

Abstract
CD11c+ cells increase greatly with islet inflammation in non-obese diabetic mice and contribute to autoimmune destruction of pancreatic beta cells. In this study, we investigated their origin and mechanism of recruitment. CD11c+ cells in inflamed islets resembled classical dendritic cells (DC) based on their transcriptional profile. However, the majority of these cells were not from the Zbtb46-dependent DC lineage. Instead, monocyte precursors could give rise to CD11c+ cells in inflamed islets. Chemokines Ccl5 and Ccl8 were persistently elevated in inflamed islets and the influx of CD11c+ cells was partially dependent on their receptor Ccr5. Treatment with islet antigen-specific regulatory T cells (Tregs) led to a marked decrease of Ccl5 and Ccl8 and a reduction of monocyte recruitment. These results implicate a monocytic origin of CD11c+ cells in inflamed islets and suggest that therapeutic Tregs directly or indirectly regulate their influx by altering the chemotactic milieu in the islets.
Introduction
Most non-lymphoid tissue dendritic cells (DCs) can be divided into CD103+ and CD11b+ subsets. The CD103+ subset arises primarily from bone marrow pre-DC precursors whereas the CD11b+ subset can arise from either pre-DCs or monocytes (1, 2). The heterogeneity of non-lymphoid tissue CD11b+ DCs is reflected in their transcriptional profile and the distinct functions they serve in the tissue (3).
In normal islets, the predominant immune cells are CD11c+CD11b+ cells that originate from colony stimulating factor 1-dependent monocyte precursors (1, 4–6). CD11c+ cells in non-obese diabetic (NOD) mouse pancreas have distinct gene expression profile and their number greatly with inflammation in the islets (7, 8). Depletion of phagocytes or CD11c+ cells resolves islet inflammation and delays diabetes development (9, 10), demonstrating the importance of these cells in T1D pathogenesis. However, more recent studies suggest the existence of different subsets of myeloid cells in inflamed islets that serve distinct functions. For example, a subset of CRIg-expressing myeloid cells are reported to be protective against diabetes progression (11) and a rare subset of Batf3-dependent CD11b+CD103+ DC are essential for disease initiation in NOD mice (12). The origin and identity of the majority of CD11c+ cells in inflamed islets remains unknown however, and is the focus of this study.
Materials and Methods
Mice
NOD.Rag2−/−, NOD.CD28−/−, NOD.CD11c-YFP.CD28−/−, NOD.BDC2.5 Thy1.1 TCR transgenic, B6, B6.Zbtb-GFP, B6.Ccr2−/−, B6.Ccr5−/−, B6.Ccr7−/−, and B6.Cx3cr1-GFP mice were housed and bred under specific pathogen-free (SPF) conditions at the University of California, San Francisco Animal Barrier Facility. The Institutional Animal Care and Use Committee approved all experiments.
Flow cytometry of islet infiltrates
Islets were isolated and dissociated as previously described (13). Peritoneal cells were collected by lavage. Splenocytes were prepared with 250μg DNase I and 800 Mandl U/ml collagenase D (Roche). Cells were stained with antibodies to CD45 (30-F11), Ly5.1 (A20), Ly5.2 (104), B220 (RA3-3A1/6.1), CD11c (N418), CD11b (M1/70), CD103 (2E7), F4/80 (BM8), CD115 (AFS98), Sirpa (P84), DCIR2(33D1) and Ly6c (HK1.4). DAPI (Invitrogen) was used to exclude dead cells. Cells were analyzed and purified using a BD FACS Aria II or Fortessa.
qRT-PCR
mRNA from purified cells or whole islets was extracted using Arcturus Pico Kit (Life Technologies). Reverse transcription was done using iScript Advanced cDNA Synthesis Kit (Bio Rad). qPCR was performed using SYBR Green mastermix (Bio Rad) and primers from QIAGEN and Bio-Rad on a Bio-Rad CFX 96.
Chemokine protein measurements
Handpicked islets were cultured overnight at 37°C in RPMI with 10% FCS at a density of 2 μl/islet. Chemokines in the supernatants or cell lysate including the supernatant were measured using ELISA (CCL8 from Bosterbio and CCL7 from Thermo Fisher) or multiplex luminex (all other chemokines, Eve Technologies).
