

ABSTRACT
We undertook this study to identify DNA methylation signatures of three systemic autoimmune rheumatic diseases (SARDs), namely rheumatoid arthritis, systemic lupus erythematosus, and systemic sclerosis, compared to healthy controls. Using a careful design to minimize confounding, we restricted our study to subjects with incident disease and performed our analyses on purified CD4+ T cells, key effector cells in SARD. We identified differentially methylated (using the Illumina Infinium HumanMethylation450 BeadChip array) and expressed (using the Illumina TruSeq stranded RNA-seq protocol) sites between cases and controls, and investigated the biological significance of this SARD signature using gene annotation databases. We recruited 13 seropositive rheumatoid arthritis, 19 systemic sclerosis, 12 systemic lupus erythematosus subjects, and 8 healthy controls. We identified 33 genes that were both differentially methylated and expressed (26 over- and 7 under-expressed) in SARD cases versus controls. The most highly overexpressed gene was CD1C (log fold change in expression = 1.85, adjusted P value = 0.009). In functional analysis (Ingenuity Pathway Analysis), the top network identified was lipid metabolism, molecular transport, small molecule biochemistry. The top canonical pathways included the mitochondrial L-carnitine shuttle pathway (P = 5E-03) and PTEN signaling (P = 8E-03). The top upstream regulator was HNF4A (P = 3E-05). This novel SARD signature contributes to ongoing work to further our understanding of the molecular mechanisms underlying SARD and provides novel targets of interest.
KEYWORDS: DNA methylation, integrative analysis, systemic autoimmune rheumatic diseases, transcriptome
Introduction
Systemic autoimmune rheumatic diseases (SARDs) are chronic, systemic inflammatory diseases characterized by self-directed inflammation.1 Individually, SARDs are relatively rare,2-4 but collectively, SARDs affect up to 5% of the population.2,5 SARDs are associated with high rates of disability, impaired health-related quality of life, premature mortality,6-10 and significant societal costs, both direct and indirect,11-14 in particular because those affected are people of work-force age. Large gaps in our understanding of SARDs remain. Defining the molecular mechanisms of SARDs is essential to improve outcomes in these chronic diseases.
Rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and systemic sclerosis (SSc) are SARDs that share demographic (the majority of affected individuals are women), clinical [arthritis, lung, and vascular (i.e., Raynaud's phenomenon) disease], serological (antinuclear antibodies4 and anti-Ro52/TRIM21 antibodies15), immunological (type I interferon signature16 and complex abnormalities in CD4+ T lymphocyte function, in particular Th17 and Treg cell subsets)17-19] and genetic similarities (e.g., MHC class II alleles, IRF5, STAT4, PTPN22 loci).17,20,21 This suggests that there may be similar biologic pathways that underlie SARDs, and research across diseases has the potential to identify novel mechanistic commonalities.
Epigenetic regulation governs gene expression and cellular function. DNA methylation is one such epigenetic mechanism. It is influenced both by inherited DNA sequences and by environmental exposures, thereby providing an important link between the environment and genetic predisposition to disease. The prevailing hypothesis for the etiopathogenesis of SARDs is that the inflammatory cascade is triggered by environmental factors in genetically susceptible hosts. Thus, dysregulated DNA methylation is an attractive mechanism by which gene and environment may interact to contribute to SARD onset. DNA methylation also represents attractive biomarkers because, compared to mRNA and most proteins, methylated DNA is quite stable over time and does not fluctuate in response to short-term stimuli.22
We undertook this study to identify cross-disease SARD signatures using a careful design to minimize confounding and an integrative approach. We restricted our study to SARD subjects with incident, mostly treatment-naïve disease and, instead of using mixed cell populations, we performed our analyses on purified CD4+ T cells, key effector cells in SARD. Then, of the differentially methylated sites between cases and controls, we identified sites that also demonstrated differential gene expression. Finally, we investigated the biological significance of this SARD signature using gene annotation databases.
Results
Study subjects
We recruited incident seropositive RA (n = 13), SSc (n = 19), SLE (n = 12), and control subjects (n = 8). Baseline characteristics are presented in Table 1.
Table 1.
Baseline characteristics of subjects and controls.
