

Abstract
Drug discovery heavily relies on cell viability studies to assess the potential toxicity of drug candidates. L-Lactate dehydrogenase (LDH) is a cytoplasmic enzyme that catalyzes the concomitant interconversions of pyruvate to L-lactate and NADH to NAD+ during glycolysis, and the reverse reactions during the Cori cycle (Decker and Lohmann-Matthes, 1988; Nachlas et al., 1960). In response to cellular damage, induced by endogenous cellular mechanisms or as a result of exogenously applied insults, LDH is released from the cytoplasm into the extracellular environment. Its stability in cell culture medium makes it a well-suited correlate for the presence of damage and toxicity in tissues and cells (Stoddart, 2011). We herein present protocols for a reproducible and validated LDH assay optimized for several cell types. In contrast to commercially available LDH assays often associated with proprietary formulations and high cost, our protocols provide ample opportunities for experiment-specific optimization with low variability and cost.
Keywords: lactate dehydrogenase, cell viability, plate reader-based assays
INTRODUCTION
Quantification of lactate dehydrogenase (LDH) is a well-established assay for cell viability that provides fast, robust and reproducible insights into the potential toxicity of experimental compounds and potential drug candidates. Experimentally, LDH activity is typically determined by utilizing a coupled enzymatic reaction, where LDH oxidizes lactate to pyruvate, which subsequently reacts with iodonitrotetrazolium chloride (INT) to form formazan. Formazan is water soluble and can be readily detected using colorimetry, measuring the absorbance at 490 nm (Decker and Lohmann-Matthes, 1988). The underlying assumption for this assay is that any increase in the amount of formazan produced in the culture supernatant is directly correlated with cell viability.
We here provide a detailed protocol for a custom LDH assay that is widely applicable to a variety of cell types, including primary cultured cells and cell lines, for the assessment of both neuroprotection and glioprotection.
STRATEGIC PLANNING
BASIC PROTOCOL 1. LDH release (LDHr) assay
The LDH release assay provides fast and reliable quantification of normalized LDH release, as a surrogate marker for cell viability. The workflow is straightforward, consisting of three steps: 1.) transfer of a 50 μl sample from the Sample Plate to the Assay Plate; 2.) addition of 50 μl Assay Buffer, followed by a 1 hr incubation; 3.) addition of 50 μl Stop Solution and measurement of absorbance at λ = 490 nm. Fig. 1 provides a simplified workflow diagram. For guidance, a sample plate layout is provided in Fig. 2. (cf. note on technical replicates in the Critical Parameters section below).
Figure 1. Workflow of the LDHr assay.

The LDHr assay follows a straightforward three-step protocol: 1.) addition of test compounds (e.g. tBHP); 2.) LDHr assay by transfer of a 50 μl sample of tissue culture supernatant to the Assay Plate and addition of 50 μl Assay Buffer; 3.) addition of 50 μl Stop Solution following a one hour incubation in the dark and subsequent measurement of absorbance at λ = 490 nm (OD490).
Figure 2. Sample plate layout.
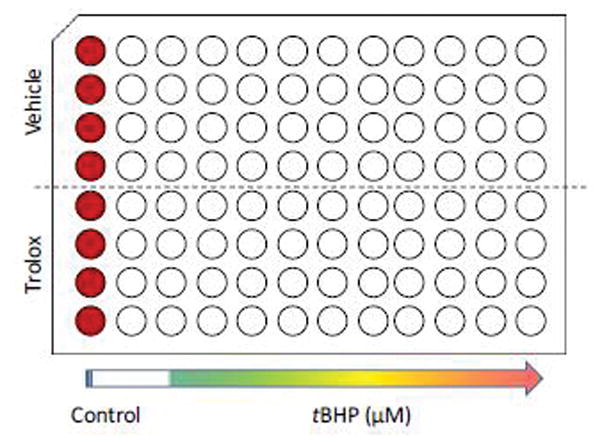
Each condition is tested in 4 technical replicates. The first column serves as control wells for the LDHt assay. The top half of the plate is reserved for pre-treatment with vehicle, whereas the bottom half will receive the test compound. tBHP in order to chemically induce oxidative stress will be applied at increasing concentrations in columns 3–12.
