;124:24–32. doi: 10.1182/blood-2013-11-540278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Z, Keel SB, Shimamura A, Liu L, Gerds AT, Li HY, Wood BL, Scott BL, Abkowitz JL. Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and del(5q) myelodysplastic syndrome. Sci Transl Med. 2016;8:338ra67. doi: 10.1126/scitranslmed.aaf3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cahan P, Li H, Morris SA, Lummertz da Rocha E, Daley GQ, Collins JJ. CellNet: Network biology applied to stem cell engineering. Cell. 2014;158:903–915. doi: 10.1016/j.cell.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen D, Coleman J, Chan E, Nicholson TP, Dai L, Sheppard PW, Patton WF. Novel cell- and tissue-based assays for detecting misfolded and aggregated protein accumulation within aggresomes and inclusion bodies. Cell Biochem Biophys. 2011;60:173–185. doi: 10.1007/s12013-010-9138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mortensen M, Simon AK. Nonredundant role of Atg7 in mitochondrial clearance during erythroid development. Autophagy. 2010;6:423–425. doi: 10.4161/auto.6.3.11528. [DOI] [PubMed] [Google Scholar]
- 30.Hu Z, Zhang J, Zhang Q. Expression pattern and functions of autophagy-related gene atg5 in zebrafish organogenesis. Autophagy. 2011;7:1514–1527. doi: 10.4161/auto.7.12.18040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159:1263–1276. doi: 10.1016/j.cell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, Passegué E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013;494:323–327. doi: 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, Stranks AJ, Glanville J, Knight S, Jacobsen SEW, Kranc KR, Simon AK. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208:455–467. doi: 10.1084/jem.20101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang Y-A, Sanalkumar R, O’Geen H, Linnemann AK, Chang C-J, Bouhassira EE, Farnham PJ, Keles S, Bresnick EH. Autophagy driven by a master regulator of hematopoiesis. Mol Cell Biol. 2012;32:226–239. doi: 10.1128/MCB.06166-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, Stallings CL. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015;528:565–569. doi: 10.1038/nature16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bestebroer J, V’Kovski P, Mauthe M, Reggiori F. Hidden behind autophagy: The unconventional roles of ATG proteins. Traffic. 2013;14:1029–1041. doi: 10.1111/tra.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez J, Malireddi RKS, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan JL, Tan H, Peng J, Kanneganti TD, Virgin HW, Green DR. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17:893–906. doi: 10.1038/ncb3192. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Heijnen HF, van Wijk R, Pereboom TC, Goos YJ, Seinen CW, van Oirschot BA, van Dooren R, Gastou M, Giles RH, van Solinge W, Kuijpers TW, Gazda HT, Bierings MB, Da Costa L, MacInnes AW. Ribosomal protein mutations induce autophagy through S6 kinase inhibition of the insulin pathway. PLOS Genet. 2014;10:e1004371. doi: 10.1371/journal.pgen.1004371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–109. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G, Cantwell J, Luu C, Cornella-Taracido I, Harrington E, Fekkes P, Lei H, Fang Q, Digan ME, Burdick D, Powers AF, Helliwell SB, D’Aquin S, Bastien J, Wang H, Wiederschain D, Kuerth J, Bergman P, Schwalb D, Thomas J, Ugwonali S, Harbinski F, Tallarico J, Wilson CJ, Myer VE, Porter JA, Bussiere DE, Finan PM, Labow MA, Mao X, Hamann LG, Manning BD, Valdez RA, Nicholson T, Schirle M, Knapp MS, Keaney EP, Murphy LO. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16:1069–1079. doi: 10.1038/ncb3053. [DOI] [PubMed] [Google Scholar]
- 42.Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 43.Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K-i, Shibata T, Kunisada T, Takahashi M, Takahashi J, Saji H, Yamanaka S. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 44.Chadwick K, Wang L, Li L, Menendez P, Murdoch B, Rouleau A, Bhatia M. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood. 2003;102:906–915. doi: 10.1182/blood-2003-03-0832. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Reprogramming of patient iPSCs.
Fig. S2. Hematopoietic differentiation efficiency of iPSC lines used in this study.
Fig. S3. In vitro characterization of DBA iPSCs.
Fig. S4. Characterization of CD34-5F cells derived from DBA iPSC lines.
Fig. S5. Chemical screening for compounds that rescue defective DBA erythropoiesis.
Fig. S6. Additional characterization of SMER28 activity.
Fig. S7. SMER28 promotes erythropoiesis through autophagy factor Atg5.
Table S1. Summary of normal and DBA iPSC lines used in this study.
Table S2. Detailed engraftment information for control and DBA CD34-5F cells.
Table S3. List of significant hits from the chemical library screen.