

Commentary
Angiotensin-(1-7) (Ang-(1-7)), an alternative product of the renin-angiotensin system (RAS), was initially identified in the circulation, brain and adrenals by our laboratory over 25 years ago and was originally considered to modulate the constrictor and pressor actions of Angiotensin II (Ang II) [1]. However, the experimental evidence to date suggests that Ang-(1-7) exhibits a wide range of protective effects apart from the regulation blood pressure [2,3]. Circulating Ang-(1-7) is derived by the direct conversion of the inactive precursor peptide Ang I by the metalloendopeptidase (MEP) neprilysin while the cellular levels of the peptide may refect processing of Ang I by another MEP -thimet oligopeptidase; both enzymes hydrolyze the Pro7-Phe8 bond of Ang I to generate Ang-(1-7) [4]. Endogenous levels of Ang-(1-7) may also originate from the direct conversion of Ang II by mono-carboxypeptidases angiotensin converting enzyme 2 (ACE2) and prolyl carboxypeptidase [4]. These latter pathways initially require the conventional processing of Ang I by ACE to generate Ang II; however, ACE is the principal pathway in the circulation to degrade Ang-(1-7) to Ang-(1-5) and explains the pronounced effect of ACE inhibitors to augment the circulating levels of Ang-(1-7) [5]. Elucidation of the processing steps and unique actions of Ang-(1-7) has ostensibly led to the divergence of the RAS into multiple functional arms that include the ACE-Ang II-AT1 receptor (AT1R) and the ACE2/MEP-Ang-(1-7)-AT7/MasR [2,3].
In contrast to the Ang II-AT1R that mediates a number of pathological events associated with an “activated RAS”, the Ang-(1-7) pathway is thought to antagonize many of cellular actions of the Ang II-AT1R axis [3]. In this regard, the pathologies typical of an activated RAS such as inflammation, fibrosis and an altered redox balance may well refect a stimulated Ang II-AT1R and an attenuated Ang-(1-7)-AT7R axis [6]. Therapeutic approaches to block the ACE-Ang II-AT1R that include ACE inhibitors (ACEIs) and AT1R antagonists (ARBs) increase endogenous levels of Ang-(1-7) [4]. Moreover, treatment with Ang-(1-7) may convey important therapeutic benefits in a range of pathologies including cancer, diabetes, and hypertension and tissue fibrosis [3]. The current review examines the cellular signaling pathways of the Ang-(1-7)-AT7/MasR axis that are associated with the anti-fibrotic actions of the peptide.
Angotensin-(1-7) Attenuates Fibrosis
Tissue fibrosis is a normal reparative process involved in the cellular response to tissue injury; however, progressive and sustained fibrosis in various clinical pathologies including heart failure, pulmonary hypertension, diabetic nephropathy, non-alcoholic liver disease, peripheral arterial disease, muscular dystrophy and diabetic retinopathy leads to an inevitable decline in organ function. Activation of the ACE-Ang II-AT1R is typically associated with fibrosis in multiple tissues that can refect an increase in blood pressure and the direct cellular effects of Ang II. In turn, blockade of the RAS by ACEIs or ARBs can be an effective approach to attenuate the progression of fibrosis. Since RAS blockade may result in enhanced levels of Ang-(1-7) and/or higher expression of the AT7/MasR an emerging number of studies have begun to directly examine the impact of exogenous Ang-(1-7) in different models of fibrosis.
