

Abstract
Deamination of adenosine in RNA to form inosine has wide ranging consequences on RNA function including amino acid substitution to give proteins not encoded in the genome. What determines which adenosines in an mRNA are subject to this modification reaction? The answer lies in an understanding of the mechanism and substrate recognition properties of adenosine deaminases that act on RNA (ADARs). Our recent publication of x-ray crystal structures of the human ADAR2 deaminase domain bound to RNA editing substrates shed considerable light on how the catalytic domains of these enzymes bind RNA and promote adenosine deamination. Here we review in detail the deaminase domain-RNA contact surfaces and present models of how full length ADARs, bearing double stranded RNA-binding domains (dsRBDs) and deaminase domains, could process naturally occurring substrate RNAs.
Keywords: ADAR, epitranscriptome, recoding, RNA editing
Introduction
Enzymatic reactions that change the structures of nucleosides in mRNA are now recognized as common and essential for proper control of gene expression [1]. When these reactions change the coding properties of the RNA, they are referred to as RNA editing [2]. The most common form of RNA editing in metazoa is adenosine deamination. Deamination of adenosine (A) yields inosine (I) which behaves as guanosine (G) in Watson-Crick base pairing and during translation (Figure 1A, Figure 1B). The effects of A to I editing are dependent on the location and sequence context of the edited A and may lead to diverse consequences such as alternative splicing of pre-mRNA [3], altered processing of pri-miRNA [4], altered target specificity of mature miRNA [5] and recoding in exons to generate proteins that differ from their genetically encoded form [6]. In addition, the presence of I in RNA has also been shown to alter RNA stability [7] and cellular localization [8]. Furthermore, dysregulated A to I editing activity has been linked to several human diseases [9] and known to be one of the causes of the inherited autoimmune disease Aicardi Goutières Syndrome [10].
Figure 1.

Domain maps of ADAR and reaction scheme for adenosine deamination. A: ADAR reaction showing intermediate. B: Schematic of Inosine-cytidine base pair with H-bonding. C: Domain map of active members of human ADAR family. Protein, domain and linker lengths to scale to demonstrate relative size (ADAR1: Z1 aa135-201, Z2 aa295-359, dsRBD1-3 aa504-569, 615-680, 727-792, Deaminase aa837-1222) (ADAR2: dsRBD1 aa78-142, dsRBD2 aa230-296, Deaminase aa317-701)
A to I editing is carried out by a family of enzymes called ADARs (Adenosine Deaminases Acting on RNA) (Figure 1C) [11]. In humans, the family comprises two active enzymes (ADAR1 and ADAR2) and the catalytically inactive ADAR3. The proteins share a modular domain structure with a C terminal deaminase domain and three (ADAR1) or two (ADAR2/ADAR3) double stranded RNA binding domains (dsRBDs). ADARs appeared in the common ancestor of metazoa [12] and although some organisms have incurred the loss of either ADAR1 or ADAR2 [13], to our knowledge all metazoa have retained at least one ADAR gene.
ADARs are dsRNA-modifying enzymes, initially discovered for their ability to “unwind” dsRNA [14]. Recent work has demonstrated the requirement for duplex RNA is not derived solely from the dsRBDs [15]. ADARs have a preference for adenosines within a certain local sequence context (e.g. 5′-UAG-3′)[16, 17]. ADARs deaminate approximately 50% of adenosines within long, perfectly matched duplex RNA but can be highly selective for specific adenosines found within more complex secondary structures common in cellular RNA [16]. In addition, most efficient editing occurs at adenosines found opposite C while A-U pairs are also commonly edited [18]. A-A and A-G mismatches are poorly tolerated by ADARs. Duplex RNA imperfections, such as mismatches, bulges, loops and hairpins in many naturally occurring ADAR substrates play important roles in determining which adenosines are efficiently edited [19].
Because ADARs are specific for adenosines within dsRNA, the edited nucleotide must be flipped into an extrahelical conformation in order to reach the active site. Such “base-flipping” mechanisms have been observed and characterized for DNA-modifying enzymes such as methyltransferases [20] and repair glycosylases [21] but until recently no structural information was available for enzymes that catalyze base-flipping in duplex RNA. Our recent publication of X-ray crystal structures of human ADAR2 deaminase domain (hADAR2d) bound to dsRNA provided an opportunity to understand ADAR’s flipping mechanism and the structural basis for ADAR selectivity (Figure 2A) [22]. The crystal structures captured a state with the reactive nucleotide, in the form of the adenosine analog 8-azanebularane (8AN), flipped out of the helix and into the zinc-containing deaminase active site. The 8AN base exists as a covalent hydrate in the active site mimicking the high energy intermediate of the A-to-I deamination reaction (Figure 1A). Thus, the conformations observed for both the RNA and protein near the active site in these structures are highly likely to be representative of how ADAR2 interacts with most substrates. Contacts more distant from the active site may be missing for some substrates (see below). Structures of deaminase domain-RNA complexes also provide information about the sites on substrate RNAs that are accessible to dsRBDs. Using our structures, we can propose dsRBD binding locations and consider how secondary structure at those locations may affect ADAR2’s ability to bind and edit certain substrate RNAs.