Materials
Reagents (in alphabetical order)
| Chemical | CAS number |
|---|---|
| Acetic Acid (glacial) | 64-19-7 |
| Hydrochloric acid | 7647-01-0 |
| Iodonitrotetrazolium Chloride (INT) | 146-68-9 |
| Lithium L-lactate | 27848-80-2 |
| 1-Methoxyphenazine methosulfate (MPMS) | 65162-13-2 |
| Beta-Nicotinotinamide Adenine Dinucleotide Sodium Salt (NAD) | 20111-18-6 |
| Tris Base (2-Amino-2-(hydroxymethyl)-1,3-propanediol) | 77-86-1 |
Supplies
96 well plates, clear
15 ml centrifuge tubes
1.5 ml microcentrifuge tubes
reagent reservoirs
Equipment
single channel and multi-channel pipettes
analytical balance
magnetic stir plate
pH meter
orbital shaker (optional)
plate reader (for measuring absorbance at 490 nm wavelength)
Protocol steps
Transfer 50 μl from each well of the Sample Plate to a new clear 96-well plate (the ‘Assay Plate’).
Prepare the Assay Reagent: combine equal volumes of Buffer A (2.5 ml) and Buffer B (2.5 ml), and 0.5 μl MPMS supplement per plate. Mix thoroughly. (Note: The Assay Reagent will be a light pink color, but the color may change if left on the bench top.)
Add 50 μl Assay Reagent to each well on the Assay Plate. Mix briefly on an orbital shaker (300–500 rpm for 15 sec).
Incubate plate in the dark for 60 min at room temperature.
Stop the reaction: add 50 μl 1M acetic acid to each well to stabilize the product. Mix briefly on an orbital shaker (300–500 rpm for 15 sec).
Record absorbance at 490 nm in a plate reader.
Normalize data to the control condition; a background correction is not required.
Step annotations
If the Sample Plate is also subjected to the LDHt assay, refer to the LDHt Basic Protocol 2.
If comparing the LDHr assay to the LDHt assay, we recommend addition of Lysis Solution at 5–10% by volume to the Assay Reagent for the LDHr assay. This is intended to control for the addition Triton X-100 in the lysis step of the LDHt assay and its effects on the LDH reaction kinetics. This is only necessary if the user is performing both an LDHr and LDHt on the same Sample Plate
If no shaker is available, swirl the plate by hand for 15 – 30 sec
Assay incubation time is directly related to the degree of color change. The LDH assay is a kinetic assay, users are advised to optimize the incubation time for the cell type to compensate for differing LDH concentrations within the sample. Our results suggest 1 hr is sufficiently below saturation to give a maximum spread between experimental wells although the LDHt assay does tend to result in higher signals. Do not incubate for less than 30 min.
Do not add excess amounts of acid. The reduced dye (purple) can spontaneously oxidize back to the initial reactant (light yellow) at pH < 4.0
The color difference between control and treated samples can be subtle and may not be visible by eye.
When comparing control vs. test compound conditions, the resulting data can be normalized to the control conditions without the need for complex mathematical manipulations, as the intrinsic LDH activity in the media will not affect the normalized values. When also performing the LDHt assay, cytotoxicity can be calculated using the following formula: % Cytotoxicity = OD490 (LDHr) / OD490 (LDHt) × 100
ALTERNATE PROTOCOL 1. Total LDH (LDHt) assay
The LDHt assay quantifies the maximal LDH activity by lysing the cells using Triton-X100. This allows for the calculation of relative cytotoxicity, by determining the ratio of LDHr/LDHt. When performing LDHt, a set of 4–8 replicate control wells should be added to the Sample Plate.
Materials
Reagents (in addition to those required for the Basic Protocol)
| Chemical | CAS number |
|---|---|
| Triton X-100 | 9002-93-1 |
Protocol steps
Add the appropriate amount of 10 × Lysis Solution to each control well used for the LDHt assay.
