

Abstract
We have repurposed (N)-methanocarba adenosine derivatives (A3 adenosine receptor (AR) agonists) to enhance radioligand binding allosterically at the human dopamine (DA) transporter (DAT) and inhibit DA uptake. We extended the structure-activity relationship of this series with small N6-alkyl substitution, 5′-esters, deaza modifications of adenine, and ribose restored in place of methanocarba. C2-(5-halothien-2-yl)-ethynyl 5′-methyl 9 (MRS7292) and 5′-ethyl 10 (MRS7232) esters enhanced binding at DAT (EC50 ∼35 nM) and at norepinephrine transporter (NET). 9 and 10 were selective for DAT compared to A3AR in the mouse, but not human. At DAT, binding of two structurally dissimilar radioligands was enhanced; NET binding of only one radioligand was enhanced; SERT radioligand binding was minimally affected. 10 was more potent than cocaine at inhibiting DA uptake (IC50 = 107 nM). Ribose analogues were weaker in DAT interaction than corresponding bicyclics. Thus, we enhanced the neurotransmitter transporters activity of rigid nucleosides while reducing A3AR affinity.
Keywords: Dopamine transporter, G protein-coupled receptor, scaffold repurposing, adenosine derivatives, norepinephrine transporter, structure activity relationship
Graphical abstract
Introduction
In the course of examining off-target activities of potent A3 adenosine receptor (AR) agonists for their novel application to the treatment of chronic neuropathic pain,1,2 we discovered a new class of allosteric ligands with low nM affinity at the human (h) dopamine (DA) transporter (DAT, SLC6A3).3,4 DAT function is inhibited by cocaine, which increases the levels of DA at the synapse by a combination of mechanisms.5 Micromolar activity of the same substituted nucleosides at the structurally related norepinephrine transporter (NET, SLC6A2) and weaker interactions with the serotonin transporter (SERT, SLC6A4) were also noted. These transporters are important drug targets for treatment of depression and other behavioral disorders and belong to the much larger class of sodium symporters, which transport diverse solute molecules across the cell membrane. X-ray structures of members of this family of transmembrane proteins have been determined, thus shedding light on the nature of their gating and substrate/inhibitor recognition.4,6,7
The medicinal chemistry of diverse classes of DAT modulators has been explored toward the goal of treating drug abuse.8 Conventional inhibitors of DAT in the CNS are psychostimulants that are of interest for the treatment of addiction to cocaine, heroin or nicotine, and for obesity, narcolepsy and ADHD, but there is a potential for their abuse as recreational drugs.9 Based on pharmacological10-12 and genetic data,13 decreased dopaminergic signaling in the mesocorticolimbic pathway is suggested to play a role in a wide range of behavioral disorders: depression, mood disorders, anhedonia, post-traumatic stress disorder (PTSD), addiction, attention deficit hyperactive disorder (ADHD), autism and pain. Antidepressant treatments that include DAT inhibition, including triple reuptake inhibitors (TRIs) that combine inhibition of SERT and NET with DAT, are being developed because of their expected higher efficacy in treating depression.12,14-16 The classical inhibitors of DAT, such as methylphenidate, are being considered as co-therapy for treating depression, but there is a risk of side effects, such as inducing mania in patients with bipolar disorder.17 However, an allosteric inhibitor of DAT that has a more subtle action might have an advantage over stimulant drugs that are orthosteric DAT inhibitors in the treatment of behavioral disorders associated with DA deficiency.
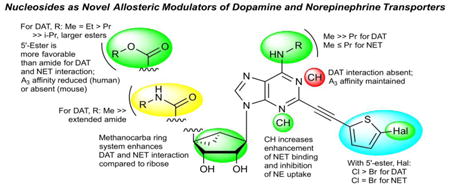
We have utilized a bicyclo[3.1.0]hexane ring (methanocarba) system in place of ribose in numerous nucleoside derivatives and noted that high affinity and selectivity at the A3AR could be achieved with the retention of full agonism.2,18,19 Because this rigid, bicyclic ring system offers the opportunity for systematic substitution at multiple positions with defined geometry, we have explored its use as a component of a repurposed scaffold: shifting adenine nucleosides from ARs to other target proteins, such as receptors and transporters.3,20 For example, (1S,2R,3S,4R,5S)-4-(2-((5-chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (MRS5980, 1, Chart 1), which displays >10,000-fold greater affinity for the hA3AR (Ki = 0.70 nM) than at other ARs, also inhibited DA uptake with an IC50 of 253 nM.3 Contrary to effects on uptake, however, 1 enhanced the binding of a tropane radioligand [125I]methyl (1R,2S,3S)-3-(4-iodophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate (RTI-55, 23)3 to hDAT with an EC50 of 35 nM. The closely related derivatives 2 and 3 differing only in the terminal aryl group enhanced binding of [125I]23 with EC50 values of 9.1 and 70 nM, respectively. Compound 1 enhanced the affinity of radioligand binding at hDAT, with 100 nM of 1 reducing the Kd of [125I]23 from 1.3 nM to 0.39 nM. We now report the synthesis and in vitro structure-activity relationship (SAR) of an expanded class of nucleosides. Results indicate that disparate nucleoside structures are effective A3AR and transporter ligands with a unique pharmacology.
Chart 1.
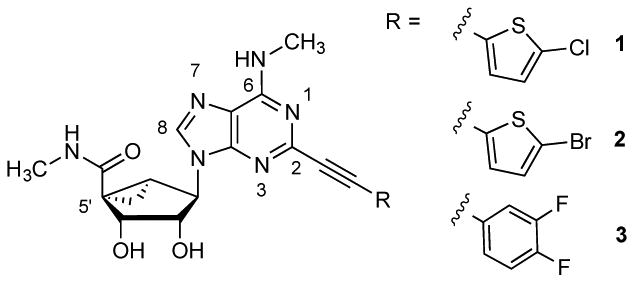
Structures of potent A3AR agonists that displayed activity at hDAT.
Results
Chemical synthesis
The class of nucleosides initially examined was based on our earlier report that (N)-methanocarba nucleosides, containing a rigid C2-arylethynyl group and 5′-carbonyl and N6-methyl substitution, allosterically modulate hDAT.3 Compounds 4,3 6 and 8 - 16, 22, and 27 - 29 were synthesized by the routes shown in Schemes 1 - 3. Known nucleosides 1 - 3, 5 and 7 were prepared as reported.2,21 The novel modifications included extended small N6- alkyl substitution (7, 28), 5′-esters (9 - 21), 5′-carboxylate (22), 5′ extended amide (8), deaza modifications of adenine (5, 6, and 13) and ribose restored in place of methanocarba (27 - 29), and combinations thereof (Table 1). For the (N)-methanocarba adenosine derivatives, 5′-ethyl ester intermediate 32 was prepared by a reported method,2 which upon Sonogashira reaction with 5-chloro or 5-bromo-2-ethynylthiophene gave compounds 33 and 34 (Scheme1). Deprotection of the acetonide group of compounds 33 and 34, with 10% aqueous TFA provided the ethyl ester derivatives 10 and 12. Compound 12 was subjected to transesterification with sodium methoxide to give compound 11. Compound 12 was saponified to provide carboxylic acid 22 or aminolyzed with ethylenediamine to yield extended amide 8. Acid derivative 22 was esterified with various alcohols in the presence of DCC and DMAP to afford 5′-esters 14 - 21.
Scheme 1.
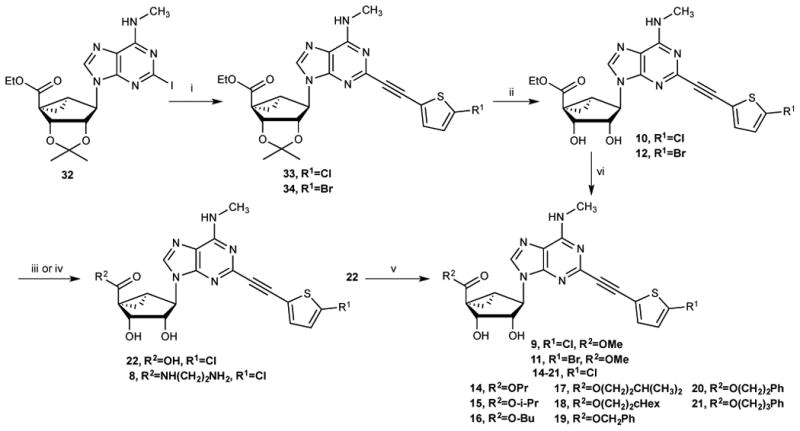
Synthesis of (N)-methanocarba 5′-esters 9-21, carboxylate 22, and extended amide 8 derivatives. Reagents and Conditions (% yields): (i) 2-Chloro or 2-bromo-5-ethynylthiophene, Pd(Ph3P)2Cl2, CuI, Et3N, DMF, rt, 82-83%; (ii) 10%TFA, MeOH, 70 °C, 90-91%; (iii) for 22: 1N NaOH, MeOH, rt, 82%; (iv) for 8: ethylenediamine, MeOH, rt, 71%; (v) for 14 - 21: ROH, DCC, DMAP, DMF, rt, 51-59%; (vi) for 9, 11: MeONa, MeOH, rt, 69%.
Scheme 3.
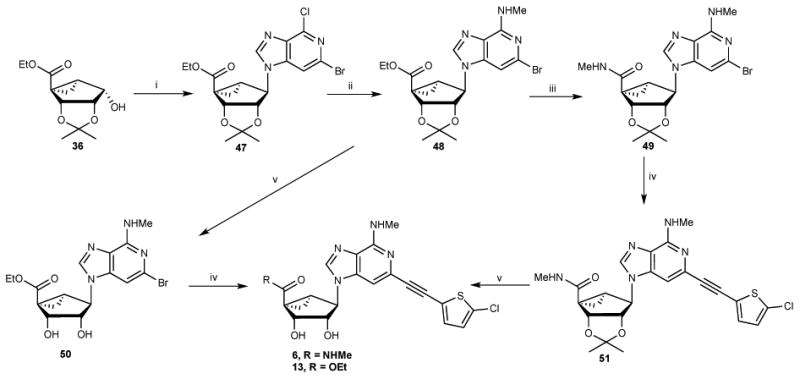
Synthesis of 3-deaza-(N)-methanocarbanucleosides 6 and 13. Reagent and Conditions (% yields): (i) 3-deaza-2-bromo-6-chloropurine, Ph3P, DIAD, THF, 69%; (ii) MeNH2.HCl, DIPEA, isopropanol, 125 °C, microwave, 76%; (iii) 40% MeNH2, MeOH, rt; (iv) 5-chloro-2-ethynylthiophene, PdCl2(Ph3P)2, CuI, DMF, 130 °C, μwave, 63%; (v) 10%TFA, MeOH, 70 °C, 90-91%.
Table 1.
Structures, initial screeing results at hDATa,b ([3H]24, unless noted) and binding affinity at A3ARd of (N)-methanocarba adenosine derivatives (1-22) and ribose derivatives (27 - 29). R2 = Cl; R3 = CH3; R4 = H; and X and Y = N, unless noted.
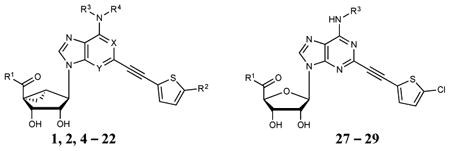
| ||||
|---|---|---|---|---|
| No. | R1, and other changes as noted | hDAT Binding Enhancement, unless noteda, % of control (n) | hDAT Binding Inhibition, unless notedb, % of control | A3AR Binding Ki, nM (species), or % at 10 μMd |
| 1d | NHCH3 | 556% | NE | 0.70±0.11 (h), 36.1±4.7 (m) |
| 2d | NHCH3, R2 = Br | 235% | NE | 0.44±0.12 (h), 43.7±2.1 (m) |
| 3d | NHCH3e | 322% | NE | 1.65±0.08 (h), 86±6 (m) |
| 4d | NHCH3, R4 = CH3 | NE | 25% | 23.5±10.6 (h), 597±63 (m) |
| 5ad | NHCH3, X = CH | c | c | 2.34±0.20 (h), 31.3±1.9 (m) |
| 5b | NHCH3, X = CH, R3 = CH2CH3 | c | c | 1.69 ± 0.44 (h), 16.2±3.0 (m) |
| 6 | NHCH3, Y = CH | 334±35% (5) | NE | 9.33 (h), 38±1% (m) |
| 7d | NHCH3, R3 = (CH2)2CH3 | 159% | NE | 1.11 ± 0.34 (h), 6.79±0.28 (m) |
| 8 | NH(CH2)2NH2 | 136±15% (5) | NE | 156±8 (h), 0% (m) |
| 9 | OCH3 | 352±50% (4) | NE | 5.38±0.03 (h), 36±1% (m) |
| 10 | OCH2CH3 | 539±106% (4) | NE | 14.5±2.3 (h), 45±5% (m) |
| 11 | OCH3, R2 = Br | 370±91% (5) | NE | 8.56±0.10 (h), 57±1% (m) |
| 12 | OCH2CH3, R2 = Br | 430±86% (3) | NE | 6.42±0.35 (h), 1800±90 (m) |
| 13 | OCH2CH3, Y = CH | 354% | NE | 169±24 (h), 18±4% (m) |
| 14 | O(CH2)2CH3 | 426±14% (2) | NE | 5.78±1.45 (h), 2810±150 (m) |
| 15 | OCH(CH3)2 | NE | 36% | 42.9±22.8 (h), 30±1% (m) |
| 16 | O(CH2)3CH3 | 285±35% (2) | NE | 17.5±1.6 (h), 54±1% (m) |
| 17 | O(CH2)2-CH(CH3)2 | 196 ±58% | NE | 24.4±2.8 (h), 32±2% (m) |
| 18 | O(CH2)2-cHex | c | c | 770±75 (h), 5±1% (m) |
| 19 | OCH2-Ph | c | c | 7.31±0.29 (h), 48±2% (m) |
| 20 | O(CH2)2-Ph | c | c | 262±44 (h), 3±2% (m) |
| 21 | O(CH2)3-Ph | c | c | 255±30 (h), 9±1% (m) |
| 22 | OH | 50±32% (4) | NE | 684±195 (h), 0% (m) |
| 27 | NHCH3 | 321±49% (4) | NE | 1.55±0.03 (h), 1170±40 (m) |
| 28 | NHCH3, R3 = (CH2)2CH3 | 84±35% (4) | NE | 6.25±1.55 (h), 597±63 (m) |
| 29 | OCH3 | 220±54% (4) | NE | 11.5±0.9 (h), 34±2 (m) |
Values indicate enhancement at 10 μM of [3H]24 binding at hDAT by PDSP, i.e. a negative percent inhibition in that screen is shown as a positive % here. Full curves for selected compounds appear in Figures 1 and 2 and in Supporting Information. Nonspecific binding was determined using 10 μM 30. Values are expressed as the mean ± SEM.
