

ABSTRACT
Early work on the control of transcription of the human immunodeficiency virus (HIV) laid the foundation for our current knowledge of how RNA Polymerase II is released from promoter-proximal pausing sites and transcription elongation is enhanced. The viral Tat activator recruits Positive Transcription Elongation Factor b (P-TEFb) and Super Elongation Complex (SEC) that jointly drive transcription elongation. While substantial progress in understanding the role of SEC in HIV gene transcription elongation has been obtained, defining of the mechanisms that govern SEC functions is still limited, and the role of SEC in controlling HIV transcription in the absence of Tat is less clear.
Here we revisit the contribution of SEC in early steps of HIV gene transcription. In the absence of Tat, the AF4/FMR2 Family member 4 (AFF4) of SEC efficiently activates HIV transcription, while gene activation by its homolog AFF1 is substantially lower. Differential recruitment to the HIV promoter and association with Human Polymerase-Associated Factor complex (PAFc) play key role in this functional distinction between AFF4 and AFF1. Moreover, while depletion of cyclin T1 expression has subtle effects on HIV gene transcription in the absence of Tat, knockout (KO) of AFF1, AFF4, or both proteins slightly repress this early step of viral transcription. Upon Tat expression, HIV transcription reaches optimal levels despite KO of AFF1 or AFF4 expression. However, double AFF1/AFF4 KO completely diminishes Tat trans-activation. Significantly, our results show that P-TEFb phosphorylates AFF4 and modulates SEC assembly, AFF1/4 dimerization and recruitment to the viral promoter.
We conclude that SEC promotes both early steps of HIV transcription in the absence of Tat, as well as elongation of transcription, when Tat is expressed. Significantly, SEC functions are modulated by P-TEFb.
KEYWORDS: eukaryotic transcription, human immunodeficiency virus (HIV), positive transcription elongation factor b (P-TEFb), RNA polymerase II, super elongation complex (SEC), Tat trans-activator, transcription elongation
Introduction
Of the steps that comprise metazoan gene transcription, elongation by RNA polymerase II (Pol II) is a key rate-limiting event that dictates overall gene expression.1-4 Accordingly, it is not surprising that disturbance of transcription elongation has been directly associated with human disorders such as cancer, developmental syndromes, and immunodeficiency.5-7 Following transcription initiation, Pol II quickly pauses between +25 to +60 nt downstream of the transcription starting sites (TSS),8 as negative transcription factors, DRB sensitivity inducing factor (DSIF), and negative elongation factor (NELF), halt Pol II and repress productive transcription elongation.9-13 Release of paused Pol II and productive transcription elongation depends on the activity of positive transcription elongation factor b (P-TEFb) and super elongation complex (SEC), which are recruited to the HIV promoter by the Tat trans-activator.14 P-TEFb is composed of a regulatory cyclin T (either CycT1, T2a, or T2b) and a Cyclin-dependent kinase 9 (Cdk9) and mediates transcription elongation by phosphorylating the C-terminal domain (CTD) of Pol II at Ser2, as well as NELF-E and Spt5 of DSIF.15-22 In cells, P-TEFb availability and activity are tightly regulated, and the kinase activity of Cdk9 is inhibited by Hexim1/2, part of the 7SK small nuclear Ribonucleo-Protein (snRNP).23-27 Viral infection, shifts cell demands for transcription elongation, triggering the release of P-TEFb from its inactive configuration by HIV Tat that with the help of SEC, extracts P-TEFb from 7SK-snRNP.28-34 In the absence of Tat, SEC and P-TEFb are recruited to gene promoters by the bromodomain-containing proteins 4 (Brd4), which tethers the host elongation machinery to acetylated chromatin.35-37 SEC also recruits P-TEFb via interactions with Med26 of the mediator,38,39 while the YEATS domain of ENL/AF9 brings SEC to chromatin via the human polymerase-associated factor complex (PAFc).40-42 Overall, depending on cell type and genes, different SEC complexes are formed, each containing one of the AF4 members and either ENL or AF9.27,43
