

Abstract
Objective
Williams syndrome (WS) is characterized by obstructive aortopathy attributable to heterozygous loss of ELN, the gene encoding elastin. Lesions are thought to result primarily from excessive smooth muscle cell (SMC) proliferation and consequent medial expansion, although an initially smaller caliber and increased stiffness of the aorta may contribute to luminal narrowing. The relative contributions of such abnormalities to the obstructive phenotype had not been defined.
Approach and Results
We quantified determinants of luminal stenosis in thoracic aortas of Eln−/− mice incompletely rescued by human ELN. Moderate obstruction was largely due to deficient circumferential growth, most prominently of ascending segments, despite increased axial growth. Medial thickening was evident in these smaller diameter elastin-deficient aortas, with medial area similar to that of larger diameter control aortas. There was no difference in cross-sectional SMC number between mutant and wild-type genotypes at multiple stages of postnatal development. Decreased elastin content was associated with medial fibrosis and reduced aortic distensibility due to increased structural stiffness but preserved material stiffness. Elastin-deficient SMCs exhibited greater contractile-to-proliferative phenotypic modulation in vitro than in vivo. We confirmed increased medial collagen without evidence of increased medial area or SMC number in a small ascending aorta with thickened media of a WS subject.
Conclusions
Deficient circumferential growth is the predominant mechanism for moderate obstructive aortic disease resulting from partial elastin deficiency. Our findings suggest that diverse aortic manifestations in WS result from graded elastin content, and SMC hyperplasia causing medial expansion may require additional elastin loss superimposed on ELN haploinsufficiency.
Keywords: aorta, smooth muscle cells, elastin, collagen, vascular remodeling
Graphical abstract
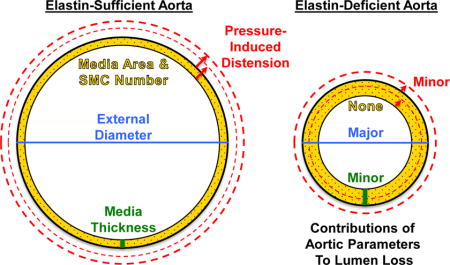
Introduction
Williams syndrome (WS) is a congenital, multisystem disorder caused by chromosomal microdeletion and heterozygous loss of 26 to 28 genes, including ELN which encodes the protein elastin [1]. Arterial abnormalities are common in WS and typically attributed to elastin deficiency since a similar phenotype manifests in Supravalvular Aortic Stenosis, a condition caused by ELN mutations [1]. The hallmark vascular lesion of WS, as the name of the associated condition implies, is stenosis of the ascending aorta just above the aortic root that contains the aortic valve. Besides focal obstruction of the proximal aorta, WS patients may manifest more diffuse obstruction within other large- and medium-sized arteries. The degree and extent of luminal stenosis varies widely from severe and focal in a minority of patients, which restricts blood flow and requires urgent treatment, to mild-to-moderate and diffuse in most subjects. In his original report of the syndrome, Williams ascribed the supravalvular stenosis partly to “constriction of the aortic wall” and partly to “hypertrophy of the media” [2]. Modern medical imaging confirms that subjects with WS have a relatively small and less compliant ascending aorta regardless of clinically apparent disease [3,4]. Gross and microscopic examination reveal further that the media is thickened, with a dysplastic appearance of irregularly arranged cells in a collagen-rich matrix [5,6] having an increased number of attenuated elastic laminae [7,8]. It is widely assumed that medial thickening arises from smooth muscle cell (SMC) hyperplasia because cells explanted from WS aortas have a higher proliferative rate during in vitro culture [9,10]. Nevertheless, SMC numbers have not been quantified and wall thickness, aortic diameter, and aortic compliance have not been compared across subjects with WS to determine their importance relative to the severity of the obstructive phenotype.
Experimental studies to elucidate mechanisms underlying WS aortopathy have been pursued in various genetic models of elastin deficiency in mice. Although these models do not incorporate all of the mutations that cause WS, extrapolation to the aortic phenotype in WS may be valid due to the fundamental role of elastin in vascular biology and mechanics. Previous work using a homozygous Eln−/− model of severe WS documented increased SMC proliferation, medial thickening, smaller aortic diameter, and marked luminal obstruction [10,11]. These elastin null mice die within a few days of birth, however, and thus are not informative about the phenotype in juveniles and adults, ages at which clinical insight is needed. This mouse model is also not directly comparable to WS where elastin is expressed by the remaining ELN allele, albeit at levels less than the expected 50% of normal [9]. On the other hand, heterozygous Eln+/− mice with approximately 50% elastin expression have a mild obstructive phenotype with only slightly smaller aortas and thinning, not thickening, of the aortic wall [12,13]. Other haploinsufficiency models with around 40% elastin expression, resulting from chromosomal microdeletions that include an Eln allele, have unchanged or mildly thickened walls in aortas of unreported size [14,15]. In contrast, partial rescue of Eln−/− mice by transgenic expression of human ELN resulting in approximately 30% elastin expression leads to a vascular phenotype similar to that seen in typical WS patients, namely, moderately smaller aortic diameter, medial thickening, and aortic stiffening [16]. This model has not been fully characterized, particularly with respect to relative contributions of various aortic parameters to the obstructive phenotype.
We quantified determinants of luminal stenosis in thoracic aortas of Eln−/− mice incompletely rescued by human ELN. Unexpectedly, we found that deficient circumferential growth and, to a lesser degree, medial thickening and diminished distension, but not increased medial area or cross-sectional SMC number, causes the moderate luminal narrowing of the aorta in these animals with partial elastin deficiency. Analyses in a WS patient with modest aortic obstruction were congruent with the animal data, thereby representing a counterexample that proves that stenosis need not arise from SMC hyperplasia.
Materials and Methods
Available in the online-only Data Supplement.
Results
Decreased External Diameter, not Increased Medial Area, Determines Aortic Luminal Stenosis Attributable to Partial Elastin Deficiency
To characterize pathological aortic remodeling in a model of WS, we first measured the dimensions and mass of the thoracic aorta in mice that were wild-type (mWT), heterozygous (mHET), or homozygous null (mNULL) for murine Eln but expressed human ELN in a bacterial artificial chromosome (hBAC). We did not use C57BL/6 mice as controls because any difference from hBAC-mNULL mice could not be unambiguously ascribed to loss of murine Eln versus gain in human ELN. Several in vivo, in situ, and ex vivo methods were used to collectively yield an overall unbiased characterization since all methods have inherent limitations (Supplemental Table I). The external diameter (width) of the ascending aorta in hBAC-mNULL mice was consistently smaller than in hBAC-mHET or hBAC-mWT mice at 3, 6, and 9 weeks of age (Fig. 1A,B). Unlike the growth of elastin-sufficient ascending aortas over time, the minimal increases in diameter of elastin-deficient vessels from 3 to 9 weeks did not reach statistical significance. Similar results were seen for the aortic arch, while differences were less, though significant, in the descending aorta (Supplemental Fig. IA,B). Strikingly, the thoracic aorta was longer in hBAC-mNULL mice (Fig. 1C,D), most prominently the ascending segment (Supplemental Fig. IC,D). The longer aortas had a more tortuous course as body length did not differ among genotypes (Supplemental Fig. IE).
