

Summary
Background
Inherited human variants that concurrently cause disorders of primary hemostasis and coagulation are uncommon. Nevertheless, rare cases of co-existent damaging variants are likely to cause more severe bleeding and may go undiagnosed.
Objective
We prospectively sought to determine pathogenic variants in a three-generational pedigree with excessive bleeding.
Patients/methods
Platelet number, size and light transmission aggregometry to multiple agonists were evaluated in pedigree members. Transmission electron microscopy determined platelet morphology and granule content. Thromboxane release studies and light transmission aggregometry in the presence or absence of prostaglandin G2 assessed specific functional defects in the arachidonic acid pathway. Whole exome sequencing (WES) and targeted nucleotide sequence analysis identified potentially deleterious variants.
Results
Pedigree members with excessive bleeding had impaired platelet aggregation with arachidonic acid, epinephrine and low-dose ADP, as well as reduced platelet thromboxane B2 release. Impaired platelet aggregation in response to 2MesADP was rescued with prostaglandin G2, a prostaglandin intermediate downstream of prostaglandin synthase-1 (PTGS-1) that aids in the production of thromboxane. WES identified a non-synonymous variant in the signal peptide of PTGS-1 (rs3842787; c.50C>T; p.Pro17-Leu) that completely co-segregated with disease phenotype. A variant in the F8 gene causing hemophilia A (rs28935203; c.5096A>T; p.Y1699F) was also identified. Individuals with both variants had more severe bleeding manifestations than characteristic of mild hemophilia A alone.
Conclusion
We provide the first report of co-existing variants in both F8 and PTGS-1 genes in a three-generation pedigree. The PTGS-1 variant was associated with specific functional defects in the arachidonic acid pathway and more severe hemorrhage.
Keywords: bleeding, blood platelet disorder, DNA sequencing, hemophilia A, platelet aggregation
Introduction
Spontaneous or excessive bleeding following minor trauma or surgery may suggest an underlying disorder of hemostasis or coagulation. Typical bleeding patterns may help to clinically distinguish disorders of primary hemostasis (a defect in formation of the platelet plug characterized by mucosal bleeding, cutaneous bleeding and menorrhagia) from disorders of coagulation (secondary hemostasis characterized more commonly by spontaneous or traumatic joint, muscle, internal organ and intracranial bleeding). Hemophilia A, an X-linked recessive disease caused by factor (F) VIII deficiency, is a common disorder of secondary hemostasis. This condition occurs in approximately 1 in 5000 male births and, although incidence rates vary, approximately 35–45% of affected individuals have severe disease (FVIII activity < 1% of normal) [1]. Individuals with mild hemophilia A (FVIII activity ≥ 6–49% of normal) represent about 25% of cases and typically only develop bleeding following trauma or surgery. Bleeding patterns in hemophilia follow closely the absolute amount of residual FVIII activity.
Disorders of primary hemostasis include defects of the vessel wall (e.g. Ehlers-Danlos syndrome), defects in proteins that mediate platelet adhesion (e.g. von Willebrand disease [VWD]) and defects in platelets themselves. These bleeding disorders can be attributed to acquired or inherited abnormalities. Inherited platelet function disorders (IPFDs) are heterogeneous and characterized by recurrent, lifelong episodes of abnormal bleeding that can range in severity from mild to life threatening [2]. Although severe inherited platelet defects, such as Bernard Soulier syndrome and Glanzmann thrombasthenia, can be diagnosed by platelet aggregation and/or flow cytometry, many IPFDs are not readily diagnosed [3]. Especially in non-syndromic cases, establishing a precise diagnosis can be challenging, as clinical bleeding assessment tools do not reliably distinguish IPFDs from other bleeding diatheses [4]. Moreover, many IPFDs may go clinically unrecognized until unmasked by trauma, surgery, antiplatelet use or other events. Therefore, a molecular diagnosis is needed to characterize cryptic or obscure genotype/phenotype relationships. Such information may also be useful to ultimately guide clinical management and improve patient care [2]. Next-generation sequencing technologies, including whole exome sequencing (WES), have emerged as important tools for identifying causative variants in IPFD patients [5–7].
Coexisting human variants that affect both platelet function and coagulation pathways are uncommon. We report for the first time the co-occurrence of pathogenic variants in both coagulation factor VIII (F8, causing mild hemophilia A) and prostaglandin-endoperoxide synthase 1 (PTGS-1, aka COX-1) genes in persons with a bleeding disorder. The variants completely co-segregated with platelet function defects and excessive bleeding of a mucosal pattern in a three-generational pedigree. Moreover, individuals with both the F8 and PTGS-1 variants had more severe hemorrhage than characteristic of mild hemophilia A alone.
Patients and methods
Platelet counting, light transmission aggregometry and thromboxane release
All subjects provided informed consent (IRB # 37022) and refrained from taking any medications or supplements possibly interfering with platelet function studies for at least 2 weeks prior to testing. Whole blood was drawn into sterile BD Vacutainer® potassium EDTA or 3.2% buffered sodium citrate vacutainer tubes for platelet counts or platelet aggregation studies, respectively. Platelet counts were performed on an AcT 10 Hematology Analyzer (Beckman Coulter, Brea, CA, USA). Plateletrich plasma (PRP) was prepared from citrated whole blood by unbraked centrifugation at 99 ×g for 20 min at room temperature. PRP counts were normalized to 250 000 platelets/μL using autologous platelet-poor plasma as the diluent. Platelet aggregation was performed by light transmission aggregometry (LTA) on a PAP-8E Platelet Aggregation Profiler (Bio/Data, Horsham, PA, USA) at baseline and using type I collagen (2 and 5 μg mL−1; Helena Laboratories, Beaumont, TX, USA), adenosine diphosphate (ADP, 3, 5 and 10 μM; Helena Laboratories), arachidonic acid (AA, 0.5 mg mL−1; Bio/Data Corporation), epinephrine (300 μM; Helena Labs), u44619 (1 μM; Cayman Chemicals, Ann Arbor, MI, USA) or ristocetin (1.5 and 0.5 mg mL−1; Biopool, Jamestown, NY, USA) [8].
