

Abstract
Background
The ability to correlate plastic changes in synaptic physiology with changes in synaptic anatomy has been very limited in the central nervous system because of shortcomings in existing methods for recording the activity of specific CNS synapses and then identifying and studying the same individual synapses on an anatomical level.
New method
We introduce here a novel approach that combines two existing methods: paired neuron electrophysiological recording and array tomography, allowing for the detailed molecular and anatomical study of synapses with known physiological properties.
Results
The complete mapping of a neuronal pair allows determining the exact number of synapses in the pair and their location. We have found that the majority of close appositions between the presynaptic axon and the postsynaptic dendrite in the pair contain synaptic specializations. The average release probability of the synapses between the two neurons in the pair is low, below 0.2, consistent with previous studies of these connections. Other questions, such as receptor distribution within synapses, can be addressed more efficiently by identifying only a subset of synapses using targeted partial reconstructions. In addition, time sensitive events can be captured with fast chemical fixation.
Comparison with existing methods
Compared to existing methods, the present approach is the only one that can provide detailed molecular and anatomical information of electrophysiologically-characterized individual synapses.
Conclusions
This method will allow for addressing specific questions about the properties of identified CNS synapses, even when they are buried within a cloud of millions of other brain circuit elements.
Keywords: Synapses, Synaptic transmission, Hippocampus, Array tomography, Paired recording
1. Introduction
Numerous attempts have been made over the years at developing methods to correlate the physiology and plasticity of synapses with their anatomy. While these attempts have allowed us to learn much about the relationship between synaptic physiology and synaptic anatomy, gaining a complete and comprehensive understanding has been elusive. This question has been particularly hard to address in the central nervous system of vertebrates because of the difficulty of recording the physiology of individual synapses and then finding those very same synapses within the cloud of millions of neighboring synapses in order to perform an anatomical analysis. One successful approach is to follow electrophysiological recordings from neuronal pairs by electron microscopy; such studies have revealed that the number of synaptic connections and their probabilities of release vary greatly depending on neuron type (reviewed in: Branco and Staras, 2009) (Buhl et al., 1997; Silver et al., 2003; Tamas et al., 1997; Deuchars and Thomson, 1995; Biro et al., 2006; Gulyas et al., 1993). However, electron microscopic reconstructions are very time-consuming, do not allow the investigation of large numbers of neuronal pairs, and, most importantly, cannot be used to study the molecular composition of the synapses in a pair. In this paper, we describe the development of a method combining paired-cell electrophysiological recording and array tomography that enables us to determine the physiological properties of a small population of synapses and then to study the anatomical and molecular characteristics of that same synapse population.
Simultaneous electrophysiology recordings from two synaptically-connected neurons have been used to study the detailed physiological properties of synaptic connections for a number of years (Pavlidis and Madison, 1999; Debanne et al., 1998; Malinow, 1991; Emond et al., 2010; Fourie et al., 2014; Montgomery and Madison, 2002; Montgomery and Madison, 2004). In this so-called ‘cell pair recording’, a current pulse-induced action potential travels down the axon of the presynaptic cell of the pair, and activates neurotransmitter release, inducing a synaptic current in the postsynaptic neuron of the pair. The main advantage of cell-pair recording is that it uniquely produces the minimal action potential-evoked synaptic response that can be unambiguously attributed to specific synapses: only those made between these individual pre- and postsynaptic neurons. Other types of minimal synaptic responses that have been employed in the past, are either not action potential evoked (Chen et al., 2004), or cannot be unambiguously attributed to specific members of the synapse population (e.g., “spontaneous” or “minimally stimulated”) (Dobrunz and Stevens, 1997; Choi et al., 2000). With cell-pair recording however, it is known that the recorded responses are action potential-dependent, and that they arise from only the synapses made from one member of the cell pair to the other. A post-hoc analysis of those synapses with array tomography adds the ability to determine anatomical and molecular properties of the physiologically-characterized synapses. The main advantage of this combination of techniques is, thus, that function and structure can be directly correlated at the very same synapses.
In this paper, we describe the methodology of using array tomography to reconstruct neurons from pair recordings, locate points of contact between axons and dendrites, verify their identity as synapses using the presence of antibody markers for synaptic proteins, and visualize the distribution of receptor subunits. The technique as we have developed it can be used to image an entire filled pair of neurons, and thus to find all of the synapses between those two neurons, or to image just part of the interaction field of those neurons to sample the properties of synapses within that field, but limited to synapses made between those two neurons.
2. Materials and methods
2.1. Preparation of and recording from organotypic slices
Hippocampal organotypic slice cultures were prepared from 5 day old C57BL/6 mice or from P7 Sprague-Dawley rats, and maintained in vitro on a plastic membrane (Millicell culture insert) for 6–10 days before whole cell recordings were performed, as previously described (Stoppini et al., 1991). The physiological recording and array tomographic methods are identical when using rats or mice, with the only significant difference being that it is easier to fully reconstruct a cell pair from a mouse because the mouse neurons have smaller dendritic arbors. To prepare a cultured slice for recording, a patch of the plastic membrane containing a slice was carefully cut out of the insert, using the point of a scalpel. The slice, sitting on top of its membrane, was immersed in ACSF containing (mM): 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1 Na2HPO4, 26.2 NaHCO3 and 11 glucose, and perfused at a rate of 2 mL/min. Recordings were made at room temperature (~21 °C). Pyramidal neurons in area CA3 were visualized with differential interference contrast microscopy (DIC) under infrared illumination for simultaneous whole cell recordings from two neurons. The use of infrared illumination improves the ability to visualize the live neurons, but the images must be viewed with a camera that can detect infrared (most commercial CCD cameras will detect infrared, as long as the filter that is commonly factory-installed in front of the lens is absent or removed). Presynaptic neurons were recorded in current clamp in whole-cell electrode configuration, using an internal solution consisting of (mM): 120 K gluconate, 40 HEPES, 5 MgCl2, 0.3 NaGTP, 2 NaATP, and 5 QX-314; pH adjusted to 7.2 with KOH. Postsynaptic neurons were also recorded in whole-cell mode, in voltage clamp with an internal solution consisting of (mM): 120 Cs gluconate, 40 HEPES, 5 MgCl2, 0.3 NaGTP, 2 NaATP, and 5 QX-314, with pH adjust to 7.2 with CsOH. To mark the neurons for later array tomography, either 0.2% Lucifer yellow (Sigma–Aldrich) or 0.1% Alexa 594 hydrazide and 0.5% neurobiotin (Vector Laboratories) was included in the internal solution. The Alexa 594 is present to allow us to see the neuron in the resin block (as in Figs. 1C or 4A), which facilitates block trimming. In earlier experiments, we put neurobiotin in the presynaptic and Lucifer yellow in the postsynaptic electrode, but later inverted this because we found that Lucifer yellow better filled the axonal processes. Neurons were recorded for 25 min before electrodes were carefully withdrawn to allow time to fill with the markers. The slice remained in the recording chamber for another 15 min before fixation.
Fig. 1.
