

ABSTRACT
The MLH1 promoter polymorphism rs1800734 is associated with MLH1 CpG island hypermethylation and expression loss in colorectal cancer (CRC). Conversely, variant rs1800734 is associated with MLH1 shore, but not island, hypomethylation in peripheral blood mononuclear cell DNA. To explore these distinct patterns, MLH1 CpG island and shore methylation was assessed in CRC cell lines stratified by rs1800734 genotype. Cell lines containing the variant A allele demonstrated MLH1 shore hypomethylation compared to wild type (GG). There was significant enrichment of transcription factor AP4 at the MLH1 promoter in GG and GA cell lines, but not the AA cell line, by chromatin immunoprecipitation studies. Preferential binding to the G allele was confirmed by sequencing in the GA cell line. The enhancer-associated histone modification H3K4me1 was enriched at the MLH1 shore; however, H3K27ac was not, indicating the shore is an inactive enhancer. These results demonstrate the role of variant rs1800734 in altering transcription factor binding as well as epigenetics at regions beyond the MLH1 CpG island in which it is located.
KEYWORDS: Chromatin immunoprecipitation, colorectal cancer, DNA methylation, DNA mismatch repair, histone modifications, mutL homolog 1, single nucleotide polymorphisms
Introduction
A single nucleotide polymorphism (SNP) in the mismatch repair (MMR) gene mutL homolog 1 (MLH1) promoter (rs1800734, MLH1-93G>A) is associated with MLH1 promoter CpG island hypermethylation and microsatellite instability colorectal cancer (MSI CRC).1,2 MSI, occurring in ∼15% of sporadic CRCs, is the change in length of repetitive microsatellite DNA sequences due to defective MMR.3-5 MSI may occur as a result of mutations in MMR genes, including MLH1, MSH2, MSH6, and PMS2, or, more frequently, from MLH1 CpG island hypermethylation.5,6 SNP rs1800734 is associated with MSI CRC risk, as well as other neoplasms including glioblastoma, gastric, lung, and ovarian cancers.7-11 It may also be a risk SNP for CRC overall.12 We previously demonstrated that the allelic variant of rs1800734 decreases the transcriptional activity of MLH1.13 Upstream of the MLH1 CpG island is a region recognized as the MLH1 shore. Shores are regions flanking CpG islands, upstream and/or downstream, by up to 2000 bp with a lower GC content than islands. We have shown that this upstream MLH1 shore incurs hypomethylation in association with variant rs1800734 genotype in peripheral blood mononuclear cell (PBMC) DNA of CRC cases and controls.14 While MLH1 also has a shore downstream of its island, we determined that it does not incur SNP-associated methylation changes in PBMC DNA.14
Due to its location in the promoter of MLH1, SNP rs1800734 likely impacts epigenetic control and transcriptional regulation beyond DNA methylation alterations, including histone modifications and transcription factor binding. Just as presence or absence of DNA methylation can alter gene expression, histones and their modifications can also alter DNA activity, in part by regulating the degree of accessibility of the DNA to transcription factors or other transcriptional machinery.15 For example, trimethylation of lysine 27 on histone H3 (H3K27me3) is associated with repressed regions of the DNA. Histones H3K4me1 and H3K27ac mark active enhancers while H3K4me3 and H3K27ac mark active promoters.16,17
In addition to these marks, transcription is regulated by a variety of transcription factors and transcriptional machinery, such as RNA polymerase II (Pol II). Transcription factors contain DNA binding domains, allowing them to bind specific DNA sequences. Disruption of this sequence, such as due to occurrence of a SNP, may prevent binding. Other types of factors exist that prevent interactions between promoters and enhancers, called insulators. Proteins such as CCCTC binding factor (CTCF) act as insulators and can prevent spreading of DNA methylation to maintain genomic regions that are free of methylation.18,19 Here, we assessed DNA methylation status, selected histone modifications, CTCF, Pol II, and transcription factor AP-4 (TFAP4/AP4) at the CpG island and shore of MLH1 and investigated how their binding is modulated by SNP genotype of rs1800734.
Results
Genotype and methylation status of CRC cell lines
A panel of five CRC cell lines was genotyped for SNP rs1800734 and subjected to methylation assessment at the MLH1 CpG island and shore region. The results are shown in Table 1. HCT 116 and LS 174T were wild type (GG), COLO 320HSR and SNU-C2B were heterozygous (GA), and HCT-15 was homozygous variant (AA) for rs1800734. All five cell lines were completely unmethylated at the CpG island, whereas shore methylation was more variable. The GG cell lines were more highly methylated (73.1-99.6%) compared to the heterozygous (23.4-28.9%) or homozygous variant (34.6%) cell lines.
Table 1.
Promoter SNP genotype and methylation of MLH1 CpG island and shore in CRC cell lines. A panel of five colorectal carcinoma cell lines was genotyped by Sanger sequencing to determine genotype of rs1800734. MethyLight was utilized to determine the percentage of fully methylated alleles at the CpG island and shore of MLH1. Average PMR (percent methylated reference) of two duplicate reactions is shown.