For thromboxane release studies, platelets were isolated from whole blood as previously described [9] and incubated for 5 min in the absence or presence of thrombin (0.1 U mL−1). Next, platelets were centrifuged (12 000 ×g for 5 min) and supernatants were assayed for thromboxane B2 using a commercial enzyme immunoassay (Cayman). The assay detects thromboxane B2 levels from 1.6–1000 pg mL−1, demonstrates 100% cross-reactivity for thromboxane B2 per manufacturer testing and is used in this field [10]. For the range of thromboxane B2 levels measured in our studies, the intra- and inter-assay variance of this assay is approximately 11.5% and 12.9%, respectively.
Rescue of the aggregation defect in affected pedigree members was conducted by stimulating washed human platelets (2 × 108) with 30 nM 2MesADP in the presence or absence of 1 μM prostaglandin G2, a prostaglandin intermediate downstream of prostaglandin synthase-1 (PTGS-1) that aides in the production of thromboxane. Platelet aggregation was measured using a lumi-aggregometer (Chrono-Log, Havertown, PA, USA) at 37 °C under stirring conditions. The change in light transmission was measured in the presence and absence of agonists and PGG2.
Unstimulated platelet lysates (5 × 108) were also assayed for the expression of proteins by immunoblot. Briefly, samples were solubilized in Laemmli buffer and electrophoresed on 10% SDS gels. Next, proteins were transferred to a polyvinylidene fluoride (PVDF) membrane and probed with antibodies specific for phosphorylated Akt (pAkt; Cell Signaling, Danvers, MA, USA), PTGS-1 (Life Technologies, Carlsbad, CA, USA), TBXAS-1 (Cayman Chemicals) and b-actin (Santa Cruz, Dallas, TX, USA, used to ensure equal loading). Enhanced chemiluminescence was used to detect expression.
Platelet transmission electron microscopy
Platelets were isolated from whole blood as previously described [9] and fixed with 2.5% glutaraldehyde in phosphate buffered saline (PBS) for 20 min. Subsequently platelets were post-fixed with 2% osmium tetroxide in water for 60 min and dehydrated in a graded acetone series before embedding in Epon-Araldite. Thin sections were cut and stained with uranyl acetate and lead citrate. Grids were examined with a JEOL JEM-1011 electron microscope at 80 kV. Images were captured with a side-mounted Advantage HR CCD camera (Advanced Microscopy Techniques, Woburn, MA, USA) [11].
Whole exome sequencing
DNA was extracted from whole blood and libraries were prepared for WES using the SureSelectXT Human All Exon V5+UTRs capture kit (Agilent Technologies, Inc., Santa Clara, CA, USA). Next, the exome capture libraries were paired-end sequenced using the Illumina HiSeq sequencing system (San Diego, CA, USA). Sequencing reads from individuals III.1 and III.4 were sequenced to a length of 125 base pairs (bp) and reads from all other individuals were sequenced to a length of 101 bp. Individuals were sequenced to an average depth ranging from 75× to 202× across all captured regions (plus a 100 bp padding) as determined by BEDTools [12]. All paired-end reads were aligned to the human reference genome (GRCh37) using the BWA-MEM aligner [13]. All read alignment information was recorded in the BAM file format with the use of SAMtools [14]. Sequencing read genomic coordinate sorting, sequencing read duplicate marking and BAM file indexing were performed using Picard Tools (http://broadinstitute.github.io/picard/). Variant calling was performed using the Genome Analysis Toolkit (GATK, version 3.3-0) [15]. Specifically, indel realignment and base-quality score recalibration were performed on the aligned reads from all six individuals indicated in Fig. 2(A). Next, HaplotypeCaller was used to generate both single nucleotide polymorphism (SNP) and indel genotype calls for each sequenced individual. Ninety-nine CEU (Utah Residents [CEPH] with Northern and Western European Ancestry) and 92 GBR (British in England and Scotland) exome sequenced individuals from the 1000 Genomes Project [16] were also included in this genotyping step in order to properly power downstream variant recalibration steps. Obtained SNP and indel variant calls then underwent variant quality score recalibration (VQSR) to identify false-positive variants calls. A VQSR truth sensitivity cut-off of 99.0 was applied to both SNPs and indels to remove false-positive variants. All steps and parameters of the GATK variant calling pipeline were performed in accordance with the GATK Best Practices workflow [17,18]. Further, variant calling was limited to the SureSelectXT Human All Exon V5+UTRs exome capture regions with a 100 bp padding on both sides of each capture region in order to ensure proper variant recalibration and accurate genotyping.
Fig. 2.
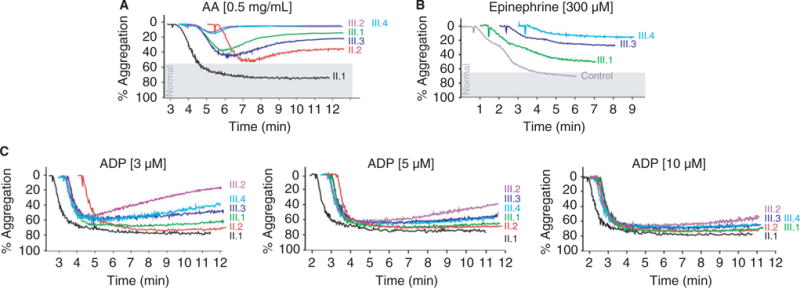
Platelet aggregation was impaired in response to arachidonic acid and epinenephrine, consistent with a defect in the arachidonic acid pathway. (A) Platelet aggregation in response to arachidonic acid (AA) was impaired in the mother (II.2) and all four children (III.1–III.4), but normal in the father (II.1). (B) Platelet aggregation in response to epinephrine was also impaired in three children (III.1, III.3 and III.4). (C) Loss of the secondary wave of platelet aggregation was also evident in the mother (II.2) and four children (III.1–III.4) in response to lower doses of adenosine diphosphate (ADP). This defect was masked at a higher dose of ADP.