Mix on an orbital shaker for at least 3 min to completely lyse cells.
Remove 50 μl from each well of the Sample Plate and transfer to a new clear 96-well plate.
Proceed starting at Step 2 of the LDHr assay (Basic Protocol)
Step annotations
For every 100 μl of supernatant remaining in each well, add 10 μl of lysis solution.
Lysis progress can be confirmed with a basic tissue culture microscope and should appear as wells without attached cells and minor amounts of rounded cell debris remaining.
SUPPORT PROTOCOL 1. Experimental setup for neuroprotection studies using primary cultured rat cortical neurons
We have validated the LDH assay protocols described herein for neuroprotection experiments. We have previously described detailed protocols for the use of primary neuronal culture for high-throughput screening experiments using plate reader-based assays (Burroughs et al., 2012). The present protocol extends these protocols to measuring LDH release.
Given the role of oxidative stress in a number of pathologies, as well as the physiology of aging, we used tert-butyl hydroperoxide as a means to chemically induce oxidative stress. In order to demonstrate feasibility for testing neuroprotective compounds, we chose the prototypic antioxidant, Trolox.
The EC50 of tBHP, i.e. the concentration at which tBHP showed the half-maximal effect, was 26.2 μM (Fig. 3A), which is similar to our previous reports for the EC50 for tBHP in primary neuronal culture when using the MTT assay (Burroughs et al., 2012). Trolox shifted the EC50 for tBHP to 37.1 μM (Fig. 3A).
Figure 3. Sample results for Trolox-mediated cytoprotection using the LDHr assay.
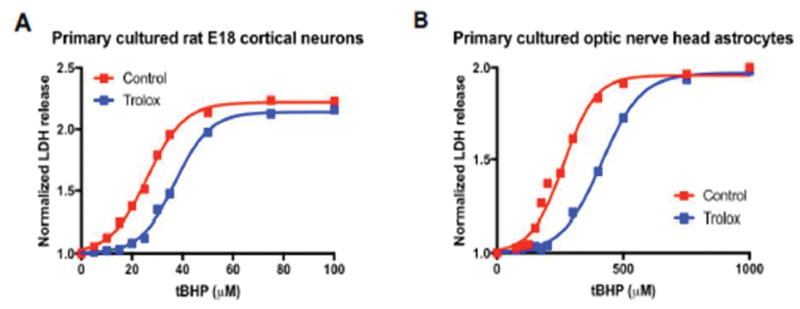
A.) Trolox protects primary cultured E18 rat cortical neurons from tBHP-induced oxidative stress, by shifting the EC50 from 26.2 μM to 37.1 μM. B.) Analogously, Trolox protects primary cultured adult rat optic nerve head astrocytes from tBHP-induced oxidative stress, by shifting the EC50 from 264.5 μM to 419.1 μM. For both experiments, data was plotted in Prism 7.0 software (GraphPad, La Jolla, CA) and analyzed by non-linear regression using a four-parameter logistic equation with variable Hill slope, performed on the mean value of the technical replicates (n = 4) to determine the mean EC50 values for tBHP for each pre-treatment condition (Control vs. Trolox).
The protocol presented here is a standard protocol that can be refined and optimized based on the user’s needs.
Materials
Reagents
| Chemical | CAS number |
|---|---|
| tert-butyl hydroperoxide (70% solution) | 75-91-2 |
| Trolox | 53188-07-1 |
Protocol steps
Prepare primary E18 rat cortical neurons using standard protocols (e.g. (Burroughs et al., 2012)) and seed at 25,000 cells per well in poly-amine- or poly-D-lysine/laminin-coated 96-well plates using a total volume of 100 μl per well.
Differentiate cells in vitro for 5 days, performing a partial media change on day 3.
Add Trolox at a final concentration of 100 μM and incubate for 1 hr.
Add tBHP (range 2.5 – 50 μM) and incubate for 16 hr.
Perform LDHr and/or LDHt assay as described in Basic Protocol and Alternate Protocol above.