Values indicate inhibition at 10 μM of [3H]24 binding at hDAT by PDSP. Full curves for selected compounds appear in the Supporting information. Nonspecific binding was determined using 10 μM 30. Values are expressed as the mean ± SEM.
Found to have negligible or no modulation of [3H]24 binding at hDAT by PDSP, i.e. % inhibition at 10 μM was between -20% and 20% (see Figures 1 and 2).
Data for hA3AR and mA3AR affinity (n = 2 – 3) of 1 – 5 and 7 are from Tosh et al.2,18 and PDSP data (except for 5b) are from Janowsky et al.3 The radioligand used for A3AR in both species was [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide 25. Values are expressed as the mean ± SEM. Nonspecific binding was determined using 10 μM adenosine-5′-N-ethyluronamide.
Structure is in Chart 1.
NE – no effect.
9-Riboside analogues were prepared as shown in Scheme 2 from the intermediate 35a.22a Protection of the 2′ and 3′-hydroxy groups provided an acetonide derivative 35b, which underwent C6 substitution with alkyl amines to give compounds 37 and 38. PDC oxidation of the 5′-hydroxy group of compounds, 37 and 38, and coupling of resulting carboxylic acids, 39 and 40, with methylamine in presence of HATU afforded the methylamide derivatives 41 and 42. Compound 39 was also condensed with MeOH in presence DCC and DMAP to give the ester derivative 43. Sonogashira coupling of compounds 41-43 with 5-chloro-2-ethynylthiophene and subsequent acid hydrolysis provided compounds 27-29.
Scheme 2.
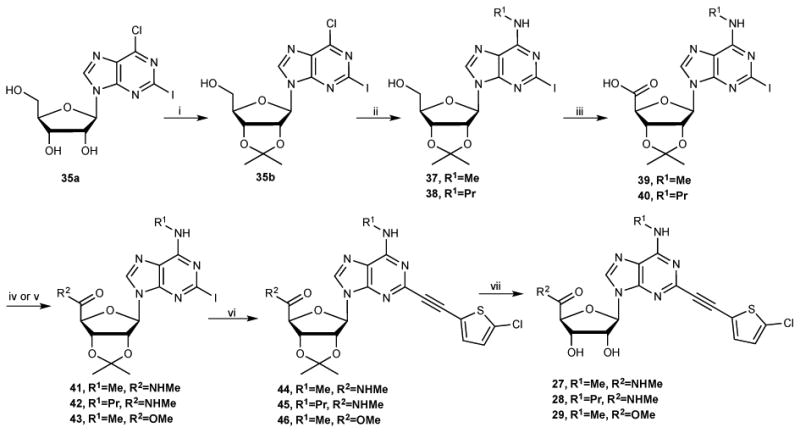
Synthesis of 5′-ester and 5′-methylamides 27-29 in the riboside series. Reagents and Conditions (% yields): (i) 2,2-dimethoxypropane, acetone, p-TSA, rt, 91%; (ii) RNH2, Et3N, MeOH, rt, 83-85%; (iii) PDC, DMF, 40 °C, 67-68%; (iv) for 41, 42: RNH2, HATU, DIPEA, DMF, rt, 68-69%; (v) for 43: MeOH, DCC, DMAP, DMF, rt, 52%; (vi) 2-chloro-5-ethynylthiophene, Pd(Ph3P)2Cl2, CuI, Et3N, DMF, rt, 82-84%; (iv) 10%TFA, MeOH, 70 °C, 90-91%.
The 3-deazaadenine derivatives (6 and 13) were synthesized from key bicyclo[3.1.0]hexane intermediate 3622b as depicted in Scheme 3. A Mitsunobu condensation of compound 35 with 3-deaza-2-bromo-6-chloropurine gave compound 47, which underwent a C6 substitution with MeNH2 under microwave conditions at 125 °C to give compound 48. Aminolysis of compound 48 with aqueous MeNH2 solution gave the methylamide 49, which was subjected to a Sonogashira coupling with 5-chloro-2-ethynylthiophene under microwave conditions at 130 °C to provide compound 51, and subsequent acid hydrolysis afforded the 3-deaza derivative 6. Acid hydrolysis of compound 48 gave the nucleoside derivative 50, which also underwent a Sonogashira coupling with 5-chloro-2-ethynylthiophene under microwave condition to afford compound 13.
Biological testing
In vitro assays
DAT binding screen
The nucleoside derivatives were screened initially by the Psychoactive Drug Screening Program (PDSP) for binding interactions at diverse receptors, ion channels and transporters (Table 1, Supporting Information).23 A fixed concentration of 10 μM was used in the primary screen for inhibition of radioligand binding at the hDAT. Because (N)-methanocarba nucleosides may enhance binding of a high affinity tropane derivative, [3H]methyl (1R,2S,3S)-3-(4-fluorophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate (WIN35,428, 24), at hDAT,3 a significant negative value for % inhibition of radioligand binding, i.e. greater binding than control, was also interpreted as a hit. The radioligands used for NET and SERT binding were [3H](R,S)-3-(2-methoxyphenoxy)-N-methyl-3-phenylpropan-1-amine (nisoxetine, 52) and [3H](R,S)-1-[3-(dimethylamino)propyl]-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile (citalopram, 53), respectively. In the secondary screen by the PDSP, compounds that inhibited binding by >50% or significantly enhanced binding were characterized in full concentration response curves.
A secondary screen of selected binding enhancers allowed a more precise comparison of their interaction with hDAT and in few cases, other transporters. Figures 1 and 2 show the full concentration response curves for % enhancement of [3H]24 binding by selected nucleosides (6, 8, 10, 11, 14, 15, 22, 27-29) in comparison to known inhibitor 30. EC50 values for 7, 8, 15, 22 and 28 were >10-6 M; 1.2 μM for 16; 400 nM for 12. Significant (>200%) enhancement of binding (% at 10 μM) was seen with compounds 6 (334%), 9 (352%), 10 (539%), 11 (370%), 12 (430%), 13 (354%), 14 (426%), 16 (285%), 27 (321%), and 29 (220%). i-Propyl (15) ester slightly inhibited binding of [3H]24 at 10 μM, while 5′-propyl ester 14 enhanced binding; thus, branching of the α-carbon in 5′-esters was not tolerated. Among extended 5′-alkyl and 5′-alkylaryl esters (17 - 21), only the i-pentyl analogue 17 significantly interacted with hDAT (196% enhancement at 10 μM). 5′-Carboxylate (22) and N6-propyl riboside (28) interacted only weakly at hDAT at 10 μM, indicated by enhancement of [3H]24 binding of <100%. 5′-N-(2-aminoethyl)-carboxamide 8 (133% enhancement of [3H]24 binding) was not comparable in efficacy to the corresponding 5′-methylamide 1 at hDAT. 5-Bromothien-2-yl 5′-ethyl ester derivative 12 appeared to be comparable to reference 5-chlorothien-2-yl derivative 1 in enhancing hDAT binding. Thus, 5-bromo is a suitable substitution for 5-chloro with respect to hDAT interaction (also evident in 2 compared to 1).3 Curiously, benzyl ester 19 inhibited binding at SERT with a Ki value of 5.6 μM, and 28 inhibited NET binding with a Ki of 3.1 μM, and none of the other newly synthesized nucleosides inhibited binding at these two transporters with Ki < 10 μM.
Figure 1.
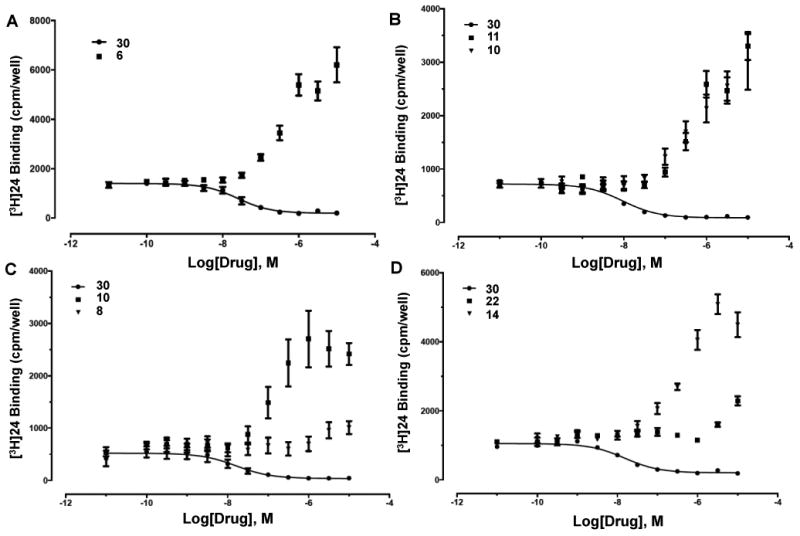
Drug-induced enhancement of [3H]24 binding to hDAT (PDSP) by (N)-methanocarba analogues (representative of 2 - 5 determinations): (A) 3-deaza-5′-methylamide 6; (B) 5′-ethyl ester 10 and 2-(5-bromothienylethynyl)-5′-methyl ester 11; (C) 2-(5-chlorothienylethynyl)-5′-N-(2-aminoethyl)-carboxamide 8 and its 5′-ethyl ester analogue 10; (D) 5′-n-propyl ester 14 and 5′-carboxylic acid 22. Comparison with binding inhibition by 1-[2-[bis(4-fluorophenyl)methoxy]ethyl]-4-(3 phenylpropyl)-piperazine (GBR12909, 30) is shown.
Figure 2.
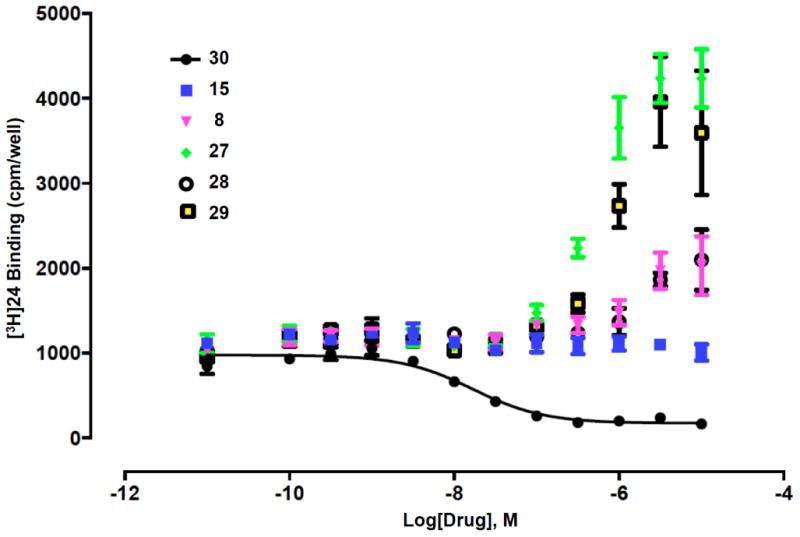
(A) Drug-induced enhancement of [3H]24 binding to hDAT (PDSP) by ribosides 27 - 29 (representative of 4 determinations) in comparison to weak enhancers (N)-methanocarba 5′-N-(2-aminoethyl)-carboxamide 8 and 5′-isopropyl ester 15. Comparison with binding inhibition by 30 is shown.
Various 1- or 3-deaza derivatives (5, 6, and 13) were compared to probe the participation of these H-bond acceptor nitrogens of adenine for recognition at hDAT. The 1-deaza modification (5a and 5b) prevented enhancement of [3H]24 binding at hDAT, but not A3AR activation.3,21 The corresponding 3-deaza 5′-methylamide 6 significantly enhanced [3H]24 binding at hDAT, but with less efficacy than 1. Thus, the N1, but not N3, nitrogen atom was essential for interaction with hDAT.
Ribosides 27 and 29 displayed % enhancement of [3H]24 binding at 10 μM of 321% and 220%, respectively, which were less than the corresponding (N) methanocarba analogues (1 and 9, with enhancement of 556% and 391%, respectively). Compound 28 inhibited NET binding with a Ki value of 3.1 μM (Supporting Information).
Detailed characterization at DAT, NET and SERT
The PDSP screen led to a selection of strongly enhancing compounds that were further characterized in greater detail at hDAT and the other transporters (e.g. binding of [125I]23 at DAT, NET and SERT and functional activity at each). Selected compounds were also characterized for their effect on binding of [3H](±)-5-(4-chlorophenyl)-3,5-dihydro-2H-imidazo[2,1-a]isoindol-5-ol (mazindol, 26). The latter detailed pharmacological parameters for the novel compounds are reported in Table 2 in comparison to compounds 1, 2, 3 and 7, which were characterized in our previous study.3
Table 2.