| Cell Line | rs1800734 Genotype | CpG shore PMR | CpG island PMR |
|---|---|---|---|
| HCT 116 | GG | 99.6% | 0% |
| LS 174T | GG | 73.1% | 0% |
| SNU-C2B | GA | 28.9% | 0% |
| COLO 320HSR | GA | 23.4% | 0% |
| HCT-15 | AA | 34.6% | 0% |
Bisulfite sequencing was also performed for three overlapping PCR amplicons in CRC cell lines to measure methylation across the entire upstream regulatory region of MLH1 including its island and shore. Amplicon A, in the shore region, was highly methylated in the two GG cell lines, HCT 116 and LS 174T, whereas the GA cell lines, SNU-C2B and COLO 320HSR, and the AA cell line, HCT-15, were hypomethylated compared to wild type at the MLH1 shore (Fig. 1). The two GG cell lines were hypermethylated (at least 50% of CpGs methylated per allele) at 100% of the sequenced alleles. The GA and AA cell lines were hypermethylated at 0–38.5% of alleles. The last three CpG sites in Amplicon A overlapped with the first three CpG sites of Amplicon B. At Amplicon B, there was some methylation at the first five CpG sites, but CpGs were unmethylated downstream in all cell lines except HCT 116. HCT 116 had a small number of clones (2/10, 20%) that were hypermethylated across Amplicon B. Amplicon B and Amplicon C overlapped by one CpG site. All five cell lines, regardless of rs1800734 SNP genotype, were unmethylated at Amplicon C in the CpG island of MLH1. These results agree with quantitative methylation results (Table 1) and suggest that in CRC cell lines methylation at the MLH1 shore is correlated with genotype of rs1800734.
Figure 1.
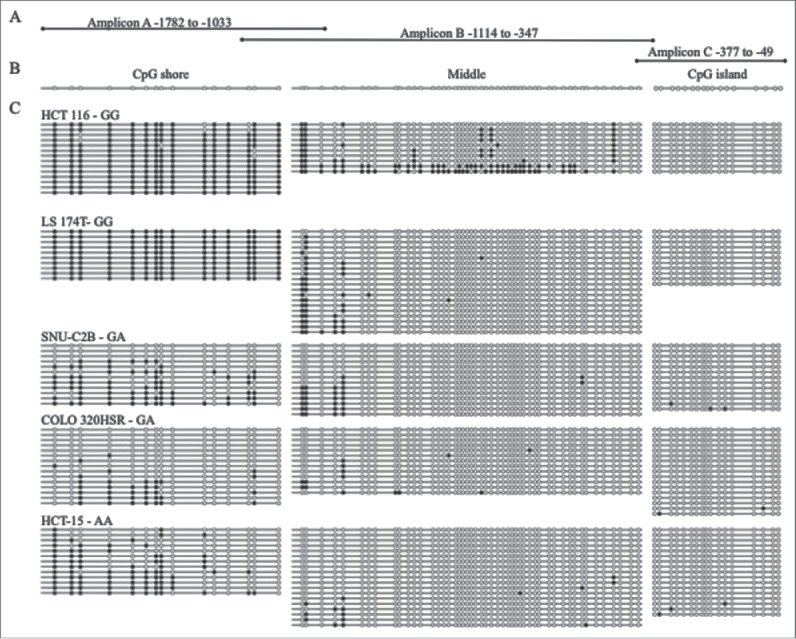
Bisulfite sequencing of MLH1 CpG island and shore in colorectal cancer cell lines. (A) Three overlapping regions upstream of MLH1 were amplified by bisulfite sequencing in cell lines: Amplicon A, Amplicon B, and Amplicon C. Amplicon A and B overlap at 3 CpGs. Amplicon B and C overlap at 1 CpG. (B) Representative unmethylated clone for each of the three amplicons, located in the CpG shore, middle region, and CpG island. Empty circles represent unmethylated CpG sites and filled in circles represent methylated CpG sites. (C) Methylation patterns in the colorectal cancer cell lines HCT 116, LS 174T, SNU-C2B, COLO 320HSR, and HCT-15 with rs1800734 genotype indicated. Each horizontal line represents a single DNA strand and circles represent individual CpG sites.
Sequence-specific binding of AP4
Chromatin immunoprecipitation (ChIP) experiments were undertaken for selected transcription factors and histone modifications in three of the cell lines that were profiled for DNA methylation at MLH1 by bisulfite sequencing: HCT 116, SNU-C2B, and HCT-15. All three cell lines display MSI and are MMR deficient without MLH1 CpG island hypermethylation.20-22 HCT 116 has a hemizygous mutation at codon 252 in MLH1, while HCT-15 has a 1 bp deletion at codon 252 and a 5 bp deletion/substitution at codon 1103.23,24 SNU-C2B is MSI but no mutations in the mismatch repair genes MLH1, MSH2, MSH6, or PMS2 have been reported.21,22
Publicly available databases and in silico transcription factor binding programs (UCSC Genome Browser, ENCODE, HOMER, Transfac, TF Bind, HaploReg) were explored to identify candidate proteins predicted to bind the wild type DNA sequence surrounding SNP rs1800734 at the G allele but not the A allele. We selected AP4 since it binds to the non-canonical E-box sequence CAGCTG containing wild type G but not the sequence CAGCTA containing variant A. ChIP for AP4 in the cell lines HCT 116 and SNU-C2B containing G allele(s) of rs1800734 resulted in enrichment at promoter amplicon P1 but not in the AA cell line HCT-15 (Fig. 2). HCT 116 had significantly higher occupancy of AP4 at the promoter region P1 than SNU-C2B (P = 0.013) and HCT-15 (P = 0.003). SNU-C2B also had significantly higher enrichment for AP4 at the MLH1 promoter than HCT-15 (P = 4.21 ×10−4). There was no enrichment at regions S1, S2, M1, or M2 as expected based on sequence specificity of AP4 binding.
Figure 2.