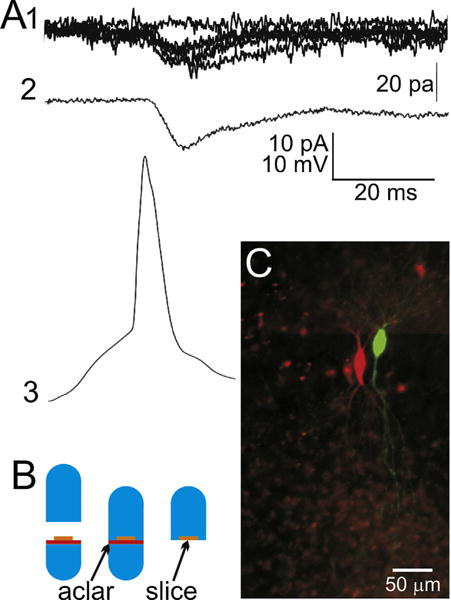
Recording of a neuron pair and preparation for array tomography. (A1) Raw individual excitatory postsynaptic currents, evoked by a presynaptic action potential (A3). Shown are 5 superimposed EPSCs plus one ‘failure’ trace for reference. (A2) An action potential-evoked EPSC that is the average of 100 consecutive non-failure trials (116 consecutive trials, 100 with successful transmission and 16 synaptic failures). (A3) An exemplar presynaptic action potential from the presynaptic neuron of this pair, temporally aligned with its postsynaptic responses. During the recording, the two cells are filled with either neurobiotin or Lucifer yellow by injection through the whole-cell recording electrode. (B) An illustration of the procedure for keeping the slice flat during embedding. A cured LRWhite resin stub (bright blue) is placed in the lower half of a gelatin capsule. An Aclar plastic punch (red) is placed on top of the resin stub, and the fixed slice (orange) is placed on top of that. The lower half of the gelatin capsule is then filled up with unpolymerized LR White resin (light blue). The top half of the capsule is used to close the capsule which is left to polymerize at 55 °C for 24 h. Once cured, the gelatin capsule is peeled off the polymerized resin block, which can then be easily broken off at the Aclar border, leaving the flat slice on the surface of the block. (C) A micro-photograph of the two cells on the gelatin capsule block face. This visibility of the cells allows for the accurate trimming of the block face in preparation for ultramicrotomy. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 4.
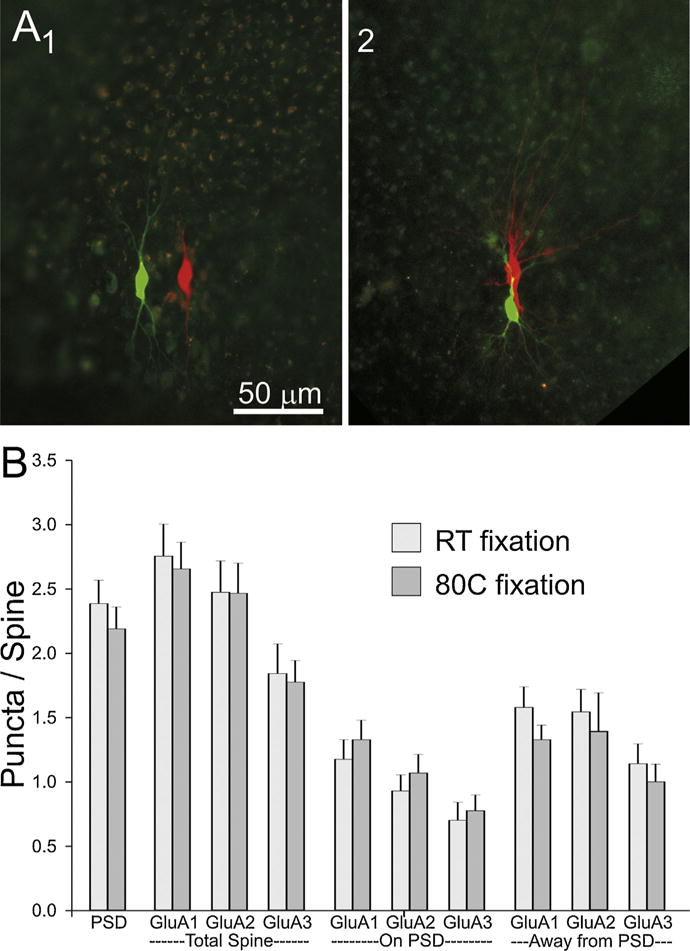
Comparison of cell pairs at two different fixation temperatures. (A) Low magnification photographs of slices fixed overnight at room temperature (1) or for 2 min at 80 °C (2). (B) A comparison of the distribution of PSD95 and AMPA receptor subunits between tissue fixed at room temperature and at 80 °C. “PSD” number of sections in which staining for PSD95 appears in an average spine. “GluAx” indicates the average number of puncta/spine. Location of GluAx puncta are broken down by content of the whole (total) spine or by whether it overlaps with the PSD95 punctum or is found in the portion of the spine away from the PSD95 punctum.
2.2. Fixation and resin embedding
Slices were removed from the recording chamber by holding the edge of the millicell membrane with a pair of Dumont forceps. With the slice still on the membrane, it was placed in a solution of 4% formaldehyde (diluted from 8% formaldehyde, EM grade, Electron Microscopy Supplies) with 2.5% sucrose in PBS in a scintillation vial. To speed tissue fixation, the slice was microwaved in a PELCO BioWave Pro microwave equipped with a PELCO ColdSpot preset to 12 °C (Micheva et al., 2010a). The procedure was to irradiate the tissue samples once at 100–150 W 1 min on/1 min off/1 min on, and then 3 times at 350–400 W 20 s on/20 s off/20 s. The fixed slice was then left in fixative at room temperature for one hour, followed by a wash in PBS buffer. The slice was then carefully teased away from the plastic membrane using the tip of a 3/0 White Sable brand nylon artist’s paintbrush. The CA3 region was cut from the slice under a dissection microscope using a #9 single edge razor blade, and then dehydrated serially in washes of 50%, 70%, 95% and 100% ethanol, each time microwaved at 350 W for 30 s. The dehydrated tissue was then infiltrated with LR White resin, medium grade (SPI Supplies), first with a mixture of ethanol and LR White (1:1) and then in three changes of 100% LR White, microwaved at 350 W for 30 s each time (Micheva et al., 2010a). The slice was then left in unpolimerized LR White resin overnight at 4 °C.
To ensure that the slice did not curl during embedding, a short stub of polymerized LR White resin was put into the bottom part of a 00 gelatin capsule to provide a flat surface. The resin stub was covered by a circle of Aclar plastic (Ted Pella), over which the slice was positioned, and then unpolymerized LR White resin was slowly added to fill the bottom part of the capsule. The top part was used to close the gelatin capsule, which was then polymerized at 55 °C for 24 h. After polymerization, the gelatin capsule was peeled off and the supporting resin stub snapped off at the Aclar border, leaving the flat polymerized slice on top of the remaining resin block (Fig. 1B) (Palmieri and Kiss, 2005; Migheli and Attanasio, 1991).
2.3. Sectioning and staining
For sectioning and staining, the face of the block was trimmed around the tissue with a razor blade to a trapezoid shape approximately 2 mm wide and 1 mm high. Ultrathin serial sections (100–200 nm) were cut using a Leica EM UC6 ultra microtome with a DiATOME Histo Jumbo knife and mounted on subbed coverslips (Micheva et al., 2010b). As the sections come off the microtome blade, they adhere edge to edge, forming a ribbon that maintains their serial order. These ribbons generally reach about 30 sections in length. The entire biological slice will typically be contained within about 20 of these ribbons (or approximately 600 sections), with the structure of the pair of neurons spanning 6 or 7 ribbons.