Affinities, potencies and efficacies of adenosine derivatives for hDAT, hNET and hSERT (n = 3-6).
| No. | hDAT | hNET | hSERT | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| [125I]23 | [3H]26 | [3H]DA | [125I]23 | [3H]26 | [3H]NE | [125I]23 | [3H]26 | [3H]5-HT | |
| binding, | binding, | uptake, | binding, | binding, | uptake, | binding, | binding, | uptake, | |
| EC50a or Ki (nM), | EC50 or Ki (nM), | IC50 (nM) | EC50 or Ki (nM), | EC50 or Ki (nM),c | IC50 (uM) | EC50 or Ki (nM),c | EC50 or Ki (nM).c | IC50(nM)c | |
| % control | % control | % control | % control | % control | % control | ||||
| binding | binding | binding | binding | binding | binding | ||||
| 1b | 35.1 ± 8.4a | 131 ± 31a | 253 ± 92 | 1180 ± 360a | >10 μM | >10 μM | >10 μM | >10 μM | >10 μM |
| 550 ± 110% | 403 ± 33% | 386 ± 73% | NE | NE | NE | ||||
| 2b | 9.1 ± 1.7a | 870 ± 130a | 229 ± 30 | 670 ± 200a | >10 μM | 6110 ± 570 | >10 μM | >10 μM | >10 μM |
| 217 ± 24% | 369 ± 31% | 285 ± 22% | NE | NE | NE | ||||
| 3b | 70 ± 26a | 128 ± 18a | 92 ± 16 | 1760 ± 640a | >10 μM | >10 μM | >10 μM | >10 μM | >10 μM |
| 690 ± 180% | 438 ± 41% | 371 ± 67% | NE | NE | NE | ||||
| 6 | 239 ± 79a | 550 ± 180a | 163 ± 42 | 740 ± 190a | >10 μM | 5060 ± 360 | >10 μM | >10 μM | >10 μM |
| 437 ± 60% | 405 ± 18% | 318 ± 22% | NE | NE | |||||
| 7b | 1120 ± 220a | ND | >8700 | 900 ± 320a | ND | ND | ND | ND | ND |
| 268 ± 32% | ND | 159 ± 1% | |||||||
| 9 | 35 ± 13a | 124 ± 21a | 127 ± 40 | 310 ± 110a | >10 μM | 3380 ± 290 | >10 μM | >10 μM | >10 μM |
| 458 ± 71% | 540 ± 33% | 375 ± 24% | NE | NE | |||||
| 10 | 34 ± 13a | 267 ± 80a | 107 ± 14 | 490 ± 120a | >8,400 | 3980 ± 330 | >10 μM | >10 μM | >10 μM |
| 434 ± 64% | 455 ± 39% | 365 ± 11% | NE | NE | |||||
| 11 | 53.5 ± 9.8a | 308 ± 20a | 246 ± 10 | 400 ± 130a | >10 μM | 3560 ± 500 | >10 μM | >10 μM | >10 μM |
| 374 ± 50% | 520 ± 100% | 293 ± 32% | NE | NE | |||||
| 14 | 294 ± 82a | 535 ± 29a | 1145 ± 52 | 980 ± 270a | >10 μM | 7900 ± 1400 | >10 μM | >10 μM | >10 μM |
| 461 ± 67% | 347 ± 60% | 302 ± 24% | NE | NE | |||||
| 29 | 127 ± 32a | 860 ± 180a | 396 ± 83 | 1520 ± 180a | >10 μM | 2690 ± 220 | >10 μM | >10 μM | >10 μM |
| 312 ± 45% | 269 ± 28% | 227 ± 18% | NE | NE | |||||
| Cocaine | 720 ± 140 | 1270 ± 530 | 274 ± 31 | 2410 ± 590 | 10,700 ± | 221 ± 13 | 657 ± 56 | 229 ± 56 | 277 ± 24 |
| NE | NE | NE | 1100 | NE | |||||
| 23 | ND | 11.0 ± 2.9 | ND | ND | 70.0 ± 8.8 | ND | ND | 1.27 ± 0.29 | ND |
| NE | NE | ||||||||
| 26 | 34.6 ± 5.7 | 74.2 ± 8.4 | 13.0 ± 0.6 | 11.3 ± 3.5 | 13.5 ± 1.5 | 0.92 ± 0.10 | 136 ± 15 | 45 ± 10 | 54 ± 11 |
| NE | NE | NE | NE | NE | |||||
The EC50 value for drugs that enhanced radioligand binding.
Data from Janowsky et al.3
Ki values are provided for drugs that inhibited binding.
NE, No enhancement of binding.
ND, Not determined.
5′-Methyl (9) and ethyl (10) esters containing a 5-chlorothienyl moiety enhanced [125I]23 binding to hDAT equipotently to a maximal level ∼450% of control binding (absence of drug), with EC50 values of ∼35 nM (Figure 3). Interestingly, 9 and 10 also enhanced [125I]23 binding to NET to a maximal level ∼370% of control, with EC50 values of 310 and 490 nM, respectively (Table 2). The effects of 9 and 10 on SERT binding of [125I]23 were small (inhibition at >10 μM). Contrary to their enhancing effects on radioligand binding to hDAT, compounds 9 and 10 were potent DA uptake inhibitors, with IC50 values of 127 and 107 nM, respectively, compared with 274 nM for cocaine and 13.0 nM for 26. Thus, the potencies of esters 9 and 10 in inhibiting DA uptake were ∼2-fold greater than the corresponding methylamide 1. Compounds 9 and 10 were less potent at inhibiting norepinephrine uptake by the NET with IC50 values of 3.38 and 3.98 μM, respectively, compared with 221 nM for cocaine and 0.92 nM for 26. The inhibitory effect of both compounds on 5HT uptake was estimated as >10 μM.
Figure 3.
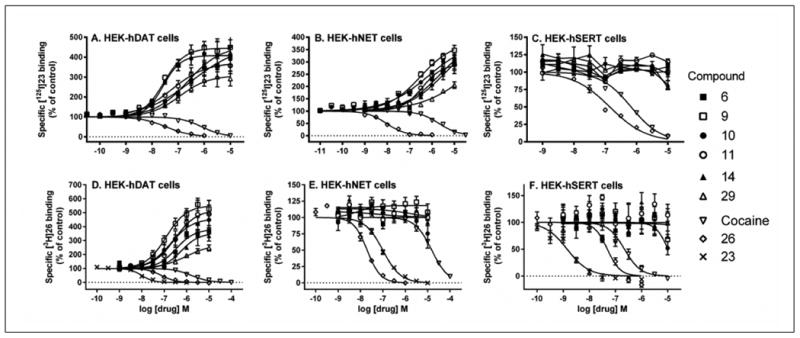
Drug-induced enhancement or inhibition of [125I]23 binding and [3H]26 binding to HEK-hDAT, HEK-hNET and HEK-hSERT cell membranes.
Compounds with intermediate potency in enhancing binding of [125I]23 at hDAT (in order of increasing EC50, nM) were: 5-bromothienyl 5′-ethyl ester 11 (53.5), ribose 5′-methyl ester 29 (127), 3-deaza 5′-methylamide 6 (239) and 5′-propyl ester 14 (294). At NET, compounds 6, 11 and 14 were roughly proportionately weaker at NET (EC50 ∼400 nM - 1 μM) than at hDAT, except the ribose derivative 29 was disproportionately weak with EC50 > 1 μM. However, the newly synthesized 5′-ester compounds tended to be more potent in enhancement of [125I]23 binding to NET than the previous set of 5′-methylamides 1 - 3.
Five nucleoside derivatives were tested in binding and functional assays at the mouse (m) DAT expressed in HEK-293 cells (Table 3).24 Cocaine was comparable in inhibiting binding at mDAT and at hDAT, but 26 was 12-fold more potent at hDAT than mDAT. The radioligand [125I]23 was less potent in binding to mDAT than hDAT with a 3-fold higher Kd value (3.9 vs 1.3 nM). Qualitatively, the nucleosides acted similarly at mDAT and hDAT in that they enhanced [125I]23 binding and inhibited [3H]DA uptake. At the fixed concentration of radioligand, the percent of maximal binding enhancement by all of the compounds in the mDAT experiments was greater than at hDAT. However, all of the adenosine derivatives were less potent at mDAT than at hDAT in both enhancing binding and inhibiting uptake. The rank order of EC50 values (decreasing affinity in binding) was 9 > 2, 10 > 1 > 6 at mDAT compared to 2 > 1, 9, 10 > 6 at hDAT. The factors for inhibition by the nucleosides of DA uptake, between hDAT and mDAT, were 7- to 26-fold. The two 5′-amide derivatives 1 and 2 displayed affinity and potency of 1 - 3 μM at mDAT. Thus, those compounds remain somewhat selective for the A3AR compared to DAT in both human and mouse. However, two nucleoside 5′-esters 9 and 10 and one 5′-amide 6, which were inactive in binding to the mA3AR (<50% inhibition at 10 μM), still displayed considerable activity (1 - 4 μM) at mDAT. Thus, moderate DAT selectivity was indicated, at least in one species.
Table 3.
Affinities, potencies and efficacies of adenosine derivatives for mDAT expressed in HEK-293 cells.
| No. | [125I]23 binding, EC50 ± SEM (nM) | Maximal % control [125I]23 binding | [3H]DA uptake, IC50 ± SEM (nM) |
|---|---|---|---|
| 1 | 2230 ± 430 | 1150 ± 60% | 2290 ± 470 |
| 2 | 1100 ± 290 | 810 ± 110% | 3260 ± 830 |
| 6 | 2760 ± 310 | 1000 ± 0% | 4300 ± 1000 |
| 9 | 810 ± 270 | 1440 ± 190% | 940 ± 130 |
| 10 | 1070 ± 320 | 1560 ± 200% | 1170 ± 150 |
| [125I]23 binding Ki ± SEM (nM) | |||
| Cocaine | 850 ± 170 | NA | 200 ± 24 |
| 26 | 404 ± 88 | NA | 13.0 ± 2.6 |
NA, not applicable.
5′-Esters 9, 10 and 29 were also compared for their effect on hDAT binding of a nontropane radioligand, [3H]26 (Figure 3). The enhancement of [3H]26 binding by riboside 5′-methyl ester 29 was weaker and less efficacious, with a 7-fold higher EC50 value and half the maximal effect, compared to the corresponding (N)-methanocarba analogue 9. Furthermore, riboside 28 inhibited binding of nontropane radlioligand [3H]52 to NET with a Ki value of 3.1 μM (data not shown).
Effects of the nucleosides on uptake of radiolabeled neurotransmitters were measured in the cell lines expressing hDAT and hNET. The order of potency in inhibiting DA uptake was: 3, 10, 9 > 1, 2, 11 > 14. The order of potency in inhibiting norepinephrine uptake was: 9, 10, 11 > 6, 2 > 1, 3, and there was no significant inhibition of 5HT uptake. Thus, 5′ esters 9 and 10 were among the most potent to inhibit uptake of both catecholamines. 10 was more potent than cocaine at inhibiting DA uptake (IC50 = 107 nM).
A3AR binding in two species
Because the reference compounds 1 - 3, 5 and 7 were highly selective agonists for the hA3AR with nM affinity and were nearly inactive at the other AR subtypes, we measured the binding affinity at recombinant hA3AR (Table 1). The nucleosides were also tested at the mouse (m) A3AR, because of the known phenomenon of species-dependent variation of affinity at this receptor.2,18 A standard agonist radioligand ([125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide 25) binding assay was used,2 and IC50 values were converted to Ki values as reported.25
Among 5′-ester derivatives, considerable affinity was observed at the hA3AR, but the mA3AR affinity was greatly reduced compared to amides. For example, 5′-methylamide 1 was 8-fold more potent than corresponding 5′-methyl ester 9 at the hA3AR, but >300-fold at the mA3AR (extrapolated Ki of 9 > 10 μM). Similarly, larger esters that contained the full complement of the adenine nitrogens 10 - 12 and 14 - 16 were weak or inactive at the mA3AR. The n-propyl ester 14 displayed a Ki value of 2.81 μM at the mA3AR.
Replacement of the adenine 3-nitrogen atom with a CH group in 6 prevented recognition at the mA3AR but not at the hA3AR, which registered only a 13-fold reduction of affinity compared to 1. In general, adenine ring-modified analogues displayed affinity at the hA3AR in the order: adenine ≥ 1-deaza > 3-deaza.
This expanded set of nucleoside analogues also provided an opportunity to compare (N)-methanocarba nucleosides directly with the corresponding ribose analogues at the A3AR in two species. In general, 2- to 5-fold greater hA3AR affinity was associated with the methanocarba analogues compared to the equivalent ribose derivatives (cf. 1 versus 27; 7 versus 28; 9 versus 29). At the mA3AR, the differences in affinity were even greater, for example, 88-fold greater affinity of bicyclic 5′-amide 7 compared to ribose 5′-amide 28. Curiously, in the ribose series, the mA3AR affinity was at least one order of magnitude higher for a 5′-ester 29 than for the conventional 5′-methylamide agonists of this receptor, i.e. 27 and 28. The same comparison in the (N)-methanocarba series showed the opposite; in contrast to the potent 5′-methylamide 1, 5′-ester 9 was nearly inactive at the mA3AR, but it still bound with high affinity at the hA3AR (Ki = 5.38 nM). Thus, the dependence of mA3AR affinity on the (N)-methanocarba modification vs. ribose was dramatically reversed between amides and esters.
In a functional assay of hA3AR-mediated inhibition of cAMP formation (n=3),18 methyl ester 9 was a low efficacy partial agonist and 3-deaza ester 13 had no significant activity with maximal efficacies (at 10 μM) of 11.6±1.0% and 3.6±3.0%, respectively, compared to reference agonist adenosine-5′-N-ethyluronamide set at 100%. However, the 3-deaza analogue 6 of full A3AR agonist 4 was also a full agonist (116.5±14.6%).