Monocyte trafficking
Monocytes were isolated from NOD.Rag2−/− bone marrow using EasySep Mouse Monocyte Isolation Kit (Stemcell). The cells were labelled with 5 μM CSFE and were retro-orbitally (r.o.) injected to recipient mice. In vivo monocyte labeling was achieved by r.o. injection of 1-μm fluorescent beads (Polysciences Inc) as previously described (14).
5-FU chimeras
Mice were injected i.p. with 150 mg/kg of 5-FU (Sigma) in sterile PBS a day before transfer of T cell depleted bone marrow.
Enumerating islet CD11c+ cells
Handpicked islets from NOD.CD11c-YFP mice were stained in 10 μM CMTMR and embedded in RPMI medium with 0.5% agarose (Invitrogen) on a coverslip. Images of islets were acquired on a custom-built 4-PMT-detector video-rate two-photon microscope using a water immersion 20x/0.95 NA objective with the aid of Micromanager Software. Data were analyzed using Imaris (Bitplane AG).
T cell transfers
FACS-purified CD4+CD62L+CD25+ Tregs and CD4+CD62L+CD25− effector T cells (Teff) from lymph nodes of NOD.BDC2.5.Thy1.1 TCR transgenic mice were expanded as previously described (15). BDC2.5 Treg treatments were administered via i.p. injection to NOD.CD28−/− mice (106 cells) at 5 to 7 weeks of age. 106 BDC2.5 Teff were i.p. injected to NOD.Rag2−/− mice to induce islet inflammation.
Statistical analysis
Statistical analyses were performed with the aid of Prism software (GraphPad).
Results and Discussion
CD11c+CD11b+ cells in inflamed islets have a cDC transcriptional signature
Autoimmune-mediated inflammation in the pancreatic islet leads to massive influx of CD11c+CD11b+ cells (13). A previous study found that a minor population of Batf3 dependent CD103+ DCs in the inflamed islets (12). However, the identity of the majority of the CD11c+ cells remains to be defined. We have examined islet CD11c+CD11b+ cells in NOD.CD28−/− mice that develop diabetes with higher penetrance and better synchrony than wildtype NOD mice due to a deficit in Tregs (16). Similarly to NOD mice (5), the majority of CD11c+ cells in inflamed islets of NOD.CD28−/− mice were a fairly uniform population of CD11b+F4/80−CD103−MHCII+Sirpa+, and dim for CD4 and DCIR2 (Fig. 1A and Supplemental Fig. 1). Thus, these cells do not resemble lymphoid tissue CD11c+CD11b+ cDC2 but more similar to monocyte-derived DCs (17).
Figure 1. Transcriptional profile of CD11c+ cells in inflamed islets.
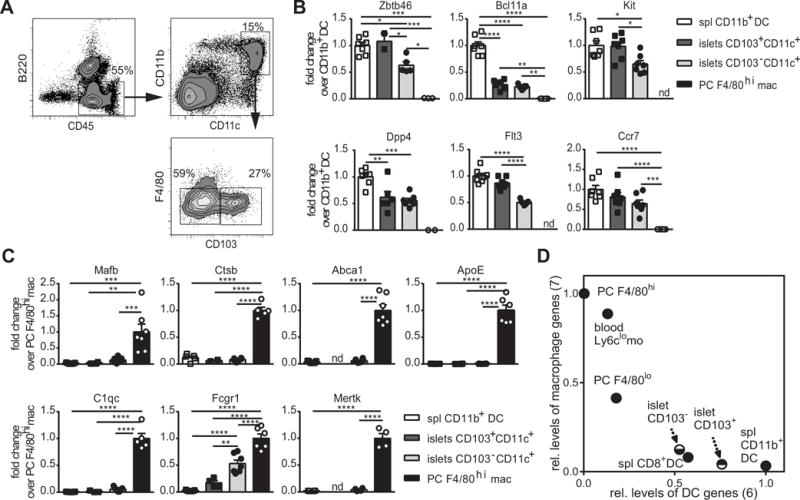
Representative plots of flow cytometric analysis of islets CD11c+ cells in 9-week-old pre-diabetic NOD.CD28−/− mice. (B and C) Fold change in cDC genes (B) and macrophage genes (C) in islet CD103+ and CD103− CD11c+ cells over the reference populations of splenic CD11b+ DC (DAPI− CD45+ B220− F4/80low CD11b+ CD11c+) and peritoneal F4/80hi macrophages (DAPI− CD115+ B220− MHCIIint F4/80hi). Each symbol represents one mouse and bars represent mean + SEM. Data are a summary of two independent experiments with at least 3 animals per experiment. One way ANOVA test with Bonferroni’s post hoc test were used to determine statistical significance (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001). (D) Index of all DC signature genes (x axis) and macrophage signature genes (y axis) as listed in B and C for islet CD103+ and CD103− CD11c+ cells in comparison to reference DC and macrophage populations (PC: peritoneal cavity, mo: monocytes, spl: spleen).