Staining and elution of the arrays was done according to the methods described in Micheva et al. (2010c). The sections were encircled with a PAP pen (Vector Laboratories), treated with a 50 mM glycine solution in Tris-buffered saline for 5 min and then treated with blocking buffer (0.1% BSA and 0.05% Tween in Tris-buffered saline) for 5 min. Solutions of rabbit anti-Lucifer yellow (1:200, Invitrogen A5750) in blocking buffer were prepared and centrifuged at 13,000 RPM for 2 min and applied to the sections overnight at 4 °C in a humidified 100 mm Petri dish. The sections were rinsed with Tris-buffered saline 3 times at 5 min each. A solution of secondary goat anti-rabbit antibody conjugated to Alexa 488 (1:150, Invitrogen A11034) and streptavidin-Alexa 594 (1:100, Invitrogen S11227) in blocking buffer was prepared and centrifuged at 13,000 RPM for 2 min. The rinsed sections were treated with this secondary antibody and streptavidin solution for 30 min at room temperature. The sections were then rinsed 3 times with Tris-buffered saline 5 min each and finally with distilled water. The sections were mounted on glass slides with SlowFade Gold Antifade Reagent with DAPI (Invitrogen) for imaging. After the sections were imaged, the antibodies were eluted to prepare for subsequent immunostaining with a different set of antibodies. For elution the mounting medium was washed away with dH2O and the sections were treated with a solution of 0.2 M NaOH and 0.02% SDS in water for 20 min. The coverslips were rinsed twice with Tris buffer 10 min each and then with distilled water. The coverslips were left at room temperature to dry, and then placed on a hot plate at 60 °C for 30 min. Subsequent rounds of antibody staining and elution were by the same methods described above.
2.4. Acquiring and aligning digital images of arrays
The mounted sections were scanned on a Zeiss Axio Imager.Z1 upright fluorescence microscope with a motorized stage and an Axiocam HR Digital Camera (Zeiss). A tiled mosaic image of all of the sections was obtained with a 10× objective using the MosaiX module of the Axiovision software (Zeiss). The MosaicPlanner software, written by Forrest Collman in the laboratory of Stephen Smith at Stanford (now at the Allen Brain Institute), was used to identify and mark the x–y coordinates of processes of the two filled neurons. The MosaicPlanner software is available for download at https://code.google.com/p/smithlabsoftware/. For high resolution images, a 5 × 8 tiled mosaic was taken at each section using these x,y coordinates with a 63×/1.4 NA Plan Apochromat objective with the MosaiX module for Axiovision. The mosaic images generated were digitally stitched together using the Axiovision MosaiX plugin using the DAPI as the reference channel. The stitched images were exported as TIFF files then aligned with ImageJ (NIH) using a custom plugin written by Forrest Collman, using the DAPI channel as a reference (https://code.google.com/p/smithlabsoftware). The images from all seven of the ribbon arrays were ordered into one large stack and aligned with the TrakEM2 plugin for ImageJ to remove non-linear warping.
2.5. Locating synapses in the digital images
Points of contact between the two neurons were identified by creating masks of both neuron image channels (Alexa 488 and Alexa 594 channels), enlarging the postsynaptic neuron mask by a few pixels with the dilate feature of ImageJ, then locating points of overlap between the enlarged postsynaptic mask and the presynaptic mask image data. The ROI manager in ImageJ was used to manually screen these points of overlap on the original image stacks of both neurons to identify only points of contact between the presynaptic axon and the postsynaptic dendrite. The arrays containing these points of contact were immunostained with Synapsin and then VGLUT1 and imaged at 63×. The stacks of images containing antibody fluorescence image data were exported as TIFF and registered to the neuron image data. Synaptograms were generated from these combined stacks at each point of contact using a custom ImageJ plugin written by Brad Busse and Nick Weiler of the Stephen Smith Lab. For some arrays, antibodies were eluted again and a new set of antibodies for AMPA receptor subunits and PSD95 was applied, and the arrays were re-imaged. This process of antibody elution, re-application, re-imaging can be performed several times on any given array. The limitation on the number of different antibodies that can be applied in any given cycle is generally set by the number of color channels that can be utilized and by the combinations of primary/secondary antibodies that are available.
2.6. Protocol variations
2.6.1. Protocol variation #1: targeted partial reconstruction of neuronal pairs
Complete reconstruction of neuronal pairs is very time consuming and not always necessary to address specific scientific questions. We have modified the present method to allow a much faster identification of synaptic contacts in a neuronal pair, by adjusting the protocol to preserve Lucifer yellow fluorescence and thus eliminate the need for antibody staining, and by reconstructing only a portion of the proximal dendritic arbor of the postsynaptic cell, where most of the synapses are located (Fig. 2 and Pavlidis and Madison, 1999).
Fig. 2.
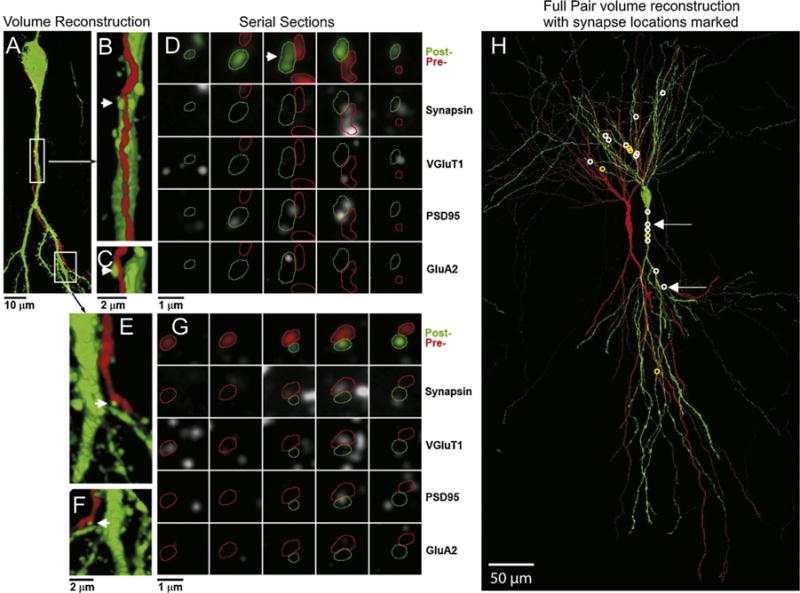
Volume reconstruction of a recorded pair of synaptically-connected CA3 neurons and location of all synapses made between that pair. (A) Partial volume reconstruction of a portion of the presynaptic dendrite (neurobiotin–streptavidin conjugated to Alexa 594, red) and the postsynaptic soma and proximal apical dendrite (anti-Lucifer yellow, green). The locations of the magnified segments in (B) and (E) are marked with boxes. The contacts between the presynaptic axon and postsynaptic dendrite in (B) and (E) (arrow) are shown in a rotated view (C and F), and as serial sections (D and G). The 5 consecutive serial sections across each row are stained with the same marker(s), indicated to the right, with each column being the same section. Panels A–C, 37 sections thick, E and F, 18 sections thick, all 200 nm. (H) The full volume reconstruction (182 sections, all 200 nm) of the entire structure of this pair of cells, with the synapses formed from the red presynaptic cell to the green postsynaptic cell, marked with a white circle. Non-synaptic contacts between the presynaptic and postsynaptic neuron are marked with yellow circles. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
To preserve Lucifer yellow fluorescence better, slices were dehydrated only to 70% ethanol. Specifically, slices were dehydrated in 50% ethanol, and then two times in 70%, ethanol, each time microwaved at 350 W for 30 s. The dehydrated tissue was then infiltrated with a mixture of ethanol and LR White (1:3) followed by three changes of 100% LR White, microwaved at 350 W for 30 s each time (Micheva et al., 2010a). The slice was then left in LR White resin overnight at 4 °C.