Figure 1.
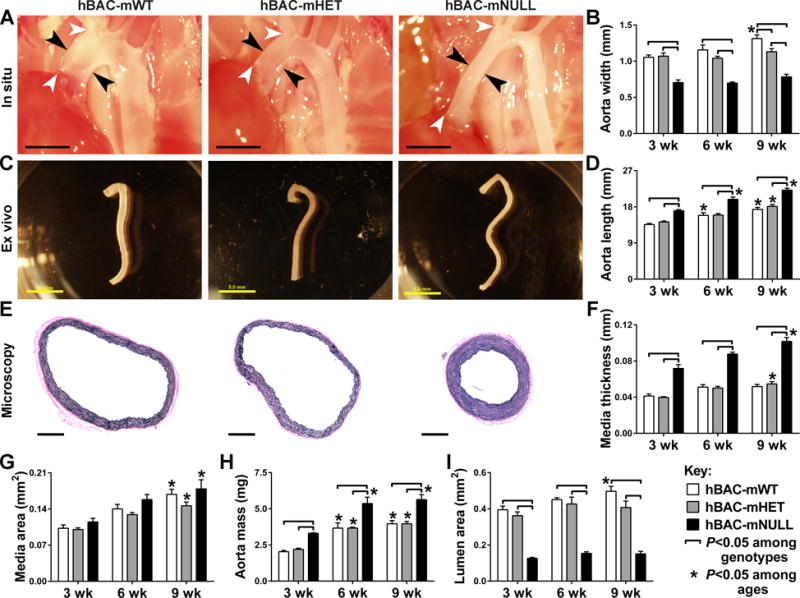
Thoracic Aorta Dimensions and Mass in hBAC-mWT (white bars), hBAC-mHET (gray bars), and hBAC-mNULL (black bars) mice. (A) Representative images of the ascending aorta in situ at 9 weeks of age with the width (i.e., external diameter) delineated between black arrows and the length between white arrows (bars: 2 mm). (B) In situ measurements of ascending aorta width at 3, 6, and 9 weeks. (C) Representative images of the thoracic aorta ex vivo at 9 weeks of age (bars: 5 mm). (D) In situ measurements of thoracic aorta length at 3, 6, and 9 weeks. (E) Representative cross-sectional images of EVG-stained ascending aorta at 9 weeks of age (bars: 500 μm). (F) Microscopic measurements of ascending aorta medial thickness at 3, 6, and 9 weeks. Measurements of (G) ascending aorta medial area, (H) thoracic aorta mass, and (I) ascending aorta lumen area at 3, 6, and 9 weeks. Data represent mean ± SEM; n = 3 (6 weeks) to 6 (3 and 9 weeks); ⎴ P < 0.05 among genotypes and *P < 0.05 among ages; two-way ANOVA.
The media of the ascending aorta was noticeably thicker in hBAC-mNULL mice (Fig. 1E,F), but medial cross-sectional area was not different (Fig. 1G) due to the smaller caliber. Similar findings, although of a lesser magnitude, were seen for medial thickness of the descending aorta (Supplemental Fig. IF), while adventitial thickness did not differ among genotypes (Supplemental Fig. IG). Despite equivalent cross-sectional areas, the mass of the thoracic aorta was greater in hBAC-mNULL mice as early as 3 weeks of age (Fig. 1H), likely a result of increased vessel length. When compared over time, the thoracic aorta progressively increased in mass in all genotypes. Other organs, such as the heart, also increased in mass over time, but did not differ among genotypes (Supplemental Fig. IIA). Since the various groups of animals were of similar body mass, aortic mass indexed to body mass was also greater in hBAC-mNULL mice (Supplemental Fig. IIB,C).
Consistent with these changes in aortic diameter and medial thickness, luminal area was markedly less in hBAC-mNULL mice (Fig. 1I). Specifically, there was a 70% loss of luminal area by 9 weeks of age; the smaller aortic diameter accounted for most of the luminal narrowing as recalculation in hBAC-mNULL mice using medial thickness values of hBAC-mWT mice would still result in a 54% reduction of luminal area. The predominance of deficient circumferential growth contributing to lumen loss is in keeping with the ~0.5 mm smaller aortic diameter but only ~0.05 mm thicker media in 9 week old hBAC-mNULL mice (where lumen diameter = external diameter − 2 × mural thickness). These data demonstrate continued aortic lengthening and medial thickening during postnatal maturation of elastin-deficient aortas, particularly of the ascending segment, with deficient circumferential growth driving the obstructive phenotype.
Partial Elastin Deficiency Associates with Increased Medial Collagen without Change in Cross-Sectional SMC Number
We quantified cellular and extracellular matrix (ECM) components of the aortic wall to determine their contributions to changes in mural mass. The total number of medial cells per cross-section did not differ among genotypes (Fig. 2A,B). Cell numbers paralleled the enlarging medial area from 3 to 9 weeks of age such that medial cell density remained unchanged (Supplemental Fig. IIIA). As expected, the elastin content of the media, measured as % positive histological staining, was consistently lower in ascending aortas of hBAC-mNULL mice (Fig. 2C,D) despite an increased number of elastic laminae (Supplemental Fig. IIIB). Similar differences of lesser magnitude were noted in the descending aorta (Supplemental Fig. IIIC). In contrast, medial collagen, localized by histological staining between the elastic laminae and SMCs, was greatly increased in ascending and descending aortas of hBAC-mNULL mice (Fig. 2E,F and Supplemental Fig. IIID). Similar results were obtained when the extent of collagen staining was measured as area of positive staining or as intensity of staining (Supplemental Fig. IIIE,F). We confirmed that sirius red labeling enhanced the characteristic birefringence of collagen under polarized light (Fig. 2E insets). Unlike in the media, levels of collagen within the adventitia were similar among genotypes at 6 and 9 weeks (Supplemental Fig. IIIG). These observations suggest compartment-specific remodeling of collagen fibers in response to elastin deficiency whose expression is primarily within the media.
Figure 2.