Iwata et al. initially reported evidence of specific Ang-(1-7) binding sites expressed in isolated cardiac fibroblasts, a key cell type involved in tissue fibrosis; the Ang-(1-7) receptor sites were sensitive to the AT7/ MasR antagonist D-Ala7-Ang-(1-7) (DALA, A779) and the nonselective angiotensin antagonist [Sar1,Tr8]-Ang II (Sarthran), but not AT1R or AT2R blockers [7]. Ang-(1-7) reduced the Ang II-dependent stimulation of 3H-proline uptake as an index of fibrosis and reduced the expression of endothelin-1 and the cytokine LIF in cardiofibroblasts, but failed to attenuate TGF-β expression, an important pro-fibrotic cytokine and downstream effector of Ang II [7]. Several reports subsequently demonstrated that exogenous administration of Ang-(1-7) attenuated the development of cardiac fibrosis in models of pressure overload induced by aortic co-arctation [8], DOCA-salt hypertension [9], chronic Ang II treatment [10,11], LNAME hypertension [12], diabetic cardiomyopathy [13] and doxorubicin-induced cardiotoxicity [14]. Importantly, treatment with the AT7/MasR antagonist DALA exacerbated the extent of Ang II-induced cardiac fibrosis and the expression of multiple cytokines including TGF-β, TNFα, MCP-1 and ICAM-1, as well the metalloproteinase inhibitors TIMP 1 and 2 suggesting that intrinsic Ang-(1-7) tone mediates the pro-fibrotic actions of Ang II within the heart [6]. The anti-fibrotic actions of Ang-(1-7) in experimental models are not limited exclusively to the heart as Ang-(1-7) administration ameliorates fibrosis in liver steatosis [15,16], pulmonary hypertension [17], pulmonary asthma [18], idiopathic pulmonary fibrosis vascular hypertension [19] muscular dystrophy [20] and both obstructive and diabetic nephropathies [21,22]. Indeed, Ang-(1-7) offered greater protective effects than the AT1R antagonist valsartan to ameliorate the diabetic nephropathy [23] and the combination of Ang-(1-7) and an ACE inhibitor was more effective than either agent alone to attenuate diabetic cardiac fibrosis [24].
Ang-(1-7) Signalling Pathways
TGF-β/SMAD
Activation of the Ang-(1-7) signal transduction pathway generally involves the MasR protein and is blocked by the specific antagonists DALA and D-Pro7-Ang-(1-7); however, AT1R, AT2R and bradykinin B2R antagonists are also reported to block some of the actions of Ang-(1-7) suggesting either a direct interaction of these receptors with the MasR or downstream effects of an activated Ang-(1-7)-MasR on these receptor systems [3,5,25,26]. In regards to tissue fibrosis, the activation of TGF-β and the SMAD transcription factor pathway are considered a key signaling event in the initiation and progression of cellular fibrosis (Figure 1). Cai et al. reported that Ang-(1-7) attenuated SMAD phosphorylation, as well as reduced collagen, CTGF and α-SMA expression in the bile-duct ligation model of liver fibrosis [15]. Acuna et al. also find that Ang-(1-7) attenuated the TGF-β/SMAD pathway in skeletal muscle of an experimental model of muscular dystrophy [20]. Moreover, Ang-(1-7) treatment was associated with a reduction in the pro-fibrotic miRNA-21 in both the skeletal muscle and in fibroblasts isolated form the mdx rat model [20]. In renal epithelial NRK-52 cells exposed to high glucose conditions, Ang-(1-7) attenuated the increase in TGF-β expression suggesting that Ang-(1-7) may directly influence the expression of the cytokine. The inhibitory effect of Ang-(1-7) on TGF-β in the NRK-52 cells was reversed by the AT7/MasR antagonist DALA [27].
Figure 1.
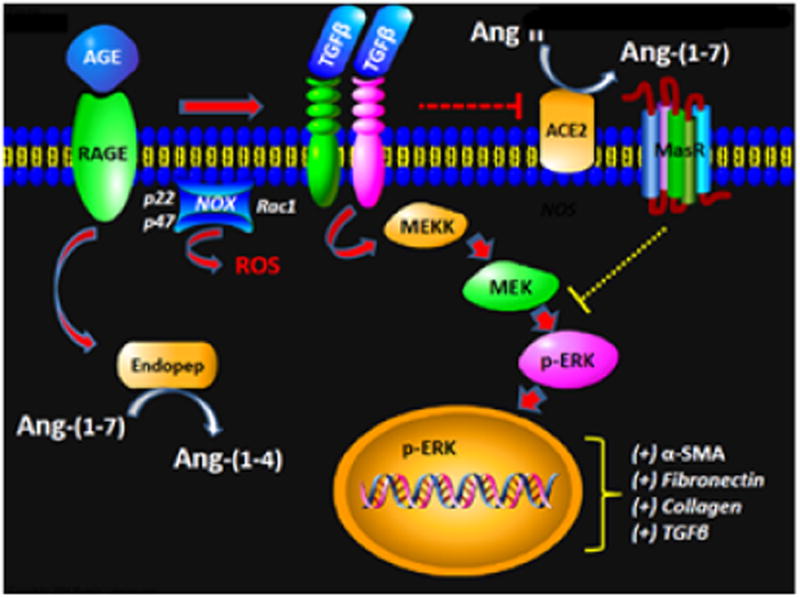