For partial reconstructions, the slice can be trimmed much more aggressively, preserving only the area that includes the postsynaptic cell body and proximal dendrites. After the slice is fixed and washed in PBS buffer, the area containing the neuronal pair is dissected out (approximately 2 by 1 mm rectangle), dehydrated and embedded in LR White resin. Because in this case the size of the tissue is very small, slice warping is not an issue and the tissue can be directly embedded in the gelatin capsule without the use of a stub of prepolymerized resin as when embedding the entire slice. After embedding and resin polymerization, the slice can be further trimmed down to less than 0.5 mm in height (the axis running parallel to the pyramidal layer), which allows the collection of much longer arrays of ultrathin sections (100 sections routinely, up to 150 in some cases). During imaging, much smaller tiled mosaics are acquired, typically 1 by 3 tiles per section, and only arrays that include the postsynaptic cell body and the immediately adjacent arrays are imaged (3–4 coverslips).
2.6.2. Protocol variation #2: increased antibody multiplexing capabilities
The complete pair reconstruction, as seen in Fig. 2, was performed on arrays of 200 nm thick sections collected on gelatin-subbed coverslips. While the use of thicker sections reduces the number of sections that have to be imaged, such sections are more susceptible to adverse effects during elution. Similarly, gelatin subbed coverslips which are very convenient for picking up sections and thus minimize the risks of losing tissue while sectioning, do not withstand multiple cycles of elution well. Typically, after 2–3 elutions, the sections may begin detaching from the coverslips, resulting in the formation of small folds that decrease image quality. Two elutions (3 imaging cycles) are sufficient for applying pre- and postsynaptic markers needed for synapse identification, but this becomes a limiting factor when a more detailed molecular characterization of the identified synapses is needed. When planning to apply a larger number of antibodies, we use arrays of thinner sections (100 nm each) collected onto carbon-coated coverslips. The thinner sections have the additional advantage of doubling the z-axis resolution. Gelatin-subbed and then carbon-coated coverslips result in a much better adhesion of the sections, allowing more than 10 serial elutions. However, carbon-coated coverslips are hydrophobic and therefore less convenient for array collection, which is why we use them only when required for a large number of elutions. Carbon coating of gelatin-subbed coverslips was performed using a Denton Bench Top Turbo Carbon Evaporator (Denton Vacuum, Moorestown, NJ). Light carbon coating was sufficient for section adhesion. Glow discharge (a standard function on many carbon evaporators) of the carbon-coated coverslips is not recommended. Even though it renders the surface of coverslips hydrophilic and thus makes them much easier to use, it also results in decreased adherence of the tissue to the coverslips.
2.6.3. Protocol variation #3: fast chemical fixation
Some of the partially reconstructed slices were fixed using a new “hot” fixation method, SNAPSHOT (Dissing-Olesen and MacVicar, 2015). This method results in rapid fixation of tissue, which is important when correlating anatomical features of the tissue with relatively fast physiological changes. In this procedure, slices are removed from the recording chamber after completion of the electrophysiological experiment, and immediately placed in fixative (4% paraformaldehyde in PBS) preheated to 80 °C. After exactly 2 min, the slice is transferred to a PBS wash at room temperature.
3. Results
3.1. Complete reconstruction and synapse mapping of neuronal pairs
The full reconstruction of the cell pair from Fig. 1, as well as examples of synaptic contacts between these two neurons, are shown in Fig. 2. The presynaptic cell was filled with neurobiotin and the postsynaptic cell with Lucifer yellow. Both dyes were visualized after sectioning, on the arrays, using an anti-Lucifer yellow antibody and a fluorescent streptavidin conjugate (streptavidin—Alexa 594), respectively. The entire aspect of the postsynaptic soma and dendrites spanned through 182 sections of 200 nm, i.e., a thickness of 36.4 μm (Fig. 1H). This volume also included the soma and a portion of the axon of the presynaptic neuron. As is typical, the entire axonal structure of the presynaptic cell extends far beyond the boundaries of this reconstructed volume (Pavlidis and Madison, 1999).
For this cell pair, 18 sites were identified where the axon of the presynaptic neuron came in close apposition to the dendrite of the postsynaptic neuron (for example Fig. 2B, C, E, and F). To test whether these appositions represent actual synaptic contacts, the corresponding arrays were stained with antibodies against the synaptic proteins synapsin and VGluT1. The results are visualized using synaptograms (Fig. 2D and G), where columns represent serial sections through a potential synapse, and rows show different antibodies or combinations of antibodies. Fourteen out of the 18 sites (white circles in Fig. 1H) were found to contain immunolabel for synapsin and VGluT1 within the presynaptic axon, thus confirming that they most likely represent synaptic contacts. Some sites were more extensively tested by subsequently applying postsynaptic markers (Fig. 2D and G; PSD95 and GluA2 antibodies). Both contacts shown in Fig. 2 contain PSD95 within their postsynaptic dendrite, but only one of them contains immunolabel for GluA2.
3.2. Release probability of synapses in a pair
The average EPSC evoked from an action potential to all of these synapses was determined to be 17 pA (Fig. 1A). Because the quantal size is known to be ~9–10 pA, this means that only about 2 synapses are transmitting in the average trial. If all of the identified 14 synaptic contacts are functional, then the average release probability for an individual synapse in this connection is below 0.2.
3.3. Partial reconstruction of neuronal pairs
Often it is sufficient to identify only a subset of synapses made between two neurons. In such cases, partial reconstructions of neuronal pairs are a much more efficient approach. This task is made easier by the observation that most synaptic contacts in pyramidal neuron pairs are located proximally (within 150 μm) to the postsynaptic cell body (present results and Pavlidis and Madison, 1999). Fig. 3 shows images from 2 partially-reconstructed pairs of synaptically-connected CA3 pyramidal neurons. We used two strategies that aim at maximizing the yield of located synaptic contacts: imaging a smaller number of sections and wider area, or larger number of sections and smaller area. In one of the cases presented in Fig. 3B, 144 serial sections of 100 nm each were imaged (1 × 3 tiled mosaic), and in the other (Fig. 3C), 367 serial sections of 100 nm each (only one tile per section) were imaged. Both strategies resulted in the identification of a comparable number of synapses (9 in 3B and 13 in 3C) and took a similar amount of time (about 1 week to find the synaptic contacts and perform 3 staining and imaging cycles). Overall, 7 neuronal pairs were partially reconstructed using either of these two strategies, allowing to identify on average 10 synapses per pair (ranging between 9 and 13 synaptic contacts per pair). In order to compare the efficiency of this strategy with the complete reconstruction presented in 3.1, for each reconstructed pair we calculated the number of fields of view (139 by 104μm) imaged per identified synapse, i.e., the number of sections multiplied by the number of tiles per section and divided by the number of synapses. With partial reconstructions, we imaged on average 33 fields of view per identified synapse, compared to 520 fields of view per synapse for the complete reconstruction. Thus, partial reconstructions of neuronal pairs considerably reduce the imaging stage of the process (15 times) and are a much more efficient approach when identification of all synaptic contacts is not necessary.