Step annotations
-
3
We recommended adding Trolox (or any other neuroprotectant) in 50 μl volume at 3× concentration (new total volume per well: 150 μl). For Trolox, prepare a 100 mM stock solution in 100% ethanol, prepared fresh for every experiment.
-
4
We recommend preparing a 100 mM stock solution of tBHP in complete media immediately prior to the experiment. Use this stock solution to prepare test solutions at 4× concentration in complete media and add 50 μl to each well (new total volume per well: 200 μl).
SUPPORT PROTOCOL 2. Experimental setup for glioprotection studies using primary cultured rat optic nerve head astrocytes
Glioprotection refers to the experimental strategy of protecting glia cells from experimentally and/or pathologically-induced damage. We have pioneered the culture of primary adult rat optic nerve head astrocytes and have previously shown the feasibility of using this specialized subtype of astrocytes for plate reader-based assays to identify novel glioprotectants (Kaja et al., 2015a; Kaja et al., 2015b).
Given the role of oxidative stress in a number of pathologies, as well as the physiology of aging, we use tert-butyl hydroperoxide as a means to chemically induce oxidative stress. In order to demonstrate feasibility for testing neuroprotective compounds, we chose the prototypic antioxidant, Trolox, analogously to the experimental design for primary neuronal culture described above.
The EC50 of tBHP was 264.5 μM (Fig. 3B), which was shifted to 414.9 μM in the presence of Trolox, in accordance with our previous observations (Kaja et al., 2015a).
The protocol presented here is a standard protocol that can be refined and optimized based on the user’s needs.
Materials
Reagents
| Chemical | CAS number |
|---|---|
| tert-butyl hydroperoxide (70% solution) | 75-91-2 |
| Trolox | 53188-07-1 |
Protocol steps
Prepare primary optic nerve head astrocytes using standard protocols (Kaja et al., 2015a) and seed at a density of 7,500 cells per well in a 96-well plate, using a total volume of 100 μl per well.
Add Trolox at a final concentration of 100 μM and incubate for 1 hr.
Add tBHP (range 50 μM – 1 mM) and incubate for 5 hr.
Perform LDHr and/or LDHt assay as described in Basic Protocol and Alternate Protocol above.
Step annotations
-
2
We recommended adding Trolox (or any other glioprotectant) in 50 μl volume at 3× concentration (new total volume per well: 150 μl). For Trolox, prepare a 100 mM stock solution in 100% ethanol, prepared fresh for every experiment.
-
3
We recommend preparing a 100 mM stock solution of tBHP in complete media immediately prior to the experiment. Use this stock solution to prepare test solutions at 4× concentration in complete media and add 50 μl to each well (new total volume per well: 200 μl).
REAGENTS AND SOLUTIONS
Recipes
Buffer A: 4mM INT (4×) in 0.2 M Tris-HCl, pH 8.2
Dissolve 0.5057 g INT in 250 ml 0.2 M Tris-HCl, pH 8.2
Mix thoroughly and filter to remove any undissolved particles. Solution should be clear to slightly yellow.
(Note: Heating or extensive time spent mixing (>15 min) can lead to the formation of a black precipitate. In that case, dispose of the solution and restart.)
Aliquot and store frozen (−20 °C) for up to 6 months.
Note: aliquots of 3 ml for a single plate or 13.5 ml for 5 plates in 15 ml conical tubes are recommended
Buffer B: 6.4 mM NAD (4×), 320 mM lithium lactate (4×), in 0.2 M Tris-HCl buffer
Dissolve 1.1 g NAD and 8.9 g lithium lactate in 250 ml 0.2 M Tris-HCl, pH 8.2
Mix thoroughly and filter to remove any undissolved particles. (The solution is clear when freshly prepared, but will turn light yellow after a few days.)
Aliquot and store frozen (−20 °C) for up to 12 months.
Note: aliquots of 3 ml for a single plate or 13.5 ml for 5 plates in 15 ml conical tubes are recommended
Note: Sodium lactate can be used as an alternative for lithium lactate.
MPMS Supplement: 150 mM MPMS in Tris buffer (10,000×)
Dissolve 100 mg MPMS in 1.98 ml 0.2 M Tris-HCl, pH 8.2
Do not filter. MPMS Supplement is a dark purple or brown solution.