Studies from the ImmGen consortium have revealed that in some nonlymphoid tissues, cells previously described as DCs are transcriptionally more closely related to macrophages than to DCs. By profiling expression levels of both cDC and macrophage signature genes in inflamed islet CD11c+ cells, we sought to more definitively determine the identity of these cells. We profiled the expression of cDC and macrophage signature genes defined by the ImmGen consortium (3, 18) in CD103+ and CD103− CD11c+ cells in inflamed islets. Both subsets expressed core cDC genes (Fig. 1B) and lacked the expression of macrophage signature genes except Fc receptor, IgG, high affinity I (Fcgr1) (Fig. 1C). We quantitatively analyzed the level of “cDC-ness” and “macrophage-ness” for each cell type by calculating the average relative expression for all DC signature genes and all macrophage signature genes, respectively. The CD11c+CD11b+ splenic DCs were given a cDC score of 1 and a macrophage score of 0; whereas the peritoneal macrophages were assigned a cDC score of 0 and a macrophage score of 1. CD11c+CD11b+ cells in inflamed islets showed a stronger alliance with cDCs than with macrophages, with the CD103+ subset demonstrating stronger ‘cDC-ness’ than the CD103− subset (Fig. 1D). Thus, at the transcriptional level, CD11c+CD11b+ cells in inflamed islets possessed a DC-like phenotype.
Monocytes, but not pre-DCs, contribute to CD11c+CD11b+ cells in inflamed islets
We next sought to determine the precursor of CD11c+ cells in inflamed islets. To this end, we developed a 5-FU bone marrow chimera approach, in which Ly5.1+ NOD.Rag2−/− mice were treated with 5-fluorouracil (5-FU) to ablate their bone marrow one day before receiving Ly5.2+ bone marrow cells as a source of DC precursors. The use of 5-FU instead of radiation preserves endogenous CD11c+ cells and architecture of lymphoid organs, thus minimally impacting peripheral immune status and diabetes onset (Supplemental Fig. 2A). One week after bone marrow reconstitution, some mice were injected with islet antigen-specific CD4+CD25− effector T cells (Teff) from BDC2.5 TCR transgenic mice to induce islet inflammation (Supplemental Fig. 2B). Ly5.2+ cells appear in the blood one week after bone marrow cell injection and can be found among CD11c+ MHCII+ cells in the islets at two weeks (Supplemental Fig. 2C). Transfer of BDC2.5 Teff resulted in a significant increase of Ly5.2+ cells in the islets, demonstrating that bone marrow could give rise to islet CD11c+ cells during inflammation (Supplemental Fig. 2D and E).
We then used this model to define the origin of CD11c+ cells in inflamed islets. Zbtb46 distinguishes cDCs from other mononuclear phagocytes, and the Zbtb46-GFP reporter mouse is a valuable tool for identifying cDCs. We reconstituted 5-FU treated Ly5.1+ NOD.Rag2−/− mice with bone marrow cells from Ly5.2+ Zbtb46-GFP reporter mice and found more than 80% of Ly5.2+ cells among islet CD11c+CD11b+ cells did not express GFP (Fig. 2A). In comparison, most of the Ly5.2+CD11c+ cells in the spleen and draining pancreatic lymph node were GFP+ (Fig. 2A). Although we have not transferred pre-DC to further validate our findings, these data suggest that the majority of CD11c+ cells in inflamed islets do not arise from pre-DC precursors despite their DC-like gene expression profile.
Figure 2. Monocyte contribution to the islet CD11c+ cell population.