All variants that passed VQSR were analyzed by the Ensembl Variant Effect Predictor tool (release 83) to determine their impact on protein coding genes [18]. All passing variants were also annotated with information from the database for non-synonymous SNPs’ functional predictions (dbNSFP) v2.9 [19]. dbNSFP contains allele frequency information and variant functional predictions from 11 different algorithms. These algorithms are composed of SIFT [20], PolyPhen2-HVAR and Poly-Phen2-HDIV [21], MutationTaster [22], LRT [23], MutationAssessor [24], FATHMM [25], PROVEAN [26], CADD [27], MetaSVM [28] and MetaLR [28]. Binary predictions of variant functionality are provided by dbNSFP for 8 of 11 algorithms. Any variants with a PolyPhen2-HDIV or PolyPhen2-HVAR score above 0.5 were considered deleterious by the respective algorithms. Any variant with a CADD score above 20 was considered to be deleterious by CADD. Variant functional predictions from all of these algorithms and allele frequency information were used to determine which variants were likely to be deleterious.
Statistical analyses
Quantitative data were examined for normality using skewness and kurtosis tests. Groups were compared using Student’s t-test or Wilcoxon rank sum and the chi-squared or Fisher’s exact test (GraphPad Prism, v6.0f, La Jolla, CA, USA). A two-tailed P-value < 0.05 was considered statistically significant.
Results
Bleeding manifestations were more severe in affected pedigree members
The proband was a 10-year-old boy with mild hemophilia A, with markedly more severe hemorrhage manifestations than observed in most individuals with his measured FVIII level (Table 1). He had spontaneous joint and gastrointestinal (GI) bleeding, epistaxis, oral mucosal bleeding and excessive surgical bleeding. Despite only having mild hemophilia A, he required regularly scheduled treatments with recombinant FVIII and desmopressin acetate (DDAVP) to control his bleeding symptoms (Table 1), similar to individuals with FVIII activity levels of approximately 0.02 IU dL−1 (2%). He had three male siblings from the same non-consanguineous parents. One of the siblings and their maternal grandfather also had mild hemophilia A and bleeding manifestations more severe than characteristic of mild hemophilia A (Table 1). A careful history revealed that the other two male siblings without hemophilia A and the children’s mother (a hemophilia A carrier) also had spontaneous excessive bleeding. No family members had immune or renal dysfunction, facial or musculoskeletal anatomic abnormalities, other features suggestive of a syndromic IPFD or required platelet transfusions. Evaluations of plasma plasminogen, PAI-1, alpha 2 antiplasmin and tissue plasminogen activator, as well as thrombin time and reptilase time, were normal, total leukocyte counts and hemoglobin levels were normal, and VWD was excluded in all family members (not shown). Inhibitor assays were negative in the family members with hemophilia A. Given the uncharacteristic severity of the proband’s bleeding history and the history of abnormal bleeding in family members without hemophilia A, we hypothesized that an undiagnosed IPFD accounted for the excessive hemorrhage noted in this family.
Table 1.
Clinical characteristics of pedigree members
| ID Gender Birth year |
Hem. A | Hemorrhage manifestations | Factor VIII (IU dL−1) | Factor VIII or DDAVP use | WBC (K μL−1) | Hgb (mg dL−1) | BAT score |
|---|---|---|---|---|---|---|---|
| I.1 Male b. 1949 |
Yes | Joint bleeding, excessive surgical bleeding | 0.11 | PRN FVIII | 5.6 | 15.7 | 8 |
| I.2 Female b. 1949 |
No | None | 1.23 | No | 4.8 | 13.0 | 4 |
| II.1 Male b. 1973 |
No | None | 1.63 | No | 5.7 | 15.3 | 12 |
| II.2 Female b. 1973 |
Carrier | Menorrhagia requiring hysterectomy, excessive surgical bleeding, PRBC transfusion | 0.55 | DDAVP | 5.8 | 13.0 | 7 |
| II.3 Female b. 1976 |
Carrier | Menorrhagia, excessive surgical bleeding, oral mucosal bleeding | 0.27 | DDAVP | 9.1 | 13.6 | 18 |
| II.4 Female b. 1982 |
Carrier | Menorrhagia | 0.38 | No | 5.2 | 15.5 | 10 |
| III.1 Male b. 1998 |
Yes | Joint bleeding, severe GI bleeding, epistaxis, excessive bleeding with minor trauma | 0.06 | DDAVP + PRN FVIII | 6.0 | 15.0 | 14 |
| III.2 Male b. 2001 |
No | Epistaxis, excessive bleeding with minor trauma, excessive surgical bleeding | 1.09 | DDAVP | 4.0 | 13.0 | 19 |
| III.3 Male* b. 2004 |
Yes | Joint bleeding, epistaxis, oral mucosal bleeding, excessive bleeding with minor trauma, excessive surgical bleeding | 0.14 | DDAVP + Scheduled FVIII | 5.4 | 12.4 | 21 |
| III.4 Male b. 2008 |
No | Epistaxis, oral mucosal bleeding | 1.29 | DDAVP | 5.7 | 12.8 | 7 |
BAT, bleeding assessment tool; WBC, white blood cells; DDAVP, desmopressin acetate.
Indicates proband.
Platelet phenotyping identified a defect in the arachidonic acid pathway
Platelet number, size, morphology and granule contents were normal in all family members (Fig. 1A and B). Platelet aggregation in response to collagen and the thromboxane mimetic U44619 was normal (Fig. 1C–E), demonstrating intact glycoprotein VI and thromboxane B2 receptor function, respectively. P2Y12 plays an important role in thromboxane synthesis when platelets are stimulated with ADP. We evaluated phosphorylation of Akt in affected pedigree members to determine whether P2Y12 receptor function was intact. Phosphorylation of Akt was normal when platelets were stimulated with 2MesADP (Figure S1A), suggesting that the P2Y12 pathway is not disrupted in these patients.