Figure 2.
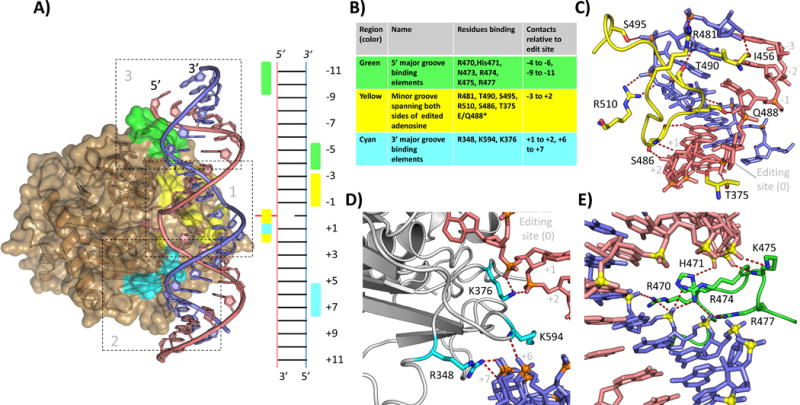
Interactions of hADAR2d with dsRNA beyond the active site. A: Overview of hADAR2d-RNA structure (5ED1, hADAR2d E488Q with Bdf2 derived 23-mer 8-azanebularane containing RNA) showing three main regions of contact. Edited strand in salmon, complementary stand in blue. Region 1 residues highlighted in yellow, region 2 in cyan, region 3 in green. Ladder diagram shows a secondary structure representation of the protein RNA contact interface (editing site shown in red flipped from the helix) B: Table summarizing the protein residues of each region and the RNA registers bound by each. C: Detail view of region 1. D. Detail view of region 2 (NOTE Fig 2C shows 5ED1 hADAR2d E488Q. E: Detail view of region 3 (NOTE Fig. 2E is from 5ED2, crystal structure of hADAR2D E488Q with hGli1 derived 23mer 8-azanebularne containing RNA. This RNA extends 1 bp farther in the 5′ direction than the Bdf2 derived RNA); (5ED1 and 5ED2 [22]).
The deaminase domain of ADAR2 contacts dsRNA over three main regions
The crystal structures of hADAR2d bound to RNA reveal a domain well suited for binding to duplex RNA [22]. Outside the active site there are three main regions of protein-RNA contact (Figure 2A, 2B). These extend across approximately 20 bp of dsRNA along one face of the double helix involving interactions with the minor groove near the edited A and the two adjacent major grooves. Below we provide an overview of these hADAR2d-RNA contacts outside the active site. For a detailed discussion of contacts made to the edited nucleotide, please see Matthews et al., 2016 [22].
Region 1, minor groove at editing site
Close to the active site, the side chain of the helix intercalating Q488 residue (E488 in wild type) accepts a hydrogen bond from the 2′-hydroxyl group of the ribose at the −1 position (5′) of the edited strand (Figure 2C). The backbone amide nitrogen and side chain oxygen of S486 hydrogen bond with the 2′- hydroxyl of the +1 and +2 positions (3′), respectively. With the contact to the 2′- hydroxyl of the edited base in the active site by T375 (not shown), these interactions define a region of four consecutive 2′ hydroxyls contacted on the edited strand. The backbone carbonyl oxygen of S486 hydrogen bonds to the 2-amino group of the 3′ guanosine (+1 position), an interaction which has been shown to play an important role in determining ADAR2’s preference for a 3′ guanosine nearest neighbor [22]. The side chain of T375 H-bonds with the phosphate between the edited base and the +1 position. The contacts to the edited strand serve to position this strand in a cleft containing the active site and stabilize the conformation of the backbone necessary for the edited adenosine to reach the catalytic center. The close approach of residues 486–491 to the minor groove allow/promote the base flipping step of the deamination mechanism. Interactions of this loop explain ADARs preference for editing at adenosines with a 5′ U or A [22]. The deep penetration of this loop into the RNA minor groove results in a significant distortion of the RNA backbone on the complementary strand. This distortion is stabilized by contacts at the 5′ phosphates of position −1, −2 and −3 via H-bond or salt bridges from the side chains of R510, S495 and R481 respectively. Finally, the backbone carbonyl oxygens of T490 and I456 H-bond with the 2′- hydroxyl groups of the −2 and −3 positions of the complementary strand. (Figure 2A, yellow; Figure 2C). The interactions in region 1 are likely to be critical for efficient deamination of most substrates.