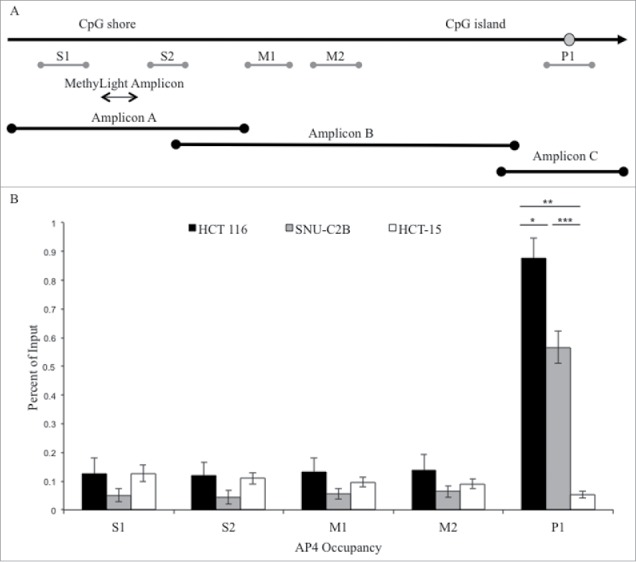
ChIP analysis of AP4 occupancy at the MLH1 CpG island and shore region. (A) Chromatin immunoprecipitation followed by qPCR was performed at five regions upstream of MLH1, located in the CpG shore (S1 and S2), middle region (M1 and M2), and promoter CpG island (P1). MethyLight and bisulfite sequencing regions interrogated are also indicated. SNP location is indicated by gray circle. (B) Experiments were performed in HCT 116 (GG), SNU-C2B (GA), and HCT-15 (AA) cell lines to compare AP4 occupancy among genotypes of SNP rs1800734. Three biological replicates of each cell line were run in triplicate and averaged after ChIP-qPCR. Error bars represent standard deviation. *P < 0.05, **P < 0.01, ***P < 0.001 by independent samples t-test.
Immunoprecipitated DNA from SNU-C2B was sequenced to confirm that enrichment was genotype-specific at the MLH1 promoter. Of the 20 alleles sequenced from the AP4 pull-down, 19 contained the G allele and one contained the A allele. Input DNA contained nearly equal numbers of G and A alleles, with 8 having the G allele and 11 having the A allele out of 19 alleles sequenced.
Histone modifications and Pol II are unchanged across genotypes of rs1800734
The histone modifications H3K4me1, H3K4me3, H3K27ac, and H3K27me3 were assessed at the MLH1 CpG island and shore at the same five regions as for AP4 (Fig. 3). H3K4me1 was enriched at the shore region of S1 and S2 compared to downstream regions M1, M2, and P1. H3K4me1 occupancy was significantly higher in SNU-C2B compared to HCT 116 at S2 (P = 0.029). There was similar enrichment for H3K4me1 in HCT-15 cells as in SNU-C2B, though this was not significantly different than HCT 116.
Figure 3.
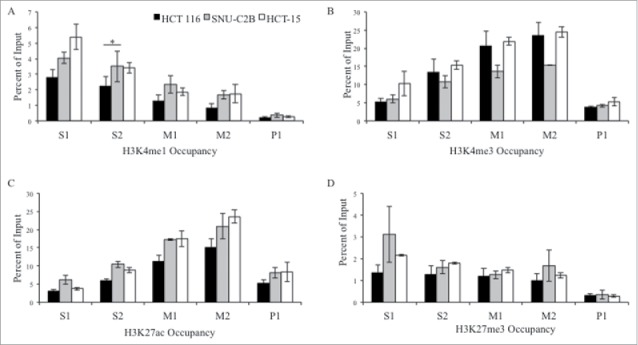
ChIP analysis of histone modifications at the MLH1 CpG island and shore. Chromatin immunoprecipitation was performed in HCT 116 (GG), SNU-C2B (GA), and HCT-15 (AA) cell lines to compare histone modifications among genotypes of SNP rs1800734. Three biological replicates of each cell line were run in triplicate and averaged after ChIP-qPCR at five regions of the MLH1 CpG island and shore: S1, S2, M1, M2, and P1. Histone modifications include: (A) H3K4me1, (B) H3K4me3, (C) H3K27ac, and (D) H3K27me3. Error bars represent standard deviation. *P < 0.05 by independent samples t-test.
H3K4me3 and H3K27ac both had similar enrichment patterns across the region, with low enrichment at promoter region P1, a peak of enrichment upstream at the M2 region, and decreasing levels of both modifications further upstream. H3K27me3 was low across the entire region tested. Presence of RNA polymerase II (Pol II) was also assessed in all three cell lines (Fig. 4). There were low levels observed for Pol II at all regions in all cell lines. Taken together, we observed a trend toward increased H3K4me1 at the MLH1 shore in cells containing variant A allele of rs1800734. Other histone modifications and factors tested, including H3K4me3, H3K27ac, H3K27me3, and Pol II, do not show such associations.
Figure 4.
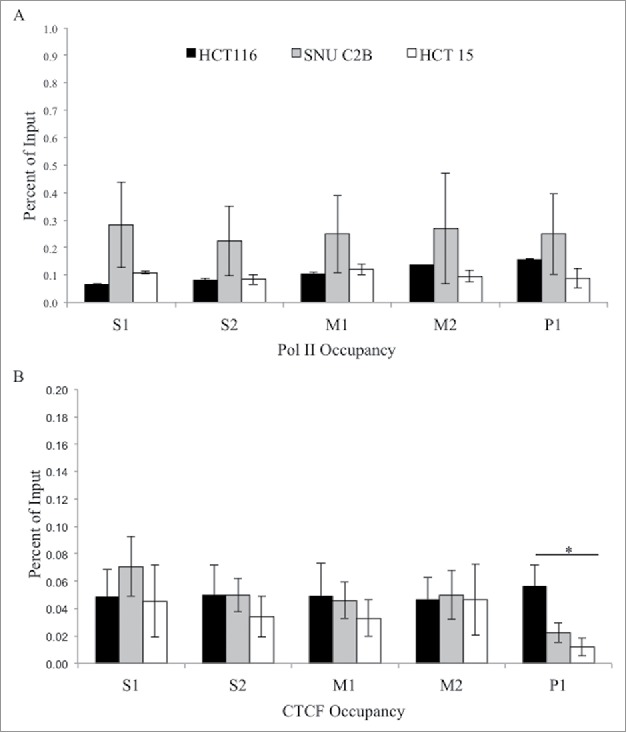
ChIP analysis of Pol II and CTCF at the MLH1 CpG island and shore. Chromatin immunoprecipitation was performed in HCT 116 (GG), SNU-C2B (GA), and HCT-15 (AA) cell lines to compare Pol II and CTCF binding among genotypes of SNP rs1800734. Three biological replicates of each cell line were run in triplicate and averaged after ChIP-qPCR at five regions of the MLH1 CpG island and shore: S1, S2, M1, M2, and P1. (A) Pol II and (B) CTCF were assessed. Error bars represent standard deviation. *P < 0.05 by independent samples t-test.