Potential scheme depicting the influence of Angiotensin-(1-7) on the AGE-TGF-β signaling pathway: Advanced glycation end products (AGE) binds to the receptor for AGE (RAGE) and induces TGF-β potentially by the generation of reactive oxygen species (ROS) through activation of the NADPH oxidase complex [NOX, p22phox (p22), p47phox (p47), Rac-1]. TGF-β stimulates the non-canonical pathway MAP kinase pathway to promote phosphorylated ERK1/2 to traffic to the nucleus and increase expression of EMT/fibrosis genes including α-smooth muscle actin (αSMA), fibronectin, collagen and TGF-β. The TGF-β pathway may reduce intrinsic Angiotensin-(1-7) [Ang-(1-7)] tone by downregulation of ACE2 and the AT7/Mas receptor (Mas), as well as increased degradation of the peptide to Ang-(1-4) through a soluble endopeptidase (Endopep). Ang-(1-7) attenuates EMT and fibrosis by inhibiting ERK1/2 phosphorylation potentially through the activation of intracellular phosphatases.
In addition to the canonical TGF-β/SMAD pathway, non-canonical pathways to promote tissue fibrosis include the TGFβ-dependent activation of MAP kinases [28]. Ang-(1-7) treatment attenuated the chronic stimulation of the MAP kinases ERK1/2, p38 and JNK associated with the amelioration of pulmonary fibrosis [17] and diabetic nephropathy [22]. We find that Ang-(1-7) abolished both ERK 1/2 phosphorylation and α-SMA expression in response to TGF-β in renal proximal tubule NRK-52 cells [29]. In this regard, the inhibitory effects of Ang-(1-7) on fibrosis may refect the activation of various cellular phosphatases including SHP-1 and the dual signalling phosphatase (DUSP, MKP-1) that inactivate a stimulated MAPK pathway [30-33].
Oxidative Stress
Alterations in the cellular redox balance also contribute to tissue fibrosis that may refect an activated TGF-β pathway. Moreover, increased oxidative stress in a positive feedback manner may promote a sustained stimulation of the TGF-β pathway [34,35]. Ang-(1-7) attenuated oxidative stress possibly through the reduction in NOX 4 expression, as well as reduced expression of the NLRP3/IL-1β in flammasone that link the anti-inflammatory actions of Ang-(1-7) to the inhibitory effects on liver fibrosis [15]. In the db/db mouse model of obesity and type 2 diabetes, the Ang-(1-7)-dependent reduction in renal fibrosis was associated with an overall reduction of oxidative stress and increased catalase activity suggesting that Ang-(1-7) stimulates scavenging pathways as well [22]. Chan et al. also find that Ang-(1-7) reduced oxidative stress and fibrosis in the kidney that was associated with the reduced expression of NOX 4, the major NADPH oxidase isoform in the kidney [36]. Indeed, Shi et al. report that Ang-(1-7) reduced the renal expression of the pro-fibrotic cytokine TGF-β, as well as the ROS-sensing proteins Nrf2 and HO-1 that may refect an overall reduction in ROS by Ang-(1-7) [36].
The stimulation of the MAPK pathway may refect an initial increase in reactive oxygen species (ROS) signaling upstream from MAPK and the reduction in oxidative stress by Ang-(1-7) may potentially attenuate MAPK stimulation of [37,38]. Consistent with this proposed pathway, Ang-(1-7) blocked Ang II-induced migration and TGF-β and collagen expression of pulmonary myofibroblasts associated with a reduction in ROS and NOX 4 expression [17]. Moreover, comparable effects to Ang-(1-7) on pulmonary myofibroblasts were achieved with the ROS scavenger tempol and the NAD(P)H oxidase inhibitors apocynin and DPI [17]. Although the intracellular sources of ROS in fibrosis are not well-defined, the role of mitochondrial ROS may constitute an additional pathway to the stimulation of TGF-β, EMT and fibrosis [37,39,40]. Indeed, we recently identified a MEP-Ang-(1-7)-AT7/MasR pathway in mitochondria isolated from the sheep kidney that may contribute to cellular redox balance and could potentially influence myofibroblast transition [41]. In a study of the mitochondrial proteonome, the Ang-(1-7) agonist AVE 0991 reduced the expression of proteins associated with inflammation and apoptosis in the kidneys of ApoE-/- knockout mice [42]. Increased oxidative stress appears to be key to the downstream activation of the MAPK and TGF-β/SMAD pathways that contribute to fibrosis.