| RA (N=13) |
SSc (N=19) |
SLE (N=12) |
Controls (N=8) |
|||||
|---|---|---|---|---|---|---|---|---|
| Mean or % | SD or N | Mean or % | SD or N | Mean or % | SD or N | Mean or % | SD or N | |
| Age, years | 55.9 | 10.1 | 56.9 | 14.1 | 37.0 | 17.0 | 52.9 | 14.9 |
| Female, % | 50.0% | 6 | 63.1% | 12 | 83.3% | 10 | 75.0% | 6 |
| Ethnicity, % | ||||||||
| White | 66.7% | 8 | 78.9% | 15 | 58.3% | 7 | 87.5% | 7 |
| Asian | 25.0% | 3 | 5.3% | 1 | — | 0 | 12.5% | 1 |
| Other | 8.3% | 1 | 15.8% | 3 | 41.7% | 5 | — | 0 |
| Smoking, % | ||||||||
| Current | 30.0% | 3 | 10.5% | 2 | 18.2% | 2 | 12.5% | 1 |
| Past | 30.0% | 3 | 31.6% | 6 | 45.5% | 5 | — | 0 |
| Never | 40.0% | 4 | 47.4% | 9 | 36.4% | 4 | 87.5% | 7 |
| Disease duration, years | 0.4 | 0.2 | 2.3 | 1.3 | 0.8 | 0.4 | ||
| Interstitial lung disease, % | 9.1% | 1 | 25.0% | 4 | — | 0 | ||
| Arthritis, % | 100.0% | 12 | 0 | 0 | 66.7% | 8 | ||
| Raynaud's, % | — | 0 | 94.7% | 18 | 16.7% | 2 | ||
| Anti-nuclear antibodies | ||||||||
| Titer ≥ 1:40, % | 100.0% | 12 | 100.0% | 19 | 100.0% | 12 | ||
| Titer ≥ 1:80, % | 66.7% | 8 | 95.7% | 18 | 91.7% | 11 | ||
| Titer ≥ 1:160, % | 50.0% | 6 | 95.7% | 18 | 91.7% | 11 | ||
| Disease specific auto-antibodies | ||||||||
| Cyclic citrullinated peptide (CCP) | 81.8% | 9 | ||||||
| Rheumatoid factor (RF) | 91.7% | 11 | ||||||
| Anti-centromere antibodies (ACA) | 26.7% | 4 | ||||||
| Anti-topoisomerase antibodies (ATA) | 26.7% | 4 | ||||||
| Anti-RNA polymerase III antibody (ARA) | 20.0% | 1 | ||||||
| DNA | 75.0% | 9 | ||||||
| Sm | 8.3% | 1 | ||||||
| Disease specific variables | ||||||||
| Limited skin disease | 31.6% | 6 | ||||||
| Diffuse skin disease | 68.4% | 13 | ||||||
SARD signature
We first looked for differentially methylated (DM) sites. From a total of 485,577 probes—although none showed statistically significant differences after using a Bonferroni correction or a FDR threshold of 0.05—there were 130 CpG probes showing evidence of deviation from the expected null distribution in the QQplot (Supplementary Fig. 1A). Specifically, the QQ-plot showed a point of inflexion at P < 0.0001. By using the Illumina annotations, we mapped these 130 CpG probes to 112 genes; these same probes did not show significant differences in methylation between disease subgroups (P > 0.2). Of 15,684 RNAseq transcripts, we identified 4791 differentially expressed (DE) sites [false discovery rate (FDR) P < 0.05].
In total, there were 33 genes that were both DM and DE (26 over- and 7 under-expressed gene; Table 2 and Fig. 1). The most highly overexpressed gene in SARD subjects compared to controls was CD1C (log fold change in expression = 1.85, adjusted P = 0.009). Another relevant DM and DE gene of interest was BCL2 (log fold change in expression = 0.63, adjusted P < 0.00014), which is known to contribute to systemic autoimmune diseases. The role of most of the top hits in SARD, however, remains unknown.
Table 2.
Top differentially methylated (DM) and expressed (DE) genes in SARD subjects compared to controls.
| Logfold change | Average expression | P value | Adjusted p value | |
|---|---|---|---|---|
| CD1C | 1.85 | −1.57 | 0.0016 | 0.0094 |
| CD36 | 1.06 | −0.39 | 0.0107 | 0.0383 |
| CALHM2 | 0.89 | 0.74 | 0.0083 | 0.0320 |
| SYNPO2 | 0.89 | −0.80 | 0.0076 | 0.0296 |
| SCD | 0.84 | 0.75 | 0.0007 | 0.0049 |
| DPYSL2 | 0.84 | 3.50 | 9.08E-05 | 0.0011 |
| SLFN12L | 0.81 | 4.10 | 2.49E-06 | 7.68E-05 |
| LIMA1 | 0.69 | 2.92 | 3.42E-05 | 0.0005 |
| CPT1A | 0.68 | 4.30 | 0.01384 | 0.0463 |
| CEP97 | 0.66 | 3.62 | 4.43E-06 | 0.0001 |
| TNRC6B | 0.65 | 7.31 | 7.25E-08 | 6.08E-06 |
| BCL2 | 0.63 | 7.32 | 0.0001 | 0.0015 |
| ACTR3 | 0.52 | 6.98 | 4.78E-06 | 0.0001 |
| PCCA | 0.44 | 2.02 | 0.0002 | 0.0023 |
| ZNF407 | 0.43 | 5.23 | 0.0007 | 0.0050 |
| MRPL48 | 0.43 | 2.26 | 0.0016 | 0.0090 |
| TFDP1 | 0.34 | 3.56 | 0.0011 | 0.0070 |
| EIF2C1 | 0.34 | 4.36 | 0.0002 | 0.0022 |
| KIF13B | 0.32 | 4.46 | 0.0004 | 0.0031 |
| NPEPPS | 0.30 | 4.46 | 3.68E-05 | 0.0006 |
| BAZ2B | 0.27 | 5.18 | 0.0133 | 0.0450 |
| HACE1 | 0.27 | 3.41 | 0.0114 | 0.0404 |
| ZMYM4 | 0.26 | 5.21 | 0.0037 | 0.0173 |
| CUL4A | 0.24 | 4.99 | 0.0007 | 0.0052 |
| ITFG1 | 0.22 | 3.84 | 0.0090 | 0.0338 |
| STK24 | 0.19 | 5.83 | 0.0091 | 0.0340 |
| YWHAG | −0.32 | 5.10 | 0.0016 | 0.0093 |
| WIPI2 | −0.32 | 4.58 | 0.0063 | 0.0261 |
| ACSL3 | −0.33 | 4.84 | 0.0146 | 0.0484 |
| ZNF552 | −0.33 | 2.47 | 0.0011 | 0.0070 |
| ATP5G2 | −0.43 | 5.22 | 0.00021 | 0.0021 |
| RNMTL1 | −0.46 | 3.29 | 2.40E-05 | 0.0004 |
| CCDC40 | −1.22 | −1.93 | 2.78E-05 | 0.0005 |
Figure 1.