Fig. 3.
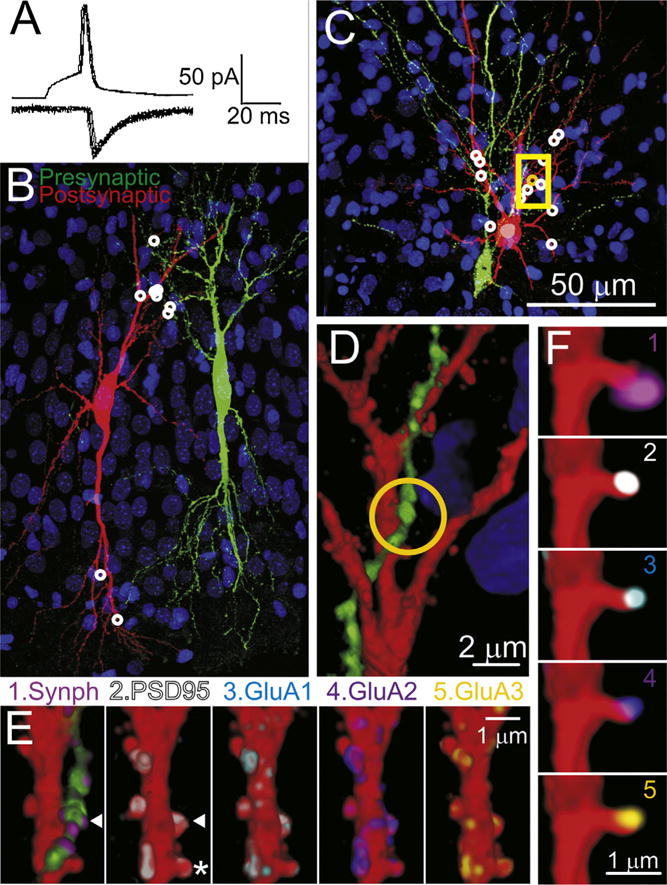
Reconstruction of synaptically-connected CA3 neurons pairs with two different fixations procedures to improve time resolution. (A) Recordings for the pre-synaptic action potentials (upper trace) and postsynaptic AMPAR-mediated synaptic currents (lower traces), taken from the cell pair illustrated in panels (C–F). Shown are 8 consecutive action potentials and corresponding postsynaptic responses. (B) A partial volume reconstruction (144 100 nm sections thick). 9 synaptic contacts (white circles) were found within this volume. This pair of cells were fixed by the standard room temperature/overnight method. (C) Another partial volume reconstruction from a pair of CA3 cells fixed at 80 °C for 2 min. This volume was reconstructed from 367 serial sections and this volume contained 13 synapses white/yellow circles. (D) A magnified view of a portion of this volume (35 sections) indicated by the yellow box in (C). (E) Magnified view of one dendritic segments (20 sections). The arrow head indicates the location of the synapse indicated by the orange circle in panel (C) and (D). E1 shows the pre- and postsynaptic injected markers (red and green) and immunostaining for synaptophysin (violet). (E2–6) show the dendrite with the presynaptic axon digitally removed from the image and immunostained for PSD95 (2), GluA1 (3), GluA2 (4) or GluA3 (5). (F) Four panels showing detail of the dendritic spine marked by an * in panel (E2), 3 sections thick, and immunostained with the same markers (1–5) as in panel (E). This spine, is viewed from behind the perspective or panel (E) (i.e., the sections stacked in reverse order). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.4. Fast chemical fixation
Some of the partially reconstructed slices were fixed using a new “hot” fixation method, SNAPSHOT (Dissing-Olesen and MacVicar, 2015). The ‘hot’ method involves immersing the brain slice in 4% paraformaldehyde pre-heated to 80 °C for 2 min. As can be seen in Fig. 3, neurons subjected to this ‘hot’ fixation method maintain their structural integrity (compare Fig. 3C and D with Fig. 3B and 2), and immunoreactivity (Fig. 3E and F). The entire fixation procedure can be completed in about 2.5 min, thus ensuring that the physiological property of interest is arrested within that time frame.
One potential issue with the high-temperature fixation is that it can quench endogenous fluorescence (Dissing-Olesen and MacVicar, 2015), which in our case is needed to guide tissue trimming before and after embedding, as well as for ultrathin sectioning. Fig. 4A shows photographs of two dissected slices after fixation and before embedding in resin. 4A1 is fixed with our standard room-temperature overnight fixation and 4A2 is fixed at 80 °C for 2 min. As can be seen, the cell pair is clearly visible in both preparations. Similarly after embedding in resin, neuron pairs in slices fixed with the two different conditions have very comparable dye fill fluorescence (Fig. 3B and C).
Another concern with hot temperature fixation is its effect on tissue antigenicity. As can be seen on Fig. 3E and F, immunolabeling of slices fixed with the “hot” method appeared to be of good quality. Quantification of the immunolabeling with the two different conditions of fixation, confirmed that the high temperature fixation does not significantly alter the antigenicity of our tissue (Fig. 4B). The number of immunofluorescent puncta per spine were not significantly different for any of the postsynaptic proteins tested (PSD-95 and each of the three AMPAR subunits: GluA1, GluA2 and GluA3).
3.5. PSD size and AMPA receptor content of dendritic spines
Because no differences were detected in the immunofluorescent staining of dendritic spines with the two fixation conditions, we pooled the data for further analysis. PSD95 is a very abundant protein of the postsynaptic density of excitatory synapses and AT immunofluorescence for PSD95 overlays more than 90% of ultra-structurally identified postsynaptic densities (Collman et al., 2015). We used PSD95 immunolabeling to estimate the size of the postsynaptic density of dendritic spines of CA3 pyramidal neurons (Fig. 5A). In 72% of dendritic spines, PSD95 immunolabeling was present on more than one section. An average of 2.34 ± 0.12 sections (or 234 nm) through a dendritic spine were immunopositive for PSD95. AMPA receptor labeling (GluA1, GluA2 and GluA3) was present in 89% of the dendritic spines and it positively correlated with the size of the PSD (R = 0.62; Fig. 5B). The majority of AMPA negative spines (71%) were of small size, with PSD95 immunolabeling on only one section. All spines with large PSDs (PSD95 immunolabeling on 4 or more sections) were immunopositive for AMPA receptors.
Fig. 5.
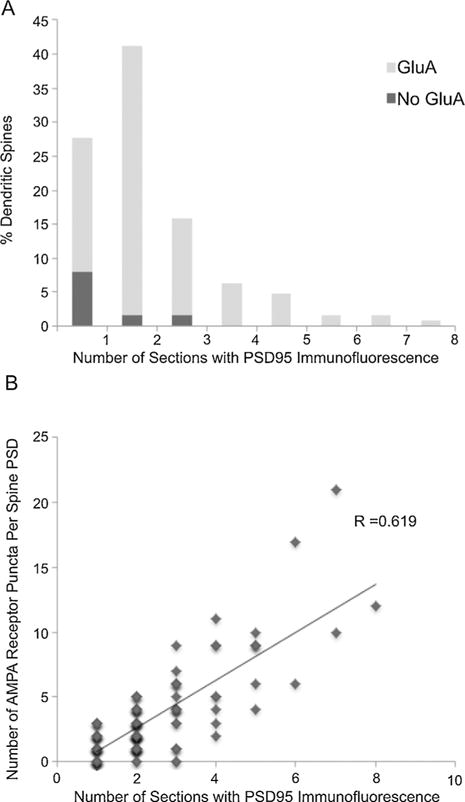
PSD size and AMPA receptor distribution at dendritic spines. (A) Size distribution of postsynaptic densities as estimated from the number of consecutive sections through a spine that are immunopositive for PSD95. The proportion of AMPA receptors immunonegative synapses is indicated in dark gray. (B) The number of GluA1, GluA2 and GluA3 immunofluorescent puncta at spines are positively correlated with PSD size (the number of consecutive sections that are immunopositive for PSD95).