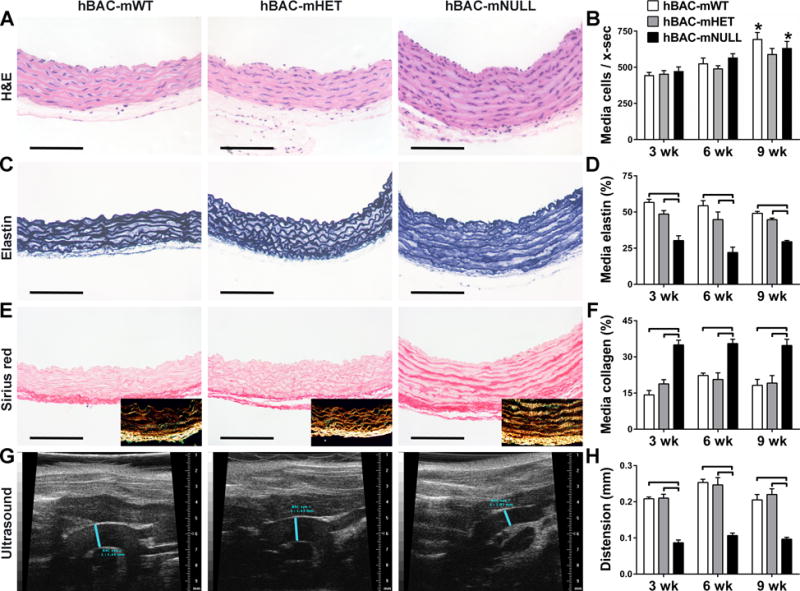
Ascending Aorta Structure and Function in hBAC-mWT (white bars), hBAC-mHET (gray bars), and hBAC-mNULL (black bars) mice. (A) Representative images of H&E stain at 9 weeks of age. (B) Number of medial cells per cross-section (x-sec) at 3, 6, and 9 weeks. (C) Representative images of elastin stain at 9 weeks of age. (D) Percent of media staining positive for elastin at 3, 6, and 9 weeks. (E) Representative images of sirius red stain (with polarized light images in insets) detecting collagen at 9 weeks of age. (F) Percent of media staining positive with sirius red at 3, 6, and 9 weeks. The histological images are oriented with the lumen above and adventitia below (bars: 100 μm). (G) Representative B-mode ultrasound images showing end-systolic diameter of the mid-ascending aorta delineated by blue lines at 9 weeks of age. (H) Aortic distension from end-diastole to end-systole at 3, 6, and 9 weeks. Data represent mean ± SEM; n = 3 (6 weeks) to 6 (3 and 9 weeks); ⎴ P < 0.05 among genotypes and *P < 0.05 among ages; two-way ANOVA.
Motivated by previous microarray results in P1 Eln−/− mice, we quantified transcript abundance for several collagens and associated molecules in the thoracic aorta of hBAC-mWT, hBAC-HET, and hBAC-mNULL mice at 3 weeks of age (Supplemental Fig. IV). There was no difference in RNA expression for the major fibrillar collagens, encoded by Col1a1 and Col3a1. Similar to findings in Eln−/− pups, aortic tissue of hBAC-mNULL mice expressed increased RNA for the short chain collagen encoded by Col8a1 and the fibrillar collagen encoded by Col11a1. There was also greater RNA expression for the Itga11 component of membrane collagen receptors (integrins), but not its Itgb1 dimerization partner. As expected, there was decreased RNA expression for murine Eln but not human ELN in the elastin-deficient genotypes and similar RNA levels for two additional housekeeping genes, Actb and Hprt. These findings suggest that medial fibrosis associated with elastin deficiency results from increased production of particular collagens.
Decreased Aortic Distension Contributes to the Obstructive Phenotype
Since medial fibrosis can affect the mechanical properties of the aorta, we assessed aortic compliance by transthoracic ultrasound and tail-cuff blood pressure. Distension (change in diameter) of the ascending aorta from end-diastole to end-systole was markedly reduced in hBAC-mNULL mice (Fig. 2G,H) and these differences remained significant when normalized by aortic diameter at diastole (Supplemental Fig. VA). Yet, the ~0.1 mm loss of distension is relatively minor in comparison to the ~0.5 mm smaller diameter as a determinant of luminal stenosis (where maximal lumen diameter = end-diastolic internal aortic diameter + systolic distension). Blood pressure at 9 weeks of age did not differ among genotypes (Supplemental Fig. VB), hence cross-sectional aortic compliance, based on changes in aortic diameter and blood pressure during the cardiac cycle, was also markedly reduced in hBAC-mNULL mice (Supplemental Fig. VC). Potential differences in ascending aortic extension (change in length) during the cardiac cycle could not be assessed as the increased length and curvature in hBAC-mNULL mice exceeded the ultrasound window.
Ultrasound also could not visualize the descending aorta, because of the air-filled lung, hence we further compared mechanical properties of the ascending and proximal descending thoracic aortic segments ex vivo using a custom computer-controlled biaxial testing device [17]. Here, we included true WT (C57BL/6) controls to determine if excess elastin in hBAC-mWT mice altered its mechanical behavior. Standard cyclic pressure-diameter and axial force-length tests over physiological levels of loading, and analyses of the associated wall stress-stretch responses, showed modest differences between WT and hBAC-mWT properties, suggesting that the addition of elastic fibers above normal did not alter the basic structural or material behaviors (Fig. 3A–D) beyond a slight increase in elastic energy storage capability (panel E). In contrast, reduction of elastic fibers in the ascending aorta in hBAC-mNULL mice resulted in marked differences in both structural and material properties (panels A–D), due in part to the reduced distensibility and extensibility under biaxial loading and evidenced well by a much reduced ability to store elastic energy upon deformation (panel F). Elastic energy stored during systole can be used by the aorta during diastole to work on the blood and augment blood flow, hence this reduction in energy storage can compromise a primary mechanical function of the aorta.
Figure 3.
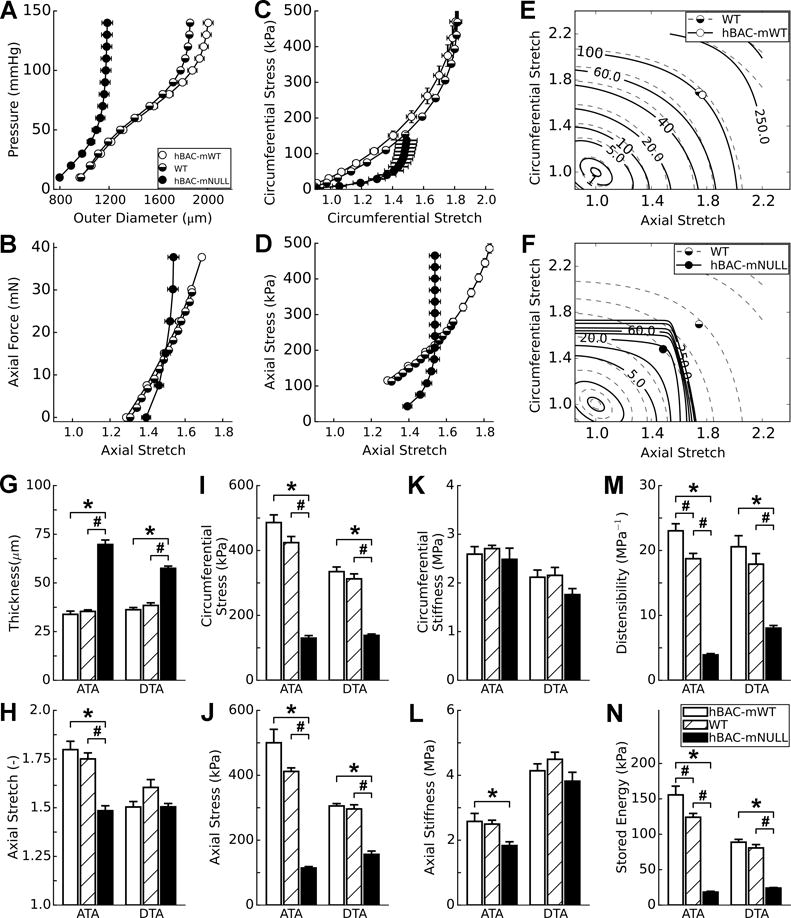
Biomechanical Phenotyping of Thoracic Aortas in hBAC-mWT (white bars), C57BL/6 WT (cross-hatched bars), and hBAC-mNULL (black bars) mice at 9 weeks of age. (A,B) Overall structural responses are revealed by cyclic pressure-diameter and axial force-stretch data whereas (C,D) basic material responses are revealed by stress-stretch data, both for the ascending aorta. (E,F) The overall mechanical functionality of the ascending aorta is revealed by isoenergetic contour plots (i.e., plots of equal energy storage for different pairs of circumferential (y-axis) and axial (x-axis) stretch), wherein symbols show systolic values at individual values of axial stretch. Shown, too, are comparisons at systolic pressure and individual axial stretches for the ascending (ATA) and descending (DTA) thoracic aorta in terms of (G) loaded wall thickness, (H) the in vivo value of axial stretch, circumferential (I) and axial (J) wall stress, circumferential (K) and axial (L) material stiffness, (M) distensibility, and (N) elastically stored energy during ex vivo loading. Data represent mean ± SEM; n = 5 (hBAC-mWT) to 7 (WT and hBAC-mNULL); *P < 0.05 vs. hBAC-mWT and #P < 0.05 vs. hBAC-mHET; one-way ANOVA.