Aliquot (5 μl in 1.5 ml microcentrifuge tubes) and store frozen (−20 °C) for up to 12 months.
Note: The solution may occasionally appear grainy, but this does not affect assay quality if the supplement is thoroughly mixed prior to addition to the Assay Reagent.
Lysis solution: 9% Triton X-100 in water (v/v)
Lysis solution: 9% Triton-X100 in water (v/v)
Add 4.5 ml Triton X-100 to 45.5 ml water.
Mix well by vortexing.
Filter for particulates.
Store refrigerated for up to 12 months.
Stop Solution: 1 M acetic acid
Transfer 57.5ml acetic acid (glacial) to a glass measuring cylinder
Add water to 1 L.
Filter if necessary.
Store at room temperature.
Diluent: 0.2 M Tris-HCl, pH 8.2
Weigh out 24.2 g Tris Base
Dissolve in 800 ml water on a magnetic stir plate
Adjust to pH 8.2 with HCl
Add water to 1L.
Filter to remove particulates.
Store refrigerated.
COMMENTARY
Background Information
The basis for the current LDH assay using a tetrazolium salt was originally described over 50 years ago (Nachlas et al., 1960), and provided a fast and reliable quantification of serum lactate dehydrogenase (Babson and Phillips, 1965). The assay was quickly adapted to automation (Capps et al., 1966) and optimized further (Babson and Babson, 1973; Raabo, 1963). Since then, LDH measurements have become routine both in clinical medicine and biomedical sciences. Clinically, quantification of LDH in serum is used to ascertain the existence and severity of acute or chronic tissue damage, and for the diagnosis and treatment monitoring for a variety of cancers. In biomedical sciences, the LDH assay has have become a standard tool in drug discovery to quantify the number of cells beginning to undergo apoptotic and necrotic processes, making it an indispensable tool to quantify the potential for neuroprotection and glioprotection of drug candidates (Stoddart, 2011; Vega-Avila and Pugsley, 2011).
Numerous commercial LDH release assay kits are available. However, these commercial solutions have numerous drawbacks, which include high cost, limited scalability, and most importantly, unknown formulations. It is well established that several chemicals inactivate LDH enzyme activity or interfere with the cellular ability to release LDH (Kendig and Tarloff, 2007), necessitating carefully-designed experimental protocols.
Our protocols presented herein are significantly more cost effective than comparable commercially available assays, providing an approximately 30–40 fold cost saving in reagent costs over commercially available assays. Given the minima labor cost for preparing stock solutions we estimate a 25-fold cost-saving over commercially available assays for LDH release.
The known formulation of our assay allows for experiment-specific optimization is fully scalable. Stock solutions can be prepared in advance, for up to thousands of plates at a time, eliminating lot to lot variability.
Importantly, we have previous validated and benchmarked our protocol against a commercially available assay in C6 astroglioma cells (Kaja et al., 2015a; Kaja et al., 2015c) and shown that our assay performs at the same level as the one of a leading commercial supplier.
Critical Parameters
1. Different cell types
Careful experimental planning is required in order to optimize the assay for various cell lines and drug administrations. The total amount and kinetics of release of LDH differ greatly between cell types, and optimizations need to be performed to establish the maximal release. Additional factors affecting assay performance include the rate of proliferation and the time required for in vitro differentiation (if applicable).
2. Drug treatments
The duration of pre-incubation with experimental drugs must be determined by taking into account the mechanism of action, ensuring maximum bioavailability of active compound.
3. Incubation with experimental stressor/insult
The application of the experimental stressor or insult intended to trigger LDH release requires careful optimization in order to achieve a sufficient window to quantify cytoprotection without confounding the assay by differences in cell proliferation. Furthermore, it is important to note that LDH may be taken back up into cells or metabolized after prolonged insults (> 24 hr). Time course experiments should be performed to determine the appropriate incubation time.