4. Discussion
4.1. Summary
We have developed a novel approach that combines paired neuron electrophysiological recording and array tomography, allowing for the detailed molecular and anatomical study of synapses with known physiological properties. The main physiological advantage of studying a minimal synaptic connection, as in paired cell recording, is that one can begin to discern the physiological properties of individual synapses. The subsequent anatomical examination of these small populations of synapses establishes the important connection between their physiology and their underlying structural and molecular properties.
An important advantage conferred to this procedure by array tomography is that it allows light level synapse identification that is much faster than using conventional EM. This ability is due to the outstanding light microscopic resolution (~100 nm) of AT, especially in the z-axis, which results from the use of ultrathin serial sections (typically 70–200 nm). The section thickness not only sets the resolution in the z-axis but also improves x–y resolution by decreasing light scattering that is a problem in thicker sections. Second, the use of such thin sections gives excellent access for immunostaining, as well as antibody elution and allows for the application of up to dozens of antibodies to the same serial arrays. In this way, points of close apposition between putative presynaptic terminals and postsynaptic spines can be examined on a section by section basis to verify whether these contacts contain pre- and postsynaptic proteins in the appropriate locations within those profiles. The high spatial resolution, as well as the ability to localize multiple markers on the same putative synaptic contact, substantially increase the reliability of synapse identification. Furthermore, the robust immunolabeling for specific synaptic compartments enables their size estimation. For example, PSD95 has been shown to be a reliable marker for postsynaptic densities of excitatory synapses (Collman et al., 2015). Because of the random 3D orientation of postsynaptic densities, the number of consecutive sections labeled with PSD95 can be used to estimate the size of spine postsynaptic densities. The estimated 234 nm PSD length in our study is very similar to the PSD size calculated using EM serial sections (242 nm maximum PSD length in serial sections through CA1 pyramidal neuron dendritic spines) (Takumi et al., 1999).
A distinguishing advantage of the present method is the ability for detailed molecular characterization of the individual synapses. As an example, we quantified the presence of AMPA receptors at dendritic spines using antibodies for different AMPA receptor subunits, GluA1, GluA2 and GluA3. The immunofluorescent AT quantification of AMPA receptors revealed the typical features described in previous EM post-embedding studies (Takumi et al., 1999; Nusser et al., 1998). AMPA receptor immunostaining was positively correlated with the size of the postsynaptic density. About 11% of dendritic spines did not have detectable AMPA receptor immunolabeling and the majority of these spines were of small size, as previously described.
It is quite possible that this approach could be combined with an optogenetic approach, in that the electrode activation of synapses, might be substituted with an optogenetic activation. However, for a combination of optogenetics and array tomography to retain the advantages of the technique as we have described it here, there would need to be a way to specifically identify the synapses that had been activated. Of course, an entire presynaptic could be optogenetically stimulated, in a similar manner to our stimulation of that cell via electrode, but in order to retain the same information, a neuron stimulated via an optogenetic dye would also have to have a voltage-sensitive dye in it as well, to verify that it had reached action potential threshold. This is certainly possible (see Abdelfattah et al., 2016), but not really any easier than simply using an electrode. A combination of optogenetics and array tomography would truly be advantageous in cases where one wanted to activate a number of given type simultaneously in order to study anatomical changes more widespread in neural networks, but even then, this would only work if one could identify the activated neurons post-hoc in the tissue arrays.
4.2. Limitations
For the initial development of this technique, cultured organotypic slices were used because they offer a greater degree of synaptic connectivity than do the more standard freshly-prepared “acute” slices of brain tissue. For example, the connection frequency of two neighboring CA3 neurons in acute slices has been found to range from 1 to 5% (personal unpublished experiments) to ~10% (Malinow, 1991), while the connection frequency for these neurons in organotypic slices is closer to 50% (Pavlidis and Madison, 1999; Montgomery et al., 2001). Organotypic hippocampal slices maintain functional synaptic properties and appropriate synaptic connections similar to acute slices (Pavlidis and Madison, 1999; Stoppini et al., 1991; Muller et al., 1993; Gahwiler et al., 1997; De Simoni et al., 2003). Unlike dissociated culture, they also maintain the ability of the glutamatergic synapses to undergo long-term potentiation and long-term depression (Pavlidis and Madison, 1999; Montgomery and Madison, 2002; Stoppini et al., 1991; Montgomery et al., 2001; Debanne et al., 1994; Muller et al., 1996; Debanne et al., 1996). However, one must always be aware that they are hyper-connected when interpreting results. We believe, for example, that organotypic slices are an excellent preparation for studying the properties inherent to synapse, but not such an advantageous preparation for experiments where a more native version of the circuit diagram is required. One other particular advantage in using organotypic slices is that synaptic plasticity, including long-term potentiation and long-term depression, can be induced in the small population of synapses isolated during cell pair recording, as has been extensively studied (Debanne et al., 1998; Malinow, 1991; Emond et al., 2010; Montgomery and Madison, 2002; Debanne et al., 1994; Debanne et al., 1996; Bolshakov and Siegelbaum, 1995; Alle et al., 2001). Array tomographic reconstruction analysis works well in native brain tissue as well, and thus this technique can also be applied to acute slices in future studies, provided that one has the time and patience to collect cell pairs at a much lower rate.
Another limitation of the presented technique is that synapse detection is performed at the light level. Traditionally synapses are best viewed under the electron microscope, where their ultra-structure can be clearly seen with very high nanometer resolution. The presence of synaptic vesicles on the presynaptic side and the opposing postsynaptic density are the typical features allowing identification of a synapse. With array tomography which lacks the resolution of electron microscopy, synapses are detected based on the spatial arrangement of pre- and postsynaptic molecular markers. However, direct comparison of light-level AT synapse detection with EM ultrastructure-based synapse detection has shown that approximately 90% of excitatory synapses are correctly identified using immunofluorescent AT (Collman et al., 2015). Because immunofluorescent AT enables the molecular characterization of synapses and is much less time-consuming compared to electron microcopy, it offers distinct advantages for studies of individual physiologically characterized synapses within brain tissue.