Whereas Fig. 3A–F show results for the ascending aorta across the full range of ex vivo biaxial loading, panels G-N compare results for the ascending and descending segments at group specific values of systolic pressure and in vivo axial stretch. Plotted in this way, the data reveal qualitatively similar results in both segments for most of the 8 geometric or mechanical metrics considered, but quantitatively stronger differences in the ascending aorta due to elastin deficiency. Importantly, these data show that the circumferential material stiffness (an intrinsic property of the wall, reflecting its microscopic composition and architecture) is preserved in the ascending aorta and differs only slightly in the descending aorta across the three groups (Fig. 3K,L). In contrast, the structural stiffness (which results from both material stiffness and wall thickness) increases dramatically with elastic fiber deficiency, consistent with the markedly decreased distensibility in panel M, primarily due to the increased thickness (panel G). Together, the increased wall thickness and lower axial stretch (panel H) served to reduce wall stresses biaxially in hBAC-mNULL vessels (panels I,J), hence revealing a biomechanical maladaptation in partial elastin deficiency. That is, arteries are thought to mechano-adapt when they maintain wall stress and wall shear stresses at homeostatic target values [17]. Like its ascending counterpart, the descending aorta also exhibited decreased elastic energy storage with elastin deficiency (Fig. 3N), again indicating a reduced mechanical functionality. In summary, while the ex vivo estimates of decreased distensibility in the ascending aorta mirror those seen in vivo, biaxial testing reveals further that diminished axial stretch preferentially affects the ascending aorta and that stiffening arises from increased wall thickness, not fundamental changes in the intrinsic material properties (Supplemental Table II summarizes all of the biomechanical data in numerical form).
SMCs Associated with Partial Elastin Deficiency Exhibit Greater Contractile-to-Proliferative Phenotypic Modulation Under In Vitro than In Vivo Conditions
Since aortic SMCs from Eln−/− mice exhibit greater proliferative rates in vitro [18] and in vivo [10,11], we investigated the growth of cultured cells with partial elastin deficiency. SMCs were derived from enzymatically-digested thoracic aortas and analyzed after 1–2 passages in the presence of growth medium. Cells from hBAC-mNULL mice proliferated more rapidly than those from both hBAC-mHET and hBAC-mWT mice (Fig. 4A,B); increased DNA replication was confirmed by a higher rate of BrdU uptake over 2 hours (Fig. 4C,D). That the replication rate and BrdU uptake in cells from hBAC-mHET mice were intermediate between hBAC-mNULL and hBAC-mWT demonstrated a graded relation. We also examined the rate of SMC proliferation in vivo by labelling with BrdU over 10 days. Increased medial cell proliferation was noted in ascending aortas of 4.5 week old hBAC-mNULL but not hBAC-mHET mice (Fig. 4G,H). Similar differences of lesser magnitude were seen in descending segments that did not reach statistical significance. Since cross-sectional medial cell numbers did not differ among genotypes, it is likely that differing rates of SMC division contributed to the axial lengthening and tortuosity of the aorta in hBAC-mNULL mice (cf. Fig 1). We also characterized the contractile phenotype of the SMCs since cultured cells from Eln−/− mice demonstrate marked phenotypic de-differentiation [18]. The expression of smooth muscle α‐actin protein and several transcripts for contractile molecules was markedly reduced in SMCs associated with partial elastin deficiency in vitro but not in vivo (Fig. 4E,F and I,J). Thus, cell culture conditions reveal additional differences in SMC responses across genotypes that are not apparent in vivo, suggesting that even modest amounts of medial elastin maintains SMC differentiation.
Figure 4.
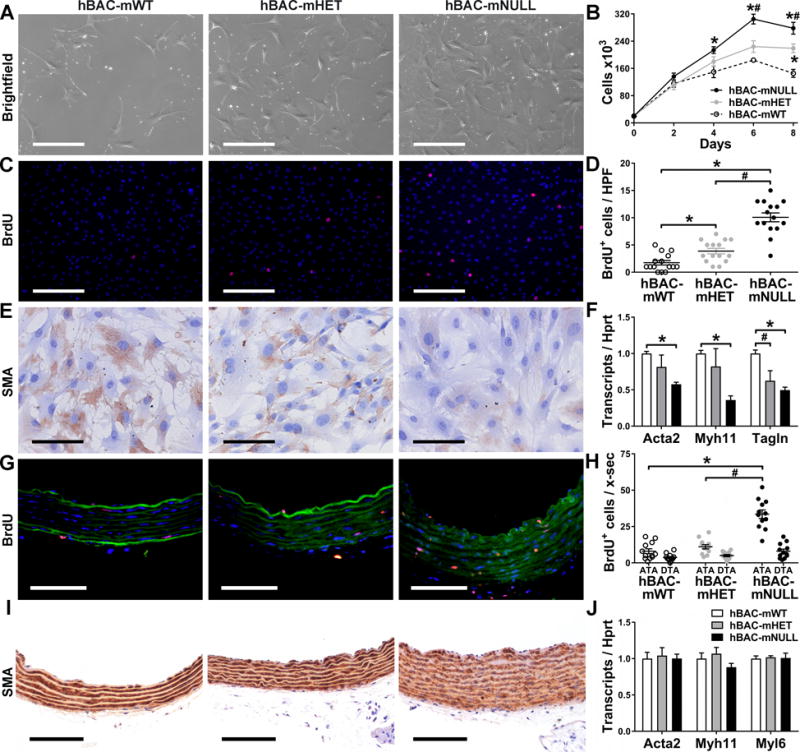
In Vitro and In Vivo Phenotypes of Aortic SMCs from hBAC-mWT (white symbols), hBAC-mHET (gray symbols), and hBAC-mNULL (black symbols) mice. SMCs were cultured from thoracic aortas of 9 week old mice. (A) Brightfield photomicrographs 2 days after plating at equal sub-confluent densities (bars: 400 μm). (B) Cell counts at 0–8 days after initial plating at 20 ×103 SMCs per well. (C) BrdU uptake (red color) by SMC nuclei (blue color) over 2 hours (bars: 400 μm) and (D) BrdU+ nuclei per high power field (HPF). (E) Smooth muscle α-actin (SMA) protein expression (brown color, bars: 100 μm). (F) Acta2, Myh11, and Tagln transcript expression relative to Hprt and normalized to mean of controls (i.e., hBAC-mWT). Thoracic aortas of 3 week old mice were also analyzed. (G) BrdU uptake (red color) by SMC nuclei (blue color) among elastic laminae (green autofluorescence) in ascending aortas after 10 days of BrdU treatment (bars: 75 μm) and (H) BrdU+ nuclei per cross-section (x-sec) of ascending (ATA) and descending (DTA) thoracic aortas. (I) SMA protein expression (brown color) of ascending aortas (bars: 100 μm). (J) Acta2, Myh11, and Myl6 transcript expression in thoracic aortas. Data represent mean ± SEM; n = 6 for cell counts in vitro, n = 15 for BrdU uptake in vitro, n = 4 for mRNA in vitro, n = 12 for BrdU uptake in vivo, and n = 6 for mRNA in vivo; *P < 0.05 vs. hBAC-mWT and #P < 0.05 vs. hBAC-mHET; one- or two-way ANOVA.