In vivo testing
Since some of these nucleosides still displayed moderate affinity at the A3AR, although diminished affinity compared to reference compound 1, several compounds were evaluated in the mouse chronic constriction injury (CCI) model of neuropathic pain27 (Figure S2, Supporting Information). This activity was shown in our previous studies to associate closely with the activity of nucleosides as A3AR agonists at peripheral and/or central sites.1,2,18-20 Mice were treated with an adenosine derivative at 7 d post-injury, i.e. the time of peak mechanoallodynia. Riboside analogues 27 and 28 (3 μmol/kg, p.o.) were fully efficacious efficacious with a long duration of action (>6 h) in increasing the paw withdrawal threshold (PWT), i.e. reducing pain.
Other off-target binding
Other off-target interactions at receptors and channels were detected for the derivatives in the PDSP screening (Figure S1, Supporting information). Compounds 11 (Ki = 258 nM), 13 (Ki = 395 nM), 15 (Ki = 430 nM) and 29 (Ki = 3.24±1.05 μM) bound to the h5HT2C serotonin receptor. By analogy to other methanocarba nucleoside derivatives,20,28 we expect these compounds to act as 5HT2C antagonists. At the α-adrenergic receptors (α-Rs) ribosides but not the corresponding methanocarba analogues bound weakly to hα2A-R (27), hα2B-R (29) or both (28) with Ki values in the 5-10 μM range. Ki values at all other α-adrenergic receptors were >10 μM. Compounds 7 (Ki = 1.44 μM) bound to the hβ3 adrenergic receptor. Compounds 13 also bound to hM5 muscarinic (Ki = 3.8 μM) and rat sigma1 (Ki = 1.04 μM) and rat sigma2 (Ki = 1.90 μM) receptors; 20 bound to sigma2 (Ki = 5.9 μM) receptors.
Discussion
Adenosine derivatives have been designed as agonists and in some cases partial agonists or antagonists of the ARs. A3AR agonists are being developed for treatment of cancer, inflammation and pain,1,29-31 but they also display off-target activities at higher concentrations.3,9 We have explored and enhanced the off-target activity of nucleosides, especially those having rigid methanocarba rings, and found them to serve as a privileged scaffold32 for a variety of targets. Targets other than neurotransmitter symporters that appear to be binding sites of moderate affinity in this chemical series are GPCRs for biogenic amines, TSPO and sigma receptors.20
The SAR in this study extends our previous findings that (N)-methanocarba adenosine derivatives enhance DAT radioligand affinity and inhibit DA uptake. Most of the compounds synthesized here contain C2-(5-halothien-2-yl)-ethynyl and N6-methyl substitution, which were determined to promote DAT interaction in the previous study.3
The three transporters for neurotransmitters examined in this study possess closely related structures, each having 12 transmembrane helices.6,7,33 The cocaine-competitive tropane ligands that display high affinity at DAT also tend to interact with NET and SERT.34 Our previous study of the off-target activity of (N)-methanocarba nucleoside derivatives at DAT was consistent with allosteric modulation of radioligand binding.3 We found similar results in this study, in which potent DAT modulation remained while the AR potency of key analogues was reduced in comparison to 1, by structural changes. Percent enhancement of binding at DAT was similar using two different tropane radioligands. These nucleosides had differing interactions with the transporters: at DAT they enhanced both [125I]23 and [3H]26 binding, at NET only [125I]23 binding was enhanced, while they had minimal effect on radioligand binding to SERT. Thus, as is characteristic of allosteric modulators, there was a probe-dependence of the interaction of these nucleosides at DAT and NET. Allosteric modulation of SERT is well validated structurally,33 and that of NET has been described; for example, a peptide inhibitor was suggested to bind to the extracellular vestibule of the transporter.35,36
Nevertheless, many of the compounds still interact with the A3AR, as evident from their binding affinity and protective effects, as agonists, in a pain model. Reference compounds 1 - 3, 5 and 7 were shown previously to be potent A3AR agonists and to reduce chronic pain in the CCI model, an effect of A3AR agonists that was shown to depend entirely on the A3AR as demonstrated using A3-/- mice.1,21 However, in this study we have characterized the simultaneous activity at transporters of various A3AR ligands of nM affinity. The potency of 1, for example, as an allosteric enhancer of hDAT binding is 50-fold weaker than its potency at the hA3AR. One objective of this study was to preserve or enhance DAT potency while decreasing A3AR affinity, so we modified the nucleosides at positions that might be predicted to lower the A3AR affinity. This was accomplished for the 5′-ester series, for which the DAT potency was maintained, and the A3AR affinity was reduced (human) or absent (mouse). 5′-Esters reduced A3AR affinity in general, compared to 5′-monoalkyl amides.37 Consistently, compound 9 was 8-fold less potent than 1 at the hA3AR, and the difference was even greater at the mA3AR. Also, the ribose 5′ binding region of ARs is limited sterically, based on X-ray structures.38 Thus, the extended, charged 5′-(2-aminoethyl)-amide 8 displayed only moderate affinity at the hA3AR and was completely inactive at the mA3AR. However, it interacted only weakly with DAT.
Furthermore, dimethylation of the exocyclic amine in 4, as noted previously,3 was more detrimental to DAT interaction than to A3AR affinity. The deaza analogues displayed differential effects at the two primary targets compared in this study. The 1-deaza (5) modification is a means of introducing A3AR selectivity compared to DAT with no loss of A3AR affinity.21 The 3-deaza (6) modification of 5′-methylamide 1 decreased A3AR affinity (13-fold lower than 1 at human and nearly inactive at mouse), while at DAT significant binding enhancement was present. The 3-deaza modification in the 5′-amide series (6) improved NET potency, but reduced DAT potency 7-fold (cf. 1). The EC50 of 6 for [125I]23 binding at DAT was only 6.8-fold higher than 1 and 4.2-fold higher for [3H]26 binding. Thus, the 1-deaza (5), but not the 3-deaza (6 and 13) modifications are a means of eliminating DAT activity in this series. We speculate that N1 might serve as an energetically important H-bond acceptor in DAT.
5′-Methyl, ethyl and n-propyl esters, but not larger esters, maintained efficacious DAT interaction. The substitution of a 5-bromo in the C2-thienylethynyl group (11) maintained NET and DAT potency in the 5′-ester series, but did not improve DAT potency as it did in the amides (cf. 2). The enhancing activity at mDAT of selected compounds was examined for a comparison with the A3AR within the same species.39 3-Deaza 5′-amide 6, 5′-methyl ester 9 and 5′-ethyl ester 10 were selective for the enhancement of binding at the mDAT with respect to the mA3AR. These compounds were nearly inactive at mA3AR, indicating that the 5′-ester group is not tolerated in general (except for bromo derivative 12 and propyl ester 14). The potency of N6-propyl derivative 7 was slightly enhanced compared to 1 at NET, but greatly reduced at DAT. Therefore, at DAT there appear to be sterically limited binding subpockets surrounding the 5′-alkylamide, 5′-alkyl ester and N6-alkyl substituents. Future studies of DAT modeling will distinguish the specific amino acids and molecular interactions involved in radioligand binding as compared to transporter function.
At the A3AR, we established importantly that the (N)-methanocarba modification enhances affinity in direct comparison to the riboside. However, there was no advantage to the DAT interaction upon restoring the ribose moiety, because the potency of ribosides was also diminished, by ∼5-fold, at the DAT and NET. Our present results establish that various 5′-ester derivatives with extended C2 substituents can display potent affinity at the hA3AR, which was not anticipated from our earlier report on 2-Cl derivatives.40 Although the ester derivatives 9-16 bound at the hA3AR with Ki values of 5 - 43 nM (13: 169 nM), two of them (9 and 13) were shown to have little or no ability to activate the receptor, suggesting that they may be A3AR antagonists. Similarly, several weakly binding adenosine 5′-ester derivatives (methanocarba series) were shown to be either partial agonist or antagonist at the hA3AR28; thus, 5′-amides but not 5′-esters tend to fully activate the hA3AR.
Additional structural modification would likely be required before this nucleoside series could be considered for therapeutic use. We reported that mixed DAT enhancer/A3AR agonist 1 at 10 mg/kg did not significantly stimulate locomotor activity in the mouse, and at 30 mg/kg reduced locomotor activity.41 The observed locomotor depression can be attributed to a peripheral action of 1 at a peripheral A3AR. Our earlier study showed that decreased physical activity with high doses A3AR agonists in the mouse correlated with histamine release by peripheral mast cells upon A3AR activation.41 Also, we predict that nucleoside 1 (MW 459, total polar surface area, tPSA = 121.9 Å2) and related congeners will display a low brain concentration that is typical of peripherally-administered nucleosides.42 Thus, in order to study the CNS effects26 of these modulators, especially 9 and 10 (both having tPSA = 119.1 Å2) that are DAT-selective in the mouse, it would be more informative to infuse them intracerebroventricularly in rodents.
Peripherally-selective inhibitors of DA and norepinephrine uptake might be useful as an emergency treatment of lung edema, to act as an inotropic agent in the heart and to increase kidney/splanchnic circulation, similar to currently used injection of biogenic amine receptor agonists, notably DA at medium doses.43-45 Alternatively, an allosteric modulator of DAT that did act centrally might have less evident behavioral stimulant effects than orthosteric inhibitors.
We have not yet studied the pharmacokinetics of 5′-esters that were shown here to be allosteric modulators of DAT. However, the in vivo stability of the corresponding 5′-ester group was shown for another member of this series of rigid (N)-methanocarba nucleosides in the context of 5HT2BR antagonists.28 Also, amide derivatives in the present set of compounds, such as orally administered 1, were shown to be bioavailable and have long lasting in vivo effects in mice and are not rapidly metabolized.2,19 These rigid nucleosides, such as 1, typically do not inhibit cytochrome P450 (5 isozymes) and are stable in the presence of liver microsomes, plasma and simulated body fluids.2 A metabolomic study of 1 indicated that the compound was well tolerated in vivo.46
In conclusion, following structural modification of our previous series of nucleoside modulators of DAT, we maintained or enhanced the activity of rigid nucleosides at DAT and NET while reducing A3AR affinity. Two nucleoside 5′-esters 9 and 10 and one 5′-amide 6 that were inactive at the mA3AR were shown to allosterically modulate mDAT in the μM range. These compounds, such as 9 and 10, will be useful as research tools for studying the structure and function of DAT and NET.
Experimental Procedures
Chemical synthesis
Materials and instrumentation
All reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). 1H NMR spectra were obtained with a Bruker 400 spectrometer using CDCl3, CD3OD and DMSO as solvents. Chemical shifts are expressed in δ values (ppm) with tetramethylsilane (δ 0.00) for CDCl3 and water (δ 3.30) for CD3OD. NMR spectra were collected with a Bruker AV spectrometer equipped with a z-gradient [1H, 13C, 15N]-cryoprobe. TLC analysis was carried out on glass sheets precoated with silica gel F254 (0.2 mm) from Sigma-Aldrich. The purity of final nucleoside derivatives was checked using a Hewlett−Packard 1100 HPLC equipped with a Zorbax SB-Aq 5 μm analytical column (50 × 4.6 mm; Agilent Technologies Inc., Palo Alto, CA). Mobile phase: linear gradient solvent system, 5 mM tetrabutylammonium dihydrogenphosphate)−CH3CN from 80:20 to 0:100 in 13 min; the flow rate was 0.5 mL/min. Peaks were detected by UV absorption with a diode array detector at 230, 254, and 280 nm. All derivatives tested for biological activity showed >95% purity by HPLC analysis (detection at 254 nm). The nucleoside products tested in biological assays do not contain any functional groups associated with assay interference compounds,47 and they display self-consistent potent structure activity relationships, with very few off-target hits in broad screening. Low-resolution mass spectrometry was performed with a JEOL SX102 spectrometer with 6-kV Xe atoms following desorption from a glycerol matrix or on an Agilent LC/MS 1100 MSD, with a Waters (Milford, MA) Atlantis C18 column. High resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine, unless noted. Observed mass accuracies are those expected based on known performance of the instrument as well as trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time-dependent drift in mass accuracy. 6-Chloro-2-iodopurine-9-riboside 35a was purchased from Matrix Scientific, Inc. (Columbia, SC). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO), Small Molecules, Inc. (Hoboken, NJ), Anichem (North Brunswick, NJ), PharmaBlock, Inc. (Sunnyvale, CA), Frontier Scientific (Logan, UT) and Tractus (Perrineville, NJ). Calculation of total polar surface area was performed using ChemDraw Professional (v. 15.1, PerkinElmer, Boston, MA).
(1S,2R,3S,4R,5S)-4-(6-((5-Chlorothiophen-2-yl)ethynyl)-4-(methylamino)-1H-imidazo[4,5-c]pyridin-1-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide (6)
Compound 6 (90%) was prepared from compound 51 following the same method as for compound 50. 1H NMR (CD3OD, 400 MHz) δ 8.11 (s, 1H), 7.20 (d, J =4.0 Hz, 1H), 7.17 (s, 1H), 6.98 (d, J =4.0 Hz, 1H), 4.81 (d, J =6.4 Hz, 1H), 4.74 (s, 1H), 3.93 (d, J =6.4 Hz, 1H), 3.10 (s, 3H), 2.87 (s, 3H), 2.25-2.21 (m, 1H), 1.94 (t, J =5.2 Hz, 1H), 1.46-1.44 (m, 1H). HRMS calculated for C21H21N5O3SCl (M + H) +: 458.1054; found 458.1058.