Fig. 1.
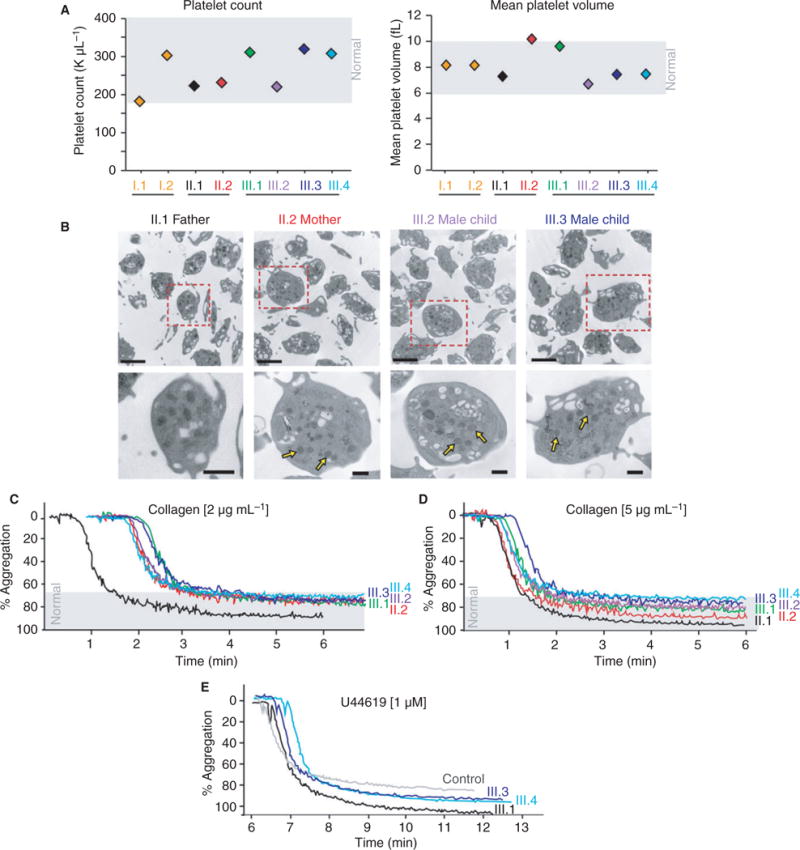
Indices of platelet number, size, morphology and aggregation to collagen and U44619 were normal in pedigree members. (A) Platelet count and size (assessed by mean platelet volume) were normal in pedigree members. Shaded gray boxes indicate the normal range. (B) Isolated platelets were assessed by thin-section transmission electron microscopy (TEM). Platelet morphology, size and granule content were normal. Representative images of platelets from the parents and two children are shown to have preserved alpha granules (bottom insets, yellow arrows). Magnification is ×30 000 and the scale bar represents 2 lm (top panels) or 500 nm (bottom insets). Platelet aggregation in response to collagen (C,D) and U44619 (E) was normal (where established, normal ranges are indicated by the gray shaded areas).
In comparison, the paternal grandfather, mother and all four children had impaired platelet aggregation to arachidonic acid (AA) and epinephrine (Fig. 2A, B, and not shown), consistent with a defect in the AA pathway. The impaired platelet aggregation to AA was reproducible and durable, as similar patterns of impaired aggregation were observed when tested in affected pedigree members at various times (not shown). We also observed a dose-dependent loss of the secondary wave of platelet aggregation in response to ADP, also consistent with an isolated defect in the AA pathway (Fig. 2C).
Whole exome sequencing identified variants in PTGS-1 and F8 that co-segregate with the phenotype of excessive bleeding
Functional platelet phenotyping by LTA suggested an IPFD in the proband, his three siblings, his mother and his maternal grandfather that involved the AA pathway (Fig. 2 and not shown). WES performed in the four male children and their parents also identified two non-synonymous variants in PTGS-1 (reference sequences NG_032900.1; NP_000953.2). PTGS-1 catalyzes the conversion of AA to thromboxane A2, resulting in platelet aggregation [29]. Given that the results of the platelet phenotyping were consistent with a specific defect in the arachidonic acid pathway (Figs 1 and 2), we examined these two variants in PTGS-1 more closely. One of the found variants in PTGS-1 (rs1236913; c.22T>C; p.Arg8Trp) was not predicted by Polyphen-2 to be damaging and did not segregate with the disease phenotype in the family. Thus, this variant was not considered as a candidate.
The second identified variant in PTGS-1 (rs3842787; c.50C>T; p.Pro17Leu) was predicted to be possibly damaging. It was also the only amino acid sequence altering variant that completely segregated with the bleeding phenotype (Fig. 3A and B) in five different genes in the AA pathway (TBXAS1, PTGS1, PTGS2, TBXA2R and P2Y12; Table 2). A logarithm of odds (LOD) calculation was performed to determine if rs3842787 significantly segregated amongst affected individuals in the pedigree. An insignificant LODmax score of 1.20 (θmax = 0.0) was obtained despite complete segregation amongst affected individuals. This is because of a lack of sufficient sample size to detect significant segregation. Just as with the variant rs1236913, SIFT was the only prediction algorithm to predict this variant as deleterious. Heterozygosity of this variant appears sufficient to cause the platelet aggregation defect (Fig. 2A and B). Principal components analysis (PCA) confirmed that all sequenced individuals are of European descent, and high quality sequencing and variant calling was achieved (Fig. 3C). Splice site and splicing enhancer prediction algorithms including Regrna 2.0 (http://regrna2.mbc.nctu.edu.tw/index.html), Human Splicing Finder (http://www.umd.be/HSF3/index.html) and Alternative Splice Site Predictor (ASSP) (http://wangcomputing.com/assp/) did not predict the presence of, or alterations in, splice sites for rs3842787. There was no evidence of consanguinity within the pedigree. Other genes involved in the AA pathway were examined. There was a silent intronic variant in TBXAS1 (rs41727) that did not segregate with disease and was not predicted to be damaging. No nucleotide variants in TBXA2R or P2RY12 were identified.