Myofibroblast Transiton
An intriguing albeit controversial aspect of fibrosis is the role of myofibroblasts derived from resident epithelial, endothelial and pericyte cells, as well as fibroblasts [37,43]. Myofibroblast transition results in a more secretory and migratory phenotype that may ultimately promote tissue fibrosis, as well as depleting the local population of normal cells. TGF-β is a prominent stimulus for myofibroblast formation, and likely contributes to myofibroblast transition elicited by other agents including Ang II, advanced glycation products (AGEs), aldosterone, and endothelin, as well as hypoxic and hyperglycemia conditions [37,38,44,45]. Treatment with Ang-(1-7) reversed the epithelial to mesenchymal or myofibroblast transition (EMT) of NRK-52 cells exposed to high glucose that was associated with reduced TGF-β expression and attenuated MAPK activation [46]. We reported that Ang-(1-7) also abolished EMT in NRK-52 cells chronically exposed to the AGE methylglyoxal albumin (MGA) [29]. Although AGE exposure increased TGF-β expression that apparently drives EMT in these cells, Ang-(1-7) failed to reverse the increase in the cellular levels of TGF-β. However, Ang-(1-7) abolished both the AGE and TGF-β induced activation of ERK 1/2 that refects downstream activation of the non-canonical TGF-β signaling pathway; both MEK and TGF-β kinase inhibitors prevented the AGE-dependent induction of EMT [29]. In contrast, treatment with an AT1R blocker did not attenuate AGE-induced EMT or ERK activation suggesting that the cellular actions of Ang-(1-7) do not refect the direct antagonism of the Ang II-AT1R axis in the NRK-52 cells [29].
RAS Expression
Finally, the progression of cellular fibrosis may potentially refect both the loss of negative feedback inhibitory pathways and the gain of positive feedback systems. In regards to the RAS, AGE-induced EMT in the renal NRK-52 cells was associated with a marked reduction in the intracellular levels of Ang-(1-7) that may result in the potential loss of Ang-(1-7) tone in these cells. AGE markedly increased the cellular metabolism of Ang-(1-7) to Ang-(1-4) through an unidentified cytosolic endopeptidase and tended to reduce the processing of Ang I to Ang-(1-7) by thimet oligopeptidase [29]. Others have shown that TGF-β or AGE exposure reduces expression of both ACE2 and the AT7/MasR, but increases the cellular components of the ACE-Ang II-AT1R axis [27,47]. In this regard, Zhou et al. recently reported that blockade of the Wnt/β-catenin axis, a key signaling pathway involved in the pro-fibrotic actions of TGF-β, abrogates the activation of the RAS components renin, angiotensinogen and the AT1R, as well as attenuates renal fibrosis and myofibroblast activation in adriamycin-induced nephropathy [48]. The role of the Wnt/β-catenin pathway on the Ang-(1-7) axis including the expression of ACE2, neprilysin and the Mas receptor is not currently known.
Conclusion
In conclusion, activation of the Ang-(1-7)-AT7/MasR axis may be a potentially important therapeutic target to attenuate fibrosis in various tissues and attenuate the progressive decline in organ function, particularly in lieu of the lack of effective approaches to inhibit fibrosis. Additional studies are clearly warranted to precisely define the signaling pathways inovled in the anti-fibrotic actions of Ang-(1-7) within distinct cell types. In this regard, an evolving area is the epigenetic response that contributes to fibrosis and the development of approaches to attenuate this mechanism [49-51]; future studies should address the extent that Ang-(1-7) impacts the epigenetic signaling mechanisms in fibrosis. The current experimental approaches have predominantly relied on the native peptide; however, Ang-(1-7) exhibits a very short half-life and is rapidly cleaved by multiple peptidases including ACE and dipeptidyl peptidase 3 (DPP 3) [52,53]. Moreover, Ang-(1-7) at higher doses may function as a partial agonist at the AT1R and contribute to rather than inhibit the progression of fibrosis. In this regard, the development of orally active and cell-penetrating Ang-(1-7) agonists that are resistant to peptidases and exhibit greater selectivity among angiotensin receptor subtypes may constitute the next step to effectively combat fibrosis.