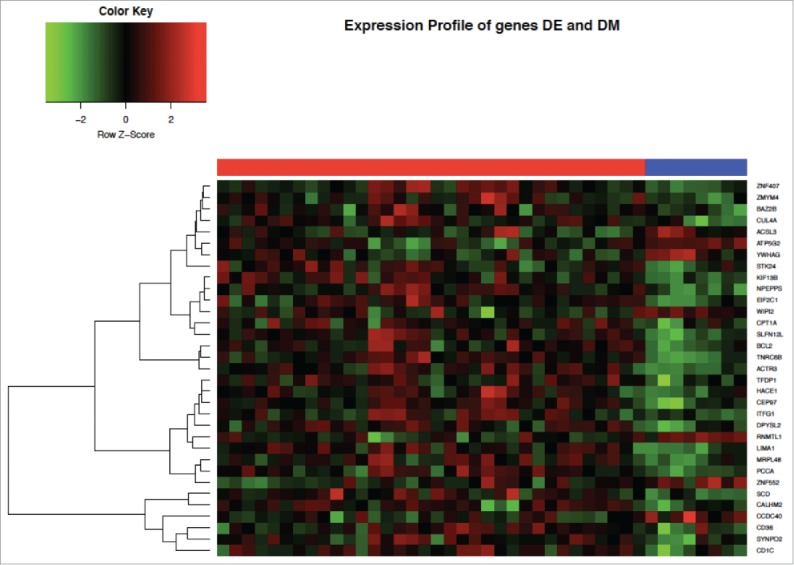
Heatmap of gene expression profiles of the top differentially methylated (DM) and expressed (DE) genes in SARD subjects compared to controls. Columns under the red bar represent SARD subjects and under the blue bar controls.
Functional analysis
For pathway analysis, it was not possible to obtain usable results based only on the 33 overlapping genes. Therefore, we decided to run pathway analysis with the 112 most significant genes from the differential methylation analysis as well as 112 genes that were most significant from the gene expression analysis.
In functional analysis (Ingenuity Pathway Analysis), the top network identified was lipid metabolism, molecular transport, small molecule biochemistry (score 28; Table 3). Other networks of interest included connective tissue disorders, developmental disorder, hereditary disorder (score 26); cellular assembly and organization, DNA replication, recombination and repair, cancer (score 23); and cancer, organismal injury and abnormalities, respiratory diseases (score 23). Top canonical pathways included mitochondrial L-carnitine shuttle pathway (P = 5E-03) and PTEN signaling (P = 8E-03; Table 4). The top upstream regulator was HNF4A (P = 3E-05; Table 5).
Table 3.
Ingenuity Pathway Analysis network analysis.
| Network | Score | Molecules in network | Focus molecules |
|---|---|---|---|
| Lipid metabolism, molecular transport, small molecule biochemistry | 28 | 14-3-3, Alp, BCR (complex), CD1C, CD36, CEBPB, CPT1A, Creb, DBH, DOHH, DPYSL2, ERK1/2, FLI1, IFN Beta, IgG, IgG1, IgG2a, Igm, Ikb, KLF2, LDL, Nr1h, PCCA, PI3K (family), Pka catalytic subunit, Ppp2c, RNASEH1, RPS6KA3, Rsk, Rxr, SCD, SLFN12L, STK24, SYNPO2, TCF | 16 |
| Endocrine System Disorders, Organismal Injury and Abnormalities, Developmental Disorder | 28 | ACBD4, ACSL3, APP, C6orf203, C7orf50, CCDC40, DGCR6/LOC102724770, DNAJB14, DNAJB7, DNAJC4, HES4, HSP90AB1, Hsp84-2, ICT1, LETM2, MAPK8IP2, ME2, MRPL2, MRPL48, MRPL54, MRPL9, MRPS18A, MTERF4, MTG1, PIGH, PTAR1, TBX22, THAP4, TSSK2, VAPA, VKORC1, YIPF5, ZBTB49, ZFPL1, ZNF784 | 16 |
| Connective Tissue Disorders, Developmental Disorder, Hereditary Disorder | 26 | 26s Proteasome, AMPK, ARL6IP5, Akt, Ap1, dpy2, CBL, CD3, CHCHD2, CSTF2, Cyclin A, FRAT1, GSK3B, Hsp27, Hsp90, MID1IP1, Mek, NFAT (complex), Nfat (family), PDGF BB, PIK3IP1, PP2A, PRKAA, PRKAG1, Pdgf (complex), SLC16A3, SOS1, STAT5a/b, Sos, TAOK1, TCR, TFDP1, VAV, ZC3HAV1, caspase | 15 |
| Cellular Assembly and Organization, DNA Replication, Recombination, and Repair, Cancer | 23 | ASH1L, ATP5G2, BAZ1B, CK1, CRY2, CSNK1G1, CUL4A, Collagen(s), DHRS12, Growth hormone, Gsk3, H3F3A/H3F3B, HDL-cholesterol, HISTONE, Histone h3, Histone h4, Hsp70, IL1, IL12 (complex), IL12 (family), Immunoglobulin, Insulin, Interferon alpha, Jnk, KIF13B, KMT5C, MRM3, NFkB (complex), P38 MAPK, PI3K (complex), RNA polymerase II, RNF25, TSG101, Tgf beta, ZNF407 | 14 |
| Cancer, Organismal Injury and Abnormalities, Respiratory Disease | 23 | ABLIM, ACTR3, AGO1, AHCYL1, CEP97, CFAP20, Cg, Ck2, EIF2B2, ERK, ETV2, FSH, Focal adhesion kinase, GTPase, HACE1, LIMA1, LOC81691, Lh, MT1X, Mapk, PAQR3, PIP4K2A, Pka, Pkc(s), Proinsulin, RASA2, Ras, SP1, SRC (family), SRPK1, TNRC6B, Vegf, YWHAG, estrogen receptor, p85 (pik3r) | 14 |
Table 4.