Region 2, major groove 3′ to editing site
A major groove interacting element (Figure 2A, cyan; Figure 2D) composed of two loops bearing residues R348 and K594 binds the charged backbone of the +6 and +7 bases of the complementary strand 3′ of the edit site. The positively charged side chain of K594 inserts into the major groove. Adjacent to the active site the side chain of K375 extends to contact phosphates of the edited strand on the +1 and +2 bases. Together with the 5′ binding loop (see below) this region defines the 20 bp region of RNA where a stable dsRNA helix is required for optimal interaction with the deaminase domain of ADAR2. In a recent publication, the effect of progressively shortening the 3′ stem of a well characterized ADAR substrate RNA was examined in vitro [23]. Editing was not significantly affected by truncations that retained at least 8 bp of duplex 3′ of the editing site. Reduction of the stem length to 6 bp resulted in an approximately 50% loss of activity while a substrate with a 4 bp stem retained less than 5% of the activity observed for the wild type RNA. Using the structures of ADAR2d this pattern of activity can be interpreted. Eight base pairs of dsRNA 3′ of the edit site should allow full contact with region 2 of the ADAR2 deaminase domain while a stem of 6 bp would partially disrupt the contacts across the 3′ major groove. A stem of only 4 bp would not support contact by either R348 or K594. Mutation of either R348 or K594 to alanine has also been shown to significantly reduce activity of hADAR2d [22]. Taken together these results reinforce the importance of region 2 to ADAR2 activity.
Region 3, major groove 5′ to editing site
A second major groove interacting loop binds 5′ to the editing site (Figure 2A, green; Figure 2E). It forms hydrogen bonds and salt bridge interactions with the negatively charged phosphodiester backbone of both strands spanning the major groove from −4 to −11 base pairs 5′ to the editing site. This loop, which consists of residues 454 through 479 in hADAR2, contains seven basic residues and is highly conserved in ADAR2 homologs across a wide range of species including mammals, insects, mollusks, fish, and reptiles [22]. The high degree of conservation suggests an important role for this loop in substrate binding and recognition for ADAR2. In our different hADAR2d-RNA structures, the backbone conformations for this loop are highly similar between R470 and Q479. Differences in protein-RNA contacts seem to be derived from slight differences in RNA positioning. In the hGLI1 RNA containing structure (5ED2), side chains of R470, R474 and R477 contact the phosphates of the complementary strand from the −6 to −4 base pairs (Figure 2C). The edited strand phosphates are bound by the side chain and backbone amide of K475 at the −9 and −10 position, respectively and the side chain of H471 contacts the phosphate of the −11 position (Figure 2C). The importance of region 3 residues on ADAR activity was recently examined by a combination of saturation mutagenesis and functional screening [24]. A saturation mutagenesis library of residues 454–462 and 469–479 within hADAR2d was screened in yeast for activity using a reporter substrate derived from the S. cerevisiae BDF2 mRNA. The results revealed several specific positions within the loop with a strong dependence on the wild type residue [24]. In the crystal structures of hADAR2 bound to dsRNA derived from the BDF2 sequence, these residues form a network of interactions through direct contact with the RNA substrate (H471, R474 and R477) or in intra-molecular interactions (F457, D469 and P472 [not shown in Figure 2E]) which stabilize the observed conformation of the loop. These results provide strong evidence that the 5′ binding loop location and sequence are well suited for interacting with dsRNA 5′ of the editing site. Analysis of these results also raised an interesting question: What is the basis for conservation of the other residues within the 5′ binding loop? Perhaps this loop can adopt alternate conformations when binding substrates with different 5′ secondary structures. Under these conditions the other conserved residues may play important roles in binding or loop stabilization. They may also participate in protein-protein interactions that were not present or relevant under the conditions used for the screening.
Conservation of binding residues within the ADAR family
A significant number of the hADAR2d residues that contact RNA in regions 1 and 2 are either conserved or highly similar in human ADAR1 and its homologs. Within region 1, residues E488, R481, T490, and R510 are completely conserved in homologs of both ADAR1 and ADAR2 from 10 species [22]. Across the same group of species, S495 differs only once, appearing as threonine in D. rerio. ADAR1 residues corresponding to hADAR2 T375 and S486 are both asparagine in the homologs in this group. Like threonine and serine, asparagine has a polar uncharged side chain capable of donating and accepting hydrogen bonds. In region 2, K594 is conserved across both ADAR1 and ADAR2 homologs while K376 appears as arginine in the ADAR1 family, a substitution which should allow contact like the salt bridge observed in the hADAR2-RNA structure.