In SEC, the AFF1-4 proteins of the AF4/FMR2 family each act as a scaffold that bridges the complex to P-TEFb, forming a bi-functional complex that synergistically triggers transcription elongation by Pol II 44-47,43,48-51 While AFF proteins form homo-dimers within SEC, they are also found as heterodimers. SECs can also form alternative complexes and include a minor complex that potentially modulates HIV latency.52 Overall, in mammals, diverse SECs are recruited by distinctive co-activators and regulate different sets of genes.53
Having a fundamental role in controlling transcription elongation, disturbance of SEC functions due to genetic alternations of its members is well documented. Of the AF4 proteins, AF4/FMR2 family members 1 and 4 are mainly implicated in HIV gene transcription control, while other members, AFF2/FRM2 and AFF3/LAF4, share similar domain organization, but are linked to other human diseases like Fragile X E (FRAXE) and Acute Lymphoblastic Leukemia.51,53 AFF members AFF1, AFF3, and AFF4 and ELL are frequent fusion partners of the mixed lineage leukemia methyltransferase (MLL) gene, and are markers for leukemia prognosis.54 MLL-SEC oncogenic chimeras are tethered to MLL target genes that control hematopoietic stem cell development, leading to mis-regulation of transcription and development of leukemia.55 Additionally, gain of function missense mutations in AFF4 have recently reported in leading to the known developmental CHOPS syndrome.7 While knowledge of the mechanisms that control eukaryotic transcription elongation and the role of SEC in this step are well established, the function of SEC when activators like Tat are absent is less defined. Moreover, knowledge of the mechanisms that govern SEC transcription activity is also limited.
In this study, we aim to better understand the role of SEC and P-TEFb in early steps of HIV transcription when Tat is not expressed. Our results show that regardless of Tat, AFF4 efficiently activates HIV gene transcription, while AFF1 activates HIV to a lesser extent. Analysis of mRNA levels, confirms these functional results, demonstrating that SEC enhances the synthesis of both short and long transcripts, with AFF4 effects are higher than those of AFF1. We also demonstrate that the distinction between AFF1 and AFF4 in their ability to activate early HIV transcription stems from their differential recruitment to the viral promoter, and from different abilities to associate with PAFc. Knockout (KO) of AFF1 or AFF4 expression slightly represses early HIV transcription in the absence of Tat. In the same conditions, KO of cyclin T1 has subtle effects on these early steps of HIV gene transcription. Upon Tat expression, HIV gene transcription reaches optimal levels despite KO of AFF1 or AFF4 expression. KO of both AFF1 and AFF4 represses HIV transcription in the absence of Tat, but, completely diminishes HIV gene transcription, when Tat is expressed. As expected, depletion of cyclin T1 expression also represses Tat transactivation. Importantly, we show that AFF4 is a substrate of P-TEFb, as inhibition of Cdk9 kinase activity by Flavopiridol, or KO of cyclin T1 expression, disrupts SEC subunit assembly, AFF1/4 dimerization, and recruitment of these proteins to the HIV promoter. Finally, AFF1 and AFF4 associate with 7SKsnRNA, and P-TEFb modulates this interaction with RNA. Our results conclude that SEC plays a key role in both early steps of HIV gene transcription, when Tat is not expressed, along with its well-established function in enhancing transcription elongation, in the presence of Tat Significantly, P-TEFb has a role in modulating SEC activity in the steps of promoter recruitment of the SEC and its assembly.