Ingenuity Pathway Analysis canonical pathways.
| Pathway | P value | Overlap | Target molecules in data set |
|---|---|---|---|
| Insulin receptor signaling | 2.25E-03 | 3.5% (5/141) | CBL, SOS1, GSK3B, EIF2B2, PRKAG1 |
| Prostate cancer signaling | 3.26E-03 | 4.3% (4/94) | TFDP1, SOS1, GSK3B, BCL2 |
| Melanocyte development and pigmentation signaling | 3.38E-03 | 4.2% (4/95) | SOS1, RPS6KA3, PRKAG1, BCL2 |
| Mitochondrial L-carnitine shuttle pathway | 5.30E-03 | 11.8% (2/17) | ACSL3, CPT1A |
| PTEN signaling | 7.50E-03 | 3.4% (4/119) | CBL, SOS1, GSK3B, BCL2 |
Table 5.
Ingenuity Pathway Analysis upstream regulators.
| Upstream regulator | Molecule type | P value | Target molecules in data set |
|---|---|---|---|
| HNF4A | Transcription regulator | 2.77E-05 | ACTR3, BAZ1B, C2orf47, CEBPB, CFAP20, CHCHD2, COMMD5, COQ6, CPT1A, GSK3B, LCMT2, MID1IP1, MRM3, MRPL2, MRPL57, MT1X, PRPF4, QTRT2, SCD, SSSCA1, STK24, TM9SF2, TMEM208, TOE1, TRMT6, TSG101, UBP1, ZSCAN18 |
| Maslinic acid | Chemical - endogenous non-mammalian | 1.44E-04 | BCL2, CEBPB, GSK3B, SOS1, YWHAG |
| CYB5R4 | Enzyme | 2.32E-04 | ACSL3, CD36, SCD |
| SCD | Enzyme | 4.29E-04 | BCL2, CD36, CEBPB, CPT1A |
| Fructus xanhii aqueous extract | Chemical - endogenous non-mammalian | 5.41E-04 | CD36, SCD |
Exploratory analyses
We performed weighted gene co-expression network analysis (WGCNA) of the methylation data comparing SARD cases to controls using the 20,000 most variable probes. The heatmap in Supplementary Fig. 2 shows correlations between the 17 identified modules and SARDs. Two modules showed promising correlations: darkorange (P < 4E-06) and orangered4 (P < 3E-04). In gene ontology (GO) analysis of the gene sets of the individual modules (Supplementary Table 1), several pathways of interest reached statistical significance (FDR < 0.05), including signaling pathways regulating pluripotency of stem cells and proteoglycans in cancer.
We also examined the WGCNA comparing SARD subjects by the presence or absence of phenotypes of interest (Supplementary Fig. 2). The orange module was negatively correlated with the presence of interstitial lung disease (P = 0.002). In GO analysis, the top pathways included fatty acid degradation, cell adhesion molecules, Epstein-Barr virus infection, and adipocytokine signaling pathway (all FDR < 0.008; Supplementary Table 2). Similarly, the darkturquoise module was negatively correlated with Raynaud's phenomenon (P = 0.006). The top pathway identified was non-alcoholic fatty liver disease (FDR < 0.004). Of note, the white module correlated strongly with age (P < 4E-05) and the top pathway was longevity regulating pathway (FDR < 0.0003).
Discussion
In this integrative analysis of the DNA methylome and transcriptome of isolated CD4+ T cells of carefully phenotyped SARD subjects, we identified 33 differentially methylated and expressed genes (27 over- and 7 under-expressed genes). Gene annotation identified multiple pathways known to be associated with SARD, thereby providing strong plausibility for the results. Of particular interest is that, in addition to genes of relevance for immune function in SARDs (e.g., previously known BCL2 and as yet largely unknown CD1C, NPEPPS, and SLFN12L), many genes and pathways identified were related to other biological functions [e.g., SYNOP2, LIMA1, KIF13B, and ZMYM4, related to the cellular cytoskeleton, and CEP97 (overexpressed) and CCDC40 (underexpressed), related to cell trafficking], providing novel cellular targets of interest.