A final consideration is the speed of the chemical fixation. Conventional methods of slice fixation by immersion in formaldehyde fixative may be too slow to capture dynamic biological events. A 4% solution of formaldehyde takes more than 1 h (1.14 h) (Dempster, 1960), to penetrate 1 mm within biological tissue at room temperature. Because the rate of formaldehyde penetration is roughly proportional to the depth within the tissue, this means that a 100 μm slice on a membrane will be completely infiltrated by the fixative in approximately 7 min. The covalent chemical reactions of formaldehyde with the tissue components however take longer (hours). We routinely use microwave irradiation to speed up fixative penetration and chemical interaction, but it still requires 1 h of fixation to achieve well reproducible results. To improve time resolution, we used a new ‘hot’ fixation method (Dissing-Olesen and MacVicar, 2015). Control experiments confirmed that this method provides a rapid (less than 2.5 min) fixation of our samples without compromising tissue structure and antigenicity or quenching dye fluorescence. High pressure freezing can ensure even faster (in the order of milliseconds) tissue fixation and has been successfully used for hippocampal slices (Studer et al., 2014). However, dissecting out and loading the sample into the high pressure freezer can take several minutes. In our hands, ‘hot’ fixation is a more convenient, faster, and reproducible approach, but for events requiring millisecond resolution, high pressure freezing strategies should be explored, so long as the problem of the time-lag inherent in loading the tissue into the freezing device can be solved in a manner compatible with performing physiological experiments.
4.3. Number of synaptic contacts and their release probability
Paired electrophysiological recordings in acute slices followed by light level or electron microscopic imaging suggest that pyramidal cells in cortex and hippocampus form between 2–8 synapses with each other (Markram et al., 1997; Feldmeyer et al., 2006; Deuchars and Thomson, 1996). Pyramidal cell pairs in organotypic slices may form a somewhat larger number of contacts. For example, in Pavlidis and Madison (1999), light-level reconstruction of two pairs of pyramidal neurons filled with neurobiotin, revealed 14 and 21 distinct close contacts between the presynaptic axon and postsynaptic dendrite. Because the resolution of light microscopy was insufficient to directly observe synaptic specializations, and no antibodies were used, these contacts could only be considered to be ‘potential synapses’. In the present study, the number of potential synaptic contacts that we identified in the fully reconstructed pair agreed closely with our previously published results (Pavlidis and Madison, 1999). We also found that almost all of the potential synapses that were identified represented actual synapses. Fourteen of the 18 places where the axon of the presynaptic neuron touched the dendrite of the postsynaptic neuron contained synaptic proteins. The average release probability of these synapses was estimated to be below 0.2, which is very similar to the value obtained using Ca2+ imaging-derived optical quantal analysis (0.14) (Holderith et al., 2012). Release probability is known to vary widely even among synapses made by the same axon (reviewed Branco and Staras, 2009). Ca2+ imaging followed by EM has demonstrated that release probability at individual synapses positively correlates with active zone size (Holderith et al., 2012). The molecular correlates of the variability of release probability can be further investigated in future studies using the method introduced here.
5. Conclusion
In conclusion, paired cell electrophysiology recordings followed by array tomography immunofluorescence imaging allows for the combined physiological and anatomical examination of identified central nervous system synapses. Unlike a typical application of array tomography which seeks to catalogue large numbers of neural elements within a volume of tissue (Kleinfeld et al., 2011; Weiler et al., 2014), this application takes the opposite approach of identifying the full extent of a single element within that tissue, in this case the most reduced element of a neural circuit, the connection made between just one neuron and one other. It also differs somewhat from a similar EM-based approach of Lichtman (Kasthuri et al., 2015; Kaynig et al., 2015) in that instead of using a ‘saturated reconstruction’ approach, we selectively label the elements we wish to study by injecting them with fluorescent markers. While the EM-based approach offers better spatial resolution, the current method has the advantage that an element (synapses between a cell pair) can be first physiologically characterized and then anatomically identified and studied. Restricting the examination of elements in this way will allow addressing specific questions about the properties of this quantum of neural circuitry, even when that ‘connectome quantum’ is buried within a cloud of millions of other brain circuit elements and their synapses.
HIGHLIGHTS.
We describe a novel combination of two established methods: paired cell electrophysiological recording and array tomography.
The combination of these two techniques allows for the isolation and anatomical study of synapses that have been characterized physiologically.
This technique can be used to find all of the synapses made between two individual neurons or a subset of those synapses.
The Physiological history of these identified synapses is known.
Acknowledgments
We would like to thank Stephen Smith, Forrest Collman, Nicholas Weiler and JoAnn Buchanan for their help and advice in developing this use of the Array Tomography technique. This work was supported by a grant from the Stanford University School of Medicine Faculty Innovation Fund, a grant from the National Institute of Mental Health (MH065541) and by the Harold and Leila Y Mathers Charitable Foundation.
Footnotes
Conflict of interest
K.D.M. has a founder’s equity interest in a company Aratome LLC, which develops array tomography for commercial purposes. There was no consultation or interaction with Aratome during the development of this method or preparation of the manuscript.
References
- Abdelfattah AS, et al. A bright and fast red fluorescent protein voltage indicator that reports neuronal activity in organotypic brain slices. J Neurosci. 2016;36:2458–2472. doi: 10.1523/JNEUROSCI.3484-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alle H, Jonas P, Geiger JR. PTP and LTP at a hippocampal mossy fiber-interneuron synapse. Proc Natl Acad Sci U S A. 2001;98:14708–14713. doi: 10.1073/pnas.251610898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biro AA, Holderith NB, Nusser Z. Release probability-dependent scaling of the postsynaptic responses at single hippocampal GABAergic synapses. J Neurosci. 2006;26:12487–12496. doi: 10.1523/JNEUROSCI.3106-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Regulation of hippocampal transmitter release during development and long-term potentiation. Science. 1995;269:1730–1734. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- Branco T, Staras K. The probability of neurotransmitter release: variability and feedback control at single synapses. Nat Rev Neurosci. 2009;10:373–383. doi: 10.1038/nrn2634. [DOI] [PubMed] [Google Scholar]
- Buhl EH, et al. Effect, number and location of synapses made by single pyramidal cells onto aspiny interneurones of cat visual cortex. J Physiol. 1997;500(Pt 3):689–713. doi: 10.1113/jphysiol.1997.sp022053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Harata NC, Tsien RW. Paired-pulse depression of unitary quantal amplitude at single hippocampal synapses. Proc Natl Acad Sci U S A. 2004;101:1063–1068. doi: 10.1073/pnas.0307149101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Klingauf J, Tsien RW. Postfusional regulation of cleft glutamate concentration during LTP at ‘silent synapses’. Nat Neurosci. 2000;3:330–336. doi: 10.1038/73895. [DOI] [PubMed] [Google Scholar]