4. Technical replicates
The LDH assay shows low experimental variability. Given the high-throughput format on 96-well plates, we routinely run 4 – 8 technical replicates for each condition (either a half column or a full column), as shown in Fig. 2. This format allows for the fast pipetting of a large number of plates using 8- and 12-channel pipets.
Troubleshooting
| Problem | Cause | Solution |
|---|---|---|
| Low or no signal | Cell density is too low | Increase seeding density |
| Test compound or media inhibit LDH activity | Assess test compound in a system with established LDH release, by quantifying both LDHr and LDHt | |
| Media may contain sodium pyruvate, a known inhibitor of LDH | Avoid media with sodium pyruvate | |
| The incubation time for the assay may be too short | Perform a kinetic run and monitor LDH release over time (cf. (Kaja et al., 2015c) | |
| Background signal too high / saturated | Cell concentration is too high | Optimize seeding densities |
| Intrinsic LDH in serum/media is high | Reduce serum concentration in media and/or reduce MPMS Supplement concentration to 1:20,000 dilution. Heat inactivation may affect the serum LDH activity. We recommend testing the endogenous activity of serum by performing cell-free serum or complete media controls. Complete media LDH activity is typically similar to the observed LDH activity of untreated cells. | |
| Target signal too high / saturated | Cell concentration is too high | Optimize seeding densities |
| Incubation time is too long | Reduce incubation time to 30 min | |
| MPMS Supplement concentration is too high | Reduce MPMS Supplement concentration to 1:20,000 dilution | |
| Dye turns yellow after addition of Stop Solution | Concentration of acetic acid is too high. The reduced dye (purple) can spontaneously oxidize back to the initial reactant (light yellow) at pH < 4.0 | Reduce the amount of Stop Solution |
Anticipated Results
After addition of and incubation in the Assay Solution, the color of samples will be dark red/purple. Higher LDH activity will result in darker color (i.e. higher absorbance). Especially when using phenol-red containing medium, differences between conditions may be hard to ascertain by eye but readily detected once measuring absorbance.
Different cell types differ in the extent of LDH activity, and the amount of LDH release in response to exogenous insults. Typically, LDHr will plateau in the range of 1.5 – 2.5-fold over normalized release in the control, unstimulated condition.
Time Considerations
The LDH assay itself only requires approx. 2 hours, when using previously prepared and aliquoted solutions. Preparation of stock solutions can be achieved in 2 – 3 hrs depending on the number of aliquots that are being prepared. Additional time is required to prepare cell culture plates for experiments, which can vary greatly between cell types.
Significance Statement.
Cell-based screening assays have taken an important place in the drug discovery pipeline as tools to account for the biological complexity of drug effects that cannot readily be studied using biochemical methods alone. Cell viability assays are employed in order to assess the potential toxicity of drug candidates or to determine the cytoprotective properties of compounds, typically in response to an induced stressor. One of the most frequently employed assays utilizes L-lactate dehydrogenase (LDH) release as a surrogate measure for cell viability. Commercially available LDH assays have several drawbacks, incl. proprietary formulations that can confound experiments. The validated custom LDH assay reported herein provides a standardized platform that can be easily modified to meet changing experimental needs.
Acknowledgments
Research reported in this publication was supported by grants from the National Eye Institute (EY022774), the National Institute on Aging (AG022550 and AG027956), the National Center for Research Resources and National Institute of General Medical Sciences (RR027093) of the National Institutes of Health (PK). This material is the result of work supported with resources and the use of facilities at the Edward Hines Jr. VA Hospital, Hines, IL. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional support by the Dr. John P. and Therese E. Mulcahy Endowed Professor in Ophthalmology (SK), Illinois Society for Prevention of Blindness (SK), the Felix and Carmen Sabates Missouri Endowed Chair in Vision Research (PK), a Challenge Grant from Research to Prevent Blindness (PK) and the Vision Research Foundation of Kansas City (PK) is gratefully acknowledged.
Footnotes
-
Detailed description of primary neuronal culture for plate reader-based assays:Burroughs, S.L., Duncan, R.S., Rayudu, P., Kandula, P., Payne, A.J., Clark, J.L., Koulen, P., and Kaja, S. 2012. Plate reader-based assays for measuring cell viability, neuroprotection and calcium in primary neuronal cultures. J Neurosci Methods 203:141–145.