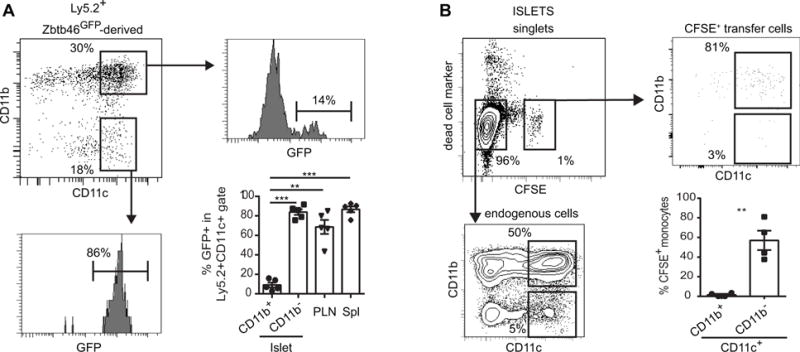
(A) 5-FU chimera mice were generated in Ly5.1+ NOD.Rag2−/− recipients using Ly5.2+ B6.Zbtb46-GFP bone marrow. Representative flow plots show the profiles of Ly5.2+ donor-derived islet-infiltrating cells (top left) and their expression of Zbtb46-GFP. Summary of the percentages of GFP+ cells among in various populations of Ly5.2+ cells (bottom right). Each symbol represents one mouse and bars represent mean + SEM. Data are a summary of 2 independent experiments. (B) Flow cytometric analysis of adoptively transferred monocytes in inflamed islets. Representative flow plots along with quantification of CD11b+ or CD11b− islet cells among CFSE+ transferred cells (bottom right) are shown. Each symbol represents one mouse and bars represent mean + SEM. Data are a summary of 2 independent experiments of two mice per experiment. Statistical analysis was performed using One way ANOVA (A) or Student t test (B, ** p<0.01, *** p<0.001, **** p<0.0001).
Steady-state islet CD11c+ cells have a monocytic origin (5). To determine whether CD11c+ cells in inflamed islets also have a monocytic origin, we transferred CFSE-labeled bone marrow CD115+ monocytes to NOD.Rag2−/− mice that had received BDC2.5 Teff cells. CFSE+ cells were readily detectable in inflamed islets 36 hours after monocyte transfer and the majority of these cells were CD11c+CD11b+CD103− (Fig. 2B and data not shown). These findings show that monocytes are recruited to inflamed islets and can give rise to islet CD11c+ cells.
Islet CD11c+ cell recruitment is partially dependent on Ccr5
Monocyte recruitment to inflamed tissues is mainly mediated through the chemokine receptor Ccr2, and roles for Ccr1 and Ccr5 have also been implicated (19). To determine chemokine ligand-receptor pair(s) involved in the recruitment of CD11c+ cells to inflamed islets, we surveyed expression of CC chemokines in inflamed islets of pre-diabetic NOD.CD28−/− mice and compared with those from non-inflamed islets in NOD.Rag2−/− mice. Although myeloid cells in NOD.Rag2−/− mice may have altered functions due to their development in the absence of the adaptive immune cells (20), islets from NOD.Rag2−/− have a similar transcriptional profile, including chemokine expression, to young NOD mice before the onset of inflammation (21).
Ccl5 and Ccl8 were elevated at multiple ages in NOD.CD28−/− islets when compared to NOD.Rag2−/− controls (Fig. 3A). In addition, Ccl5 and Ccl8 protein were upregulated in culture supernatants of NOD.CD28−/− islets when compared to NOD.Rag2−/− islets (Fig. 3B).
Figure 3. Role of chemokines in CD11c+ cell recruitment to inflamed islets.