(1S,2R,3S,4R,5S)-N-(2-Aminoethyl)-4-(2-((5-chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxamide (8)
Ethylenediamine (1.5 mL) was added to a solution of compound 10 (13 mg, 0.027 mmol) in MeOH (0.1 mL) and stirred for 2d at room temperature. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH:NH4OH = 5:1:0.1) to give the compound 8 (9 mg, 71%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.12 (s, 1H), 7.33 (d, J = 4.0 Hz, 1H), 7.04 (d, J = 4.0 Hz, 1H), 5.08 (d, J = 6.8 Hz, 1H), 4.83 (s, 1H), 4.05 (d, J = 6.8 Hz, 1H), 3.60-3.49 (m, 2H), 3.19-3.09 (m, 3H), 2.19-2.16 (m, 1H), 1.94 (t, J = 4.8 Hz, 1H), 1.49-1.46 (m, 1H). HRMS calculated for C21H23N7O3SCl (M + H) +: 488.1272; found 488.1273.
Methyl (1S,2R,3S,4R,5S)-4-(2-((5-chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (9)
DCC (32 mg, 0.15 mmol), DMAP (2.5 mg, 0.02 mmol) and MeOH (10 μL, 0.15 mmol) was added to a solution of compound 22 (23 mg, 0.05 mmol) in DMF (1 mL) and stirred at room temperature overnight. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 30:1) to afford the compound 9 (12 mg, 51%) as a colorless syrup.1H NMR (CD3OD, 400 MHz) δ 8.03 (s, 1H), 7.33 (d, J = 4.0 Hz, 1H), 7.02 (d, J = 4.0 Hz, 1H), 5.22 (d, J = 6.8 Hz, 1H), 4.84 (s, 1H), 4.09 (d, J = 6.8 Hz, 1H), 3.79 (s, 3H), 3.13 (br s, 3H), 2.23-2.20 (m, 1H), 1.92 (t, J = 5.2 Hz, 1H), 1.66-1.62 (m, 1H). HRMS calculated for C20H19N5O4SCl (M + H) +: 460.0846; found 460.0850.
Ethyl (1S,2R,3S,4R,5S)-4-(2-((5-chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (10)
A solution of compound 33 (103 mg, 0.2 mmol) in methanol (3 mL) and 10% trifluromethane sulfonic acid (3 mL) was heated at 70 °C for 4 h. Solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 30:1) to give the compound 10 (86 mg, 91%) as a colorless syrup.1H NMR (CD3OD, 400 MHz) δ 8.08 (s, 1H), 7.35 (d, J = 4.0 Hz, 1H), 7.04 (d, J = 4.0 Hz, 1H), 5.21 (d, J = 6.8 Hz, 1H), 4.84 (s, 1H), 4.27-4.22 (m, 2H), 4.12 (d, J = 6.4 Hz, 1H), 3.14 (br s, 3H), 2.23-2.20 (m, 1H), 1.91 (t, J = 5.2 Hz, 1H), 1.67-1.63 (m, 1H), 1.29 (t, J = 7.2 Hz, 3H). HRMS calculated for C21H21N5O4SCl (M + H) +: 474.1003; found 474.1007.
Methyl (1S,2R,3S,4R,5S)-4-(2-((5-bromothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (11)
MeONa (7.6 mg, 0.14 mmol) was added to a solution of compound 12 (25 mg, 0.048 mmol) in MeOH (3 mL) and stirred at room temperature overnight. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (hexane:ethylacetate = 1:1) to give the methyl ester derivative 11 (16 mg, 69%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.02 (s, 1H), 7.28 (d, J = 4.0 Hz, 1H), 7.14 (d, J = 4.0 Hz, 1H), 5.22 (d, J = 7.2 Hz, 1H), 4.83 (s, 1H), 4.09 (d, J = 7.2 Hz, 1H), 3.78 (s, 3H), 3.13 (br s, 3H), 2.23-2.20 (m, 1H), 1.92 (t, J = 5.2 Hz, 1H), 1.65-1.61 (m, 1H). HRMS calculated for C20H19N5O4SBr (M + H) +: 504.0341; found 504.0345.
Ethyl (1S,2R,3S,4R,5S)-4-(2-((5-bromothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (12)
Compound 12 (90%) was prepared from compound 34 following the same method as for compound 10. 1H NMR (CD3OD, 400 MHz) δ 8.02 (s, 1H), 7.28 (d, J = 4.0 Hz, 1H), 7.14 (d, J = 4.0 Hz, 1H), 5.20 (d, J = 6.6 Hz, 1H), 4.84 (s, 1H), 4.27-4.22 (m, 2H), 4.13 (d, J = 6.6 Hz, 1H), 3.13 (br s, 3H), 2.23-2.20 (m, 1H), 1.92 (t, J = 5.2 Hz, 1H), 1.66-1.63 (m, 1H), 1.29 (t, J = 7.2 Hz, 3H). HRMS calculated for C21H21N5O4SBr (M + H) +: 518.0498; found 518.0494.
Ethyl (1S,2R,3S,4R,5S)-4-(6-((5-chlorothiophen-2-yl)ethynyl)-4-(methylamino)-1H-imidazo[4,5-c]pyridin-1-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (13)
Compound 13 (71%) was prepared from compound 50 following the same method as for compound 51. 1H NMR (CD3OD, 400 MHz) δ 8.03 (s, 1H), 7.19 (d, J =4.0 Hz, 1H), 7.11 (s, 1H), 6.98 (d, J =4.0 Hz, 1H), 5.02 (d, J =7.6 Hz, 1H), 4.70 (s, 1H), 4.30-4.25 (m, 2H), 4.02 (d, J =6.4 Hz, 1H), 3.10 (s, 3H), 2.30-2.26 (m, 1H), 1.95 (t, J = 5.2 Hz, 1H), 1.74-1.71 (m, 1H), 1.33 (t, J = 7.2 Hz, 3H). HRMS calculated for C22H22N4O4SCl (M + H) +: 473.1050; found 473.1056.
n-Propyl (1S,2R,3S,4R,5S)-4-(2-((5-chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (14)
Compound 14 (53%) was prepared from compound 22 following the same method as for compound 9. 1H NMR (CD3OD, 400 MHz) δ 8.01 (s, 1H), 7.33 (d, J = 4.0 Hz, 1H), 7.02 (d, J = 4.0 Hz, 1H), 5.21 (d, J = 6.8 Hz, 1H), 4.83 (s, 1H), 4.18-4.11 (m, 3H), 3.13 (br s, 3H), 2.24-2.20 (m, 1H), 1.92 (t, J = 5.2 Hz, 1H), 1.72-1.63 (m, 3H), 0.94 (t, J = 7.2 Hz, 1H). HRMS calculated for C22H23N5O4SCl (M + H) +: 488.1159; found 488.1162.
Isopropyl (1S,2R,3S,4R,5S)-4-(2-((5-chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (15)
Compound 15 (52%) was prepared from compound 22 following the same method as for compound 9. 1H NMR (CD3OD, 400 MHz) δ 7.99 (s, 1H), 7.33 (d, J = 4.0 Hz, 1H), 7.02 (d, J = 4.0 Hz, 1H), 5.20 (d, J = 6.4 Hz, 1H), 5.14-5.08 (m, 1H), 4.82 (s, 1H), 4.13 (d, J = 5.2 Hz, 1H), 3.13 (br s, 3H), 2.21-2.18 (m, 1H), 1.90 (t, J = 4.8 Hz, 1H), 1.66-1.62 (m, 1H), 1.27 (d, J = 8.0 Hz, 6H). HRMS calculated for C22H23N5O4SCl (M + H) +: 488.1159; found 488.1156.
Butyl (1S,2R,3S,4R,5S)-4-(2-((5-chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (16)
Compound 16 (53%) was prepared from compound 22 following the same method as for compound 9. 1H NMR (CD3OD, 400 MHz) δ 8.01 (s, 1H), 7.33 (d, J = 4.0 Hz, 1H), 7.02 (d, J = 4.0 Hz, 1H), 5.22 (d, J = 6.4 Hz, 1H), 4.84 (s, 1H), 4.24-4.17 (m, 2H), 4.12 (d, J = 6.4 Hz, 1H), 3.13 (br s, 3H), 2.23-2.19 (m, 1H), 1.91 (t, J = 5.2 Hz, 1H), 1.69-1.62 (m, 3H), 1.43-1.34 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H). HRMS calculated for C23H25N5O4SCl (M + H) +: 502.1316; found 502.1308.
Isopentyl (1S,2R,3S,4R,5S)-4-(2-((5-chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (17)
Compound 17 (51%) was prepared from compound 22 following the same method as for compound 9. 1H NMR (CD3OD, 400 MHz) δ 8.01 (s, 1H), 7.34 (d, J = 4.0 Hz, 1H), 7.02 (d, J = 4.0 Hz, 1H), 5.22 (d, J = 6.8 Hz, 1H), 4.83 (s, 1H), 4.29-4.21 (m, 2H), 4.12 (d, J = 6.4 Hz, 1H), 3.14 (br s, 3H), 2.22-2.18 (m, 1H), 1.91 (t, J = 5.2 Hz, 1H), 1.68-1.62 (m, 2H), 1.59-1.54 (m, 2H), 0.90 (d, J = 6.0 Hz, 6H). HRMS calculated for C24H27N5O4SCl (M + H) +: 516.1472; found 516.1467.
2-Cyclohexylethyl (1S,2R,3S,4R,5S)-4-(2-((5-chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (18)
Compound 18 (54%) was prepared from compound 22 following the same method as for compound 9. 1H NMR (CD3OD, 400 MHz) δ 8.02 (s, 1H), 7.34 (d, J = 4.0 Hz, 1H), 7.03 (d, J = 4.0 Hz, 1H), 5.24 (d, J = 7.6 Hz, 1H), 4.83 (s, 1H), 4.30-4.17 (m, 2H), 4.13 (d, J = 6.4 Hz, 1H), 3.13 (br s, 3H), 2.21-2.18 (m, 1H), 1.90 (t, J = 5.2 Hz, 1H), 1.70-1.61 (m, 6H), 1.58-1.53 (m, 2H), 1.32-1.26 (m, 2H), 1.19-1.15 (m, 2H), 0.94-0.85 (m, 2H). HRMS calculated for C27H31N5O4SCl (M + H) +: 556.1785; found 556.1786.
Benzyl (1S,2R,3S,4R,5S)-4-(2-((5-chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (19)
Compound 19 (59%) was prepared from compound 22 following the same method as for compound 9. 1H NMR (CD3OD, 400 MHz) δ 7.96 (s, 1H), 7.38-7.15 (m, 6H), 6.98 (d, J = 4.0 Hz, 1H), 5.24 (d, J = 4.8 Hz, 1H), 4.83 (s, 1H), 4.11 (d, J = 6.4 Hz, 1H), 3.57 (t, J = 6.8 Hz, 1H), 3.13 (br s, 3H), 2.68 (t, J = 8.0 Hz, 1H), 2.26-2.23 (m, 1H), 1.95 (t, J = 5.2 Hz, 1H), 1.70-1.66 (m, 1H). HRMS calculated for C26H23N5O4SCl (M + H) +: 536.1159; found 536.1153.
2-Phenylethyl (1S,2R,3S,4R,5S)-4-(2-((5-chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (20)
Compound 20 (58%) was prepared from compound 22 following the same method as for compound 9. 1H NMR (CD3OD, 400 MHz) δ 7.93 (s, 1H), 7.30 (d, J = 4.0 Hz, 1H), 7.18-7.00 (m, 5H), 6.99 (d, J = 4.0 Hz, 1H), 5.19 (d, J = 6.8 Hz, 1H), 4.78 (s, 1H), 4.46-4.42 (m, 1H), 4.35-4.29 (m, 1H), 4.10 (d, J = 6.8 Hz, 1H), 3.14 (br s, 3H), 2.95 (t, J = 6.4 Hz, 2H), 2.07-2.03 (m, 1H), 1.86 (d, J = 5.2 Hz, 1H), 1.60-1.56 (m, 1H). HRMS calculated for C27H25N5O4SCl (M + H) +: 550.1316; found 550.1308.
3-Phenylpropyl (1S,2R,3S,4R,5S)-4-(2-((5-chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (21)
Compound 21 (59%) was prepared from compound 22 following the same method as for compound 9. 1H NMR (CD3OD, 400 MHz) δ 8.03 (s, 1H), 7.30 (d, J = 4.0 Hz, 1H), 7.22-7.11 (m, 5H), 7.01 (d, J = 4.0 Hz, 1H), 5.25 (d, J = 6.4 Hz, 1H), 4.83 (s, 1H), 4.27-4.11 (m, 3H), 3.13 (br s, 3H), 2.67 (t, J = 7.6 Hz, 2H), 2.18-2.14 (m, 1H), 2.02-1.95 (m, 2H), 1.90 (t, J = 5.2 Hz, 1H), 1.65-1.61 (m, 1H). HRMS calculated for C28H27N5O4SCl (M + H) +: 564.1472; found 564.1463.
(1S,2R,3S,4R,5S)-4-(2-((5-Chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylic acid (22)
2N NaOH solution (1.5 mL) was added to a solution of compound 10 (29.2 mg, 0.06 mmol) in MeOH (1.5 mL) and stirred for 1h at room temperature. Reaction mixture was neutralized with aceteic acid and evaporated under vacuum. The residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH:TFA= 10:1:0.1) to give the compound 22 (22 mg, 82%) as colorless syrup. 1H NMR (DMSO, 400 MHz) δ 8.12 (s, 1H), 7.98 (s, 1H), 7.48 (d, J = 4.0 Hz, 1H), 7.22 (d, J = 4.0 Hz, 1H), 4.92 (d, J = 6.0 Hz, 1H), 4.65 (s, 1H), 4.06-4.04 (m, 1H), 2.95 (br s, 3H), 2.03-2.00 (m, 1H), 1.69 (t, J = 4.8 Hz, 1H), 1.48-1.44 (m, 1H). HRMS calculated for C19H17N5O4SCl (M + H) +: 446.0690; found 446.0694.