As mentioned previously, the 5′ binding loop of region 3 is highly conserved in homologs of ADAR2. Importantly, ADAR1 and its homologs have a highly conserved sequence which differs significantly from the ADAR2 loop [22]. The ADAR1 loop is longer, contains fewer basic residues and a conserved phenylalanine where the ADAR2 loop interacts with the major groove. It is likely that differences in substrate selectivity observed for ADAR1 and ADAR2 arise, at least in part, from differences in how these loops bind RNA. The high similarity of the binding residues in regions 1 and 2 to their ADAR1 counterparts suggest that the RNA binding in the homologous regions of the ADAR1 catalytic domain will be similar to that observed for ADAR2. The interactions 5′ of the editing site are potentially quite different. There is evidence to suggest that the catalytic domains of ADAR1 (hADAR1d) and ADAR2 (hADAR2d) have different preferences for secondary structure 5′ of the editing site [16]. In vitro experiments using the 5-HT2CR pre-mRNA (Figure 3A) found the F site and an adenosine opposite it′s 3′ neighbor to be the most efficient sites for hADAR2d[16]. Both sites are imbedded in a long duplex region, ideal for fully contacting region 3. The same experiment identified multiple adenosines with short (3–5 bp) stems in the 5′ direction followed by loops which are edited by hADAR1d but are completely unedited by full length hADAR2 or hADAR2d [16]. Adenosines in hairpin loops of similar structure are found in the pre-mRNAs of NEIL1 [6] and AZIN1 [25] have also been shown to be good substrates for ADAR1 but not for ADAR2. Predicted secondary structure for these RNA′s place the edited adenosine 5 to 6 nucleotides from a hairpin loop in the 5′ direction (see below and Figure 3B, 3C) suggesting that ADAR1 may be optimized to bind this type of short stem loop structure, as has been previously observed [26]. The catalytic domain of ADAR2 may be better adapted to binding extended duplex. Further biochemical studies and structural information are needed to explore the role of the 5′ binding loop in determining ADAR editing preferences.
Figure 3.

Predicted secondary structures of ADAR substrates. Editing sites highlighted in red. A: 5-HT2CR [16]. B: Neil1 [6]. C: Azin1 [25]. D: BDF2 [17]. E: hGLI1 [17]. F: pre-miRNA-142 [4]. G: GluR-B Q/R [32]. H: GluR-B R/G [31]. I: GluR-B R/G 15 mer [32].
ADAR substrates may lack contacts observed in the crystal structures
By mapping the regions of hADAR2d-RNA contact to a secondary structure representation of a duplex (Figure 2A) we can compare how the secondary structures of well characterized ADAR substrates deviate from the RNAs in the crystal structures. The RNAs used for crystallization of the hADAR2d-RNA complexes were derived from the human Gli1 and S. cerevisiae Bdf2 mRNAs [17]. The parent RNAs were recently identified as substrates deaminated with very high efficiency by ADAR2 even in the absence of its dsRBDs [17]. These RNAs contain an adenosine within the optimal local sequence 5′-U-A-G-3′ and have a cytosine opposite the editing site in the form of an A-C mismatch (Figure 3D, 3E). Both substrates also contain stems predicted to contain at least eight nucleotides of primarily duplex RNA 5′ of the editing site. Where mismatches/loops do occur within the 5′ stem, they are short and symmetrical and thus should be expected to preserve the helical axis and groove widths as a reasonable approximation of an A form duplex. It is likely the combination of ideal local sequence and the ability to interact across all three regions of RNA contact observed in the recent crystal structures that allows the Bdf2 and Gli1 mRNAs to serve as efficient substrates for ADAR2d.
Comparison can be made to predicted secondary structure of other known ADAR substrates. One interesting class of substrates of to consider are micro-RNA (miRNA) precursors. The effect of A-to-I editing on the processing and target selection of miRNAs has been an area of active research for over 10 years [4, 5]. The requirement for miRNA precursors to contain hairpin stem loop structures with 20–24 bp of primarily dsRNA means these RNAs may also serve as efficient substrates for ADAR enzymes. For example, in vitro deamination of pri-mir-142 (Figure 3F) by ADAR2 leads to A-to-I editing at 10 adenosines in its stem loop structure[4], most of which contain enough adjacent duplex structure to interact within all three regions of RNA contact in hADAR2d[4].
Many other known substrates of ADAR2 would be unable to bind to all three contact regions in the deaminase domain. For example, the GluR-B Q/R and R/G edit sites are predicted to contain only five bp of duplex RNA 5′ of the editing site[27], not enough to fully engage the contacts in region 3 (Figure 3G, 3H). The 5HT2CR D-site contains a 5 nt loop on the complementary strand which occupies the −1 to −5 nucleotide position (region 1) (Figure 3A). Asymmetric loops of this size are known to induce significant kinks in the helical axis [28, 29] and could interfere with any of the interactions observed 5′ of the editing site in the crystal structures. Many substrates, including the GluR B Q/R, site also contain adenosines flanked by sub-optimal nearest neighbors and/or base paired with U rather than in the optimal A-C mismatch [30]. In cases where editing sites lack some feature of an “optimal” deaminase domain substrate, dsRBD binding to adjacent duplex structure can provide added binding affinity, effectively increasing the local concentration of the catalytic domain to facilitate editing at that site. The combination of binding from dsRBDs and the deaminase domain must provide enough binding energy to allow the base flipping loop to intercalate residue E488, stabilize the distortion surrounding the flipped base and overcome the stress induced by the close approach of G489 to the 5′ nearest neighbor base pair. Indeed, biochemical studies with models of the GluR B R/G site support this idea [31, 32]. Although these RNAs are poor substrates for ADAR2’s deaminase domain because they lack the region 3 contact surface, they react efficiently with a protein composed of the ADAR2 deaminase domain and dsRBD2[32] (see below).