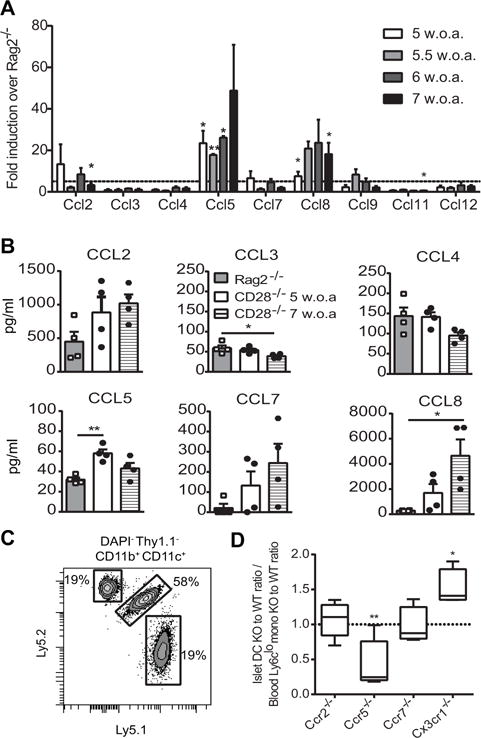
(A) Expression of chemokine mRNA in NOD.CD28−/− islets was compared to that in NOD.Rag2−/− islets. Dashed line represents a 5-fold increase in NOD.CD28−/− islets over NOD.Rag2−/− islets. Bars depict mean fold-change + SEM from 3 independent experiments with 4–5 mice pooled per age group in each experiment (w.o.a., weeks of age). Statistical analysis for each time point was performed using one sample t test with hypothetical value of 1 for NOD.Rag2−/− group. (B) Chemokine protein concentrations in islet supernatants from NOD.Rag2−/− and NOD.CD28−/− mice are shown. Representative data from 3 independent experiments with each data point representing an individual mouse. Bars depict mean + SEM. Statistical analysis was performed using Oneway ANOVA. (C and D) Mixed bone marrow chimeras in NOD.Rag2−/−.Ly5.1/Ly5.2 recipient mice were prepared with 50:50 mix of bone marrow from Ly5.1 wild-type mice and Ly5.2 chemokine receptor knockout mice. (C) Representative flow plot pre-gated on DAPI−Thy1.1−CD11c+CD11b+ islet cells showing contribution of endogenous (Ly5.1+Ly5.2+) and donor bone marrow-derived cells on day 14 after transfer. (D) The ratio of knockout to wild-type CD11c+ cells was calculated using flow cytometry data and normalized to the ratio of knockout to wild-type donor monocytes in the blood. No difference in chimerism between blood and islets would be a value of 1 (dashed line). Boxes and whiskers depict the relative chimerism of 4–8 mice per group from 5 independent experiments. Statistical analysis was performed using one-sample t test comparing the ratio of knockout to wild-type CD11c+ cells in islets normalized to the ratio of knockout to wild-type blood monocyte precursors in the same mouse to the theoretical mean of 1 (*p < 0.05, **p < 0.01).
To determine the requirement of specific chemokine receptors for recruiting CD11c+ cells to inflamed islets, we injected 5-FU-treated NOD.Rag2−/− mice with a 50/50 mixture of wild-type and chemokine receptor-deficient bone marrow cells (Fig. 3C) and tracked their migration to inflamed islets after BDC2.5 Teff transfer. Chemokine receptors tested included Ccr2, Ccr5, Ccr7, and Cx3cr1, all of which have been previously implicated in monocyte and DC recruitment.
Ccr2 is preferentially expressed by Ly6chi monocytes and has been shown to mediate recruitment of monocyte-derived DC to inflamed colon and lung by binding to Ccl2 (19). However, we found that Ccr2−/− cells had no deficiency in entering inflamed islets (Fig. 3D). Similarly, Ccr7 was not required for in CD11c+ cell recruitment into inflamed islets. Cx3cr1 is expressed on Ly6Clo patrolling monocytes in the blood and CD11c+ cells in non-inflamed islets (5). In our experiments, Cx3cr1−/− cells were more efficiently recruited into the inflamed islets than wild-type cells (Fig. 3D).
Previous data on the role of Ccr5 in the pathogenesis of diabetes in NOD mice are mixed. Anti-Ccr5 antibody reduces insulitis and delays diabetes (22) whereas Ccr5-deficient NOD show accelerated diabetes (23). Ccr5 is the receptor for Ccl5 and Ccl8 (24), two chemokines persistently elevated in inflamed islets. In mice that received a mixture of wild-type and Ccr5−/− bone marrow, we found higher proportions of Ccr5−/− cells among blood Ly6clo monocytes but the same contribution of wild-type and Ccr5−/− cells to other blood leukocyte populations, suggesting a selective buildup of Ccr5−/− Ly6clo monocytes in the blood (data not shown). Despite this, wild-type cells preferentially gave rise to CD11c+ cells in the inflamed islets. Thus, Ccr5−/− cells were at a significant disadvantage to give rise to islet CD11c+ cells (Fig. 3D). It is noteworthy that some Ccr5−/− cells entered the inflamed islets, suggesting redundant mechanisms or partial compensation for this process. Ccr1 can also bind to Ccl5 and Ccl8 (25) and may explain the mixed results on the role of Ccr5 in previous reports and partial effects we observed in this study. Altogether, these data suggest a role for Ccr5, but not Ccr2, Ccr7, or Cx3cr1 in the recruitment of CD11c+ cells to inflamed islets. It is important to note that these bone marrow chimera experiments used B6 donors that lack the intrinsic NOD autoimmune predisposition and NOD pancreatic CD11c+ cells were reported to have lower expression of Ccr5 (7); thus, validation using donors on the NOD background is warranted.