Results
AFF4 and AFF1 of SEC activate early HIV gene transcription in the absence of Tat
While the role of SEC in enhancing HIV transcription elongation, when Tat is expressed is well-established, knowledge of its function in promoting early steps of HIV transcription, when Tat is absent, is less defined. To elucidate the role of SEC in activating early HIV gene transcription, we monitored levels of HIV transcription upon over-expressing the AF4/FMR2 family members, AFF1 or AFF4, which act as scaffold of the SEC, in cells that stably express an integrated luciferase reporter gene under the regulation of the HIV-LTR promoter (HEK-LTR-Luc). HIV transcription was monitored in the presence or absence of Tat (Fig. 1A). Our functional analysis showed that in the absence of Tat, AFF4 strongly activated basal HIV gene transcription (×15 fold), while at the same conditions, AFF1 was less efficient, and its effects on Tat-independent transcription reached only ×4 fold above basal levels (Fig. 1A). To monitor effects of Tat on AFF1, or AFF4-mediated activation of HIV transcription, HEK-LTR-Luc cells were co-transfected with either AFF1, AFF4, and with HIV HA-Tat (Fig. 1A). Tat optimally enhanced HIV gene transcription (×25 fold). However, both AFF1/4 proteins displayed moderate contribution to the overall Tat transactivation, and slightly elevated levels of HIV Tat-transactivation—up to ×30 fold above basal levels. (Fig. 1A). Significantly, while Tat expression elevated AFF4-mediated activation of HIV gene transcription only ×2 fold, it had higher effects on AFF1-mediated activation—×6 fold. We assume that this result is due to the relatively initial low activation ability of AFF1 on the HIV promoter when Tat is not expressed. Upon Tat expression, HIV transcription reaches its highest levels and overexpression of SEC cannot elevate these levels. We conclude that for optimal HIV gene activation, Tat is critical. SEC also promotes early steps of HIV gene transcription—when Tat is not expressed. As AFF1 activation of HIV transcription is relatively lower compared with AFF4, enhancement effects of Tat on AFF1-mediated HIV gene transcription are higher, compared with those on the activation by AFF4 (Fig. 1A).
Figure 1.
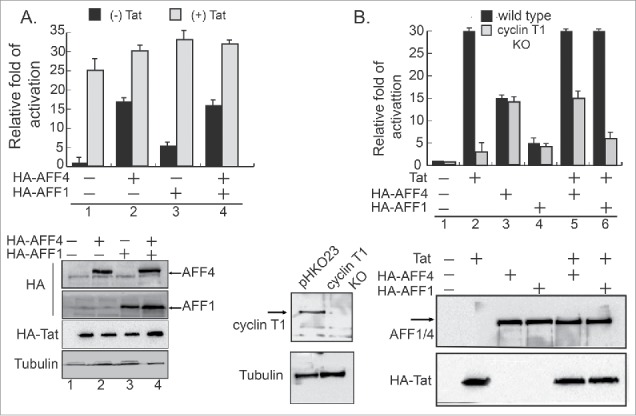
AF4/FMR2 family members 1 and 4 of SEC activate early HIV gene transcription in the absence of Tat. (A) AFF1 and AFF4 enhance HIV transcription in the absence of Tat. HEK-LTR-Luc cells stably expressing integrated HIV-luciferase were transfected either with HA-AFF4 or HA-AFF1 expressing plasmids. Cells were harvested 48 hours post transfection and their luciferase readouts were analyzed according to the manufacture protocol and normalized to protein levels. To monitor effects of Tat, cells were also transfected with HA-Tat and luciferase readings were monitored according to standard protocols. Data is a representative of three independent experiments and error bars represent standard deviation. Bottom panel presents western blot analysis for the expression levels of Tat, AFF4 and AFF1. (B) HIV transcriptional activation by AFF4 and AFF1 are independent of P-TEFb. HEK-LTR-Luc cells were depleted of cyclin T1 expression using CRISPR/Cas9-sgRNA lentiviruses. Cells expressing Cas9 and the empty sgRNA (pHKO23) were used as control. Wild type or cyclin KO cells were transfected with either HA-AFF1 or HA-AFF4, and their luciferase readings were monitored 48 hours post transfection and normalized to protein levels. Experiments were performed in the presence or absence of transiently expressed HA-Tat. Results are presented relatively to readings in cells that express LTR-Luc alone—set to 1, and are a representative of three independent experiments. Error bars represent standard deviation. Bottom panel shows western blot analysis confirming KO of cyclin T1, overexpression of HA-AFF1, and HA-AFF4 and HA-Tat.