(2S,3S,4R,5R)-5-(2-((5-Chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-3,4-dihydroxy-N-methyltetrahydrofuran-2-carboxamide (27)
Compound 27 (91%) was prepared from compound 44 following the same method as for compound 10. 1H NMR (DMSO, 400 MHz) δ 8.56 (d, J = 4.4 Hz, 1H), 8.50 (s, 1H), 8.14 (d, J = 4.0 Hz, 1H), 7.49 (d, J =4.0 Hz, 1H), 7.23 (d, J = 4.0 Hz, 1H), 5.96 (d, J = 7.6 Hz, 1H), 5.76 (d, J = 4.0 Hz, 1H), 5.57 (d, J = 6.4 Hz, 1H), 4.60-4.56 (m, 1H), 4.33 (s, 1H), 4.17-4.14 (m, 1H), 2.97 (br s, 3H), 2.78 (s, 3H). HRMS calculated for C18H18N6O4SCl (M + H) +: 449.0799; found 449.0806.
(2S,3S,4R,5R)-5-(2-((5-Chlorothiophen-2-yl)ethynyl)-6-(n-propylamino)-9H-purin-9-yl)-3,4-dihydroxy-N-methyltetrahydrofuran-2-carboxamide (28)
Compound 28 (90%) was prepared from compound 45 following the same method as for compound 10. 1H NMR (CD3OD, 400 MHz) δ 8.27 (s, 1H), 7.31 (d, J =4.0 Hz, 1H), 7.03 (d, J =4.0 Hz, 1H), 5.98 (d, J =7.6 Hz, 1H), 4.78-4.75 (m, 1H), 4.50 (s, 1H), 4.32 (d, J =6.0 Hz, 1H), 3.59 (br s, 3H), 1.77-1.71 (m, 2H), 1.05 (t, J =7.6 Hz, 3H). HRMS calculated for C20H22N6O4SCl (M + H) +: 477.1112; found 477.1115.
Methyl (2S,3S,4R,5R)-5-(2-((5-Chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-carboxylate (29)
PdCl2(PPh3)2 (1.77 mg, 0.002 mmol), CuI (1.0 mg, 0.001 mmol), 2-chloro-5-ethynylthiophene (10.8 mg, 0.075 mmol) and triethylamine (17 μL, 0.12 mmol) was added to a solution of compound 43 (6 mg, 0.012 mmol) in anhydrous DMF (0.6 mL), and stirred at room temperature overnight. Solvent was evaporated under vacuum, and the residue was roughly purified on flash silica gel column chromatography (hexane:ethylacetate = 1:1) to give the compound 46, which was subsequently stirred with 10% TFA (1 mL) and MeOH (1 mL) at 70 °C for 2 h. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 50:1) to afford the compound 29 (3.8 mg, 68%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.58 (s, 1H), 7.31 (d, J =4.0 Hz, 1H), 7.02 (d, J =4.0 Hz, 1H), 6.20 (d, J =4.8 Hz, 1H), 4.64-4.60 (m, 2H), 4.48 (m, 1H), 3.84 (s, 3H), 3.14 (br s, 3H). HRMS calculated for C18H17N5O5SCl (M + H) +: 450.0639; found 450.0645.
Ethyl (3aR,3bS,4aS,5R,5aS)-5-(2-((5-chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (33)
PdCl2(PPh3)2 (23.3 mg, 0.033 mmol), CuI (3.1 mg, 0.033 mmol), 2-chloro-5-ethynylthiophene (142 mg, 0.99 mmol) and triethylamine (0.23 mL, 1.66 mmol) was added to a solution of compound 32 (83 mg, 0.16 mmol) in anhydrous DMF (3 mL) and stirred at room temperature overnight. Solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (hexane:ethylacetate = 1:1) to give the compound 33 (71 mg, 83%) as a brownish glassy syrup. 1H NMR (CD3OD, 400 MHz) δ 8.11 (s, 1H), 7.37 (d, J = 4.0 Hz, 1H), 7.03 (d, J = 4.0 Hz, 1H), 5.89 (d, J = 6.8 Hz, 1H), 5.02 (s, 1H), 4.82 (d, J = 6.0 Hz, 1H), 4.29-4.19 (m, 2H), 3.12 (br s, 3H), 2.31-2.27 (m, 1H), 1.71-1.67 (m, 1H), 1.57-1.54 (m, 4H), 1.29 (s, 3H), 1.23 (t, J = 7.2 Hz, 3H). HRMS calculated for C24H25N5O4SCl (M + H) +: 514.1316; found 514.1318.
Ethyl (3aR,3bS,4aS,5R,5aS)-5-(2-((5-bromothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (34)
Compound 34 (82%) was prepared from compound 32 following the same method as for compound 33. 1H NMR (CD3OD, 400 MHz) δ 8.11 (s, 1H), 7.33 (d, J = 4.0 Hz, 1H), 7.15 (d, J = 4.0 Hz, 1H), 5.89 (d, J = 7.2 Hz, 1H), 5.02 (s, 1H), 4.83 (d, J = 7.2 Hz, 1H), 4.28-4.18 (m, 2H), 3.14 (br s, 3H), 2.31-2.27 (m, 1H), 1.71-1.67 (m, 1H), 1.57-1.54 (m, 4H), 1.29 (s, 3H), 1.23 (t, J = 7.2 Hz, 3H). HRMS calculated for C24H25N5O4SBr (M + H) +: 558.0811; found 558.0814.
((3aR,4R,6R,6aR)-6-(6-Chloro-2-iodo-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methanol (35b)
To a suspension of compound 35a (1 g, 2.42 mmol) in acetone (50 mL), 2,2-dimethoxypropane (1.5 mL, 12.1 mmol) and p-TSA was added and stirred at room temperature for 5 h. After completion of starting material, the reaction mixture was neutralized by addition of solid NaHCO3. Reaction mixture was filtered and the filtrate was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (hexane:ethyl acetate= 1:1) to give the compound 35b (0.996 g, 91%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.71 (s, 1H), 6.27 (d, J = 2.8 Hz, 1H), 5.33 (d, J = 6.4 Hz, 1H), 5.03 (d, J = 6.4 Hz, 1H), 4.41-4.38 (m, 1H), 3.38-3.71 (m, 2H), 1.62 (s, 3H), 1.41 (s, 3H). HRMS calculated for C13H15N4O4ICl (M + H) +: 452.9827; found 452.9829.
((3aR,4R,6R,6aR)-6-(2-Iodo-6-(methylamino)-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro [3,4-d][1,3]dioxol-4-yl)methanol (37)
Methylamine hydrochloride (880 mg, 13 mmol) and triethylamine (3.6 mL, 26.1 mmol) was added to a solution of compound 35b (1.18 g, 2.61 mmol) in methanol (25 mL) and stirred at room temperature overnight. The reaction mixture was evaporated under vacuum, and the residue was purified on flash column chromatography (hexane:ethyl acetate = 1:2) to give the desired product 37 (968 mg, 83%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.13 (s, 1H), 6.09 (d, J = 3.6 Hz, 1H), 5.24 (d, J = 6.0 Hz, 1H), 5.02 (d, J = 6.0 Hz, 1H), 4.36-4.34 (m, 1H), 3.03 (br s, 3H), 1.62 (s, 3H), 1.39 (s, 3H). HRMS calculated for C14H19N5O4I (M + H) +: 448.0482; found 448.0489.
((3aR,4R,6R,6aR)-6-(2-Iodo-6-(n-propylamino)-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro [3,4-d][1,3]dioxol-4-yl)methanol (38)
Compound 38 (85%) was prepared from compound 35b following the same method as for compound 37. 1H NMR (CD3OD, 400 MHz) δ 8.15 (s, 1H), 6.10 (d, J = 3.2 Hz, 1H), 5.24 (d, J = 6.0 Hz, 1H), 5.03 (d, J = 6.0 Hz, 1H), 4.36-4.34 (m, 1H), 3.79-3.71 (m, 2H), 3.52-3.49 (m, 2H), 1.72-1.62 (m, 2H), 1.62 (s, 3H), 1.40 (s, 3H), 1.01 (t, J = 7.6 Hz, 3H). HRMS calculated for C16H23N5O4I (M + H) +: 476.0716; found 476.0734.
(3aS,4S,6R,6aR)-6-(2-Iodo-6-(methylamino)-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro [3,4-d][1,3]dioxole-4-carboxylic acid (39)
PDC (530 mg, 1.4 mmol) was added to a solution of compound 37 (105 mg, 0.23 mmol) in dry DMF (2.5 mL) and heated at 40 °C overnight. After completion of starting material, water (10 mL) was added into the reaction mixture and extracted with ethyl acetate (3×10 mL). Combined organic layer was washed with brine (15 mL), dried, filtered, evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 35:1) to give the compound 39 (73 mg, 68%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.08 (s, 1H), 6.29 (s, 1H), 5.66 (d, J = 6.0 Hz, 1H), 5.46 (d, J = 6.0 Hz, 1H), 4.75 (d, J = 1.6 Hz, 1H), 3.04 (br s, 3H), 1.58 (s, 3H), 1.43 (s, 3H). HRMS calculated for C14H17N5O5I (M + H) +: 462.0274; found 462.0265.
(3aS,4S,6R,6aR)-6-(2-Iodo-6-(n-propylamino)-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro [3,4-d][1,3]dioxole-4-carboxylic acid (40)
Compound 40 (67%) was prepared from compound 38 following the same method as for compound 39. 1H NMR (CD3OD, 400 MHz) δ 8.10 (s, 1H), 6.29 (s, 1H), 5.65 (d, J = 6.0 Hz, 1H), 5.46 (d, J = 6.0 Hz, 1H), 4.76 (d, J = 1.6 Hz, 1H), 3.49 (br s, 2H), 1.71-1.65 (m 2H), 1.58 (s, 3H), 1.43 (s, 3H), 1.00 (d, J = 7.6 Hz, 3H). HRMS calculated for C16H21N5O5I (M + H) +: 490.0587; found 490.0583.
(3aS,4S,6R,6aR)-6-(2-Iodo-6-(methylamino)-9H-purin-9-yl)-N,2,2-trimethyltetrahydrofuro [3,4-d][1,3]dioxole-4-carboxamide (41)
MeNH2 (48 μL, 2M solution in THF) and DIPEA (14 μL, 0.084 mmol) were added to a solution of compound 39 (30 mg, 0.065 mmol) and HATU (32 mg, 0.078 mmol) in dry DMF (1.3 mL). The reaction mixture was stirred at room temperature overnight. Solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 35:1) to give the compound 41 (21 mg, 68%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.05 (s, 1H), 6.27 (d, J =1.2 Hz, 1H), 5.51 (d, J = 6.2 Hz, 1H), 5.35 (d, J = 6.2 Hz, 1H), 4.63 (d, J = 2.0 Hz, 1H), 3.05 (br s, 3H), 2.49 (s, 3H), 1.59 (s, 3H), 1.41 (s, 3H). HRMS calculated for C15H20N6O4I (M + H) +: 475.0591; found 475.0600.
(3aS,4S,6R,6aR)-6-(2-Iodo-6-(n-propylamino)-9H-purin-9-yl)-N,2,2-trimethyltetrahydrofuro [3,4-d][1,3]dioxole-4-carboxamide (42)
Compound 42 (69%) was prepared from compound 40 following the same method as for compound 41. 1H NMR (CD3OD, 400 MHz) δ 8.05 (s, 1H), 6.27 (d, J = 1.6 Hz, 1H), 5.51 (d, J = 6.0 Hz, 1H), 5.35 (d, J = 6.0 Hz, 1H), 4.63 (d, J = 2.0 Hz, 1H), 3.49 (br s, 2H), 2.48 (s, 3H), 1.71-1.66 (m, 2H), 1.59 (s, 3H), 1.41 (s, 3H), 1.00 (t, J = 7.6 Hz, 3H). HRMS calculated for C17H24N6O4I (M + H) +: 503.0904; found 503.0912.
Methyl (3aS,4S,6R,6aR)-6-(2-iodo-6-(n-propylamino)-9H-purin-9-yl)-2,2-dimethyltetrahydro furo[3,4-d][1,3]dioxole-4-carboxylate (43)
DCC (24 mg, 0.117 mmol), DMAP (2.38 mg, 0.019 mmol) and MeOH (10 μL, 0.19 mmol) was added to a solution of compound 39 (18 mg, 0.039 mmol) in DMF (0.8 mL) and stirred at room temperature overnight. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 35:1) to afford the compound 43 (9.6 mg, 52%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.06 (s, 1H), 6.29 (s, 1H) 5.65 (d, J = 6.0 Hz, 1H), 5.45 (d, J = 6.0 Hz, 1H), 4.81 (s, 1H), 3.59 (s, 3H), 3.05 (br s, 3H), 1.57 (s, 3H), 1.43 (s, 3H). HRMS calculated for C15H19N5O5I (M + H) +: 476.0431; found 476.0433.
(3aS,4S,6R,6aR)-6-(2-((5-Chlorothiophen-2-yl)ethynyl)-6-(methylamino)-9H-purin-9-yl)-N,2,2-trimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carboxamide (44)
Compound 44 (84%) was prepared from compound 41 following the same method as for compound 33. 1H NMR (CD3OD, 400 MHz) δ 8.22 (s, 1H), 7.35 (d, J =4.0 Hz, 1H), 7.02 (d, J =4.0 Hz, 1H), 6.31 (d, J =2.0 Hz, 1H), 5.51 (d, J =6.0 Hz, 1H), 5.39 (d, J =6.0 Hz, 1H), 4.66 (d, J =2.0 Hz, 1H), 3.12 (br s, 3H), 2.52 (s, 3H), 1.62 (s, 3H), 1.42 (s, 3H). HRMS calculated for C21H22N6O4SCl (M + H) +: 489.1112; found 489.1120.