Lack of CTCF at MLH1 CpG island in variant rs1800734 cell line
Through exploration of publicly available databases and in silico transcription factor binding programs we found a predicted binding site for CTCF located in between the CpG island and shore of MLH1, located in region M2 of the five regions tested by ChIP-qPCR. Yet, there was only modest yet comparable CTCF binding in cell lines at regions S1, S2, M1, and M2 irrespective of SNP genotype (Fig. 4). At region P1 there was significantly higher enrichment for CTCF in HCT 116 compared to HCT-15 (P = 0.04). Although the enrichment at P1 was low, this may indicate differential binding of CTCF at the MLH1 promoter CpG island, with decreased binding in the rs1800734 homozygous variant HCT-15 cell line.
Discussion
Despite the fact that the majority of SNPs are located in noncoding regions of the genome, their diverse roles in disease pathogenesis, including CRC, are steadily becoming established through both experimental and computational methods.25-30 Here, we have demonstrated that various epigenetic and regulatory modifications are associated with variant genotype of the MLH1-93G>A promoter SNP rs1800734. It plays a role in modifying DNA methylation at the MLH1 shore in CRC cell lines. We have also demonstrated, for the first time, significantly diminished binding of the transcription factor AP4 in cell lines lacking the wild type G allele, which may play a role in the decrease in transcriptional activity previously reported.13 There may also be decreased binding of the insulator protein CTCF at the promoter region in cell lines lacking the G allele. The enhancer histone mark H3K4me1 appears to be increased at the MLH1 shore, especially in cell lines carrying variant alleles. Interestingly, the other histone modifications tested of enhancers, active regions, and repressed regions do not differ according to genotype and/or methylation status.
A number of noncoding SNPs identified through genome-wide association studies (GWAS) have been shown to change consensus sequences, which affects binding of transcription factors, thus altering enhancer or promoter activity. For example, a prostate cancer risk SNP at 6q22.1 leads to increased binding of transcription factor HOXB13, increased transcription of RFX6, and increased deposition of the H3K4me2 mark.25 As well, variant rs6983267 at the 8q24 risk locus leads to increased binding of transcription factor TCF7L2 in CRC cell lines, causing interactions with the MYC promoter.31 Similarly, we have previously demonstrated significant decreases in transcriptional activity for the variant allele of rs1800734 compared to wild type in CRC, normal colon, and endometrial cancer cell lines.13 Our group and others have also identified, though EMSA, the binding of a certain factor(s) to the G allele but not the variant A allele in CRC cell lines.11,13 However, the precise nature of these factors was not established. In this study, we have shown the presence of AP4 enrichment directly at the SNP in cell lines with one or two G alleles. AP4 also may be present in a dose-dependent manner, as the wild type cell line HCT 116 has significantly higher enrichment for AP4 compared to the heterozygous line SNU-C2B. Preferential binding to the G allele compared to the A allele in the heterozygous SNU-C2B cell line was also confirmed by clonal sequencing. However, this binding may also potentially be due to other differences in regulation of AP4, MLH1, or other factors between the two cell lines. Binding of transcription factors and/or RNA polymerase II has been shown to block or inhibit deposition of DNA methylation.32-34 Thus, lack of AP4 binding at the promoter variant SNP may lead to decreased transcriptional activity, possibly recruitment of other factors, and over time, increased DNA methylation. AP4 may act together with MYC, which has a binding motif CACGAG located 15 bp downstream of rs1800734, to activate transcription of MLH1.35 Future experiments to measure co-localization of MYC and AP4 in this region in a sequence-specific manner would further serve to elucidate this. A proposed model for this series of events is demonstrated in Fig. 5.
Figure 5.
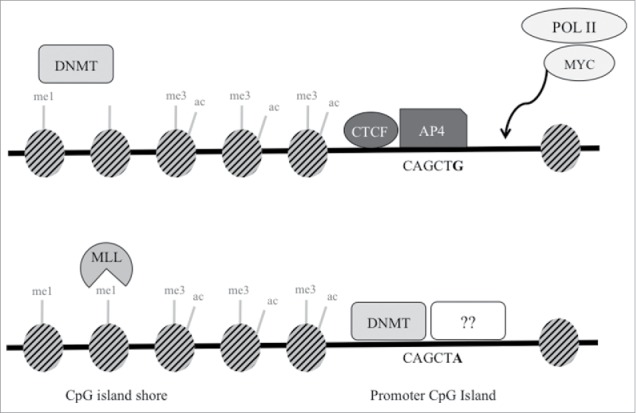
Proposed schematic model of transcription factors and epigenetic regulation at SNP rs1800734. In cells with wild type G allele (top), AP4 transcription factor binds its consensus sequence, potentially interacting with MYC and Pol II to promote transcription of MLH1. Though not demonstrated through experimental results in this study, DNA methyltransferases (DNMT) may maintain DNA methylation at the shore upstream. DNMTs may be prevented from methylating the CpG island in part due to presence of CTCF. In cells with the variant A allele (bottom), AP4 does not bind, which may decrease promoter transcriptional activity. Without the presence of AP4 (or possibly CTCF), DNMTs may methylate the exposed CpG island. This may lead to decreased methylation at the CpG shore and increased H3K4me1, which is deposited by MLL proteins. Other currently unidentified factors may also bind and repress the region further.