Small and Fibrotic Ascending Aorta without Evidence for Increased Medial Area or SMC Number in WS Subject with Subclinical Aortic Disease
We then studied clinical specimens to assess the relevance of our unexpected histomorphological findings in elastin-deficient mice. Aorta specimens from WS patients with mild or moderate disease are difficult to obtain as surgical repair is not indicated and postmortem examinations are rare. We nevertheless obtained the incomplete thoracic aorta from an adult WS subject with no history of cardiovascular disease who died suddenly of unknown cause. The ascending aorta had a diffusely smaller diameter but uniformly thicker media of similar cellularity compared to that of three age- and sex-matched subjects (Fig. 5A–F). Calculations based on aortic diameter, wall thickness, and cell density showed that the WS and referent individuals had comparable medial areas and number of SMCs per cross-section. Histological analyses of the aortas performed in a single batch allowed comparisons of ECM components among specimens. As previously noted [5,6], elastin was markedly decreased while collagen was markedly increased in the ascending aorta of the WS patient compared with the referent subjects (Fig. 5G–J). These ECM abnormalities were far less or not apparent in the descending aorta from the same WS patient, despite persistently smaller diameter and thickening of this segment (Supplemental Fig. VI). Comparatively, the phenotypic characteristics of this WS subject with typical modest aortic obstruction were similar to those seen in hBAC-mNULL mice, namely decreased aortic external diameter, thickened media without an increase in medial area or number of SMCs, medial fibrosis, and predilection of the ascending segment to the disease process. This example, counter to dogma, confirms that medial SMC hyperplasia need not contribute to the mild-moderate obstructive phenotype.
Figure 5. Analysis of Human WS and Referent Ascending Aortas.
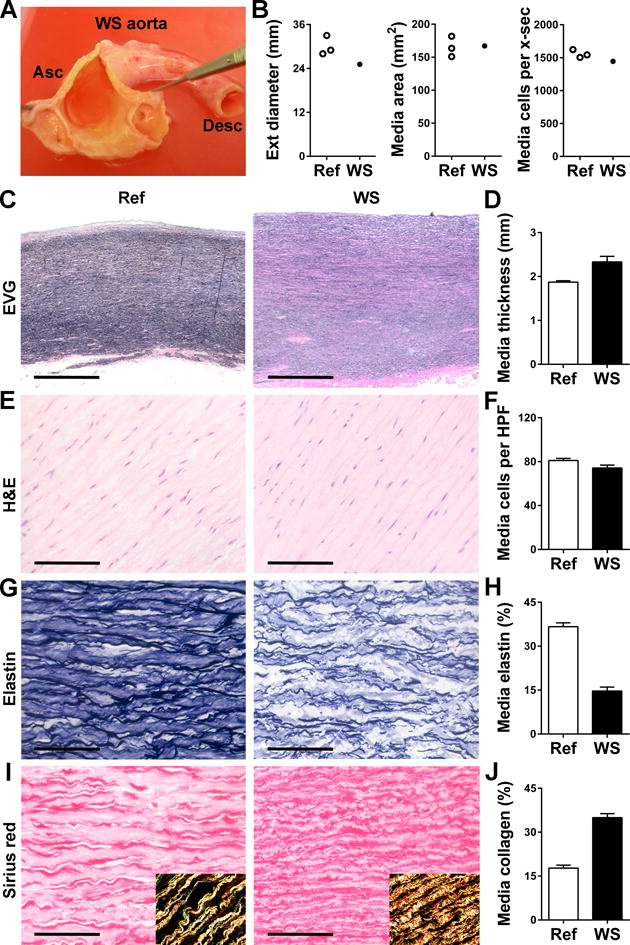
(A) Gross examination of the ascending (Asc) aorta from an adult with WS opened to reveal uniformly thickened wall without discrete supravalvular stenosis and rudimentary membranes at the sinotubular junction above the coronary ostia. (B) Measurements of external (ext) diameter, medial area, and total number of medial cells per cross-section (x-sec) in WS specimen and 3 referent (ref) ascending aortas from age- and sex-matched subjects. (C) Representative images of EVG-stained cross-sections (bars: 1 mm). (D) Medial thickness measured at 4 separate points. (E) Representative images of H&E stain (bars: 100 μm). (F) Medial cell density determined in 9 high power fields (HPF). (G) Representative images of elastin stain (bars: 100 μm). (H) Percent of media staining positive for elastin in 9 high power fields. (I) Representative images of sirius red stain (bars: 100 μm) with polarized light images in insets confirming collagen detection. (J) Percent of media staining positive for collagen in 9 high power fields. Data represent mean ± SEM; n = 4–9 replicates from 1 WS specimen vs. 3 referent ascending aortas.
Discussion
Our study reveals that deficient circumferential growth is the predominant cause of moderate, diffuse aortic obstruction in hBAC-mNULL mice without accompanying changes in medial area or cross-sectional SMC number. Morphological abnormalities are most evident in the ascending aorta and associate with a medial accumulation of collagen that preserves intrinsic material stiffness despite an overall decrease in aortic compliance due to medial thickening. Similar histopathological findings manifest in the ascending aorta of an adult WS patient with modest aortic obstructive disease.