To date, there are few studies examining DNA methylation abnormalities in relation to SARD risk in circulating immune cells on a genome-wide basis. Early studies were limited by methodological issues, including heterogeneity of cell samples studied and low-resolution approaches.23,24 The largest study reported to date examined whole-blood samples of 354 highly selected rheumatoid arthritis (RA) patients and 337 controls using the Illumina HumanMethylation450 BeadChip array.25 Genome-wide genotyping was also performed looking for genotype-methylation-phenotype relationships using standard approaches to test for mediation and, thereby, infer causality. Ten differentially methylated positions (DMPs) that appeared to mediate the genetic risk for RA were identified. The associations were replicated in monocytes in an independent cohort of 12 case-control pairs. Three DMPs were found to have methylation changes in the same direction as in whole-blood at a significance of P < 0.05 but with larger effect sizes. The authors hypothesized that, at least for these sites, monocytes were more proximal to the pathogenic cell of interest. The other sites identified in whole-blood but not in monocytes may point to epigenetic dysregulation in other circulating immune cells, in particular CD4+ T cells, which are known to have key roles in the pathogenesis of RA. To date, this remains untested.
There have been few cross-SARD analyses of DNA methylation. Lei et al. studied global DNA methylation of CD4+ T cells from 30 patients with SLE (10), SSc (10), and dermatomyositis (10), and 12 controls.26 They reported hypomethylation of SLE and SSc patients compared to controls, but not between dermatomyositis and controls. Poor resolution of the approach to measure DNA methylation may have limited the findings.
A few studies have examined gene expression patterns across multiple autoimmune diseases to highlight commonalities and differences.27 Higgs et al. identified a type I IFN gene signature in the whole-blood of five diseases, namely RA, SLE, and SSc, as well as dermatomyositis and polymyositis.28 Tuller et al. reported that commonalities in gene expression patterns were stronger between closely related diseases (e.g., Crohn's disease and ulcerative colitis) but absent between very different diseases (e.g., juvenile rheumatoid arthritis and type 1 diabetes).29 This study underscores the potential of cross-disease research, but also the pitfalls of studying diseases that are too different from each other.
A recent meta-analysis of 4 publicly available gene expression data sets including 277 SARD samples (54 SLE, 33 RA, and 190 Sjogren syndrome) and 94 controls identified a gene expression signature composed of 371 differentially expressed genes in SARD compared to controls (184 overexpressed and 187 underexpressed genes).30 Functional analysis showed that overexpressed genes were involved mainly in immune and inflammatory responses, mitotic cell cycles, cytokine-mediated signaling pathways, apoptotic processes, type I interferon-mediated signaling pathways, and responses to viruses. Underexpressed genes were involved primarily in inhibition of protein synthesis. The authors concluded that, in addition to validating genes previously reported as significant biomarkers for individual diseases, their study identified novel genes and provided new clues to the shared pathological state underlying SARD. However, their data was not without limitations, in particular, the fact that the data was derived from subjects with established disease, on various treatment modalities, and from mixed cell populations, thereby possibly confounding the results.
In Ingenuity Pathway Analysis, HNF4A was identified as the top upstream regulator. HNF4A encodes the hepatocyte nuclear factor 4 alpha (HNF4α) protein, a nuclear transcription factor that binds DNA as a homodimer. HNF4A is part of a complex regulatory network in the liver and pancreas for glucose homeostasis. The encoded protein also controls the expression of several genes, including hepatocyte nuclear factor 1 alpha, a transcription factor that regulates the expression of several hepatic genes. Mutations in this gene have been associated with monogenic autosomal dominant non-insulin-dependent diabetes mellitus.31 Interestingly, single nucleotide polymorphisms in the HNF4A loci have been found to be associated with C-reactive protein levels at a genome-wide significance.32 HNF4a was also previously identified as a regulatory hub in a protein-protein interaction map in a genome-wide DNA methylation study of CD4+ T cells from patients with SLE.33
Mitochondrial L-carnitine shuttle pathway was identified as a top canonical pathway. Mitochondrial dysfunction is one of the hallmarks of aging and age-related diseases,34 which include autoimmune diseases. In addition, mitochondrial ‘damage’-associated molecular patterns (DAMPs) have been shown to be capable of activating innate immunity.35 The role of DAMPs and mitochondrial-associated molecular patterns in the pathogenesis of SARD is increasingly being recognized.36,37
We recognize that our top results did not meet thresholds for statistical significance controlling the family-wise error rate in this study. However, the QQ-plot of the differential methylation analysis was strongly indicative of a set of probes deviating from the null hypothesis (Supplementary Fig. 1). For gene expression, we implemented the commonly used false discovery rate (FDR) threshold to select genes of interest. Nevertheless, given the uniqueness of these data, these results will require replication. We note also that, among our probes showing differential methylation, there are 5 that may map to multiple genomic locations (probes in the genes STMN3, ZNF552, SLFN12L, CUL4A, and NPEPPS) and, hence, these results would also need to be replicated carefully.
This study is not without limitations, in particular, the small sample size. In addition, 6/12 SLE subjects were on corticosteroids and/or immunosuppressants. However, that represents a small proportion of the overall sample and a sensitivity analysis adjusting for treatment exposure yielded results highly consistent with the primary results (data not shown). The strengths of the study include the study design (selection of subjects with new onset disease, use of cell-sorted CD4+ T cells, and integrative methylome/transcriptome analysis). Although we acknowledge that the results need to be replicated in a larger independent data set and that functional studies will be required to understand underlying mechanisms, this study makes a meaningful contribution to ongoing work to further our understanding of the molecular mechanisms underlying SARD.