Islet antigen-specific Tregs attenuate CD11c+ cell recruitment to inflamed islets
Adoptive transfer of BDC2.5 Tregs protects against diabetes development, both in NOD and NOD.CD28−/− mice (15, 26). We next determined the impact of Treg therapy on the accumulation of CD11c+ cells in inflamed islets. Treg treatment suppressed Ccl2, Ccl5, Ccl7, Ccl8, and Ccl9 mRNA expression in inflamed islets (Fig. 4A). Of these, Ccl5 showed the most dramatic reduction (84%), followed by Ccl8 (78%). When examined at the protein level, lysates from islets of Treg treated NOD.CD28−/− mice had less Ccl5 than those from islets of age-matched controls, while Ccl2 was unchanged (Fig. 4B). Moreover, Treg treatment let to a significant reduction in islet CD11c+ cell numbers when compared to untreated age-matched controls (Fig. 4C and D).
Figure 4. Impact of Tregs on CD11c+ cell recruitment to inflamed islets.
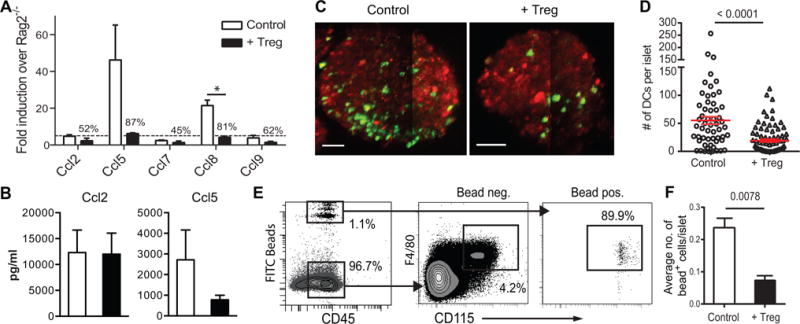
(A) Chemokine mRNA from pooled islets from NOD.Rag2−/− and female NOD.CD28−/− mice that had been treated or not with BDC2.5 Tregs 2 weeks earlier were analyzed. Results are from 2 independent experiments with at least 4 mice pooled per group. (B) Chemokine proteins in islet lysates of Treg-treated and untreated controls were analyzed. Results are from 2 independent experiments with at least 2 mice pooled per group. (C) Maximal projection images depicting representative CMTMR-labelled islets (red) from untreated (left) and BDC2.5 Treg-treated (right) NOD.CD28−/−.CD11c-YFP (green) mice taken on a two-photon microscope. Scale bars represent 50 μm. (D) Quantification of the numbers of YFP+ cells per islet. Each symbol represents one islet from a total of 4 mice per group imaged in 2 independent experiments. (E, F) BDC2.5 Treg-treated or non-treated NOD.CD28−/− mice received i.v. injection of fluorescent latex beads to label blood monocytes. Blood (E) and islet (F) FITC bead+ cells were analyzed using flow cytometry 3 days later. Data is a representative of two independent experiments with 3 mice per group in each experiment. Statistically significant differences are indicated by p values above the pair compared using Student t test (*p < 0.05). All other differences are not statistically significant.
To determine if reduction of islet CD11c+ cells was due to decreased islet CD11c+ cell recruitment after Treg treatment, we utilized an in vivo monocyte labeling method to track the recruitment of monocyte precursors to the islets (14). Fluorescently labeled latex beads were injected directly into the bloodstream of Treg-treated or age-matched NOD.CD28−/− mice. The beads were taken up by blood monocytes (Fig. 4E), thus providing a means to track their trafficking into inflamed islets. In mice that received BDC2.5 Tregs 2 weeks prior to bead injection, significantly fewer bead+ CD11c+ cells were detected in islets (Fig. 4F). Together, these findings show that Treg treatment leads to a reduction in inflammatory chemokines, decreased recruitment of CD11c+ cells, and reduced CD11c+ cell accumulation in inflamed islets. Previously we have shown Treg therapy rapidly suppresses IFNγ production by intra-islet CD4+ and CD8+ T cells within days (27). By comparison, the effect of Tregs on CD11c+ cells was more gradual and took one to two weeks to manifest (data not shown). Inhibition of IFNγ production may be an upstream event that leads to down-regulation of inflammatory chemokines in the islets, as Ccl5 can be regulated by IFNγ (28).