The lack of medial expansion or increase in cross-sectional SMC number in thoracic aortas of hBAC-mNULL mice is unexpected. Previous studies of murine models of severe WS and clinical specimens from WS patients with severe disease, requiring surgery or resulting in postmortem gross and microscopic examinations, unequivocally demonstrate a thicker aortic wall, specifically, a thicker media containing more elastic laminae [5–11]. Since each elastic lamina is contiguous to a layer of SMCs, many assume that a thicker media contains more SMCs and that SMC proliferation plays an important role in the aortic narrowing in WS patients. Yet, prior studies of WS patient material have not quantified medial area or counted SMC numbers, and their conclusions have often been extrapolated to less severe disease typical of WS. In contrast, our data in hBAC-mNULL mice supports the contention that excessive SMC proliferation is not a significant contributor to moderate (likely early stage) stenosis of aortas with partial elastin deficiency (~30% expression levels). Instead, early increases in SMC proliferation may drive the observed lengthening of the aorta, particularly the ascending segment. Lengthening, rather than thickening, could arise despite a predominant circumferential orientation of SMCs if the daughter cells orient side-by-side following mitosis. Whereas switching of the SMC “mitotic axis” from circumferential to axial appears to be mechano-adaptive in response to increased longitudinal tension [19], the decreased axial stretch in the hBAC-mNULL ascending aorta was maladaptive in that biaxial wall stress and energy storage decreased well below normal levels. Off-loading of axial stress can also induce arterial tortuosity, with increased SMC proliferation balanced by apoptosis reported to maintain the number of cells per artery length unchanged [20]. A reduction in axial stress and increase in tortuosity is consistent with that observed in the hBAC-mNULL mice. That decreases in axial stretch were greatest in the ascending aorta where proliferation rates were highest suggests further the need to understand regional differences in proliferation, which have also been reported in an angiotensin II infusion model of hypertension in wild-type mice [21].
The prominent reduction in external ascending aortic diameter, with a thickened media, in hBAC-mNULL mice is similarly apparent in representative images of Eln−/− pups [10,11]. In contrast, diameter is only modestly decreased and the media not thickened in the aorta of hBAC-mHET and heterozygous Eln+/− mice [12,13]. Cross-sectional SMC number is similarly unchanged in hBAC-mHET mice (~80% elastin expression) and although SMC numbers and medial area were not calculated in studies of Eln+/− mice (50% elastin expression), its smaller aorta with a thinner media is unlikely to contain more cells [12,13]. The adult WS patient that we studied had not been diagnosed with aortic obstruction, but the ascending aorta has a diameter over 2 standard deviations less than the population mean [22] and a media thicker than any comparable age- and sex-matched control [23]. Similar to findings in the hBAC-mNULL mice, neither SMC density nor medial area of this elastin-deficient ascending aorta is greater than in the three referent specimens. Even though we did not enumerate SMCs in the axial direction to account for differences in medial volume, this case of subclinical disease demonstrates that increased cross-sectional number of SMCs is not characteristic of all WS aortas. This finding is consistent with physical arguments. Since the diameter of the aorta is over 20x the thickness of the media in wild-type mice and humans [23], a progressively greater proportional change in medial thickness, with inward growth, would be needed to predominate over decreases in aortic diameter as a determinant of lumen loss (cf. Supplemental Appendix I and Fig. VII).
A unique aspect of our analysis is the comparison of multiple potential determinants of lumen loss to assess their significance at several times during postnatal development, thus distinguishing deficient circumferential growth from inward remodeling of mature vessels. Our data reveal that a smaller external diameter is the predominant mechanism for luminal stenosis in hBAC-mNULL mice and in a WS patient with moderate aortic obstructive disease. Clinical imaging confirms the diameter of the ascending aorta is uniformly smaller in WS [3], and its growth is restricted in a subset of pediatric patients who underwent serial examination [24,25]. Vessel size is also a relevant factor in the assessment of medial thickness as a determinant of lumen loss. For instance, if an aorta fails to grow in diameter, or shrinks without any change in mass, the wall will necessarily thicken without an increase in SMCs or medial area. Failure of outward growth implies that relative thickening of the media encroaches inward and narrows the lumen. An alternative interpretation is that a thicker media in a smaller vessel does not represent proportional vascular remodeling and that the medial area and number of SMCs are excessive even if not increased compared to larger control aortas. However, a hypertrophic vascular remodeling program is not predicted from a mechanical perspective. Circumferential and axial wall stresses are lower in hBAC-mNULL mice than in controls as blood pressure by tail-cuff is unchanged, aortic diameter is reduced, wall thickness is increased, and the axial stretch less. This suggests that the vessel wall does not grow or remodel appropriately because of the disruption in elastin architecture [26]. Even if medial area and SMC numbers are inappropriately high, the degree of medial thickening in the hBAC-mNULL mice is a minor factor for lumen loss compared to decreased aortic diameter. There are other examples of changes in vessel size confounding the interpretation of changes in medial thickness during disease pathogenesis. Acquired aneurysms of the ascending aorta have an opposite phenotype to that of WS, namely, increased aortic diameter and decreased medial thickness. This was assumed by many to indicate a loss of SMCs in the development of aneurysms. Histomorphological analysis demonstrates, however, that medial area and SMC numbers are often increased on account of the larger aortic diameter [23]. Thus, a full assessment of aortic disease requires analysis of the complete vessel, not an isolated portion. In addition to absolute measurements of aortic dimensions, studies should ideally index aorta diameter to body size, particularly as individuals with WS are characterized by short stature. Another approach is to reference the diseased aorta to a relatively non-diseased segment, the aortic root in the case of WS [3].
Besides changes in vessel wall compartments and vessel size, loss of aortic compliance may adversely impact hemodynamics. Aortic stiffening in hBAC-mNULL mice was initially documented ex vivo by reduced distensibility with associated central hypertension [16]. In contrast, peripheral blood pressure was not statistically higher in our hBAC-mNULL mice, although all genotypes displayed mild hypertension (mean blood pressure 106-113 mmHg) and tail-cuff values do not always reflect invasive central measurements. In keeping with similar hemodynamics among genotypes, we did not find differences in cardiac mass or limited early survival as described for the originating colony [16]. These discrepancies may relate to genetic drift of our founder animals or our further back-breeding to the C57BL/6 background. We also did not monitor animals older than 9 weeks of age to determine if hypertension or cardiac hypertrophy subsequently develops. We did confirm structural stiffening in vivo and ex vivo, with the added finding of nearly preserved material stiffness. Aortic stiffening, without or with hypertension, is well documented in children and adults with WS, although there is considerable variability depending on the technique and vessel examined. Echocardiographic analysis of the ascending aorta demonstrates decreased compliance in all WS patients compared with controls [4]. In contrast, measurements of arterial stiffening in terms of pulse wave velocity from the carotid artery through the descending thoracic and abdominal aorta to femoral artery, a path that does not include the ascending aorta, found substantial overlap among WS patients and controls, albeit with a significant difference in means between the two groups [27]. Studies of arteries other than the aorta (e.g., carotids) have found normal or even paradoxically reduced arterial stiffness in WS [28,29]. We suggest that the most affected segment (i.e., ascending aorta in WS) should be included in diagnostic measures of arterial stiffening. Although collagen is increased in ascending aortas of hBAC-mNULL mice, material stiffness is preserved on ex vivo analysis. These findings indicate that collagen accumulation compensates for elastin deficiency and that decreased aortic distensibility results from increased structural stiffness (essentially thickness times material stiffness). Our histomorphological analyses of the ECM delineate media from adventitia, the latter being the site of greatest collagen concentration within the vessel wall which could mask local differences. An overall increased level of collagen was previously reported in ascending aortas of the same mouse strain based on biochemical techniques [16]. The reported difference of 20% in hydroxyproline content of whole aortas is less than the 100% difference we detected in medial collagen levels by histology, possibly due to a dominant adventitial contribution that changed little. Additionally, our findings of similar levels of Col1a1 and Col3a1 transcripts on qRT-PCR analysis of whole thoracic aortas may reflect dominant synthesis by adventitial fibroblasts. Induction of Col8a1 and Col11a1 transcripts, previously associated with vascular remodeling [30,31], is perhaps readily detectable with a global analytical technique due to low basal expression. Altered expression of particular collagens may impact the transduction of mechanical stimuli from ECM to SMCs and result in disordered growth of the aorta in keeping with recent observations that integrin signaling is necessary for luminal stenosis in Eln−/− mice [32].