While the variant of interest, rs1800734, has not been discovered through GWAS studies, it has been identified as a risk SNP for CRC in a large number of individuals, including 10,409 cases and 6,965 controls.12 However, a subsequent study was unable to replicate these findings.36 Though there is controversy surrounding the overall role of this SNP in cancer susceptibility, it has been consistently shown that rs1800734 is a risk SNP for the MSI subtype of CRC.1,2 In the five cell lines selected for bisulfite sequencing analysis, we observed SNP-associated MLH1 shore hypomethylation, which was previously observed in PBMC DNA of CRC cases and controls.14 The exact mechanism of MLH1 shore hypomethylation remains to be elucidated. Potentially, MLH1 is part of the subset of genes with hypermethylation of their CpG shores alongside unmethylated CpG islands that have high transcriptional activity and a more transcriptionally permissive state, found across a variety of normal tissues and cancer types.37 A lack of AP4 binding at the promoter may decrease transcriptional activity, modifying the balance of methylation in the region. Absence of CTCF may also be responsible for the dysregulation and spreading of DNA methylation from the MLH1 shore downstream to the CpG island. Potentially, CTCF is present at the MLH1 promoter in wild type cells, and a loss or lack of CTCF in variant-containing cells is in part responsible for changing methylation patterns at the CpG island and shore.
An interesting finding of these results is the fact that three of the histone marks tested (H3K4me3, H3K27ac, H3K27me3) have genotype-independent enrichment levels across all five regions of the CpG island and shore despite having variable methylation patterns and SNP genotypes. Thus, DNA methylation changes appear to be largely independent of histone modifications present at the MLH1 region. Perhaps greater methylation differences are required in order for histone changes to occur, for example 0 vs. 100%, rather than more subtle and variable levels in between the two extremes. Alternatively, this may indicate that DNA methylation and/or transcription factor binding are more critical for regulation of this locus than histone modifications.
H3K4me1 was also examined in HCT 116, SNU-C2B, and HCT-15 cell lines. H3K4me1 is a marker of enhancer regions when found concurrently with H3K27ac, among other factors such as P300.38 H3K4me1 showed highest enrichment at the shore compared to further downstream at the promoter. However, H3K27ac was not similarly enriched at the shore. The presence of H3K4me1 without H3K27ac indicates that the MLH1 shore region may be considered an inactive or potentially ‘poised’ enhancer.17,38 Depending on the cell type, developmental, or regulatory cues, this region may act as an enhancer, but does not appear to be active in these three cells lines.
A limitation of this study is that the cell lines used in ChIP experiments were selected based on genotype of one SNP. Though all three cell lines have microsatellite instability but no MLH1 CpG island hypermethylation, each cell line differs in mutational spectra and thus may have differentially altered epigenetic and/or transcriptional machinery. In order to decrease the variability between cell lines and to experimentally establish the association between genotype, AP4 binding, and MLH1 expression, the CRISPR/Cas9 system could be used to create all three possible genotypes of rs1800734 in the same starting cell line. Yet, using cell lines to study the effects of a single nucleotide change may not necessarily reflect the mechanisms occurring within primary tumors from CRC patients. While we have previously demonstrated genotype-associated DNA methylation changes in patient specimens, experiments have not yet addressed the potential binding of AP4, CTCF, or histone modifications in tumors.1,2,14 Future investigation of these proteins in cases and/or controls, specifically binding of AP4, would be of value. We also did not take into account other SNPs located near rs1800734 in linkage disequilibrium (LD) with it. For example, in our previous studies we had shown comparable associations with CpG island and shore methylation and SNP genotype of two additional SNPs downstream of rs1800734 in strong LD, namely rs749072 and rs13098279 located 61 kb and 198 kb downstream of rs1800734, respectively.2,14
Colorectal cancer is both a genetic and an epigenetic disease and genetic changes may disrupt epigenetic and transcriptional regulation, with important consequences, specifically at MLH1 as we have shown here. We have comprehensively studied the epigenetic effects that a single nucleotide change in the MLH1 promoter can confer. Variant SNP genotype is associated with hypomethylation of the MLH1 shore. Despite methylation differences seen among CRC cell lines stratified by genotypes, the histone modifications assessed do not incur similar changes. This variant also alters the binding site of AP4 leading to diminished binding of this transcription factor. These results explore the functional epigenetic regulation and molecular mechanisms occurring at the important MLH1 region in CRC, shedding new light on the epigenetic concept of CpG shores and how DNA variants play a role in epigenetics and cancer susceptibility. If this example of genetic-epigenetic interaction is applied to the whole genome, there are clearly multitudes of ways in which the genome and epigenome may interact. Further studies of such interactions will lead to a better understanding of the processes and changes incurred by the genome and epigenome under both normal circumstances and cancer development.
Materials and methods
Cell lines
The colorectal carcinoma cell lines COLO 320HSR, HCT-15, HCT 116, LS 174T, and SNU-C2B were purchased from American Type Culture Collection. HCT 116 cells were cultured in McCoy's 5A Medium Modified. LS 174T cells were cultured in Eagle's Minimum Essential Medium. COLO 320HSR, HCT-15, and SNU-C2B cells were cultured in RPMI-1640. All cell culture media were supplemented with 10% fetal bovine serum. All cell lines were maintained in a humidified incubator at 37°C with 5% CO2.