(3aS,4S,6R,6aR)-6-(2-((5-Chlorothiophen-2-yl)ethynyl)-6-(n-propylamino)-9H-purin-9-yl)-N,2,2-trimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carboxamide (45)
Compound 45 (82%) was prepared from compound 42 following the same method as for compound 33. 1H NMR (CD3OD, 400 MHz) δ 8.22 (s, 1H), 7.35 (d, J =4.0 Hz, 1H), 7.02 (d, J =4.0 Hz, 1H), 6.31 (d, J =2.0 Hz, 1H), 5.50 (d, J =6.0 Hz, 1H), 5.38 (d, J =6.0 Hz, 1H), 4.66 (d, J =2.0 Hz, 1H), 3.57 (br s, 2H), 2.52 (s, 3H), 1.75-1.70 (m, 2H), 1.62 (s, 3H), 1.42 (s, 3H), 1.03 (d, J =7.6 Hz, 3H). HRMS calculated for C23H26N6O4SCl (M + H) +: 517.1425; found 517.1422.
Ethyl(3aR,3bS,4aS,5R,5aS)-5-(6-Bromo-4-chloro-1H-imidazo[4,5-c]pyridin-1-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (47)
DIAD (0.22 mL, 1.13 mmol) was added to a solution of triphenylphosphine (0.296 g, 1.13 mmol) and 3-deaza-2-bromo-6-chloro-purine (0.197 g, 0.845 mmol) in dry THF (4 mL) at 0 °C and after addition it was stirred at room temperature for 10 min. A solution of compound 36 (0.137 g, 0.566 mmol) in THF (2 mL) was added to the reaction mixture, and stirred overnight at room temperature. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (hexane:ethyl acetate=2:1) to give the compound 47 (0.178 g, 69%) as a colorless foamy solid. 1H NMR (CD3OD, 400 MHz) δ 8.41 (s, 1H), 7.74 (s, 1H), 5.74 (d, J =7.2 Hz, 1H), 4.92 (s, 1H), 4.80 (d, J =6.8 Hz, 1H), 3.79-3.71 (m, 2H), 2.52-2.48 (m, 1H), 1.87-1.83 (m, 1), 1.66 (t, J = 5.2 Hz, 1H), 1.54 (s, 3H), 1.34 (t, J = 7.2 Hz, 1H), 1.29 (s, 3H). HRMS calculated for C18H20N3O4Br (M + H) +: 456.0326; found 456.0329.
Ethyl (3aR,3bS,4aS,5R,5aS)-5-(6-Bromo-4-(methylamino)-1H-imidazo[4,5-c]pyridin-1-yl)-2,2-dimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxylate (48)
MeNH2.HCl (78.32 mg, 1.16 mmol) and DIPEA (0.4 mL, 2.32 mmol) was added to a solution of compound 47 (106 mg, 0.232 mmol) in isopropanol (2.5 mL) tube and heated at 125 °C under microwave condition in a sealed for 2.5 hrs. The reaction mixture was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (hexane:ethylacetate = 1:1) to give the compound 48 (80 mg, 76%) as a syrup. 1H NMR (CD3OD, 400 MHz) δ 7.98 (s, 1H), 6.83 (s, 1H), 5.70 (d, J =7.2 Hz, 1H), 4.81 (s, 1H), 4.70 (d, J =6.8 Hz, 1H), 4.33-4.25 (m, 2H), 3.06 (s, 3H), 2.43-2.39 (m, 1H), 1.85-1.81 (m, 1H), 1.59 (t, J = 5.2 Hz, 1H), 1.54 (s, 3H), 1.35 (t, J = 7.2 Hz, 3H), 1.28 (s, 3H). HRMS calculated for C19H24N4O4Br (M + H) +: 451.0981; found 451.0977.
(3aR,3bS,4aS,5R,5aS)-5-(6-Bromo-4-(methylamino)-1H-imidazo[4,5-c]pyridin-1-yl)-N,2,2-trimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxamide (49)
40% Methylamine solution (aqueous, 2.0 mL) was added to a solution of compound 48 (55 mg, 0.12 mmol) in methanol (3 mL) and stirred at room temperature overnight. Solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH=40:1) to give the compound 49 (37g, 71%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 8.01 (s, 1H), 6.88 (s, 1H), 5.56 (d, J =7.2 Hz, 1H), 4.95 (s, 1H), 4.65 (d, J =6.8 Hz, 1H), 3.05 (s, 3H), 2.86 (s, 3H), 2.37-2.33 (m, 1H), 1.69-1.65 (m, 1H), 1.55 (s, 3H), 1.50 (d, J =5.2 Hz, 1H), 1.28 (s, 3H). HRMS calculated for C18H23N5O3Br (M + H) +: 436.0984; found 436.0987.
Ethyl (1S,2R,3S,4R,5S)-4-(6-Bromo-4-(methylamino)-1H-imidazo[4,5-c]pyridin-1-yl)-2,3-dihydroxybicyclo[3.1.0]hexane-1-carboxylate (50)
10% TFA (2 mL) was added to a solution of compound 48 (22 mg, 0.048 mmol) in MeOH (2 mL) and heated at 70 °C for 3 h. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 30:1) to give the compound 50 (18 mg, 91%) as a colorless syrup. 1H NMR (CD3OD, 400 MHz) δ 7.91 (s, 1H), 6.95 (s, 1H), 5.02 (d, J =6.8 Hz, 1H), 4.63 (s, 1H), 4.30-4.24 (m, 2H), 4.02 (d, J =6.4 Hz, 1H), 3.06 (s, 3H), 2.26-2.23 (m, 1H), 1.94 (t, J = 5.2 Hz, 1H), 1.73-1.69 (m, 1H), 1.33 (t, J = 7.2 Hz, 3H). HRMS calculated for C16H20N4O4Br (M + H) +: 411.0668; found 411.0667.
(3aR,3bS,4aS,5R,5aS)-5-(6-((5-Chlorothiophen-2-yl)ethynyl)-4-(methylamino)-1H-imidazo[4,5-c]pyridin-1-yl)-N,2,2-trimethyltetrahydrocyclopropa[3,4]cyclopenta[1,2-d][1,3]dioxole-3b(3aH)-carboxamide (51)
PdCl2(PPh3)2 (10.3 mg, 0.014 mmol), CuI (1.4 mg, 0.007 mmol), 2-chloro-5-ethynylthiophene (63 mg, 0.44 mmol) and triethylamine (0.1 mL, 0.73 mmol) was added to a solution of compound 49 (32 mg, 0.073 mmol) in anhydrous DMF (1.2 mL) and was heated in a sealed tube at 130 °C under microwave condition. Solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 40:1) to give the compound 51 (23 mg, 63%) as a yellow syrup. 1H NMR (CD3OD, 400 MHz) δ 8.14 (s, 1H), 7.22 (d, J =4.0 Hz, 1H), 7.03 (s, 1H), 6.98 (d, J =4.0 Hz, 1H), 5.59 (d, J =7.2 Hz, 1H), 5.01 (s, 1H), 4.69 (d, J =6.8 Hz, 1H), 3.10 (s, 3H), 2.86 (s, 3H), 2.39-2.37 (m, 1H), 1.71-1.67 (m, 1H), 1.56 (s, 3H), 1.52 (t, J =5.2 Hz, 1H), 1.28 (s, 3H). HRMS calculated for C24H25N5O3SCl (M + H) +: 498.1367; found 498.1360.
Pharmacological procedures
Cell lines and radioligand binding
Initial screening at hDAT and other drug targets was performed by the PDSP. Ki determinations using 96-well plates and binding profiles in a broad screen of receptors and channels were generously provided by the National Institute of Mental Health's Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. For experimental details please refer to the PDSP web site http://pdsp.med.unc.edu/ and click on “Binding Assay” on the menu bar. The cell line used for preparing membranes for hDAT binding is HEK cells stably expressing hDAT, grown in DMEM medium containing 350 μg/mL geneticin and 10% fetal bovine serum. The buffer used for binding and washing was 10 mM HEPES, 135 mM NaCl, 5 mM KCl, 0.8 mM MgCl2, 1 mM EGTA, pH 7.4, room temperature. The Kd of the radioligand 24 was determined to be 11.1±1.4 nM. The reference ligand 30 had a Ki value of 3.04±0.21 nM. The radioligands used by PDSP for NET and SERT binding were [3H]52 and [3H]53, respectively.
The detailed comparison of binding enhancement at human DAT, NET and SERT were performed as described.3,48 Membranes from human embryonic kidney (HEK)-293 cells stably expressing the recombinant hDAT (HEK-hDAT), hSERT (HEK-hSERT), or hNET (HEK-hNET) were used. Transporter binding assays were performed in Krebs-HEPES assay buffer (25 mM HEPES, 122 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 2.5 mM CaCl2, 1 μM pargyline, 100 μM tropolone, 2 mg glucose/mL, 0.2 mg ascorbic acid/mL, pH 7.4). Membranes were preincubated at room temperature for 10 min with the test compound before radioligand (either [125I]23, 40-80 pM final concentration, or [3H]26) was added. Concentrations of [3H]26 were 1-3 nM for DAT and NET and 10-13 nM for SERT. The assay mixture was then incubated radioligand for 90 min at room temperature in the dark before filtration. Nonspecific binding was defined using 5 μM mazindol (HEK-hDAT and HEK-NET) or 5 μM imipramine (HEK-hSERT). In a typical hDAT binding experiment, the basal specific binding was ∼2900 cpm, and the maximal stimulated specific binding was 7000-8000 cpm.
The full length coding region of the mouse DA transporter (mDAT) cDNA in PCMV6 vector (OriGene, Rockville, MD) was prepared using a Qiagen (Chatsworth, CA) miniprep kit following transformation of One–Shot Top 10 chemically competent Escherichia coli (Invitrogen, Carlsbad, CA). HEK-293 cells were transfected with 10 μg mDAT cDNA using Lipofectamine 2000 (Invitrogen). Cells were plated in DMEM supplemented with 10% Fetal Clone I (HyClone, South Logan, UT), then selected for G418 resistance (600 μg/ml G418). Forty-eight surviving colonies were chosen and plated in the above media with 300 μg/ml G418, 10 units/ml penicillin and 10 μg/ml streptomycin (HyClone). Clonal lines that had high levels of specific [3H]DA (20 nM) uptake, defined as the difference between uptake in the absence and presence of mazindol (5 μM), were tested using [125I]23 saturation binding assays (0.05 nM –16.6 nM) and competitive [125I]23 (40 pM) binding against mazindol and cocaine (1 nM-10 μM). The HEK-mDAT line used for the binding and uptake assays herein had a Kd value in [125I]23 binding of 3.9 ± 1.8 nM and a Bmax value of 2410 ± 630 fmol/mg protein. In the binding enhancement experiments, the fixed concentration of [125I]23 was 45-50 pM. In a typical mDAT binding experiment, the basal specific binding was 750-900 cpm, and the maximal stimulated specific binding was 7000-8000 cpm.
A3AR affinity was measured in two species as described using [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide as radioligand.2,18,19 Protein was determined as reported.49
In all the binding experiments, EC50 values of Ki values were calculated using GraphPad Prism software (San Diego, CA). Values are expressed as mean ± SEM.
Uptake assays
Assays of uptake of [3H]-neurotransmitters (DA, norepinephrine, or 5HT; each 20 nM final concentration) in the same transporter-expressing HEK cell lines as above, used as intact and detached cells, were preformed as described.3 The assay used Krebs-HEPES assay buffer and was incubated for 10 min at 25 °C before termination by filtration. Values are expressed as mean ± SEM.
Supplementary Material
Acknowledgments
We acknowledge support from the NIH Intramural Research Program (NIDDK, ZIA DK031117-28); National Cancer Institute (R01CA169519) and National Heart Lung Institute (R01HL077707). We thank John Lloyd (NIDDK) for mass spectral determinations and Robert O'Connor (NIDDK) for NMR spectra. We thank Dr. Bryan L. Roth (Univ. North Carolina at Chapel Hill) and National Institute of Mental Health's Psychoactive Drug Screening Program (Contract # HHSN-271-2008-00025-C) for screening data. We also thank the NIH National Institute on Drug Abuse (NIDA)/VA Interagency Agreement #ADA12013; the Methamphetamine Abuse Research Center (P50 DA018165-06), and the Department of Veterans Affairs Research Career Scientist and Merit Review Programs.
Abbreviations
- AR
adenosine receptor
- CCI
chronic constriction injury
- DA
dopamine
- DAT
dopamine transporter (SLC6A3)
- DCC
dicyclohexylcarbodiimide
- DHTB
dihydrotetrabenezine
- DMAP
4-(dimethylamino)pyridine
- DMF
dimethylformamide
- DIPEA
diisoproylethylamine
- DIAD
diisopropyl azodicarboxylate
- GPCR
G protein-coupled receptor
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HEK
human embryonic kidney 293
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HRMS
high resolution mass spectroscopy
- 5HT
serotonin
- mazindol
(±)-5-(4-chlorophenyl)-3,5-dihydro-2H-imidazo[2,1-a]isoindol-5-ol
- METH
methamphetamine
- NET
norepinephrine transporter (SLC6A2)
- nisoxetine
(R,S)-3-(2-methoxyphenoxy)-N-methyl-3-phenylpropan-1-amine
- PDC
pyridinium dichromate
- PDSP
Psychoactive Drug Screening Program
- SERT
serotonin transporter (SLC6A4)
- TSPO
translocator protein
- p-TSA
p-toluenesulfonic acid
- VMAT2
vesicular monoamine transporter 2 (SLC18A2)
Footnotes
Supporting Information Available: NMR analysis and mass spectra of selected synthesized compounds, additional synthetic schemes and procedures, and off-target (PDSP) screening results.