Fig. 3.
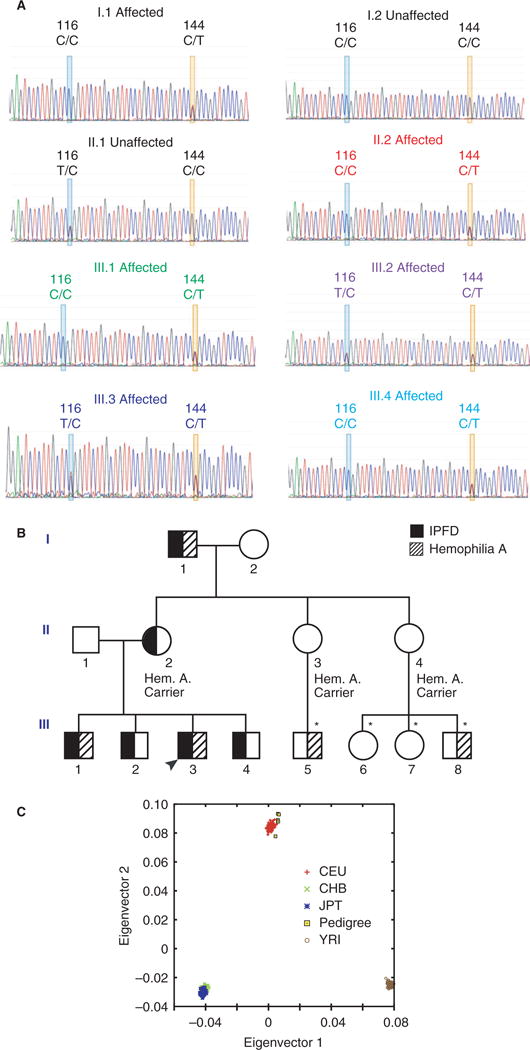
Phenotype and genotype of pedigree. (A) Sanger sequence chromatographs showing the prostaglandin synthase-1 (PTGS-1) variants in eight pedigree members spanning three generations. Pedigree members with impaired platelet aggregation and excessive hemorrhage all had heterozygosity of the PTGS-1 c.50C>T variant (P17L, rs3842787; yellow box). A second known variant in PTGS-1 (c.22T>C, rs1236913; blue box) was also present in some family members but did not segregate with the platelet disorder phenotype. (B) Pedigree for affected family. Black arrowhead indicates proband. Filled symbols represent individuals with an inherited platelet function disorder (IPFD) and hashed symbols represent individuals with hemophilia A. WES was performed on the following six pedigree members: II.1, II.2, III.1, III.2, III.3 and III.4 (*not sequenced). (C) Principle components analysis (PCA) of the exome-sequenced individuals from the analyzed pedigree. PCA was performed on variants from these individuals and from select populations of the 1000 Genomes phase 3 dataset (CEU, Utah Residents [CEPH] with Northern and Western European Ancestry; JPT, Japanese in Tokyo, Japan; CHB, Han Chinese in Beijing, China; YRI, Yoruba in Ibadan, Nigeria.). This analysis shows that all sequenced individuals are of European descent and high-quality sequencing and variant calling was achieved.
Table 2.
The number of variants that meet each filter criteria used to identify a variant causative for the observed phenotype
| Total variants | 177 125 |
| Variants shared only by affected individuals | 9666 |
| Coding variants shared only by affected individuals | 835 |
| Coding variants in the AA pathway shared only by affected individuals | 1 |
AA, arachidonic acid.
Whole exome sequencing identified a known missense variant in the F8 gene (reference sequences NG_011403.1; NP_000123.1) in pedigree members with hemophilia A (rs28935203; c.5096A>T; p.Tyr1699Phe) [30].
Impaired thromboxane release in affected pedigree members
As affected pedigree members demonstrated impaired aggregation to AA, but not collagen, and the variant occurred in a signaling peptide domain of PTGS-1, we examined thromboxane B2 release in affected pedigree members. Platelets were isolated from a subset of pedigree members (n = 3) as well as age- and gender-matched controls (n = 7) without a platelet disorder or abnormal bleeding. Sanger sequencing showed that none of these control subjects had the PTGS-1 c.50C>T variant. Thromboxane B2 release was significantly reduced in affected pedigree members compared with age- and gender-matched controls and the unaffected father within the pedigree (Fig. 4A). Impaired platelet aggregation was rescued with prostaglandin G2, a prostaglandin intermediate downstream of PTGS-1 that aids in the production of thromboxane (Fig. 4B and C). Prostaglandin G2 also rescued impaired platelet aggregation due to aspirin (Figure S2). Platelet protein expression of PTGS-1 and TBXAS-1 did not differ in affected pedigree members compared with controls (Figure S1B).
Fig. 4.
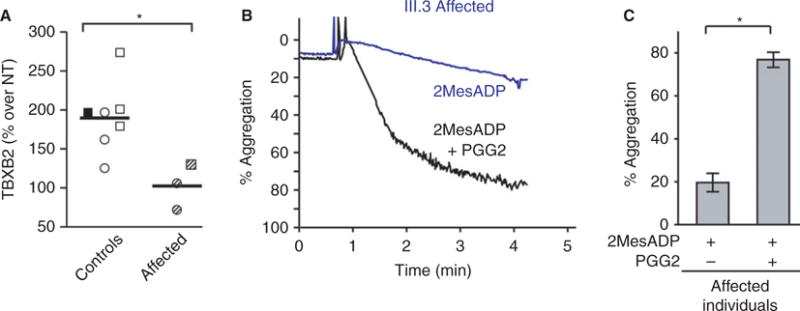
Affected pedigree members demonstrated functional impairments in prostaglandin synthase-1 (PTGS-1) activity that could be rescued by the downstream intermediate prostaglandin G2. (A) Thromboxane release was significantly lower in affected pedigree members compared with healthy controls. Platelets were isolated from pedigree members and age- and gender-matched controls. Platelets were then stimulated with thrombin (0.1 U mL−1) and supernatants were collected and assayed for thromboxane release (TBXB2). Compared with the unaffected father (black filled box) and control subjects (white boxes, n = 6), affected pedigree members (mother and two children, shaded boxes) had significantly lower thromboxane release. All samples were normalized to no-treatment (NT) conditions. (B) Representative curve demonstrating impaired aggregation in an affected child (III.3) in response to 2MesADP (30 nM) and subsequent rescue of platelet aggregation with prostaglandin G2, a prostaglandin intermediate downstream of PTGS-1 that aides in the production of thromboxane (1 lM). (C) Impaired platelet aggregation in affected pedigree members (n = 5) was rescued with prostaglandin G2 (1 μM), demonstrating that pathways downstream of PTGS-1 are intact (*P < 0.05).