Acknowledgments
Funding: These studies were supported in part by grants from the National Institute of Health grants (HL-56973, HL-51952, HD-084227, HD-047584 and HD-017644 and the American Heart Association (AHA-151521 and AHA-355741). An unrestricted grant from the Farley-Hudson Foundation (Jacksonville, NC), Groskert Heart Fund and the Wake Forest Venture Fund is also acknowledged.
Footnotes
Conflict of Interests: None declared
References
- 1.Chappell MC, Brosnihan KB, Diz DI, Ferrario CM. Identification of angiotensin-(1-7) in rat brain. Evidence for differential processing of angiotensin peptides. J Biol Chem. 1989;264:16518–16523. [PubMed] [Google Scholar]
- 2.Chappell MC. Emerging evidence for a functional angiotesin-converting enzyme 2-angiotensin-(1-7) mas receptor axis; more than regulation of blood pressure? Hypertension. 2007;50:596–599. doi: 10.1161/HYPERTENSIONAHA.106.076216. [DOI] [PubMed] [Google Scholar]
- 3.Santos RA. Angiotensin-(1-7) Hypertension. 2014;63:1138–1147. doi: 10.1161/HYPERTENSIONAHA.113.01274. [DOI] [PubMed] [Google Scholar]
- 4.Chappell MC. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute? Am J Physiol Heart Circ Physiol. 2016;310:H137–152. doi: 10.1152/ajpheart.00618.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chappell MC, Marshall AC, Alzayadneh EM, Shaltoutm HA, Diz DI. Update on the Angiotensin converting enzyme 2-Angiotensin (1-7)-MAS receptor axis: fetal programing, sex differences, and intracellular pathways. Front Endocrinol (Lausanne) 2014;4:201. doi: 10.3389/fendo.2013.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meng W, Zhao W, Zhao T, Liu C, Chen Y, et al. Autocrine and paracrine function of Angiotensin 1-7 in tissue repair during hypertension. Am J Hypertens. 2014;27:775–782. doi: 10.1093/ajh/hpt270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwata M, Cowling RT, Gurantz D, Moore C, Zhang S, et al. Angiotensin-(1-7) binds to specific receptors on cardiac fibroblasts to initiate antifibrotic and antitrophic effects. Am J Physiol Heart Circ Physiol. 2005;289:H2356–2363. doi: 10.1152/ajpheart.00317.2005. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Qian C, Roks AJ, Westermann D, Schumacher SM, et al. Circulating rather than cardiac angiotensin-(1-7) stimulates cardioprotection after myocardial infarction. Circ Heart Fail. 2010;3:286–293. doi: 10.1161/CIRCHEARTFAILURE.109.905968. [DOI] [PubMed] [Google Scholar]
- 9.Grobe JL, Mecca AP, Mao H, Katovich MJ. Chronic angiotensin-(1-7) prevents cardiac fibrosis in DOCA-salt model of hypertension. Am J Physiol Heart Circ Physiol. 2006;290:H2417–2423. doi: 10.1152/ajpheart.01170.2005. [DOI] [PubMed] [Google Scholar]
- 10.Grobe JL, Mecca AP, Lingis M, Shenoy V, Bolton TA, et al. Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1-7) Am J Physiol Heart Circ Physiol. 2007;292:H736–742. doi: 10.1152/ajpheart.00937.2006. [DOI] [PubMed] [Google Scholar]
- 11.Mercure C, Yogi A, Callera GE, Aranha AB, Bader M, et al. Angiotensin (1-7) blunts hypertensive cardiac remodeling by a direct effect on the heart. Circ Res. 2008;103:1319–1326. doi: 10.1161/CIRCRESAHA.108.184911. [DOI] [PubMed] [Google Scholar]
- 12.Benter IF, Yousif MHM, Anim JT, Cojocel C, Diz DI. Angiotensin-(1-7) prevents development of severe hypertension and end-organ damage in spontaneously hypertensive rats treated with L-NAME. Am J Physiol Heart Circ Physiol. 2006;290:H684–H691. doi: 10.1152/ajpheart.00632.2005. [DOI] [PubMed] [Google Scholar]