Patients and methods
Study subjects and ethical considerations
Study subjects were recruited from ongoing RA, SSc, and SLE research cohorts based at McGill University, Montreal, Canada. Ethics approval for this study was obtained from McGill University and every study subject signed an informed consent. All subjects had new onset disease, defined as less than 1 year since diagnosis. All RA and SSc subjects were treatment naïve. Of the 12 SLE subjects included, 6 were either on corticosteroids and/or immunosuppressants (4 on corticosteroids, 1 on methotrexate, and 5 on mycophenolate) at the time of sampling.
Cell purification
Forty milliliters of blood were obtained from each study subject and processed fresh within 4 hours of being drawn. CD4+ T cells were positively selected [anti-CD4 microbeads (Miltenyi Biotec) and auto-MACS] and their purity assessed with flow cytometric analysis. Only samples with a purity >95% were used for sequencing.
Sequencing
Genome-wide DNA methylation of CD4+ T cells was assessed using the Illumina Infinium HumanMethylation450 BeadChip array. Genome-wide gene expression was carried out using Illumina TruSeq stranded RNA-seq protocol, allowing strand-specific analyses of the gene expression levels.
Data processing, normalization, filtering, clustering, and heatmap
The methylation data from the Illumina HumanMethylation450 BeadChip were normalized with funtooNorm,38 which was specifically designed to normalize data from the HumanMethylation450 array while retaining important inter-cell-type differences. Since the samples were cell-sorted, we did not apply an explicit correction for cell-type mixture. However, we did adjust the methylation data for age and sex, then calculated the first 2 principal components of the residuals, and adjusted the data for these factors to account for additional confounding not captured by funtooNorm. Given the sample size, we decided to include only the first two principal components. These data were then used for all analyses of DNA methylation.
Gene expression raw read count values were obtained using htseq-count v. 0.5.3p9. We used the Bioconductor package edgeR to calculate normalization factors to scale the raw library sizes. We them applied the voom transformation from the Bioconductor package limma, which transforms count data to log2-counts per million and estimates the mean-variance relationship to compute appropriate observation-level weights.39 We removed 7689 genes where the total raw count was below 10 in all samples.
Analysis of differentially methylated and differentially expressed sites
Analysis of variance was used to explore differences between the SARDs methylation profiles and controls. Methylation values were transformed using a logit transformation. We constructed 3 orthogonal contrasts, one comparing SARDs to controls, as well as two additional orthogonal contrasts between disease subgroups, and tested against residual variation. Statistical significance was assessed with several definitions including a Bonferroni corrected threshold, FDR < 0.05, and whether the P values were smaller than a point of inflexion in the QQ-plots of the P values.
Using limma and the variance stabilization function, eBayes, we tested for DE RNAseq transcripts between SARD cases and controls at 15,684 genes retained for analysis after removing those with very low expression.40 Significance was assessed with Bonferroni corrections and FDR < 0.05. Illumina annotation data were used to identify which genes are close to the methylation probes, so that we could then identify genes with interesting results for both expression and methylation.
Functional analysis
In order to derive biological significance from the list of DM and DE genes, we performed functional analysis using Ingenuity Pathway Analysis and GO. Since there were relatively few genes that were both DM and DE, we expanded the gene list to include the genes above the point of inflexion of the DM QQ plot (Supplementary Fig. 1), of which there were 112, and a similar number of the top DE genes.
Exploratory analyses
In order to explore the full potential of our data set and the potential of cross-disease research, we undertook weighted gene co-expression network analysis (WGCNA)41 of the methylation data. WCGNA is an unsupervised clustering method that identifies modules or clusters in a way that favors a scale-free network clustering pattern (that is, an uneven distribution of connectedness where some hub elements are highly connected and others are linked to only a small number of other elements). We first compared all SARD cases vs. controls. Thereafter, we compared subjects by selected disease phenotypes (arthritis, interstitial lung disease, and Raynaud's phenomenon). We removed probes with multiple mappings and probes located on the X chromosome, leaving 340,236 probes. The data were further filtered for this analysis by selecting the 20,000 most variable probes. GO analysis was conducted to identify relevant biological processes represented in the modules of interest.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to acknowledge the Ludmer Center for Neuroinformatics and Mental Health for their institutional support.
Funding
This study was funded by a Lady Davis Research Institute Clinical Pilot Project award and was also in part from a CIHR grant (Canadian Institutes of Health Research #EP1-120608).