Discussion
Here we report the identification of an autosomal dominant, non-synonymous exonic sequence variant in PTGS-1 (rs3842787; c.50C>T; p.Pro17Leu) in a three-generational pedigree. Most male members of the pedigree (Fig. 3B) also had a variant in F8 (rs28935203; c.5096A>T; p.Tyr1699Phe) causing mild hemophilia A. To our knowledge, this is the first time that the co-occurrence of these two pathogenic variants in both F8 and PTGS-1 genes in persons with a bleeding disorder has been identified. In affected pedigree members with only the IPFD and in pedigree members with both the IPFD and mild hemophilia A (Fig. 3B), there was excessive hemorrhage. We did observe mucosal patterns of bleeding in pedigree members with the PTGS-1 variant, but without hemophilia A, consistent with the typical source of bleeding in patients with an IPFD (Table 1). Consistent with their increased bleeding severity, individuals with the PTGS-1 c.50C>T variant required treatment with DDAVP and, for individuals with hemophilia A, FVIII replacement therapy (Table 1). In these pedigree members with the PTGS-1 variant, DDAVP has been partially effective in controlling bleeding symptoms in the individuals with and without FVIII deficiency. This provides a potentially useful set of cases that, although requiring more clinical investigation, may help treat bleeding in other patients with the PTGS-1 c.50C>T variant. DDAVP also has ameliorated the need for some of the FVIII infusions (which are more difficult to administer) in the individuals with FVIII deficiency. No patient has thus far needed platelet transfusion since the diagnosis of platelet function disorder was made. Two pedigree members had the PTGS-1 c.50C>T variant without hemophilia. These individuals also had clinically significant bleeding, suggesting that the variant itself may result in a clinical bleeding phenotype. We speculate that it is unlikely that both variants cause the phenotype in question as a result of the two variants being completely unlinked (r2 = 0.004).
In the ExAC database (http://www.exac.broadinstitute.org), the population frequency of the PTGS-1 c.50C>T variant ranges from 0.014 (south Asians) to as high as 0.14 (Africans). In European populations from the ExAC database (the ancestry of our pedigree), the population frequency is 0.068, consistent with estimates of 0.080 in Utah residents from the CEPH collection (http://hapmap.ncbi.nlm.nih.gov). As such, we postulate that this genetic disorder may be more common than clinically recognized, perhaps because of its minor bleeding phenotype. In addition, we cannot exclude the possibility that in some individuals carrying this variant, there may be incomplete penetrance or increased bleeding may not readily occur. Nevertheless, albeit a potentially rare scenario, when this variant is present in combination with a second disorder (hemophilia A in this pedigree), thereby affecting both primary and secondary hemostasis, the bleeding severity may be compounded. Recognition of this IPFD, although relatively uncommon, may have important clinical implications for the prevention and treatment of bleeding, especially during trauma or surgery.
PTGS-1 catalyzes the formation of prostaglandin G2 and H2 from AA. Once these immediate metabolites are formed, TBXAS-1 converts prostaglandin H2 into thromboxane A2, resulting in platelet aggregation. The PTGS-1 c.50C>T variant has been associated with greater inhibition of prostaglandin H2 formation by aspirin [31] and an increased risk of bleeding following elective coronary angiography and surgery [32,33]. Consistent with the known enzymatic activity of PTGS-1, platelet aggregation in response to AA and thromboxane release was significantly lower in members of our pedigree with the PTGS-1 c.50C>T variant (Figs 2A and 4A). We also demonstrated that the platelet aggregation defect in affected pedigree members was rescued with the addition of prostaglandin G2, a prostaglandin intermediate downstream of PTGS-1 that produces thromboxane (Fig. 4B and C). Although not a direct measure of prostaglandin production and signaling, these data demonstrating normal aggregation to ADP in the presence of prostaglandin G2 are consistent with a deficit in the production of prostaglandin G2 after stimulation with ADP and arachidonic acid. Because PTGS-1 activity is necessary to convert arachidonic acid to prostaglandin G2, these data further confirm a deficiency in PTGS-1 activity in this pedigree. The PTGS-1 c.50C>T variant causes replacement of a neutral proline with a hydrophobic leucine upstream of the signal peptide region of PTGS-1. This variant has been shown to be in complete linkage disequilibrium with an upstream promoter variant (c.-842A>G) [31,32]. As such, it has been postulated that the G allele may result in the creation of an AP2 transcription factor binding site, thus altering PTGS-1 activity [31]. In addition, alterations in the signal peptide may alter protein localization inside the platelet, reducing its ability to interact with arachidonic acid. Nevertheless, exactly how this might affect PTGS-1 activity is unknown and should be investigated in future experimental model systems.