- 13.Hao PP, Yang JM, Zhang MX, Zhang K, Chen YG, et al. Angiotensin-(1-7) treatment mitigates right ventricular fibrosis as a distinctive feature of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2015;308:H1007–H1019. doi: 10.1152/ajpheart.00563.2014. [DOI] [PubMed] [Google Scholar]
- 14.Rahimi N, Dayanir V, Lashkari K. Receptor chimeras indicate that the vascular endothelial growth factor receptor-1 (VEGFR-1) modulates mitogenic activity of VEGFR-2 in endothelial cells. J Biol Chem. 2000;275:16986–16992. doi: 10.1074/jbc.M000528200. [DOI] [PubMed] [Google Scholar]
- 15.Cai SM, Yang RQ, Li Y, Ning ZW, Zhang LL, et al. Angiotensin-(1-7) Improves Liver Fibrosis by Regulating the NLRP3 Inflammasome via Redox Balance Modulation. Antioxid Redox Signal. 2016;24:795–812. doi: 10.1089/ars.2015.6498. [DOI] [PubMed] [Google Scholar]
- 16.Lubel JS, Herath CB, Tchongue J, Grace J, Jia Z, et al. Angiotensin-(1-7), an alternative metabolite of the renin-angiotensin system, is up-regulated in human liver disease and has antifibrotic activity in the bile-duct-ligated rat. Clin Sci (Lond) 2009;117:375–386. doi: 10.1042/CS20080647. [DOI] [PubMed] [Google Scholar]
- 17.Meng Y, Yu CH, Li W, Li T, Luo W, et al. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis protects against lung fibrosis by inhibiting the MAPK/NF-ΰB pathway. Am J Respir Cell Mol Biol. 2014;50:723–736. doi: 10.1165/rcmb.2012-0451OC. [DOI] [PubMed] [Google Scholar]
- 18.El-Hashim AZ, RennoWM, Raghupathy R, Abduo HT, Akhtar S, et al. Angiotensin-(1-7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-kappaB-dependent pathways. Br J Pharmacol. 2012;166:1964–1976. doi: 10.1111/j.1476-5381.2012.01905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carver KA, Smith TL, Gallagher PE, Tallant EA. Angiotensin-(1-7) prevents angiotensin II-induced fibrosis in cremaster microvessels. Microcirculation. 2015;22:19–27. doi: 10.1111/micc.12159. [DOI] [PubMed] [Google Scholar]
- 20.Acuña MJ, Pessina P, Olguin H, Cabrera D, Vio CP, et al. Restoration of muscle strength in dystrophic muscle by angiotensin-1-7 through inhibition of TGF-Î2 signalling. Hum Mol Genet. 2014;23:1237–1249. doi: 10.1093/hmg/ddt514. [DOI] [PubMed] [Google Scholar]
- 21.Kim CS, Kim IJ, Bae EH, Ma SK, Lee J, et al. Angiotensin-(1-7) Attenuates Kidney Injury Due to Obstructive Nephropathy in Rats. PLoS One. 2015;10:e0142664. doi: 10.1371/journal.pone.0142664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mori J, Patel VB, Ramprasath T, Abo Alrob O, DesAulniers J, et al. Angiotensin 1-7 mediates renoprotection against diabetic nephropathy by reducing oxidative stress, inflammation, and lipotoxicity. Am J Physiol Renal Physiol. 2014;306:F812–F821. doi: 10.1152/ajprenal.00655.2013. [DOI] [PubMed] [Google Scholar]
- 23.Zhang K, Meng X, Li D, Yang J, Kong J, et al. Angiotensin (1-7) attenuates the progression of streptozotocin-induced diabetic renal injury better than angiotensin receptor blockade. Kidney Int. 2015;87:359–369. doi: 10.1038/ki.2014.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hao P, Yang J, Liu Y, Zhang M, Zhang K, et al. Combination of angiotensin-(1-7) with perindopril is better than single therapy in ameliorating diabetic cardiomyopathy. Sci Rep. 2015;5:8794. doi: 10.1038/srep08794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yousif MHM, Benter IF, Diz DI, Chappell MC. Angiotensin-(1-7)-dependent vasorelaxation of the renal artery exhibits unique angiotensin and bradykinin receptor selectivity. Peptide in press. 2017 doi: 10.1016/j.peptides.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Villela D, Leonhardt J, Patel N, Joseph J, Kirsch S, et al. Angiotensin type 2 receptor (AT2R) and receptor Mas: a complex liaison. Clin Sci (Lond) 2015;128:227–234. doi: 10.1042/CS20130515. [DOI] [PubMed] [Google Scholar]