Our observations, together with reported phenotypes in the literature, suggest that the relationship between aortic abnormalities and elastin expression is not linear (Table 1). Instead, phenotypic differentiation may require a threshold level of elastin deficiency and then changes monotonically with further loss of elastin. Gene interaction effects could modify decreased elastin content resulting from haploinsufficiency and additional losses may accrue from proteolytic degradation or damage by mechanical fatigue. This hypothesis provides new insight into the variable aortic obstruction phenotype of WS, but also generates several unanswered questions that warrant investigation in future studies (cf. Supplemental Appendix II). Implications for therapy are that anti-proliferative pharmacological agents may not be successful in reversing the underlying pathology of decreased aortic external diameter and mechanisms for deficient aortic growth in conditions of elastin deficiency must be elucidated to identify new treatment strategies. The progressive maladaptive remodeling attributable to elastin deficiency also suggests that the therapeutic target evolves, which may be an underappreciated principle important for personalized medicine and requiring assessments of where in the natural history a particular patient is.
Table 1.
Distinct Aortic Pathology Manifests at Different Grades of Elastin Deficiency
| Eln−/− mice | hBAC-mNULL mice | Eln+/− mice | hBAC-mHET mice | |
|---|---|---|---|---|
| Elastin expression | 0% | ~30% | ~50% | ~80% |
| Lumen area | Markedly decreased | Moderately decreased | Mildly decreased | Mildly decreased |
| External diameter | NQ, appears decreased | Moderately decreased | Mildly decreased | Mildly decreased |
| Distensibility | NQ, appears decreased | Moderately decreased | Mildly decreased | Unchanged |
| Media thickness | NQ, appears increased | Moderately increased | Mildly decreased | Unchanged |
| Media area | Markedly increased | Unchanged | Mildly decreased | Unchanged |
| SMC number | Markedly increased | Unchanged | NQ, likely decreased | Unchanged |
| SMC differentiation | Moderately less | Unchanged | NQ, likely unchanged | Unchanged |
| References | 10, 11, 32 | this study, 16 | 7, 10, 12, 13, 16 | this study, 16 |
The mechanisms of obstructive aortopathy in WS have been explored in various murine models of graded elastin deficiency. Lumen loss of the aorta may result from increased media thickness relative to aortic diameter, inward medial expansion driven by SMC hyperplasia, decreased circumferential growth, and reduced compliance. Decreased lumen area and reduced external diameter is common to all grades of elastin deficiency. Reduced distensibility associates with elastin expression ≤ 50%, increased medial thickness characterizes both moderate and severe elastin deficiency, whereas medial expansion, SMC hyperplasia, and SMC dedifferentiation are restricted to severe elastin deficiency alone. Of note, media thickness is mildly increased in haploinsufficient DD mice [15], and diminished compliance is evident in hBAC-mHET aortas at supraphysiological pressures [16]. NQ = not quantified, though conclusions are based on representative images or extrapolation from related variables.
Supplementary Material
Highlights.
Moderate aortic obstruction in mice with partial elastin deficiency and in a Williams syndrome subject with subclinical disease results primarily from deficient circumferential growth of the aorta, not increased medial area or cross-sectional SMC number.
Aberrant ECM expression, SMC anomalies, and pathological vascular remodeling are more evident in the ascending than descending aorta and under in vitro than in vivo conditions.
Excessive SMC proliferation causing obstructive lesions in WS may require additional loss of elastin superimposed on ELN haploinsufficiency.
Acknowledgments
The authors thank Dr. Barbara Pober for helpful scientific discussions and critical reading of the manuscript.
Funding Sources
This work was supported by the Kiev Foundation, the Williams Syndrome Association (G.T.), and the NIH: HL105297 and HL086418 (J.D.H.) and HL105314 and HL053325 (R.P.M.).
Abbreviations
- ECM
extracellular matrix
- hBAC
bacterial artificial chromosome encoding human ELN
- mHET
heterozygous for murine Eln
- mNULL
null for murine Eln
- mWT
wild-type for murine Eln
- SMC
smooth muscle cell
- WS
Williams syndrome
Footnotes
Disclosures
There are no conflicts of interest.
References
- 1.Pober BR, Johnson M, Urban Z. Mechanisms and treatment of cardiovascular disease in Williams-Beuren syndrome. J Clin Invest. 2008;118:1606–15. doi: 10.1172/JCI35309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams JC, Barratt-Boyes BG, Lowe JB. Supravalvular aortic stenosis. Circulation. 1961;24:1311–8. doi: 10.1161/01.cir.24.6.1311. [DOI] [PubMed] [Google Scholar]
- 3.Hallidie-Smith KA, Karas S. Cardiac anomalies in Williams-Beuren syndrome. Arch Dis Child. 1988;63:809–13. doi: 10.1136/adc.63.7.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salaymeh KJ, Banerjee A. Evaluation of arterial stiffness in children with Williams syndrome: Does it play a role in evolving hypertension? Am Heart J. 2001;142:549–55. doi: 10.1067/mhj.2001.116763. [DOI] [PubMed] [Google Scholar]
- 5.O’Connor WN, Davis JB, Jr, Geissler R, Cottrill CM, Noonan JA, Todd EP. Supravalvular aortic stenosis. Clinical and pathologic observations in six patients. Arch Pathol Lab Med. 1985;109:179–85. [PubMed] [Google Scholar]
- 6.van Son JA, Edwards WD, Danielson GK. Pathology of coronary arteries, myocardium, and great arteries in supravalvular aortic stenosis. Report of five cases with implications for surgical treatment. J Thorac Cardiovasc Surg. 1994;108:21–8. [PubMed] [Google Scholar]
- 7.Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest. 1998;102:1783–7. doi: 10.1172/JCI4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dridi SM, Foucault Bertaud A, Igondjo Tchen S, Senni K, Ejeil AL, Pellat B, Lyonnet S, Bonnet D, Charpiot P, Godeau G. Vascular wall remodeling in patients with supravalvular aortic stenosis and Williams Beuren syndrome. J Vasc Res. 2005;42:190–201. doi: 10.1159/000085141. [DOI] [PubMed] [Google Scholar]