-
Detailed description of primary optic nerve head astrocyte culture for plate reader-based assays, incl. examples of kinetic analysis LDHr:Kaja, S., Payne, A.J., Naumchuk, Y., Levy, D., Zaidi, D.H., Altman, A.M., Nawazish, S., Ghuman, J.K., Gerdes, B.C., Moore, M.A., and Koulen, P. 2015a. Plate reader-based cell viability assays for glioprotection using primary rat optic nerve head astrocytes. Exp Eye Res 138:159–166.
LITERATURE CITED
- Babson AL, Babson SR. Kinetic colorimetric measurement of serum lactate dehydrogenase activity. Clinical chemistry. 1973;19:766–769. [PubMed] [Google Scholar]
- Babson AL, Phillips GE. A rapid colorimetric assay for serum lactic dehydrogenase. Clin Chim Acta. 1965;12:210–215. doi: 10.1016/0009-8981(65)90032-x. [DOI] [PubMed] [Google Scholar]
- Burroughs SL, Duncan RS, Rayudu P, Kandula P, Payne AJ, Clark JL, Koulen P, Kaja S. Plate reader-based assays for measuring cell viability, neuroprotection and calcium in primary neuronal cultures. J Neurosci Methods. 2012;203:141–145. doi: 10.1016/j.jneumeth.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capps RD, 2nd, Batsakis JG, Briere RO, Calam RR. An automated colorimetric (tetrazolium salt) assay for serum lactate dehydrogenase. Clinical chemistry. 1966;12:406–413. [PubMed] [Google Scholar]
- Decker T, Lohmann-Matthes ML. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods. 1988;115:61–69. doi: 10.1016/0022-1759(88)90310-9. [DOI] [PubMed] [Google Scholar]
- Kaja S, Payne AJ, Naumchuk Y, Levy D, Zaidi DH, Altman AM, Nawazish S, Ghuman JK, Gerdes BC, Moore MA, Koulen P. Plate reader-based cell viability assays for glioprotection using primary rat optic nerve head astrocytes. Exp Eye Res. 2015a;138:159–166. doi: 10.1016/j.exer.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaja S, Payne AJ, Patel KR, Naumchuk Y, Koulen P. Differential subcellular Ca2+ signaling in a highly specialized subpopulation of astrocytes. Exp Neurol. 2015b;265:59–68. doi: 10.1016/j.expneurol.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaja S, Payne AJ, Singh T, Ghuman JK, Sieck EG, Koulen P. An optimized lactate dehydrogenase release assay for screening of drug candidates in neuroscience. J Pharmacol Toxicol Methods. 2015c;73:1–6. doi: 10.1016/j.vascn.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendig DM, Tarloff JB. Inactivation of lactate dehydrogenase by several chemicals: implications for in vitro toxicology studies. Toxicology in vitro : an international journal published in association with BIBRA. 2007;21:125–132. doi: 10.1016/j.tiv.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachlas MM, Margulies SI, Goldberg JD, Seligman AM. The determination of lactic dehydrogenase with a tetrazolium salt. Anal Biochem. 1960;1:317–326. doi: 10.1016/0003-2697(60)90029-4. [DOI] [PubMed] [Google Scholar]
- Raabo E. Determination of serum lactic dehydrogenase by the tetrazolium salt method. Scand J Clin Lab Invest. 1963;15:233–238. doi: 10.3109/00365516309079738. [DOI] [PubMed] [Google Scholar]
- Stoddart MJ. Cell Viability Assays: Introduction. In: Stoddart MJ, editor. Mammalian Cell Viability: Methods and Protocols. Humana Press; New York City: 2011. pp. i–xi. [Google Scholar]
- Vega-Avila E, Pugsley MK. An overview of colorimetric assay methods used to assess survival or proliferation of mammalian cells. Proc West Pharmacol Soc. 2011;54:10–14. [PubMed] [Google Scholar]