The strengths of our study include the inclusion of three generations within our pedigree, comprehensive platelet functional studies, the use of WES to exclude other potentially causative sequence variants, and the concordant segregation of genotype and disease. Together, these findings provide robust evidence implicating the PTGS-1 c.50C>T variant as the causative variant in affected pedigree members with impaired platelet aggregation to AA and excessive bleeding. In contrast, others were unable to identify an association between the PTGS-1 c.50C>T variant and either increased bleeding or altered cyclooxygenase activity [34,35]. These discordant results may be a result of several factors, including incomplete phenotyping and limitations in assays used to measure platelet function. In addition, the co-segregation of the PTGS-1 c.50C>T variant in several affected family members increased our ability to detect its phenotypic effect. The co-existence of mild hemophilia A together with the PTGS-1 variant in our cohort delineated a bleeding phenotype that individually (e.g. PTGS-1 variant only) may not have been readily clinically apparent.
Despite the uniform co-segregation of genotype and phenotype in our pedigree, we cannot completely exclude the possibility of inadvertently overestimating the significance of the PTGS-1 c.50C>T variant and further large studies are needed to confirm our findings. Although we were unable to detect any differences in the protein expression of PTGS-1 in subjects with the PTGS-1 c.50C>T variant, this result may be due to limitations in precise protein expression quantification by immunoblot analysis. In some pedigree members, we were unable to obtain samples for platelet phenotyping and genotyping (III.5–III.8, Fig. 3B).
The optimal treatment of patients with clinical bleeding attributed to an inherited platelet function disorder is currently incompletely understood. For more severe platelet defects, such as Glanzmann’s thrombesthenia, intermittent platelet transfusion and/or recombinant activated FVII (rFVIIa, NovoSeven) have been shown to reduce the severity of bleeding symptoms. For mild bleeding, DDAVP and antifibrinolytic therapy have been utilized in case reports, but no randomized, controlled trials have been performed to establish efficacy. The recognition of these variants may help clinicians identify those individuals with bleeding symptoms who are at increased risk of post-traumatic or surgical bleeding and encourage the use of platelet transfusion as a primary prophylaxis therapy or when bleeding is encountered.
Conclusions
We provide the first report of co-existing variants in both F8 and PTGS-1 genes in a three-generation pedigree. The PTGS-1 c.50C>T variant was associated with specific, functional defects in the arachidonic acid pathway and more severe hemorrhage.
Supplementary Material
Fig. S1. (A) Protein expression of PTGS-1 and thromboxane synthetase-1 (TBXAS-1) did not differ in affected pedigree members (II.2, III.1, III.3 and III.4) compared with an unaffected pedigree member (the father, II.1) and two control subjects (CTRL). (B) Phosphorylation of Akt (pAkt S473) in platelets activated with 2mesADP (30 nM for 3 min) was also normal in affected pedigree members (II.2, III.1, III.3, and III.4) and the unaffected father (II.1), as compared with controls (CTRL, n = 2), consistent with intact P2Y12 and P2Y1 receptor function. The lower lanes show the β-actin loading control.
Fig. S2. (A) In washed platelets isolated from an unaffected healthy subject taking aspirin (81 mg), platelet aggregation to 2mesADP (30 nM) is impaired. (B) This defect in platelet aggregation caused by pharmacologic inhibition of PTGS-1 can be rescued by the addition of prostaglandin G2.
Essentials.
Co-existent damaging variants are likely to cause more severe bleeding and may go undiagnosed.
We determined pathogenic variants in a three-generational pedigree with excessive bleeding.
Bleeding occurred with concurrent variants in prostaglandin synthase-1 (PTGS-1) and factor VIII.
The PTGS-1 variant was associated with functional defects in the arachidonic acid pathway.
Acknowledgments
We thank C. Parker for his editorial input and D. Lim for her excellent artistic preparation of the figures. We would like to acknowledge the substantial contributions by B. Kanth Manne for the immunoblot assays and by K. J. Smock for performing and interpreting platelet aggregation studies. This work was funded by the following grants: HL066277, HL112311, and HL092161 are funded by NHLBI, AG040631 is funded by NIA, GM103806 is funded by NIGMS. Canadian Institutes of Health Research (CIHR) MOP-81208 and MOP-259952 (WHK). This material is the result of work supported with resources and the use of facilities at the George E. Wahlen VA Medical Center, Salt Lake City, UT, USA. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Footnotes
See also Johnsen JM. All too common: bleeding and genetic variation. This issue, pp 2227–9.
Addendum
D. Nance, S. A. Mereby, R. A. Campbell, N. D. Tolley, J. W. Rowley, J. M. Downie, A. S. Weyrich, and M. T. Rondina were responsible for the conception and design of the study. D. Nance, M. T. Rondina, G. A. Zimmerman, R. A. Campbell, N. D. Tolley, J. W. Rowley, J. M. Downie, W. Kahr, L. B. Jorde, and A. S. Weyrich were responsible for analysis and/or interpretation of data. D. Nance, M. T. Rondina, L. B. Jorde, W. H. Kahr, A. S. Weyrich, and G. A. Zimmerman were responsible for the critical writing or revising intellectual content. D. Nance, R. A. Campbell, J. W. Rowley, J. M. Downie, W. H. Kahr, S. Mereby, N. D. Tolley, J. W. Rowley, G. A. Zimmerman, A. S. Weyrich, and M. T. Rondina gave final approval of the manuscript to be published.