Cell line genotyping
DNA from cell lines was isolated using QIAamp Blood Mini Kit (Qiagen, Cat No. 51106). DNA (40 ng per cell line) was amplified by PCR to amplify the region surrounding the MLH1 promoter SNP rs1800734. The MLH1 gene on chromosome 3 spans from Chr3:36,993,350-37,050,846 (GenBank, GRCh38) and rs1800734 is located at Chr3:36,993,455. The flanking DNA sequence surrounding rs1800734, including 50 bp upstream and downstream is as follows: 5’-AATCAATAGCTGCCGCTGAAGGGTGGGGCTGGATGGCGTAAGCTACAGCT[G/A]AAGGAAGAACGTGAGCACGAGGCACTGAGGTGATTGGCTGAAGGCACTTC-3’. Primers are listed in Table S1. PCR products were sequenced by Sanger sequencing at The Center for Applied Genomics (TCAG) DNA Sequencing Facility, The Hospital for Sick Children, Toronto, Canada. An external forward primer was used for Sanger sequencing (Table S1).
MethyLight
Methylation analysis by the semi-quantitative real-time PCR-based MethyLight assay was performed to amplify regions in the MLH1 shore and island. The shore amplicon spanned from -1499 to -1382 relative to the MLH1 translation initiation site (TIS, the adenine residue of ATG start codon from which rs1800734/MLH1-93G>A is measured) and the island amplicon spanned from -277 to -193 relative to the TIS. DNA was extracted from cell lines using QIAamp Blood Mini Kit and was subjected to bisulfite modification with the EZ DNA Methylation-Gold Kit according to manufacturer's protocol (Zymo Research, Cat No. D5006). Region-specific primers and probe for the island and shore were used, and ALU-C4 primers and probe were used as control. Probes contained a 5’ fluorescent reporter dye and a 3’ quencher dye. Primer and probe sequences are shown in Table S1. Percent methylated reference (PMR) was calculated using the following calculation: [Gene of Interest/ALU-C4]sample/[Gene of Interest/ALU-C4]CpGenome × 100%, where CpGenome represents commercially available fully methylated CpGenome Universal Methylated DNA (EMD Millipore, Cat No. S7821). Samples were analyzed in duplicate in 96-well plates on the 7500 Real-Time PCR System thermocycler (Applied Biosystems, Waltham, MA).
Bisulfite sequencing
DNA was extracted from cell lines using QIAamp Blood Mini Kit and was bisulfite modified using EZ DNA Methylation-Gold Kit. Three overlapping regions spanning the MLH1 promoter CpG island and adjacent shore region were amplified by PCR in bisulfite modified DNA from each cell line. Amplicon A, within the MLH1 shore, spanned from -1782 to -1033 bp relative to the MLH1 TIS. Amplicon B spanned from -1114 to -347 relative to the MLH1 TIS. Amplicon C, spanning the CpG island, covered -377 to -49 relative to the MLH1 TIS and contained rs1800734. Primers are listed in Table S1. PCR products for each reaction were purified with ChargeSwitch PCR Clean Up Kit (Invitrogen, Cat No. CS12000). Molecular cloning of amplicons was performed using pGEM-T Easy Vector System I (Promega, Cat No. A1360) with Max Efficiency DH5α Competent Cells (Invitrogen, Cat No. 18258012). Plasmid DNA was prepared using QIAprep Spin MiniPrep Kit (Qiagen, Cat No. 27106) and sequenced by Sanger sequencing at TCAG. At least ten clones were utilized for Sanger sequencing at TCAG for each region in each cell line.
Chromatin immunoprecipitation
ChIP experiments were performed in triplicate on three successive passages of HCT-15, HCT 116, and SNU-C2B cells following protocols from the EZ-Magna ChIP A/G Chromatin Immunoprecipitation Kit (EMD Millipore, Cat No. 17–10086). Histone H3 (ab61251), H3K4me1 (ab8895), H3K4me3 (ab8580), H3K27ac (ab4729), H3K27me3 (ab6002) and CTCF (ab70303) antibodies were purchased from Abcam. Normal Mouse IgG and RNA polymerase II, clone CTD4H8 (Pol II) antibodies were provided in the EZ-Magna ChIP A/G kit. AP4 (HPA001912) antibody was purchased from Sigma-Aldrich. Histone H3 and Normal Mouse IgG antibodies were used as positive and negative controls, respectively, for each experiment. Positive and negative control primers were also used for each antibody for quality control. ChIP-qPCR was performed in triplicate for each reaction at five regions upstream of MLH1 with the QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems, Waltham, MA). The five regions interrogated contained two regions in the MLH1 shore (called S1 and S2), two regions in between the island and shore (called M1 and M2), and one region in the promoter CpG island (called P1). The location of the ChIP-qPCR amplicons relative to the MLH1 TIS are as follows: S1: -1649 to -1525, S2: -1335 to -1232, M1: -1056 to -938, M2: -871 to -740, and P1: -197 to -70. The list of primers used for ChIP-qPCR at the MLH1 region is listed in Table S1.
Confirmation of genotype from ChIP experiments
Immunoprecipitated DNA from the ChIP experiments for AP4 and input from SNU-C2B cells was amplified by PCR using the same primers utilized for cell line genotyping. PCR products for AP4 and input were purified with ChargeSwitch PCR Clean Up Kit. Molecular cloning of amplicons was performed using pGEM-T Easy Vector System with Max Efficiency DH5α Competent Cells. The same protocol was utilized as for bisulfite sequencing, described previously, except DNA was not bisulfite modified. Plasmid DNA was prepared using QIAprep Spin MiniPrep Kit and at least 19 clones were sequenced by Sanger sequencing at TCAG for each reaction.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We would like to thank Dr. Ken J. Kron for his assistance in selection of candidate transcription factors.
Funding
This work was supported by the University of Toronto (Doctoral Completion Award, Dr. Rajalakshmi S. Dittakavi and Dr. Prema M. Rao Graduate Award, Fellowship Award), Sinai Health System (Lunenfeld-Tanenbaum Research Institute Studentships from Mount Sinai Hospital) and Canadian Institutes of Health Research (Interdisciplinary Health Research Team Program Studentship).