Early steps of HIV transcription activation by AFF1/4 are independent of P-TEFb
To further investigate the role of P-TEFb in SEC-mediated activation of early HIV transcription, we monitored HIV transcription by AFF4 or AFF1 in cells where cyclin T1 expression was depleted (KO) by Cas9/CRISPR (Fig. 1B). Depletion of protein expression was confirmed both by western blot analysis (WB) (Fig. 1B), as well as by PCR and sequencing. Our results indicate that KO of cyclin T1 expression slightly repressed early HIV transcription, when Tat is not expressed. We confirmed that in the absence of Tat, AFF1 is less efficient than AFF4 in activating HIV promoter. Tat expression further elevated AFF1 or AFF4-mediated activation, but to similar levels of Tat transactivation without AFF1/4. Upon cyclin T1 KO, Tat transactivation was significantly repressed, confirming the key role of P-TEFb/cyclin T1 in HIV Tat transactivation (Fig. 1B). Significantly, KO of cyclin T1 expression had no effects on the activation of HIV transcription by AFF1 or AFF4, when Tat is not expressed. Upon Tat expression, KO of cyclin T1 abolished Tat contribution of AFF1/4-mediated HIV gene activation, bringing transcription levels to those with AFF1 or AFF4 activation alone. Our results confirm that cyclin T1 is a key player in Tat transactivation, but is not involved in early steps of HIV gene activation by AFF1 or AFF4 of SEC, when Tat is not expressed (Fig. 1B).
Effects of AFF1 and AFF4 depletion of expression on HIV transcription in the presence or absence of Tat
We also monitored the involvement of AFF4 and AFF1 in early steps of HIV transcription, when Tat is not expressed in Jurkat cells that stably express the HIV-LTR (J-LTR-Luc) (Fig. 2A). The expression of either cyclin T1, AFF4, or AFF1 scaffolds of SEC was depleted, by transducing cells with lentivirus that drive the expression of the specific CRISPR/Cas9/sgRNA. Depletion of protein expression was confirmed by WB and single clones were also sequenced to verify gene KO (Fig. 2B and Fig. S3). Our results show that KO of AFF1 or AFF4 expression repressed early steps of HIV transcription—when Tat is not expressed (×2–3-fold). In these conditions, KO of cyclin T1 had no effects on HIV gene transcription. To investigate the effects of Tat on transcription activation in cells that are depleted of cyclin T1, AFF1, or AFF4 expression, wild-type or KO J-LTR-Luc cells were further transduced with lentivirus expressing Tat (HIV-Tat-BFP), monitoring HIV transcription by analyzing luciferase output. Our results indicate that despite KO of either AFF4 or AFF1 expression, HIV Tat transactivation still reached optimal levels (Fig. 2). As expected, KO of cyclin T1 diminished Tat transactivation in Jurkat-LTR-Luc cells. Interestingly, expression of both AFF1 and AFF4 was also depleted, and activation of HIV transcription was monitored in the presence or absence of Tat. Our results show that upon KO of both AF4 proteins, early steps of transcription, in the absence of Tat, were slightly repressed, while activation of the HIV promoter by Tat was completely diminished. Importantly, effects of KO of AFF1 or AFF4 were specific to the HIV LTR promoter, as transcription from other promoters, like the EF1α or the PGK promoter, was relatively not affected despite KO of AFF4/1 (Fig. 2C).
Figure 2.