Disclosure of Conflict of Interests
The authors state that they have no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
References
- 1.Carcao MD. The diagnosis and management of congenital hemophilia. Semin Thromb Hemost. 2012;38:727–34. doi: 10.1055/s-0032-1326786. [DOI] [PubMed] [Google Scholar]
- 2.Gresele P, Harrison P, Bury L, Falcinelli E, Gachet C, Hayward CP, Kenny D, Mezzano D, Mumford AD, Nugent D, Nurden AT, Orsini S, Cattaneo M. Diagnosis of suspected inherited platelet function disorders: results of a worldwide survey. J Thromb Haemost. 2014;12:1562–9. doi: 10.1111/jth.12650. [DOI] [PubMed] [Google Scholar]
- 3.Cox K, Price V, Kahr WH. Inherited platelet disorders: a clinical approach to diagnosis and management. Expert Rev Hematol. 2011;4:455–72. doi: 10.1586/ehm.11.41. [DOI] [PubMed] [Google Scholar]
- 4.Lowe GC, Lordkipanidze M, Watson SP, UK GAPP study group Utility of the ISTH bleeding assessment tool in predicting platelet defects in participants with suspected inherited platelet function disorders. J Thromb Haemost. 2013;11:1663–8. doi: 10.1111/jth.12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watson SP, Lowe GC, Lordkipanidze M, Morgan NV, GAPP consortium Genotyping and phenotyping of platelet function disorders. J Thromb Haemost. 2013;11(Suppl 1):351–63. doi: 10.1111/jth.12199. [DOI] [PubMed] [Google Scholar]
- 6.Peyvandi F, Kunicki T, Lillicrap D. Genetic sequence analysis of inherited bleeding diseases. Blood. 2013;122:3423–31. doi: 10.1182/blood-2013-05-505511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leo VC, Morgan V, Bem D, Jones ML, Lowe GC, Lordkipanidze M, Drake S, Simpson MA, Gissen P, Mumford A, Watson SP, Daly ME, UK GAPP Study Group Use of next-generation sequencing and candidate gene analysis to identify underlying defects in patients with inherited platelet function disorders. J Thromb Haemost. 2015;13:643–50. doi: 10.1111/jth.12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smock KJ, Rodgers GM. Laboratory evaluation of aspirin responsiveness. Am J Hematol. 2010;85:358–60. doi: 10.1002/ajh.21674. [DOI] [PubMed] [Google Scholar]
- 9.Rondina MT, Schwertz H, Harris ES, Kraemer BF, Campbell RA, Mackman N, Grissom CK, Weyrich AS, Zimmerman GA. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J Thromb Haemost. 2011;9:748–58. doi: 10.1111/j.1538-7836.2011.04208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiorucci S, Santucci L, Wallace JL, Sardina M, Romano M, del Soldato P, Morelli A. Interaction of a selective cyclooxygenase-2 inhibitor with aspirin and NO-releasing aspirin in the human gastric mucosa. Proc Natl Acad Sci USA. 2003;100:10937–41. doi: 10.1073/pnas.1933204100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahr WH, Lo RW, Li L, Pluthero FG, Christensen H, Ni R, Vaezzadeh N, Hawkins CE, Weyrich AS, Di Paola J, Landolt-Marticorena C, Gross PL. Abnormal megakaryocyte development and platelet function in Nbeal2(−/−) mice. Blood. 2013;122:3349–58. doi: 10.1182/blood-2013-04-499491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:13033997. 2013 [Google Scholar]
- 14.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The 1000 Genomes Project Consortium. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;11:11.10.1–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013;34:E2393–402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 21.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 23.Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–61. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, Day IN, Gaunt TR. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24:2125–37. doi: 10.1093/hmg/ddu733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamberg M, Svensson J, Samuelsson B. Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci USA. 1975;72:2994–8. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Higuchi M, Kazazian HH, Jr, Kasch L, Warren TC, McGinniss MJ, Phillips JA, 3rd, Kasper C, Janco R, Antonarakis SE. Molecular characterization of severe hemophilia A suggests that about half the mutations are not within the coding regions and splice junctions of the factor VIII gene. Proc Natl Acad Sci USA. 1991;88:7405–9. doi: 10.1073/pnas.88.16.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halushka MK, Walker LP, Halushka PV. Genetic variation in cyclooxygenase 1: effects on response to aspirin. Clin Pharmacol Ther. 2003;73:122–30. doi: 10.1067/mcp.2003.1. [DOI] [PubMed] [Google Scholar]
- 32.Motovska Z, Kvasnicka J, Hajkova J, Kala P, Simek S, Bobcikova P, Petr R, Bilkova D, Poloczek M, Miklik R, Fischerova M, Maly M, Widimsky P. Platelet gene polymorphisms and risk of bleeding in patients undergoing elective coronary angiography: a genetic substudy of the PRAGUE-8 trial. Atherosclerosis. 2010;212:548–52. doi: 10.1016/j.atherosclerosis.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 33.Yagmur E, Weiskirchen R, Schedel A, Bugert P. PTGS1 compound heterozygosity impairs gene expression and platelet aggregation and is associated with severe bleeding complications. Thromb Haemost. 2013;110:1083–5. doi: 10.1160/TH13-02-0110. [DOI] [PubMed] [Google Scholar]
- 34.Pettinella C, Romano M, Stuppia L, Santilli F, Liani R, Davi G. Cyclooxygenase-1 haplotype C50T/A-842G does not affect platelet response to aspirin. Thromb Haemost. 2009;101:687–90. [PubMed] [Google Scholar]
- 35.Lee CR, Bottone FG, Jr, Krahn JM, Li L, Mohrenweiser HW, Cook ME, Petrovich RM, Bell DA, Eling TE, Zeldin DC. Identification and functional characterization of polymorphisms in human cyclooxygenase-1 (PTGS1) Pharmacogenet Genomics. 2007;17:145–60. doi: 10.1097/01.fpc.0000236340.87540.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (A) Protein expression of PTGS-1 and thromboxane synthetase-1 (TBXAS-1) did not differ in affected pedigree members (II.2, III.1, III.3 and III.4) compared with an unaffected pedigree member (the father, II.1) and two control subjects (CTRL). (B) Phosphorylation of Akt (pAkt S473) in platelets activated with 2mesADP (30 nM for 3 min) was also normal in affected pedigree members (II.2, III.1, III.3, and III.4) and the unaffected father (II.1), as compared with controls (CTRL, n = 2), consistent with intact P2Y12 and P2Y1 receptor function. The lower lanes show the β-actin loading control.
Fig. S2. (A) In washed platelets isolated from an unaffected healthy subject taking aspirin (81 mg), platelet aggregation to 2mesADP (30 nM) is impaired. (B) This defect in platelet aggregation caused by pharmacologic inhibition of PTGS-1 can be rescued by the addition of prostaglandin G2.