- Collman F, et al. Mapping synapses by conjugate light-electron array tomography. J Neurosci. 2015;35:5792–5807. doi: 10.1523/JNEUROSCI.4274-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simoni A, Griesinger CB, Edwards FA. Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J Physiol. 2003;550:135–147. doi: 10.1113/jphysiol.2003.039099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Gahwiler BH, Thompson SM. Asynchronous pre- and postsynaptic activity induces associative long-term depression in area CA1 of the rat hippocampus in vitro. Proc Natl Acad Sci U S A. 1994;91:1148–1152. doi: 10.1073/pnas.91.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Gahwiler BH, Thompson SM. Cooperative interactions in the induction of long-term potentiation and depression of synaptic excitation between hippocampal CA3-CA1 cell pairs in vitro. Proc Natl Acad Sci U S A. 1996;93:11225–11230. doi: 10.1073/pnas.93.20.11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Gahwiler BH, Thompson SM. Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J Physiol. 1998;507(Pt 1):237–247. doi: 10.1111/j.1469-7793.1998.237bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster WT. Rates of penetration of fixing fluids. Am J Anat. 1960;107:59–72. doi: 10.1002/aja.1001070105. [DOI] [PubMed] [Google Scholar]
- Deuchars J, Thomson AM. Innervation of burst firing spiny interneurons by pyramidal cells in deep layers of rat somatomotor cortex: paired intracellular recordings with biocytin filling. Neuroscience. 1995;69:739–755. doi: 10.1016/0306-4522(95)00288-t. [DOI] [PubMed] [Google Scholar]
- Deuchars J, Thomson AM. CA1 pyramid-pyramid connections in rat hippocampus in vitro: dual intracellular recordings with biocytin filling. Neuroscience. 1996;74:1009–1018. doi: 10.1016/0306-4522(96)00251-5. [DOI] [PubMed] [Google Scholar]
- Dissing-Olesen L, MacVicar BA. Fixation and immunolabeling of brain slices: SNAPSHOT method. Curr Protoc Neurosci. 2015;71:1 23 1–1 23 12. doi: 10.1002/0471142301.ns0123s71. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Emond MR, et al. AMPA receptor subunits define properties of state-dependent synaptic plasticity. J Physiol. 2010;588:1929–1946. doi: 10.1113/jphysiol.2010.187229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer D, Lubke J, Sakmann B. Efficacy and connectivity of intracolumnar pairs of layer 2/3 pyramidal cells in the barrel cortex of juvenile rats. J Physiol. 2006;575:583–602. doi: 10.1113/jphysiol.2006.105106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourie C, Kiraly M, Madison DV, Montgomery JM. Paired whole cell recordings in organotypic hippocampal slices. J Vis Exp. 2014;5:1958. doi: 10.3791/51958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahwiler BH, Capogna M, Debanne D, McKinney RA, Thompson SM. Organotypic slice cultures: a technique has come of age. Trends Neurosci. 1997;20:471–477. doi: 10.1016/s0166-2236(97)01122-3. [DOI] [PubMed] [Google Scholar]
- Gulyas AI, et al. Hippocampal pyramidal cells excite inhibitory neurons through a single release site. Nature. 1993;366:683–687. doi: 10.1038/366683a0. [DOI] [PubMed] [Google Scholar]
- Holderith N, et al. Release probability of hippocampal glutamatergic terminals scales with the size of the active zone. Nat Neurosci. 2012;15:988–997. doi: 10.1038/nn.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasthuri N, et al. Saturated reconstruction of a volume of neocortex. Cell. 2015;162:648–661. doi: 10.1016/j.cell.2015.06.054. [DOI] [PubMed] [Google Scholar]
- Kaynig V, et al. Large-scale automatic reconstruction of neuronal processes from electron microscopy images. Med Image Anal. 2015;22:77–88. doi: 10.1016/j.media.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinfeld D, et al. Large-scale automated histology in the pursuit of connectomes. J Neurosci. 2011;31:16125–16138. doi: 10.1523/JNEUROSCI.4077-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R. Transmission between pairs of hippocampal slice neurons: quantal levels, oscillations, and LTP. Science. 1991;252:722–724. doi: 10.1126/science.1850871. [DOI] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Roth A, Sakmann B. Physiology and anatomy of synaptic connections between thick tufted pyramidal neurones in the developing rat neocortex. J Physiol. 1997;500(Pt 2):409–440. doi: 10.1113/jphysiol.1997.sp022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, O’Rourke N, Busse B, Smith SJ. Array tomography: rodent brain fixation and embedding. Cold Spring Harb Protoc. 2010a;2010 doi: 10.1101/pdb.prot5523. pdb rot5523. [DOI] [PubMed] [Google Scholar]
- Micheva KD, O’Rourke N, Busse B, Smith SJ. Array tomography: production of arrays. Cold Spring Harb Protoc. 2010b;2010 doi: 10.1101/pdb.prot5524. pdb rot5524. [DOI] [PubMed] [Google Scholar]
- Micheva KD, O’Rourke N, Busse B, Smith SJ. Array tomography: immunostaining and antibody elution. Cold Spring Harb Protoc. 2010c;2010 doi: 10.1101/pdb.prot5525. pdb rot5525. [DOI] [PubMed] [Google Scholar]
- Migheli A, Attanasio A. Two methods for flat embedding sections in LR white cut from paraffin blocks. Biotech Histochem. 1991;1:89–92. doi: 10.3109/10520299109110557. [DOI] [PubMed] [Google Scholar]
- Montgomery JM, Madison DV. State-dependent heterogeneity in synaptic depression between pyramidal cell pairs. Neuron. 2002;33:765–777. doi: 10.1016/s0896-6273(02)00606-2. [DOI] [PubMed] [Google Scholar]
- Montgomery JM, Madison DV. Discrete synaptic states define a major mechanism of synapse plasticity. Trends Neurosci. 2004;27:744–750. doi: 10.1016/j.tins.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Montgomery JM, Pavlidis P, Madison DV. Pair recordings reveal all-silent synaptic connections and the postsynaptic expression of long-term potentiation. Neuron. 2001;29:691–701. doi: 10.1016/s0896-6273(01)00244-6. [DOI] [PubMed] [Google Scholar]
- Muller D, Buchs PA, Stoppini L. Time course of synaptic development in hippocampal organotypic cultures. Dev Brain Res. 1993;71(1):93–100. doi: 10.1016/0165-3806(93)90109-n. [DOI] [PubMed] [Google Scholar]
- Muller D, et al. PSA-NCAM is required for activity-induced synaptic plasticity. Neuron. 1996;17:413–422. doi: 10.1016/s0896-6273(00)80174-9. [DOI] [PubMed] [Google Scholar]
- Nusser Z, et al. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron. 1998;21:545–559. doi: 10.1016/s0896-6273(00)80565-6. [DOI] [PubMed] [Google Scholar]
- Palmieri M, Kiss JZ. A novel technique for flat-embedding cryofixed plant specimens in LR white resin. Microsc Res Tech. 2005;68:80–84. doi: 10.1002/jemt.20237. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Madison DV. Synaptic transmission in pair recordings from CA3 pyramidal cells in organotypic culture. J Neurophysiol. 1999;81:2787–2797. doi: 10.1152/jn.1999.81.6.2787. [DOI] [PubMed] [Google Scholar]
- Silver RA, Lubke J, Sakmann B, Feldmeyer D. High-probability uniquantal transmission at excitatory synapses in barrel cortex. Science. 2003;302:1981–1984. doi: 10.1126/science.1087160. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Studer D, et al. Capture of activity-induced ultrastructural changes at synapses by high-pressure freezing of brain tissue. Nat Protoc. 2014;9:1480–1495. doi: 10.1038/nprot.2014.099. [DOI] [PubMed] [Google Scholar]
- Takumi Y, Ramirez-Leon V, Laake P, Rinvik E, Ottersen OP. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci. 1999;2:618–624. doi: 10.1038/10172. [DOI] [PubMed] [Google Scholar]
- Tamas G, Buhl EH, Somogyi P. Fast IPSPs elicited via multiple synaptic release sites by different types of GABAergic neurone in the cat visual cortex. J Physiol. 1997;500(Pt 3):715–738. doi: 10.1113/jphysiol.1997.sp022054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler NC, Collman F, Vogelstein JT, Burns R, Smith SJ. Synaptic molecular imaging in spared and deprived columns of mouse barrel cortex with array tomography. Sci Data. 2014;1:140046. doi: 10.1038/sdata.2014.46. [DOI] [PMC free article] [PubMed] [Google Scholar]