How can RNA binding and deaminase domains bind simultaneously?
dsRBD binding is required for editing of some substrates
It is well established that many ADAR substrates require dsRBDs for efficient editing [16, 33, 34] and the restrictions placed on dsRBD binding sites by bulges and loops in RNA is thought to play an important role in ADAR specificity [35]. How then could ADAR2 dsRBDs bind concurrently with the deaminase domain? ADAR dsRBDs are approximately 65 amino acid domains which follow the typical α–β–β–β–α fold observed for other dsRBDs[34]. They bind dsRNA along one face, contacting two minor grooves and the intervening major groove (Figure 4)[34]. ADAR2 dsRBD1 and dsRBD2 are separated by 93 amino acids of unknown structure while the linker between dsRBD2 and the catalytic domain consists of 22 amino acids.
Figure 4.
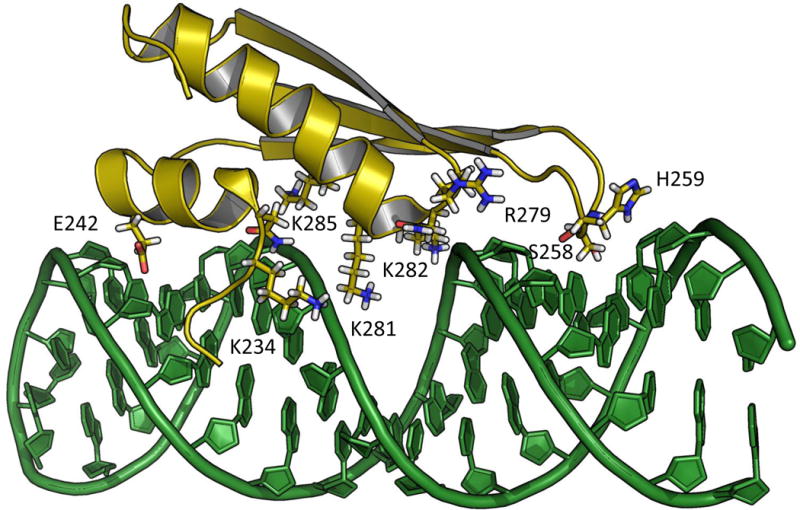
NMR solution structure (2L2K) of hADAR2 dsRBD2 in complex with GluR-B R/G derived hairpin RNA (2L2K [38]).
The role of dsRBDs in editing at the GluR B R/G site has been extensively studied [35, 36] [32, 34, 37]. Models of hADAR2 dsRBD2 bound to a GluR B R/G hairpin RNA have been presented based on NMR data that identified an RNA sequence specific contact between S258 of dsRBD2 and the 2-amino group of the guanosine immediately 3′ to the R/G editing site [38]. Based on this interaction, it was proposed that the guanosine-specific contact of dsRBD2 provided the structural basis for ADAR2’s observed 3′ nearest neighbor preference. Our crystal structures of hADAR2d bound to dsRNA indicated that the 3′ nearest neighbor preference can be explained by the contact made by S486 of the deaminase domain. Overlaying the crystal structure of the deaminase domain bound to RNA and the dsRBD2-RNA NMR structure creates an impossible clash between the two domains (Figure 5A). The interaction of the catalytic domain and RNA in this region is critical for base flipping [22, 39] which is essential to achieve the RNA conformation required for catalysis. Thus, this analysis indicates that, if dsRBD2 and the deaminase domain bind concurrently, dsRBD2 must bind at a location different from that suggested by the NMR data.
Figure 5.
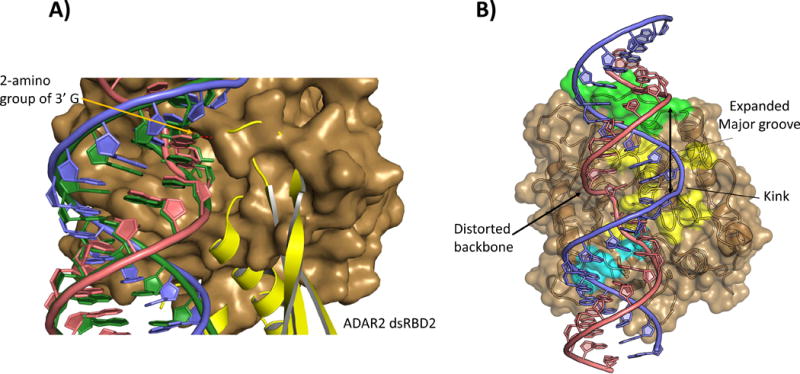
A: Overlay of ADAR2 deaminase domain(brown surface):BDF2 dsRNA (blue and salmon) crystal structure with solution structure of dsRBD2(yellow cartoon) bound to a GluR B R/G derived RNA (Green). Superimposing the guanosine contacted by S258 in the NMR structure of dsRBD2 (2L2K) with the 3′ guanosine of the ADAR2d:dsRNA crystal structures reveals that the two binding models cannot exist simultaneously. B: The phosphodiester backbone of the edited strand is significantly distorted for several bases 5′ to the flipped nucleotide. On the opposite face of the helix the complementary strand is kinked and the major groove is expanded.