References
- 1.McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med 2006; 3(8):e297; PMID:16942393; https://doi.org/ 10.1371/journal.pmed.0030297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol 1997; 84(3):223-243; PMID:9281381; https://doi.org/ 10.1006/clin.1997.4412 [DOI] [PubMed] [Google Scholar]
- 3.Cooper GS, Stroehla BC. The epidemiology of autoimmune diseases. Autoimmun Rev 2003; 2(3):119-125; PMID:12848952; https://doi.org/ 10.1016/S1568-9972(03)00006-5 [DOI] [PubMed] [Google Scholar]
- 4.Kavanaugh A, Tomar R, Reveille J, Solomon DH, Homburger HA. Guidelines for clinical use of the antinuclear antibody test and tests for specific autoantibodies to nuclear antigens. American College of Pathologists. Arch Pathol Lab Med 2000; 124(1):71-81; PMID:10629135; https://doi.org/ 10.1043/0003-9985 (2000)124<0071:GFCUOT>2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- 5.Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, Liang MH, Kremers HM, Mayes MD, Merkel PA, et al.. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheumatism 2008; 58(1):15-25; PMID:18163481; https://doi.org/19342954 10.1002/art.23177 [DOI] [PubMed] [Google Scholar]
- 6.Sokka T. Long-term outcomes of rheumatoid arthritis. Curr Opin Rheumatol 2009; 21(3):284-90; PMID:19342954; https://doi.org/ 10.1097/BOR.0b013e32832a2f02 [DOI] [PubMed] [Google Scholar]
- 7.Steen VD, Medsger TA Jr. The value of the Health Assessment Questionnaire and special patient-generated scales to demonstrate change in systemic sclerosis patients over time. Arthritis Rheum 1997; 40(11):1984-91; PMID:9365087; https://doi.org/ 10.1002/art.1780401110 [DOI] [PubMed] [Google Scholar]
- 8.Mayes MD, Lacey JV Jr, Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, Schottenfeld D. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum 2003; 48(8):2246-55; PMID:12905479; https://doi.org/ 10.1002/art.11073 [DOI] [PubMed] [Google Scholar]
- 9.Uramoto KM, Michet CJ Jr, Thumboo J, Sunku J, O'Fallon WM, Gabriel SE. Trends in the incidence and mortality of systemic lupus erythematosus, 1950-1992. Arthritis Rheum 1999; 42(1):46-50; PMID:9920013; https://doi.org/ 10.1002/1529-0131(199901)42:1%3c46::AID-ANR6%3e3.0.CO;2-2 [DOI] [PubMed] [Google Scholar]
- 10.Marie I, Hachulla E, Hatron PY, Hellot MF, Levesque H, Devulder B, Courtois H. Polymyositis and dermatomyositis: short term and longterm outcome, and predictive factors of prognosis. J Rheumatol 2001; 28(10):2230-7; PMID:11669162 [PubMed] [Google Scholar]
- 11.Clarke AE, Zowall H, Levinton C, Assimakopoulos H, Sibley JT, Haga M, Shiroky J, Neville C, Lubeck DP, Grover SA, et al.. Direct and indirect medical costs incurred by Canadian patients with rheumatoid arthritis: a 12 year study. J Rheumatol 1997; 24(6):1051-60; PMID:9195508 [PubMed] [Google Scholar]
- 12.Bernatsky S, Hudson M, Panopalis P, Clarke A, Pope J, LeClercq S, St Pierre Y, Canadian Scleroderma Research Group, M B . The cost of systemic sclerosis. Arthritis Rheumatism 2008; 61(1):119-23; PMID:19116974; https://doi.org/21362757 10.1002/art.24086 [DOI] [PubMed] [Google Scholar]
- 13.Bernatsky S, Panopalis P, Pineau CA, Hudson M, St Pierre Y, Clarke AE. Healthcare costs of inflammatory myopathies. J Rheumatol 2011; 38(5):885-8; PMID:21362757; https://doi.org/ 10.3899/jrheum.101083 [DOI] [PubMed] [Google Scholar]
- 14.Clarke AE, Esdaile JM, Bloch DA, Lacaille D, Danoff DS, Fries JF. A Canadian study of the total medical costs for patients with systemic lupus erythematosus and the predictors of costs. Arthritis Rheumatism 1993; 36:1548-59; PMID:8240431; https://doi.org/ 10.1002/art.1780361109 [DOI] [PubMed] [Google Scholar]
- 15.Hudson M, Pope J, Mahler M, Tatibouet S, Steele R, Baron M, Canadian Scleroderma Research Group (CSRG). Fritzler M. Clinical significance of antibodies to Ro52/TRIM21 in systemic sclerosis. Arthritis Res Therapy 2012; 14(2):R50; PMID:22394602; https://doi.org/22324944 10.1186/ar3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higgs BW, Zhu W, Richman L, Fiorentino DF, Greenberg SA, Jallal B, Yao Y. Identification of activated cytokine pathways in the blood of systemic lupus erythematosus, myositis, rheumatoid arthritis, and scleroderma patients. Int J Rheumatic Dis 2012; 15(1):25-35; PMID:22324944; https://doi.org/ 10.1111/j.1756-185X.2011.01654.x [DOI] [PubMed] [Google Scholar]
- 17.Cho JH, Gregersen PK. Genomics and the multifactorial nature of human autoimmune disease. N Engl J Med 2011; 365(17):1612-23; PMID:22029983; https://doi.org/ 10.1056/NEJMra1100030 [DOI] [PubMed] [Google Scholar]
- 18.Davidson A, Diamond B. Autoimmune diseases. N Engl J Med 2001; 345(5):340-50; PMID:11484692; https://doi.org/ 10.1056/NEJM200108023450506 [DOI] [PubMed] [Google Scholar]
- 19.Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell 2007; 130(1):25-35; PMID:17632054; https://doi.org/ 10.1016/j.cell.2007.06.033 [DOI] [PubMed] [Google Scholar]