In summary, this work demonstrates in a model of autoimmune diabetes that CD11c+ cells in inflamed islets were mostly derived from monocytes despite their DC-like phenotype. These cells were attracted to the inflamed islets in part via Ccr5, likely in response to elevated Ccl5 and Ccl8. Therapeutic Tregs decreased Ccl5 and Ccl8 expression and CD11c+ cell recruitment to the islets, pointing to the possible role of this process in propagating autoimmune pathology.
Supplementary Material
Acknowledgments
We thank R. Guerrero-Moreno, J. Wang, V Dang, N. Lescano for mouse husbandry.
This work is supported by grants from NIH, DK08231 (QT) and P30DK063720 (MG) and a UCSF PBBR grant.
References
- 1.Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, Price J, Yin N, Bromberg J, Lira SA, Stanley ER, Nussenzweig M, Merad M. The origin and development of nonlymphoid tissue CD103+ DCs. The Journal of Experimental Medicine. 2009;206:3115–3130. doi: 10.1084/jem.20091756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Satpathy AT, Wu X, Albring JC, Murphy KM. Re(de)fining the dendritic cell lineage. Nature immunology. 2012;13:1145–1154. doi: 10.1038/ni.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, Hashimoto D, Chow A, Price J, Greter M, Bogunovic M, Bellemare-Pelletier A, Frenette PS, Randolph GJ, Turley SJ, Merad M, C. Immunological Genome Deciphering the transcriptional network of the dendritic cell lineage. Nature immunology. 2012;13:888–899. doi: 10.1038/ni.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calderon B, Suri A, Miller MJ, Unanue ER. Dendritic cells in islets of Langerhans constitutively present beta cell-derived peptides bound to their class II MHC molecules. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6121–6126. doi: 10.1073/pnas.0801973105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yin N, Xu J, Ginhoux F, Randolph GJ, Merad M, Ding Y, Bromberg JS. Functional specialization of islet dendritic cell subsets. Journal of immunology (Baltimore, Md: 1950) 2012;188:4921–4930. doi: 10.4049/jimmunol.1103725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calderon B, Carrero JA, Ferris ST, Sojka DK, Moore L, Epelman S, Murphy KM, Yokoyama WM, Randolph GJ, Unanue ER. The pancreas anatomy conditions the origin and properties of resident macrophages. The Journal of Experimental Medicine. 2015 doi: 10.1084/jem.20150496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beumer W, Welzen-Coppens JM, van Helden-Meeuwsen CG, Gibney SM, Drexhage HA, Versnel MA. The gene expression profile of CD11c+ CD8alpha- dendritic cells in the pre-diabetic pancreas of the NOD mouse. PLoS One. 2014;9:e103404. doi: 10.1371/journal.pone.0103404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melli K, Friedman RS, Martin AE, Finger EB, Miao G, Szot GL, Krummel MF, Tang Q. Amplification of autoimmune response through induction of dendritic cell maturation in inflamed tissues. J Immunol. 2009;182:2590–2600. doi: 10.4049/jimmunol.0803543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nikolic T, Geutskens SB, van Rooijen N, Drexhage HA, Leenen PJ. Dendritic cells and macrophages are essential for the retention of lymphocytes in (peri)-insulitis of the nonobese diabetic mouse: a phagocyte depletion study. Laboratory investigation; a journal of technical methods and pathology. 2005;85:487–501. doi: 10.1038/labinvest.3700238. [DOI] [PubMed] [Google Scholar]
- 10.Saxena V, Ondr JK, Magnusen AF, Munn DH, Katz JD. The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse. Journal of immunology (Baltimore, Md: 1950) 2007;179:5041–5053. doi: 10.4049/jimmunol.179.8.5041. [DOI] [PubMed] [Google Scholar]