References
- 1.Raptis S, Mrkonjic M, Green RC, Pethe VV, Monga N, Chan YM, Daftary D, Dicks E, Younghusband BH, Parfrey PS et al.. MLH1 -93G>A promoter polymorphism and the risk of microsatellite-unstable colorectal cancer. J Natl Cancer Inst 2007; 99(6):463-74; PMID:17374836; https://doi.org/ 10.1093/jnci/djk095 [DOI] [PubMed] [Google Scholar]
- 2.Mrkonjic M, Roslin NM, Greenwood CM, Raptis S, Pollett A, Laird PW, Pethe VV, Chiang T, Daftary D, Dicks E et al.. Specific variants in the MLH1 gene region may drive DNA methylation, loss of protein expression, and MSI-H colorectal cancer. PLoS One 2010; 5(10):e13314; PMID:20967208; https://doi.org/ 10.1371/journal.pone.0013314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thibodeau SN, French AJ, Cunningham JM, Tester D, Burgart LJ, Roche PC, McDonnell SK, Schaid DJ, Vockley CW, MIchels VV et al.. Microsatellite instability in colorectal cancer: Different mutator phenotypes and the principal involvement of hMLH1. Cancer Res 1998; 58(8):1713-8; PMID:9563488 [PubMed] [Google Scholar]
- 4.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nat Lett 1993; 363:558-61; PMID:8505985; https://doi.org/ 10.1038/363558a0 [DOI] [PubMed] [Google Scholar]
- 5.Boland C, Thibodeau S, Hamilton S, Sidranksy D, Eshleman J, Burt R, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN et al.. A national cancer institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998; 58(22):5248-57; PMID:9823339 [PubMed] [Google Scholar]
- 6.Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010; 138(6):2073-87; PMID:20420947; https://doi.org/24325908 10.1053/j.gastro.2009.12.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez-Hernandez I, Perdomo S, Santos-briz A, Garcia JL, Gomez-Moreta JA, Cruz JJ, Gonzalez-Sarmiento R. Analysis of DNA repair gene polymorphisms in glioblastoma. Gene 2014; 536(1):79-83; PMID:24325908; https://doi.org/ 10.1016/j.gene.2013.11.077 [DOI] [PubMed] [Google Scholar]
- 8.Zhu H, Li X, Zhang X, Chen D, Li D, Ren J, Gu H, Shu Y, Wang D. Polymorphisms in mismatch repair genes are associated with risk and microsatellite instability of gastric cancer, and interact with life exposures. Gene 2016; 579(1):52-7; PMID:26724419; https://doi.org/ 10.1016/j.gene.2015.12.050 [DOI] [PubMed] [Google Scholar]
- 9.Niu L, Li S, Liang H, Li H. The hMLH1 −93G>A polymorphism and risk of ovarian cancer in the chinese population. PLoS One 2015; 10(8):e0135822; PMID:26275295; https://doi.org/ 10.1371/journal.pone0135822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lo YL, Hsiao CF, Jou YS, Chang GC, Tsai YH, Su WC, Chen KY, Chen YM, Huang MS, Hsieh WS et al.. Polymorphisms of MLH1 and MSH2 genes and the risk of lung cancer among never smokers. Lung Cancer 2011; 72(3):280-6; PMID:21093954; https://doi.org/ 10.1016/j.lungcan.2010.10.009 [DOI] [PubMed] [Google Scholar]
- 11.Miyakura Y, Tahara M, Lefor AT, Yasuda Y, Sugano K. Haplotype defined by the MLH1-93G/A polymorphism is associated with MLH1 promoter hypermethylation in sporadic colorectal cancers. BMC Res Notes 2014; 7:835; PMID:25421847; https://doi.org/ 10.1186/1756-0500-7-835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whiffin N, Broderick P, Lubbe SJ, Pittman AM, Penegar S, Chandler I, Houlston RS. MLH1-93G >A is a risk factor for MSI colorectal cancer. Carcinogenesis 2011; 32(8):1157-61; PMID:21565826; https://doi.org/ 10.1093/carcin/bgr089 [DOI] [PubMed] [Google Scholar]
- 13.Perera S, Mrkonjic M, Rawson JB, Bapat B. Functional effects of the MLH1-93G>A polymorphism on MLH1/EPM2AIP1 promoter activity. Oncol Rep 2011; 25(3):809-15; PMID:21206982; https://doi.org/ 10.3892/or.2010.1129 [DOI] [PubMed] [Google Scholar]
- 14.Savio AJ, Lemire M, Mrkonjic M, Gallinger S, Zanke BW, Hudson TJ, Bapat B. MLH1 Region Polymorphisms Show a Significant Association with CpG Island Shore Methylation in a Large Cohort of Healthy Individuals. PLoS One 2012; 7(12):e51531; PMID:23240038; https://doi.org/ 10.1371/journal.pone.0051531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol 2013; 20(3):259-66; PMID:23463310; https://doi.org/ 10.1038/nsmb.2470 [DOI] [PubMed] [Google Scholar]
- 16.Kimura H. Histone modifications for human epigenome analysis. J Hum Genet 2013; 58(7):439-45; PMID:23739122; https://doi.org/21632746 10.1038/jhg.2013.66 [DOI] [PubMed] [Google Scholar]
- 17.Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 2011; 21(8):1273-83; PMID:21632746; https://doi.org/ 10.1101/gr.122382.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000; 405(6785):482-5; PMID:10839546; https://doi.org/ 10.1038/35013100 [DOI] [PubMed] [Google Scholar]
- 19.Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 2000; 405(6785):486-9; PMID:10839547; https://doi.org/ 10.1038/35013106 [DOI] [PubMed] [Google Scholar]