We have created new models which we believe agree with available structural and biochemical evidence (Figure 6). In in vitro deamination assays on GluR B R/G substrates, ADAR2 truncations lacking dsRBD1 or dsRBD2 retain approximately 30% and 10% of wild type editing, respectively [34]. The truncation lacking both dsRBDs (e.g. hADAR2d) showed no editing at the R/G site[34]. Interestingly, a truncation containing dsRBD2 and the catalytic domain (hADAR2-R2D) still retained editing activity on a model GluR B R/G substrate of just 15 base pairs (Figure 3I) with five bp 5′ and nine bp 3′ of the editing site [32]. Together these results suggest that dsRBD2 can bind a portion of the 15 bp duplex and provide sufficient binding affinity to facilitate the distortion of the RNA backbone and base flipping required for editing.
Figure 6.

Proposed models of hADAR2d and dsRBD2 bound to dsRNA. (5ED1, hADAR2d E488Q bound to Bdf2 derived 8-azanebularane containing 23 bp RNA and 2L2K, NMR solution structure of hADAR2 dsRBD2 bound to GluR-B R/G derived hairpin RNA.) A: dsRBD2 binding to the distal face, RNA duplex from 2L2K shown in yellow highlights the similarity in helix geometry on the distal face of the RNA bound to hADAR2d to the RNA bound by dsRBD2. B: Model of how dsRBD2 might bind RNA 3′ of editing site with extended duplex 10 bp beyond Bdf2 derived 23mer. C: Model of how dsRBD2 might bind RNA 5′ of editing site with extended duplex 10 bp beyond Bdf2 derived 23mer.
Models for concurrent binding of dsRBD and deaminase domains
Given that the 15 bp GluR B R/G substrate would lie entirely within the region of duplex present in the solved structures of ADAR2d-RNA complexes, an interesting question arises; Do the RNAs in these structures harbor a suitable binding site for a dsRBD? Sterics obviously limit access to regions of the duplex not directly bound by the deaminase domain. Outside of the observed protein:RNA interface there is some significant distortion of the dsRNA from A form geometry. As the edited strand leaves the active site in the 5′ direction, its backbone is distorted for several base pairs with a widening of the major groove and a deviation from the plane of the strands across the adjacent grooves. It seems unlikely that a dsRBD could bind at this site as the RNA geometries deviate significantly from those found in known dsRBD:RNA complexes [40–42]. At approximately four nucleotides in the 5′ direction from the editing site the RNA resembles an A′ form helix with a major groove expanded from the canonical A form [43]. Multiple structures (X-ray and NMR solution) of dsRBD-RNA complexes (including ADAR2 dsRBD2) have shown dsRBDs binding across expanded A′ type major grooves [38, 40–42]. The groove distances in this region fit well with those observed in the ADAR2 dsRBD2-RNA complex [38] and superposition of the dsRBD2-RNA structure with the ADAR2d-dsRNA crystal structure (Figure 6A) indicates the structure of the RNA approximately 1/3 of a turn from the deaminase domain appears very close to the RNA bound by dsRBD2 in the available solution structures.
This represents an intriguing model for simultaneous binding of hADAR2d and dsRBD2. Interaction of hADAR2d induces a widening of the major groove on the face of the helix opposite the editing site and dsRBD2 binding also occurs with a widening of the major groove within its binding site. The RNA conformation induced by the binding of these two domains can be complementary and binding of one domain may serve to remodel the RNA for binding of the other. We should note that the distance between the N terminus of the deaminase domain and the C terminus of dsRBD2 in the model presented in Figure 6A is farther than could be accommodated by the amino acids not present in either structure. It may be possible for the N-terminal portion of the deaminase domain to adopt an alternative conformation to allow for this binding mode. Spanning such a distance with a 21 amino acid linker (N297-L316) is clearly possible and similar to the linker connecting the palm and thumb domains in DNA polymerase IV [44].
Many efficient ADAR2 substrates, such as the 5HT2cR D site (Figure 3A), have significant helix defects within the RNA duplex bound by the catalytic domain that would prevent simultaneous binding as shown in Figure 6A. For these structures, alternate binding sites for dsRBDs may be available adjacent to the editing site that are not possible to model with the RNA observed in the ADAR2d-RNA structures alone. For instance, if we consider RNA duplexes with an additional 10 bp added in either the 3′ direction (Figure 6B) or the 5′ direction (Figure 6C), simultaneous dsRBD2-deaminase domain binding could be easily accommodated. Such binding modes for dsRBD2 may be possible when the overlapping binding site (Figure 6A) is blocked by helix defects. Also, depending on the substrate, there may be one preferred binding mode for dsRBD2 or several complexes of relatively similar affinity that could support deamination at an editing site.