- 9.Urbán Z, Riazi S, Seidl TL, Katahira J, Smoot LB, Chitayat D, Boyd CD, Hinek A. Connection between elastin haploinsufficiency and increased cell proliferation in patients with supravalvular aortic stenosis and Williams-Beuren syndrome. Am J Hum Genet. 2002;71:30–44. doi: 10.1086/341035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W, Li Q, Qin L, Ali R, Qyang Y, Tassabehji M, Pober BR, Sessa WC, Giordano FJ, Tellides G. Rapamycin inhibits smooth muscle cell proliferation and obstructive arteriopathy attributable to elastin deficiency. Arterioscler Thromb Vasc Biol. 2013;33:1028–35. doi: 10.1161/ATVBAHA.112.300407. [DOI] [PubMed] [Google Scholar]
- 11.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–80. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 12.Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, Minkes RK, Blumer KJ, Kovacs A, Kelly DP, Li DY, Starcher B, Mecham RP. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest. 2003;112:1419–28. doi: 10.1172/JCI19028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagenseil JE, Nerurkar NL, Knutsen RH, Okamoto RJ, Li DY, Mecham RP. Effects of elastin haploinsufficiency on the mechanical behavior of mouse arteries. Am J Physiol Heart Circ Physiol. 2005;289:H1209–17. doi: 10.1152/ajpheart.00046.2005. [DOI] [PubMed] [Google Scholar]
- 14.Goergen CJ, Li HH, Francke U, Taylor CA. Induced chromosome deletion in a Williams-Beuren syndrome mouse model causes cardiovascular abnormalities. J Vasc Res. 2011;48:119–29. doi: 10.1159/000316808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campuzano V, Segura-Puimedon M, Terrado V, Sánchez-Rodríguez C, Coustets M, Menacho-Márquez M, Nevado J, Bustelo XR, Francke U, Pérez-Jurado LA. Reduction of NADPH-oxidase activity ameliorates the cardiovascular phenotype in a mouse model of Williams-Beuren Syndrome. PLoS Genet. 2012;8:e1002458. doi: 10.1371/journal.pgen.1002458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirano E, Knutsen RH, Sugitani H, Ciliberto CH, Mecham RP. Functional rescue of elastin insufficiency in mice by the human elastin gene: implications for mouse models of human disease. Circ Res. 2007;101:523–31. doi: 10.1161/CIRCRESAHA.107.153510. [DOI] [PubMed] [Google Scholar]
- 17.Ferruzzi J, Bersi MR, Humphrey JD. Biomechanical phenotyping of central arteries in health and disease: Advantages of and methods for murine models. Annl Biomed Eng. 2013;41:1311–30. doi: 10.1007/s10439-013-0799-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, Li DY. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130:411–23. doi: 10.1242/dev.00223. [DOI] [PubMed] [Google Scholar]
- 19.Dajnowiec D, Sabatini PJ, Van Rossum TC, Lam JT, Zhang M, Kapus A, Langille BL. Force-induced polarized mitosis of endothelial and smooth muscle cells in arterial remodeling. Hypertension. 2007;50:255–60. doi: 10.1161/HYPERTENSIONAHA.107.089730. [DOI] [PubMed] [Google Scholar]
- 20.Jackson ZS, Dajnowiec D, Gotlieb AI, Langille BL. Partial off-loading of longitudinal tension induces arterial tortuosity. Arterioscler Thromb Vasc Biol. 2005;25:957–62. doi: 10.1161/01.ATV.0000161277.46464.11. [DOI] [PubMed] [Google Scholar]
- 21.Owens AP, 3rd, Subramanian V, Moorleghen JJ, Guo Z, McNamara CA, Cassis LA, Daugherty A. Angiotensin II induces a region-specific hyperplasia of the ascending aorta through regulation of inhibitor of differentiation 3. Circ Res. 2010;106:611–9. doi: 10.1161/CIRCRESAHA.109.212837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolak A, Gransar H, Thomson LE, Friedman JD, Hachamovitch R, Gutstein A, Shaw LJ, Polk D, Wong ND, Saouaf R, Hayes SW, Rozanski A, Slomka PJ, Germano G, Berman DS. Aortic size assessment by noncontrast cardiac computed tomography: normal limits by age, gender, and body surface area. JACC Cardiovasc Imaging. 2008;1:200–9. doi: 10.1016/j.jcmg.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Tang PC, Coady MA, Lovoulos C, Dardik A, Aslan M, Elefteriades JA, Tellides G. Hyperplastic cellular remodeling of the media in ascending thoracic aortic aneurysms. Circulation. 2005;112:1098–105. doi: 10.1161/CIRCULATIONAHA.104.511717. [DOI] [PubMed] [Google Scholar]
- 24.Wren C, Oslizlok P, Bull C. Natural history of supravalvular aortic stenosis and pulmonary artery stenosis. J Am Coll Cardiol. 1990;15:1625–30. doi: 10.1016/0735-1097(90)92837-r. [DOI] [PubMed] [Google Scholar]
- 25.Radford DJ, Pohlner PG. The middle aortic syndrome: an important feature of Williams’ syndrome. Cardiol Young. 2000;10:597–602. doi: 10.1017/s1047951100008878. [DOI] [PubMed] [Google Scholar]
- 26.Humphrey JD, Milewicz DM, Tellides G, Schwartz MA. Cell biology. Dysfunctional mechanosensing in aneurysms. Science. 2014;344:477–9. doi: 10.1126/science.1253026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kozel BA, Danback JR, Waxler JL, Knutsen RH, de Las Fuentes L, Reusz GS, Kis E, Bhatt AB, Pober BR. Williams syndrome predisposes to vascular stiffness modified by antihypertensive use and copy number changes in NCF1. Hypertension. 2014;63:74–9. doi: 10.1161/HYPERTENSIONAHA.113.02087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aggoun Y, Sidi D, Levy BI, Lyonnet S, Kachaner J, Bonnet D. Mechanical properties of the common carotid artery in Williams syndrome. Heart. 2000;84:290–3. doi: 10.1136/heart.84.3.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lacolley P, Boutouyrie P, Glukhova M, Daniel Lamaziere JM, Plouin PF, Bruneval P, Vuong P, Corvol P, Laurent S. Disruption of the elastin gene in adult Williams syndrome is accompanied by a paradoxical reduction in arterial stiffness. Clin Sci (Lond) 2002;103:21–9. doi: 10.1042/cs1030021. [DOI] [PubMed] [Google Scholar]
- 30.Lopes J, Adiguzel E, Gu S, Liu SL, Hou G, Heximer S, Assoian RK, Bendeck MP. Type VIII collagen mediates vessel wall remodeling after arterial injury and fibrous cap formation in atherosclerosis. Am J Pathol. 2013;182:2241–53. doi: 10.1016/j.ajpath.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhasin M, Huang Z, Pradhan-Nabzdyk L, Malek JY, LoGerfo PJ, Contreras M, Guthrie P, Csizmadia E, Andersen N, Kocher O, Ferran C, LoGerfo FW. Temporal network based analysis of cell specific vein graft transcriptome defines key pathways and hub genes in implantation injury. PLoS One. 2012;7:e39123. doi: 10.1371/journal.pone.0039123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Misra A, Sheikh AQ, Kumar A, Luo J, Zhang J, Hinton RB, Smoot L, Kaplan P, Urban Z, Qyang Y, Tellides G, Greif DM. Integrin β3 inhibition is a therapeutic strategy for supravalvular aortic stenosis. J Exp Med. 2016;213:451–63. doi: 10.1084/jem.20150688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.