- 20.Ahmed D, Eide PW, Eilertsen IA, Danielsen SA, Eknæs M, Hektoen M, et al.. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013; 2:e71; PMID:24042735; https://doi.org/ 10.1038/oncsis.2013.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ku JL, Park JG. Biology of SNU cell lines. Cancer Res Treat 2005; 37(1):1-19; PMID:19956504; https://doi.org/24755471 10.4143/crt.2005.37.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mouradov D, Sloggett C, Jorissen RN, Love CG, Li S, Burgess AW, Arango D, Strausberg RL, Buchanan D, Wormald S, et al.. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res 2014; 74(12):3238-47; PMID:24755471; https://doi.org/ 10.1158/0008-5472.CAN-14-0013 [DOI] [PubMed] [Google Scholar]
- 23.Boyer JC, Umar A, Risinger JI, Lipford JR, Kane M, Yin S, Barrett JC, Kolodner RD, Kunkel TA. Microsatellite instability, mismatch repair deficiency, and genetic defects in human cancer cell lines. Cancer Res 1995; 55(24):6063-70; PMID:8521394 [PubMed] [Google Scholar]
- 24.Papdopolous N, Nicolaides NC, Liu B, Parsons R, Lengauer C, Palombo F. Mutations of GTBP in genetically unstable cells. Science 1995; 268(5219):1915-7; PMID:7604266 [DOI] [PubMed] [Google Scholar]
- 25.Spisak S, Lawrenson K, Fu Y, Csabai I, Cottman RT, Seo JH, Haiman C, Han Y, Lenci R, Li Q et al.. CAUSEL: an epigenome- and genome-editing pipeline for establishing function of noncoding GWAS variants. Nat Med 2015; 21(11):1357-63; PMID:26398868; https://doi.org/ 10.1038/nm.3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Tian W. Explaining the disease phenotype of intergenic SNP through predicted long range regulation. Nucleic Acids Res 2016; 44(18):8641-54; PMID:27280978 ; https://doi.org/ 10.1093/nar/gkw519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemire M, Qu C, Loo LWM, Zaidi SHE, Wang H, Berndt SI, Bézieau S, Brenner H, Campbell PT, Chan AT et al.. A genome-wide association study for colorectal cancer identifies a risk locus in 14q23.1. Hum Genet 2015; 134(11-12):1249-62; PMID:26404086; https://doi.org/ 10.1007/s00439-015-1598-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biancolella M, Fortini BK, Tring S, Plummer SJ, Mendoza-Fandino GA, Hartiala J, Hitchler MJ, Yan C, Schumacher FR, Conti DV et al.. Identification and characterization of functional risk variants for colorectal cancermapping to chromosome 11q23.1. Hum Mol Genet 2014; 23(8):2198-209; PMID:24256810; https://doi.org/ 10.1093/hmg/ddt584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butter F, Davison L, Viturawong T, Scheibe M, Vermeulen M, Todd JA, Mann M. Proteome-wide analysis of disease-associated SNPs that show allele-specific transcription factor binding. PLoS Genet 2012; 8(9):e1002982; PMID:23028375; https://doi.org/ 10.1371/journal.pgen.1002982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tak YG, Farnham PJ. Making sense of GWAS: using epigenomics and genomeengineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics Chromatin 2015; 8:57; PMID:26719772; https://doi.org/ 10.1186/s13072-015-0050-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, Doddapaneni H, Beckwith CA, Chan JA, Hills A, Davis M et al.. The 8q24 cancer risk variant rs6983267 demonstrates long-range interaction with MYC in colorectal cancer. Nat Genet 2009; 41(8):882-4; PMID:19561607 ; https://doi.org/ 10.1038/ng.403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gebhard C, Benner C, Ehrich M, Schwarzfischer L, Schilling E, Klug M, Dietmaier W, Thiede C, Holler E, Andreesen R et al.. General transcription factor binding at CpG islands in normal cells correlates with resistance to de novo DNA methylation in cancer cells. Cancer Res 2010; 70(4):1398-407; PMID:20145141; https://doi.org/ 10.1158/0008-5472.CAN-09-3406 [DOI] [PubMed] [Google Scholar]
- 33.Takeshima H, Yamashita S, Shimazu T, Niwa T, Ushijima T. The presence of RNA polymerase II, active or stalled, predicts epigenetic fate of promoter CpG islands. Genome Res 2009; 19(11):1974-82; PMID:19652013; https://doi.org/ 10.1101/gr.093310.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blattler A, Farnham PJ. Cross-talk between site-specific transcription factors and DNA methylation states. J Biol Chem 2013; 288(48):34287-94; PMID:24151070; https://doi.org/ 10.1074/jbc.R113.512517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jackstadt R, Röh S, Neumann J, Jung P, Hoffmann R, Horst D, Berens C, Bornkamm GW, Kirchner T, Menssen A et al.. AP4 is a mediator of epithelial-mesenchymal transition and metastasis in colorectal cancer. J Exp Med 2013; 210(7):1331-50; PMID:23752226; https://doi.org/ 10.1084/jem.20120812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen H, Shen Z, Hu Y, Xiao Q, Bei D, Shen X, Ding K. Association between MutL homolog 1 polymorphisms and the risk of colorectal cancer: a meta-analysis. J Cancer Res Clin Oncol 2015; 141(12):2147-58; PMID:25986311; https://doi.org/ 10.1007/s00432-015-1976-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edgar R, Tan PP, Portales-Casamar E, Pavlidis P. Meta-analysis of human methylomes reveals stably methylated sequences surrounding CpG islands associated with high gene expression. Epigenetics Chromatin 2014; 7(1):28; PMID:25493099; https://doi.org/ 10.1186/1756-8935-7-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet 2014; 15(4):272-86; PMID:24614317; https://doi.org/ 10.1038/nrg3682 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.