- 11.Fu W, Wojtkiewicz G, Weissleder R, Benoist C, Mathis D. Early window of diabetes determinism in NOD mice, dependent on the complement receptor CRIg, identified by noninvasive imaging. Nat Immunol. 2012;13:361–368. doi: 10.1038/ni.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferris Stephen T, Carrero Javier A, Mohan James F, Calderon B, Murphy Kenneth M, Unanue Emil R. A Minor Subset of Batf3-Dependent Antigen-Presenting Cells in Islets of Langerhans Is Essential for the Development of Autoimmune Diabetes. Immunity. 2014;41:657–669. doi: 10.1016/j.immuni.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melli K, Friedman RS, Martin AE, Finger EB, Miao G, Szot GL, Krummel MF, Tang Q. Amplification of autoimmune response through induction of dendritic cell maturation in inflamed tissues. Journal of immunology (Baltimore, Md: 1950) 2009;182:2590–2600. doi: 10.4049/jimmunol.0803543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tacke F, Ginhoux F, Jakubzick C, van Rooijen N, Merad M, Randolph GJ. Immature monocytes acquire antigens from other cells in the bone marrow and present them to T cells after maturing in the periphery. The Journal of experimental medicine. 2006;203:583–597. doi: 10.1084/jem.20052119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. The Journal of experimental medicine. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 17.Dong MB, Rahman MJ, Tarbell KV. Flow cytometric gating for spleen monocyte and DC subsets: differences in autoimmune NOD mice and with acute inflammation. Journal of Immunological Methods. 2016;432:4–12. doi: 10.1016/j.jim.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua W-JJ, Hansen TH, Turley SJ, Merad M, Randolph GJ, C. Immunological Genome Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nature immunology. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nature reviews. Immunology. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moulin V, Andris F, Thielemans K, Maliszewski C, Urbain J, Moser M. B lymphocytes regulate dendritic cell (DC) function in vivo. Journal of Experimental Medicine. 2000;192:475–482. doi: 10.1084/jem.192.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carrero JA, Calderon B, Towfic F, Artyomov MN, Unanue ER. Defining the transcriptional and cellular landscape of type 1 diabetes in the NOD mouse. PloS one. 2012;8 doi: 10.1371/journal.pone.0059701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carvalho-Pinto C, García MII, Gómez L, Ballesteros A, Zaballos A, Flores JM, Mellado M, Rodríguez-Frade JMM, Balomenos D, Martínez-A C. Leukocyte attraction through the CCR5 receptor controls progress from insulitis to diabetes in non-obese diabetic mice. European journal of immunology. 2004;34:548–557. doi: 10.1002/eji.200324285. [DOI] [PubMed] [Google Scholar]
- 23.Solomon M, Balasa B, Sarvetnick N. CCR2 and CCR5 chemokine receptors differentially influence the development of autoimmune diabetes in the NOD mouse. Autoimmunity. 2010;43:156–163. doi: 10.3109/08916930903246464. [DOI] [PubMed] [Google Scholar]
- 24.Blanpain C, Migeotte I, Lee B, Vakili J, Doranz BJ, Govaerts C, Vassart G, Doms RW, Parmentier M. CCR5 Binds Multiple CC-Chemokines: MCP-3 Acts as a Natural Antagonist. Blood. 1999;94:1899–1905. [PubMed] [Google Scholar]
- 25.Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. 2012;36:705–716. doi: 10.1016/j.immuni.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T Cells, Expanded with Dendritic Cells Presenting a Single Autoantigenic Peptide, Suppress Autoimmune Diabetes. The Journal of Experimental Medicine. 2004;199:1467–1477. doi: 10.1084/jem.20040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahne AE, Klementowicz JE, Chou A, Nguyen V, Tang Q. Therapeutic regulatory T cells subvert effector T cell function in inflamed islets to halt autoimmune diabetes. The Journal of Immunology. 2015;194:3147–3155. doi: 10.4049/jimmunol.1402739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wen X, Kudo T, Payne L, Wang X, Rodgers L, Suzuki Y. Predominant interferon-γ-mediated expression of CXCL9, CXCL10, and CCL5 proteins in the brain during chronic infection with Toxoplasma gondii in BALB/c mice resistant to development of toxoplasmic encephalitis. Journal of Interferon and Cytokine Research. 2010;30:653–660. doi: 10.1089/jir.2009.0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.