Conclusions and outlook
In summary, high resolution crystal structures of the human ADAR2 deaminase domain bound to RNA identified three regions of RNA-contact surface on the protein outside the active site that result in an interface that extends over 20 bp of duplex RNA. Comparison of the duplexes used in ADAR-RNA crystallography to known, naturally occurring ADAR substrates suggests the three regions may not all be needed for binding to each ADAR substrate and binding of dsRBDs likely compensates for missing interactions to the deaminase domain. In addition, the availability of high-resolution structures of deaminase domain-RNA and dsRBD-RNA complexes allowed us to present models for complexes that include both the ADAR2 deaminase domain and dsRBD2. These models are valuable since they provide the basis for additional experiments to define fully the binding sites for dsRBDs in ADAR-RNA complexes relevant to catalysis at specific editing sites. For instance, our models suggest specific minor groove contact points for dsRBD2, particularly for the overlapping binding site where binding is limited to only a few binding registers (Figure 6A). The importance of the proposed sites can now be tested experimentally. For instance, chemically modified RNA substrates that sterically occlude binding at that location can be used in deamination assays [33]. Such experiments are currently underway in our laboratory. Finally, this analysis highlights the need for additional structural studies of full length ADAR proteins bound to RNA, including structures with human ADAR1.
Acknowledgments
P.A.B. acknowledges the National Institutes of Health for financial support in the form of grant GM061115. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Abbreviations
- ADAR
adenosine deaminase acting on RNA
- dsRBD
double stranded RNA binding domain
References
- 1.Chen K, Zhao BS, He C. Nucleic Acid Modifications in Regulation of Gene Expression. Cell Chem Biol. 2016;23:74–85. doi: 10.1016/j.chembiol.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grosjean H. Fine-Tuning of RNA Functions by Modification and Editing Berlin Heidelberg. Springer; 2005. [Google Scholar]
- 3.Mazloomian A, Meyer IM. Genome-wide identification and characterization of tissue-specific RNA editing events in D. melanogaster and their potential role in regulating alternative splicing. RNA Biol. 2015;12:1391–401. doi: 10.1080/15476286.2015.1107703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang W, Chendrimada TP, Wang Q, Higuchi M, et al. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui Y, Huang T, Zhang X. RNA editing of microRNA prevents RNA-induced silencing complex recognition of target mRNA. Open Biol. 2015;5:150126. doi: 10.1098/rsob.150126. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Yeo J, Goodman RA, Schirle NT, David SS, et al. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc Natl Acad Sci USA. 2010;107:20715–9. doi: 10.1073/pnas.1009231107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scadden ADJ. The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nat Struct Mol Biol. 2005;12:489–96. doi: 10.1038/nsmb936. [DOI] [PubMed] [Google Scholar]
- 8.Zhou J, Wang Q, Chen L-L, Carmichael GG. On the mechanism of induction of heterochromatin by the RNA-binding protein vigilin. RNA. 2008;14:1773–81. doi: 10.1261/rna.1036308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slotkin W, Nishikura K. Adenosine-to-inosine RNA editing and human disease. Genome Med. 2013;5:105. doi: 10.1186/gm508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rice GI, Kasher PR, Forte GM, Mannion NM, et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–8. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–46. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grice LF, Degnan BM. The origin of the ADAR gene family and animal RNA editing. BMC Evol Biol. 2015;15:4. doi: 10.1186/s12862-015-0279-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin Y, Zhang W, Li Q. Origins and evolution of ADAR-mediated RNA editing. IUBMB Life. 2009;61:572–8. doi: 10.1002/iub.207. [DOI] [PubMed] [Google Scholar]
- 14.Bass BL, Weintraub H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell. 1988;55:1089–98. doi: 10.1016/0092-8674(88)90253-x. [DOI] [PubMed] [Google Scholar]
- 15.Phelps KJ, Tran K, Eifler T, Erickson AI, et al. Recognition of duplex RNA by the deaminase domain of the RNA editing enzyme ADAR2. Nucleic Acids Res. 2015;43:1123–32. doi: 10.1093/nar/gku1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eggington JM, Greene T, Bass BL. Predicting sites of ADAR editing in double stranded RNA. Nat Commun. 2011;2:319. doi: 10.1038/ncomms1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eifler T, Pokharel S, Beal PA. RNA-Seq analysis identifies a novel set of editing substrates for human ADAR2 present in Saccharomyces cerevisiae. Biochemistry. 2013;52:78577869. doi: 10.1021/bi4006539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong SK, Sato S, Lazinski DW. Substrate recognition by ADAR1 and ADAR2. RNA. 2001;7:846–58. doi: 10.1017/s135583820101007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lehmann KA, Bass BL. The importance of internal loops within RNA substrates of ADAR1. J Mol Biol. 1999;291:1–13. doi: 10.1006/jmbi.1999.2914. [DOI] [PubMed] [Google Scholar]