- 27.Chou CH, Chuang LY, Lu CY, Guh JY. Interaction between TGF-β and ACE2-Ang-(1-7)-Mas pathway in high glucose-cultured NRK-52E cells. Mol Cell Endocrinol. 2013;366:21–30. doi: 10.1016/j.mce.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alzayadneh EM, Chappell MC. Angiotensin-(1-7) abolishes AGE-induced cellular hypertrophy and myofibroblast transformation via inhibition of ERK1/2. Cell Signal. 2014;26:3027–3035. doi: 10.1016/j.cellsig.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gava E, Samad-Zadeh A, Zimpelmann J, Bahramifarid N, Kitten GT, et al. Angiotensin-(1-7) activates a tyrosine phosphatase and inhibits glucose-induced signalling in proximal tubular cells. Nephrol Dial Transplant. 2009;24:1766–1773. doi: 10.1093/ndt/gfn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCollum LT, Gallagher PE, Ann TE. Angiotensin-(1-7) attenuates angiotensin II-induced cardiac remodeling associated with upregulation of dual-specificity phosphatase 1. Am J Physiol Heart Circ Physiol. 2012;302:H801–H810. doi: 10.1152/ajpheart.00908.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sampaio WO, Henrique de CC, Santos RA, Schiffrin EL, Touyz RM. Angiotensin-(1-7) counterregulates angiotensin II signaling in human endothelial cells. Hypertension. 2007;50:1093–1098. doi: 10.1161/HYPERTENSIONAHA.106.084848. [DOI] [PubMed] [Google Scholar]
- 33.Su Z, Zimpelmann J, Burns KD. Angiotensin-(1-7) inhibits angiotensin II-stimulated phosphorylation of MAP kinases in proximal tubular cells. Kidney Int. 2006;69:2212–2218. doi: 10.1038/sj.ki.5001509. [DOI] [PubMed] [Google Scholar]
- 34.Rhyu DY, YangY, Ha H, Lee GT, Song JS, et al. Role of reactive oxygen species in TGF-beta1-induced mitogen- activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. 2005;16:667–675. doi: 10.1681/ASN.2004050425. [DOI] [PubMed] [Google Scholar]
- 35.Samarakoon R, Dobberfuhl AD, Cooley C, Overstreet JM, Patel S, et al. Induction of renal fibrotic genes by TGF-Î21 requires EGFR activation, p53 and reactive oxygen species. Cell Signal. 2013;25:2198–2209. doi: 10.1016/j.cellsig.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 36.ShiY, Lo CS, Padda R, Abdo S, Chenier I, et al. Angiotensin-(1-7) prevents systemic hypertension, attenuates oxidative stress and tubulointerstitial fibrosis, and normalizes renal angiotensin-converting enzyme 2 and Mas receptor expression in diabetic mice. Clin Sci (Lond) 2015;128:649–663. doi: 10.1042/CS20140329. [DOI] [PubMed] [Google Scholar]
- 37.Tennakoon AH, Izawa T, Kuwamura M, Yamate J. Pathogenesis of Type 2 Epithelial to Mesenchymal Transition (EMT) in Renal and Hepatic Fibrosis. Clin Med. 2015;5 doi: 10.3390/jcm5010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhan M, Kanwar YS. Hierarchy of molecules in TGF-Î21 signaling relevant to myofibroblast activation and renal fibrosis. Am J Physiol Renal Physiol. 2014;307:F385–387. doi: 10.1152/ajprenal.00338.