References
- 1.Little JW, Ford A, Symons-Liguori AM, Chen Z, Janes K, Doyle T, Xie J, Luongo L, Tosh DK, Maione S, Bannister K, Dickenson A, Vanderah TW, Porreca F, Jacobson KA, Salvemini D. Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain. 2015;138:28–35. doi: 10.1093/brain/awu330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tosh DK, Finley A, Paoletta S, Moss SM, Gao ZG, Gizewski E, Auchampach J, Salvemini D, Jacobson KA. In vivo phenotypic screening for treating chronic neuropathic pain: Modification of C2-arylethynyl group of conformationally constrained A3 adenosine receptor agonists. J Med Chem. 2014;57:9901–9914. doi: 10.1021/jm501021n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janowsky A, Eshleman A, Tosh DK, Jacobson KA. Rigid adenine nucleoside derivatives as novel modulators of the human sodium symporters for dopamine and other neurotransmitters. J Pharmacol Exp Ther. 2016;357:24–35. doi: 10.1124/jpet.115.229666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LeVine MV, Cuendet MA, Khelashvili G, Weinstein H. Allosteric mechanisms of molecular machines at the membrane: Transport by sodium-coupled symporters. Chem Rev. 2016;116:6552–6587. doi: 10.1021/acs.chemrev.5b00627. [DOI] [PubMed] [Google Scholar]
- 5.Vaughan RA, Foster JD. Mechanisms of dopamine transporter regulation in normal and disease states. Trends Pharmacol. 2013;34:489–496. doi: 10.1016/j.tips.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H, Goehring A, Wang KH, Penmatsa A, Ressler R, Gouaux E. Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature. 2013;503:141–145. doi: 10.1038/nature12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang KH, Penamasta A, Gouaux E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature. 2015;521:322–327. doi: 10.1038/nature14431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newman AH, Kulkarni S. Probes for the dopamine transporter: New leads toward a cocaine-abuse therapeutic—A focus on analogues of benztropine and rimcazole. Med Res Rev. 2002;22:429–464. doi: 10.1002/med.10014. [DOI] [PubMed] [Google Scholar]
- 9.Carroll FI, Howard JL, Howell LL, Fox BS, Kuhar MJ. Development of the dopamine transporter selective RTI-336 as a pharmacotherapy for cocaine abuse. AAPS J. 2006;8:E196–E203. doi: 10.1208/aapsj080124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Enman NM, Arthur K, Ward SJ, Perrine SA, Unterwald EM. Anhedonia, reduced cocaine reward, and dopamine dysfunction in a rat model of posttraumatic stress disorder. Biol Psychiatry. 2015;78:871–879. doi: 10.1016/j.biopsych.2015.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakrikar D, Mazei-Robison MS, Mergy MA, Richtand NW, Han Q, Hamilton PJ, Bowton E, Galli A, Veenstra-VanderWeele J, Gill M, Blakely RD. Attention deficit/hyperactivity disorder-derived coding variation in the dopamine transporter disrupts microdomain targeting and trafficking regulation. J Neurosci. 2012;32:5385–5397. doi: 10.1523/JNEUROSCI.6033-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller LL, Leitl MD, Banks ML, Blough BE, Negus SS. Effects of the triple monoamine uptake inhibitor amitifadine on pain-related depression of behavior and mesolimbic dopamine release in rats. Pain. 2015;156:175–184. doi: 10.1016/j.pain.0000000000000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu S, Bellvé KD, Fogarty KE, Haley E, Melikian HE. Ack1 is a dopamine transporter endocytic brake that rescues a trafficking-dysregulated ADHD coding variant. Proc Natl Acad Sci U S A. 2015;112:15480–15485. doi: 10.1073/pnas.1512957112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma H, Santra S, Dutta A. Triple reuptake inhibitors as potential next-generation antidepressants: a new hope? Future Med Chem. 2015;7:2385–2406. doi: 10.4155/fmc.15.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santra S, Sharma H, Vedachalam S, Antonio T, Reith M, Dutta A. Development of potent dopamine-norepinephrine uptake inhibitors (DNRIs) based on a (2S,4R,5R)-2-benzhydryl-5-((4-methoxybenzyl)amino)tetrahydro-2H-pyran-4-ol molecular template. Bioorg Med Chem. 2015;23:821–828. doi: 10.1016/j.bmc.2014.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lavretsky H, Reinlieb M, Cyr NS, Siddarth P, Ercoli LM, Senturk D. Combined citalopram and methylphenidate improved treatment response compared to either drug alone in geriatric depression: a randomized double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172:561–569. doi: 10.1176/appi.ajp.2014.14070889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malhi GS, Byrow Y, Bassett D, Boyce P, Hopwood M, Lyndon W, Mulder R, Porter R, Singh A, Murray G. Stimulants for depression: On the up and up? Aust N Z J Psychiatry. 2016;50:203–207. doi: 10.1177/0004867416634208. [DOI] [PubMed] [Google Scholar]
- 18.Tosh DK, Deflorian F, Phan K, Gao ZG, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Structure-guided design of A3 adenosine receptor-selective nucleosides: Combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions. J Med Chem. 2012;55:4847–4860. doi: 10.1021/jm300396n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tosh DK, Padia J, Salvemini D, Jacobson KA. Efficient, large-scale synthesis and preclinical studies of MRS5698, a highly selective A3 adenosine receptor agonist that protects against chronic neuropathic pain. Purinergic Signalling. 2015;11:371–387. doi: 10.1007/s11302-015-9459-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paoletta S, Tosh DK, Salvemini D, Jacobson KA. Structural probing of off-target G protein-coupled receptor activities within a series of adenosine/adenine congeners. PLoS ONE. 2014;9:e97858. doi: 10.1371/journal.pone.0097858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tosh DK, Crane S, Chen Z, Paoletta S, Gao ZG, Gizewski E, Auchampach JA, Salvemini D, Jacobson KA. Rigidified A3 adenosine receptor agonists: 1-Deaza modification maintains high in vivo efficacy. ACS Med Chem Lett. 2015;6:804–808. doi: 10.1021/acsmedchemlett.5b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Bressi JC, Choe J, Hough MT, Buckner FS, VanVoorhis WC, Verlinde CL, Hol WG, Gelb MH. Adenosine analogues as inhibitors of Trypanosoma brucei phosphoglycerate kinase: elucidation of a novel binding mode for a 2-amino-N6-substituted adenosine. J Med Chem. 2000;43:4135–4150. doi: 10.1021/jm000287a. [DOI] [PubMed] [Google Scholar]; Joshi BV, Melman A, Mackman RL, Jacobson KA. Synthesis of ethyl (1S,2R,3S,4S,5S)-2,3-O-(isopropylidene)-4-hydroxy-bicyclo[3.1.0]hexane-carboxylate from L-ribose: A versatile chiral synthon for preparation of adenosine and P2 receptor ligands. Nucleosides, Nucleotides Nucleic Acids. 2008;27:279–291. doi: 10.1080/15257770701845253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, Simeons FR, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Wetsel WC, Gilbert IH, Roth BL, Hopkins AL. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu X, Gu HH. Molecular cloning of the mouse dopamine transporter and pharmacological comparison with the human homologue. Gene. 1999;233:163–170. doi: 10.1016/s0378-1119(99)00143-2. [DOI] [PubMed] [Google Scholar]
- 25.Cheng YC, Prusoff WH. Relationship between inhibition constant (K1) and concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic-reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 26.Beardsley PM, Shelton KL. Prime-, stress-, and cue-induced reinstatement of extinguished drug-reinforced responding in rats: cocaine as the prototypical drug of abuse. Curr Protocols Neurosci. 2012:1–40. doi: 10.1002/0471142301.ns0939s61. Chapter 9 (Unit 9.39) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 28.Tosh DK, Ciancetta A, Warnick E, Crane S, Gao ZG, Jacobson KA. Structure-based scaffold repurposing for G protein-coupled receptors: Transformation of adenosine derivatives into 5HT2B/5 HT2C serotonin receptor antagonists. J Med Chem. 2016;59:11006–11026. doi: 10.1021/acs.jmedchem.6b01183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fishman P, Cohen S. The A3 adenosine receptor (A3AR): therapeutic target and predictive biological marker in rheumatoid arthritis. Clin Rheumatol. 2016 doi: 10.1007/s10067-016-3202-4. [DOI] [PubMed] [Google Scholar]
- 30.Borea PA, Gessi S, Merighi S, Varani K. Adenosine as a multi-signalling guardian angel in human diseases: When, where and how does it exert its protective effects? Trends Pharmacol Sci. 2016;37:419–434. doi: 10.1016/j.tips.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 31.Janes K, Symons-Liguori AM, Jacobson KA, Salvemini D. Identification of A3 adenosine receptor agonists as novel non-narcotic analgesics. Br J Pharmacol. 2016;173:1253–1267. doi: 10.1111/bph.13446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Welsch ME, Snyder SA, Stockwell BR. Privileged scaffolds for library design and drug discovery. Curr Opin Chem Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coleman JA, Green EM, Gouaux E. X-ray structures and mechanism of the human serotonin transporter. Nature. 2016;532:334–339. doi: 10.1038/nature17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat Neurosci. 2008;11:780–789. doi: 10.1038/nn.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mortensen OV, Kortagere S. Designing modulators of monoamine transporters using virtual screening techniques. Front Pharmacol. 2015;6:223. doi: 10.3389/fphar.2015.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paczkowski FA, Sharpe IA, Dutertre S, Lewis RJ. chi-Conotoxin and tricyclic antidepressant interactions at the norepinephrine transporter define a new transporter model. J Biol Chem. 2007;282:17837–17844. doi: 10.1074/jbc.M610813200. [DOI] [PubMed] [Google Scholar]
- 37.Bruns RF. Adenosine receptor activation in human fibroblasts: nucleoside agonists and antagonists Can. J Physiol Pharmacol. 1980;58:673–691. doi: 10.1139/y80-110. [DOI] [PubMed] [Google Scholar]
- 38.Tosh DK, Phan K, Gao ZG, Gakh A, Xu F, Deflorian F, Abagyan R, Stevens RC, Jacobson KA, Katritch V. Optimization of adenosine 5′-carboxamide derivatives as adenosine receptor agonists using structure-based ligand design and fragment-based searching. J Med Chem. 2012;55:4297–4308. doi: 10.1021/jm300095s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bruss M, Wieland A, Bonisch H. Molecular cloning and functional expression of the mouse dopamine transporter. J Neural Transm. 1999;106:657–662. doi: 10.1007/s007020050187. [DOI] [PubMed] [Google Scholar]
- 40.Melman A, Wang B, Joshi BV, Gao ZG, de Castro S, Heller CL, Kim SK, Jeong LS, Jacobson KA. Selective A3 adenosine receptor antagonists derived from nucleosides containing a bicyclo[3.1.0]hexane ring system. Bioorg Med Chem. 2008;16:8546–8556. doi: 10.1016/j.bmc.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carlin JL, Tosh DK, Xiao C, Piñol RA, Chen Z, Salvemini D, Gavrilova O, Jacobson KA, Reitman ML. Peripheral adenosine A3 receptor activation causes regulated hypothermia in mice that is dependent on central histamine H1 receptors. J Pharmacol Exp Ther. 2016;356:474–482. doi: 10.1124/jpet.115.229872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaddelee MP, Read KD, Cleypool CG, IJzerman AP, Danhof M, de Boer AG. Brain penetration of synthetic adenosine A1 receptor agonists in situ: role of the rENT1 nucleoside transporter and binding to blood constituents. Eur J Pharm Sci. 2005;24:59–66. doi: 10.1016/j.ejps.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 43.a) De Backer D, Biston P, Devriendt J, Madl C, Chochrad D, Aldecoa C, Brasseur A, Defrance P, Gottignies P, Vincent JL for the SOAP II Investigators. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362:779–789. doi: 10.1056/NEJMoa0907118. [DOI] [PubMed] [Google Scholar]; b) Kamiya M, Sato N, Nozaki A, Akiya M, Okazaki H, Takahashi Y, Mizuno K, Shimizu W. Renal effects of added low-dose dopamine in acute heart failure patients with diuretic resistance to natriuretic peptide. J Cardiovasc Pharmacol. 2015;65:282–288. doi: 10.1097/FJC.0000000000000193. [DOI] [PubMed] [Google Scholar]
- 44.Adir Y, Sznajder JI. Regulation of lung edema clearance by dopamine. Isr Med Assoc J. 2003;5:47–50. [PubMed] [Google Scholar]
- 45.Willems JL, Buylaert WA, Lefebvre RA, Bogaert MG. Neuronal dopamine receptors on autonomic ganglia and sympathetic nerves and dopamine receptors in the gastrointestinal system. Pharmacol Rev. 1985;37:165–216. [PubMed] [Google Scholar]
- 46.Fang ZZ, Tosh DK, Tanaka N, Wang H, Krausz KW, O'Connor R, Jacobson KA, Gonzalez FJ. Metabolic mapping of A3 adenosine receptor agonist MRS5980. Biochem Pharmacol. 2015;97:215–223. doi: 10.1016/j.bcp.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 48.Eshleman AJ, Carmolli M, Cumbay M, Martens CR, Neve KA, Janowsky A. Characteristics of drug interactions with recombinant biogenic amine transporters expressed in the same cell type. J Pharmacol Exp Ther. 1999;289:877–885. [PubMed] [Google Scholar]
- 49.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.