- 20.Klimasauskas S, Kumar S, Roberts RJ, Cheng X. Hhal methyltransferase flips its target base out of the DNA helix. Cell. 1994;76:357–69. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 21.Fromme JC, Banerjee A, Huang SJ, Verdine GL. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY andenine DNA glycosylase. Nature. 2004;427:652–6. doi: 10.1038/nature02306. [DOI] [PubMed] [Google Scholar]
- 22.Matthews MM, Thomas JM, Zheng Y, Tran K, et al. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat Struct Mol Biol. 2016;23:426–33. doi: 10.1038/nsmb.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukuda M, Oyama Y, Nishitarumizu A, Omura M, et al. Identification of an RNA element for specific coordination of A-to-I RNA editing on HTR2C pre-mRNA. Genes Cells. 2015;20:834–46. doi: 10.1111/gtc.12272. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Beal PA. Probing RNA recognition by human ADAR2 using a high-throughput mutagenesis method. Nucleic Acids Res. 2016;44:9872–9880. doi: 10.1093/nar/gkw799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Li Y, Lin CH, Chan TH, et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med. 2013;19:209–16. doi: 10.1038/nm.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herbert A, Rich A. The role of binding domains for dsRNA and Z-DNA in the in vivo editing of minimal substrates by ADAR1. Proc Natl Acad Sci USA. 2001;98:12132–7. doi: 10.1073/pnas.211419898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rueter S, Emeson R. Modification and Editing of RNA, Chapter 19: Adenosine-to-inosine Conversion in mRNA. ASM press; 1998. [Google Scholar]
- 28.Harjes E, Kitamura A, Zhao W, Morais MC, et al. Structure of the RNA claw of the DNA packaging motor of bacteriophage ϕ29. Nucleic Acids Res. 2012;40:9953–63. doi: 10.1093/nar/gks724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Q, Kim N-K, Peterson RD, Wang Z, et al. Structurally conserved five nucleotide bulge determines the overall topology of the core domain of human telomerase RNA. Proc Natl Acad Sci USA. 2010;107:18761–8. doi: 10.1073/pnas.1013269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stephens OM, Haudenschild BL, Beal PA. The Binding Selectivity of ADAR2’s dsRBMs Contributes to RNA-Editing Selectivity. Chem & Biol. 2004;11:1239–50. doi: 10.1016/j.chembiol.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 31.Haudenschild BL, Maydanovych O, Veliz EA, Macbeth MR, et al. A transition state analogue for an RNA-editing reaction. J Am Chem Soc. 2004;126:11213–9. doi: 10.1021/ja0472073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macbeth MR, Lingam AT, Brenda LSS. Evidence for auto-inhibition by the N terminus of hADAR2 and activation by dsRNA binding. RNA. 2004;10:1563–71. doi: 10.1261/rna.7920904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stephens OM, Haudenschild BL, Beal PA. The binding selectivity of ADAR2’s dsRBMs contributes to RNA-editing selectivity. Chem & Biol. 2004;11:1–20. doi: 10.1016/j.chembiol.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 34.Stefl R, Xu M, Skrisovska L, Emeson RB, et al. Structure and specific RNA binding of ADAR2 double-stranded RNA binding motifs. Structure (London, England : 1993) 2006;14:345–55. doi: 10.1016/j.str.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 35.Ohman M, Källman AM, Bass BL. In vitro analysis of the binding of ADAR2 to the pre-mRNA encoding the GluR-B R/G site. RNA (New York, NY) 2000;6:687–97. doi: 10.1017/s1355838200000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaikaran DCJ, Collins CH, MacMillan AM. Adenosine to inosine editing by ADAR2 requires formation of a ternary complex on the GluR-B R/G Site. J Biol Chem. 2002;277:37624–9. doi: 10.1074/jbc.M204126200. [DOI] [PubMed] [Google Scholar]
- 37.Wong S, Sato S, Lazinski DW. Substrate recognition by ADAR1 and ADAR2. RNA. 2001;7:846–58. doi: 10.1017/s135583820101007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stefl R, Oberstrass FC, Hood JL, Jourdan M, et al. The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell. 2010;143 doi: 10.1016/j.cell.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuttan A, Bass BL. Mechanistic insights into editing-site specificity of ADARs. Proc Natl Acad Sci USA. 2012;109:E3295–304. doi: 10.1073/pnas.1212548109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryter JM, Schultz SC. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 1998;17:7505–13. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jayachandran U, Grey H, Cook AG. Nuclear factor 90 uses an ADAR2-like binding mode to recognize specific bases in dsRNA. Nucleic Acids Res. 2016;44:1924–36. doi: 10.1093/nar/gkv1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blaszczyk J, Gan J, Tropea JE, Court DL, et al. Noncatalytic assembly of ribonuclease III with double-stranded RNA. Structure. 2004;12:457–66. doi: 10.1016/j.str.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 43.Tolbert BS, Miyazaki Y, Barton S, Kinde B, et al. Major groove width variations in RNA structures determined by NMR and impact of 13C residual chemical shift anisotropy and 1H–13C residual dipolar coupling on refinement. J Biomol NMR. 2010;47:205–19. doi: 10.1007/s10858-010-9424-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kottur J, Sharma A, Gore KR, Narayanan N, et al. Unique structural features in DNA polymerase IV enable efficient bypass of the N2 adduct induced by the nitrofurazone antibiotic. Structure. 2015;23:56–67. doi: 10.1016/j.str.2014.10.019. [DOI] [PubMed] [Google Scholar]