- 20.Cotsapas C, Voight BF, Rossin E, Lage K, Neale BM, Wallace C, Abecasis GR, Barrett JC, Behrens T, Cho J, et al.. Pervasive sharing of genetic effects in autoimmune disease. PLoS Genetics 2011; 7(8):e1002254; PMID:21852963; https://doi.org/ 10.1371/journal.pgen.1002254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hudson M, Rojas-Villarraga A, Coral-Alvarado P, Lopez-Guzman S, Mantilla RD, Chalem P, Baron M, Anaya JM. Polyautoimmunity and familial autoimmunity in systemic sclerosis. J Autoimmunity 2008, 31(2):156-9; PMID:18644698; https://doi.org/ 10.1016/j.jaut.2008.05.002 [DOI] [PubMed] [Google Scholar]
- 22.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 2010, 11(3):191-203; PMID:20125086; https://doi.org/ 10.1038/nrg2732 [DOI] [PubMed] [Google Scholar]
- 23.Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, Berdasco M, Fraga MF, O'Hanlon TP, Rider LG, et al.. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Re 2010; 20(2):170-9; PMID:20028698; https://doi.org/ 10.1101/gr.100289.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin SY, Hsieh SC, Lin YC, Lee CN, Tsai MH, Lai LC, Chuang EY, Chen PC, Hung CC, Chen LY, et al.. A whole genome methylation analysis of systemic lupus erythematosus: hypomethylation of the IL10 and IL1R2 promoters is associated with disease activity. Genes Immunity 2012; 13(3):214-20; PMID:22048455; https://doi.org/ 10.1038/gene.2011.74 [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, Reinius L, Acevedo N, Taub M, Ronninger M, et al.. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol 2013; 31(2):142-7; PMID:23334450; https://doi.org/ 10.1038/nbt.2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lei W, Luo Y, Lei W, Luo Y, Yan K, Zhao S, Li Y, Qiu X, Zhou Y, Long H, et al.. Abnormal DNA methylation in CD4+ T cells from patients with systemic lupus erythematosus, systemic sclerosis, and dermatomyositis. Scand J Rheumatol 2009; 38(5):369-74; PMID:19444718; https://doi.org/ 10.1080/03009740902758875 [DOI] [PubMed] [Google Scholar]
- 27.Silva GL, Junta CM, Mello SS, Garcia PS, Rassi DM, Sakamoto-Hojo ET, Donadi EA, Passos GA. Profiling meta-analysis reveals primarily gene coexpression concordance between systemic lupus erythematosus and rheumatoid arthritis. Ann N Y Acad Sci 2007; 1110:33-46; PMID:17911418; https://doi.org/ 10.1196/annals.1423.005 [DOI] [PubMed] [Google Scholar]
- 28.Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, Brohawn P, Kiener PA, Richman L, Fiorentino D, et al.. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis 2011; 70(11):2029-36; PMID:21803750; https://doi.org/ 10.1136/ard.2011.150326 [DOI] [PubMed] [Google Scholar]
- 29.Tuller T, Atar S, Ruppin E, Gurevich M, Achiron A. Common and specific signatures of gene expression and protein-protein interactions in autoimmune diseases. Genes Immunity 2013; 14(2):67-82; PMID:23190644; https://doi.org/ 10.1038/gene.2012.55 [DOI] [PubMed] [Google Scholar]
- 30.Toro-Dominguez D, Carmona-Saez P, Alarcon-Riquelme ME. Shared signatures between rheumatoid arthritis, systemic lupus erythematosus and Sjogren's syndrome uncovered through gene expression meta-analysis. Arthritis Res Ther 2014; 16(6):489; PMID:25466291; https://doi.org/ 10.1186/s13075-014-0489-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamagata K FH, Oda N, Kaisaki PJ, Menzel S, Cox NJ, Fajans SS, Signorini S, Stoffel M, Bell GI. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature 1996; 384:458-60; PMID:8945471; https://doi.org/ 10.1038/384458a0 [DOI] [PubMed] [Google Scholar]
- 32.Dehghan A, Dupuis J, Barbalic M, Bis JC, Eiriksdottir G, Lu C, Pellikka N, Wallaschofski H, Kettunen J, Henneman P, et al.. Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation 2011; 123(7):731-8; PMID:21300955; https://doi.org/ 10.1161/CIRCULATIONAHA.110.948570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeffries M, Dozmorov M, Tang Y, Merrill J, Wren J, Sawalha A. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics 2011; 6(5):593-601; PMID:21436623; https://doi.org/ 10.4161/epi.6.5.15374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013; 153(6):1194-217; PMID:23746838; https://doi.org/ 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464(7285):104-7; PMID:20203610; https://doi.org/ 10.1038/nature08780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harris HE, Andersson U, Pisetsky DS: HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev 2012; 8(4):195-202; PMID:22293756; https://doi.org/ 10.1038/nrrheum.2011.222 [DOI] [PubMed] [Google Scholar]
- 37.Pisetsky DS. The role of mitochondria in immune-mediated disease: the dangers of a split personality. Arthritis Res Ther 2016; 18:169; PMID:27424174; https://doi.org/ 10.1186/s13075-016-1063-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oros Klein K, Grinek S, Bernatsky S, Bouchard L, Ciampi A, Colmegna I, Fortin JP, Gao L, Hivert MF, Hudson M, et al.. funtooNorm: an R package for normalization of DNA methylation data when there are multiple cell or tissue types. Bioinformatics 2016 Feb 15; 32(4):593-5; PMID:26500152; https://doi.org/24485249 10.1093/bioinformatics/btv615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014; 15(2):R29; PMID:24485249; https://doi.org/ 10.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015; 43(7):e47; PMID:25605792; https://doi.org/ 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9:559; PMID:19114008; https://doi.org/ 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.