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim SM, Kim YG, Jeong KH, Lee SH, Lee TW, et al. Angiotensin II-induced mitochondrial Nox4 is a major endogenous source of oxidative stress in kidney tubular cells. PLoS One. 2012;7:e39739. doi: 10.1371/journal.pone.0039739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee DY, Wauquier F, Eid AA, Roman LJ, Ghosh-Choudhury G, et al. Nox4 NADPH oxidase mediates peroxynitrite-dependent uncoupling of endothelial nitric-oxide synthase and fibronectin expression in response to angiotensin II: role of mitochondrial reactive oxygen species. J Biol Chem. 2013;288:28668–28686. doi: 10.1074/jbc.M113.470971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson BA, Nautiyal M, Gwathmey TM, Rose JC, Chappell MC. Evidence for a Mitochondrial Angiotensin-(1-7) System in the Kidney. Am J Physiol Renal Physiol. 2015;310:F637–F645. doi: 10.1152/ajprenal.00479.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suski M, Olszanecki R, Stachowicz A, Madej J, Bujak-Giżycka B, et al. The influence of angiotensin-(1-7) Mas receptor agonist (AVE 0991) on mitochondrial proteome in kidneys of apoE knockout mice. Biochim Biophys Acta. 2013;1834:2463–2469. doi: 10.1016/j.bbapap.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 43.Inoue T, Umezawa A, Takenaka T, Suzuki H, Okada H. The contribution of epithelial-mesenchymal transition to renal fibrosis differs among kidney disease models. Kidney Int. 2015;87:233–238. doi: 10.1038/ki.2014.235. [DOI] [PubMed] [Google Scholar]
- 44.Xu X, Tan X, Tampe B, Sanchez E, Zeisberg M, et al. Snail Is a Direct Target of Hypoxia-inducible Factor 1alpha (HIF1alpha) in Hypoxia-induced Endothelial to Mesenchymal Transition of Human Coronary Endothelial Cells. J Biol Chem. 2015;290:16653–16664. doi: 10.1074/jbc.M115.636944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou L, Xue H, Yuan P, Ni J, Yu C, et al. Angiotensin AT1 receptor activation mediates high glucose-induced epithelial-mesenchymal transition in renal proximal tubular cells. Clin Exp Pharmacol Physiol. 2010;37:e152–e157. doi: 10.1111/j.1440-1681.2010.05421.x. [DOI] [PubMed] [Google Scholar]
- 46.Zhou L, Xue H, Wang Z, Ni J, Yao T, et al. Angiotensin-(1-7) attenuates high glucose-induced proximal tubular epithelial-to-mesenchymal transition via inhibiting ERK1/2 and p38 phosphorylation. Life Sci. 2012;90:454–462. doi: 10.1016/j.lfs.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 47.Cao W, Xu J, Zhou ZM, Wang GB, Hou FF, et al. Advanced oxidation protein products activate intrarenal renin-angiotensin system via a CD36-mediated, redox-dependent pathway. Antioxid Redox Signal. 2013;18:19–35. doi: 10.1089/ars.2012.4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou L, Li Y, Hao S, Zhou D, Tan RJ, et al. Multiple genes of the renin-angiotensin system are novel targets of Wnt/Î2-catenin signaling. J Am Soc Nephrol. 2015;26:107–120. doi: 10.1681/ASN.2014010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Comer BS, Ba M, Singer CA, Gerthoffer WT. Epigenetic targets for novel therapies of lung diseases. Pharmacol Ther. 2015;147:91–110. doi: 10.1016/j.pharmthera.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:220–229. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tzouvelekis A, Kaminski N. Epigenetics in idiopathic pulmonary fibrosis. Biochem Cell Biol. 2015;93:159–170. doi: 10.1139/bcb-2014-0126. [DOI] [PubMed] [Google Scholar]
- 52.Cruz-Diaz N, Wilson BA, Pirro NT, Brosnihan KB, Marshall AC, et al. Identification of dipeptidyl peptidase 3 as the Angiotensin-(1-7) degrading peptidase in human HK-2 renal epithelial cells. Peptides. 2016;83:29–37. doi: 10.1016/j.peptides.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilson BA, Marshall AC, Alzayadneh EM, Chappell MC. The ins and outs of angiotensin processing within the kidney. Am J Physiol Regul Integr Comp Physiol. 2014;307:R487–489. doi: 10.1152/ajpregu.00177.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]