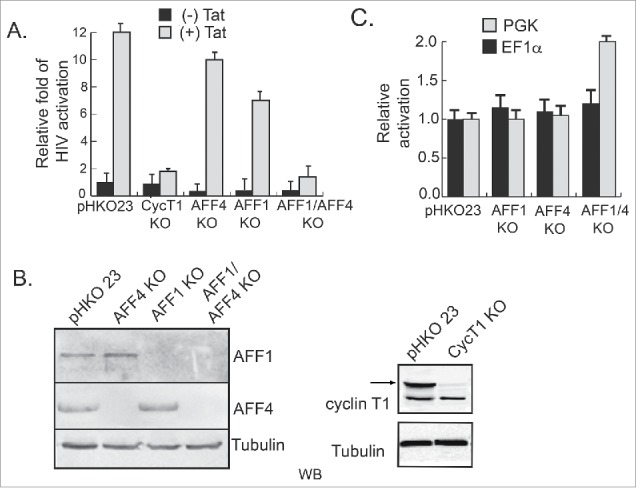
Effects of AFF1 and AFF4 depletion of expression on HIV transcription in the presence or absence of Tat. Jurkat-LTR-luciferase cells (J-LTR-Luc) that stably express an integrated luciferase reporter gene under the control of the HIV promoter, were depleted of AFF1, AFF4, or cyclin T1 expression, using lentivirus driving the expressing of Cas9 and the corresponding sgRNA. Depletion of both AFF1 and AFF4 expression was obtained by transducing puromycin-resistant AFF4 KO cells with AFF1-Cas9/sgRNA that harbors Blastocydin. Depletion of protein expression was confirmed by western blot using specific endogenous antibodies (panel B). Lentiviruses expressing Cas9 and the empty sgRNA (pHKO23) were used to generate a control cell with no sgRNA. HIV gene transcription in the presence or absence of Tat was monitored in J-LTR-Luc wild type and KO cells. Tat was introduced to cells with lentiviruses that code for Tat-BFP (Blue Fluorescent Protein). Luciferase levels were monitored 24 hours post infection and data are presented relatively to Luc readouts in J-LTR-Luc cells infected with pHKO23 lentivirus and set to 1. Luciferase readings are a representative of three independent experiments. Error bars represent standard deviation. (C) Depletion of AFF1 and AFF4 expression does not affect gene activation from PGK or EF1α promotors. Jurkat cells, where AFF1; AFF4; or AFF1/4 expression is KO were transduced with lentivirus expressing either the PGK-luciferase or the EF1α-DsRed reporter promoters. Cells were harvested 48 hour post transduction and subjected to luciferase analysis in the case of PGK-luciferase, or FACS, when EF1α-DsRed reporter was used. Data is presented relative to control cells that express only-pHKO23-CAs9—with no sgRNA and a representative of three independent experiments where error bars represent standard deviation.
P-TEFb modulates SEC assembly and phosphorylates AFF4
AFF1/4 each serves as a scaffold for the assembly of SEC subunits along with P-TEFb. We tested if P-TEFb plays a role in SEC assembly. To this aim, SEC complex assembly was monitored in cells where P-TEFb kinase activity was modulated either by Flavopiridol, or through knocking down the expression of cyclin T1 (Fig. 3A+B). In Flavopiridol treated cells, SEC assembly was disrupted, reducing protein incorporation of mainly ELL2 and Cdk9 into the complex with HA-AFF4 (Fig. 3A). Overexpressing Cdk9 also promoted SEC assembly, primarily for ELL2 (Fig. 3A). SEC assembly was also analyzed in HEK-LTR-Luc cells where cyclin T1 expression was KO using CRISPR/Cas9 approach (Fig. 3B). HA-AFF4 was overexpressed in wild-type or cyclin T1 KO cells, followed by immuno-precipitation (IP) with anti-HA IgG and WB to monitor incorporation of SEC subunits into the complex. Our WB analysis confirmed that cyclin T1 expression was KO. In cyclin T1 KO cells, SEC assembly was indeed modulated. ELL2 incorporation into SEC/AFF4 complex was slightly decreased, but to a lesser extent when seen in Flavopiridol treated cells. Similarly, upon HA-AFF4-IP, CdK9 incorporation into the SEC was also diminished in cyclin T1 KO cells, confirming that cyclin T1 may have a regulatory role in mediating SEC assembly and it also contributes to Cdk9 protein stability and incorporation into the SEC. Interestingly, levels of cyclin T2 expression were also reduced, implying that cyclin T2 might also incorporated into the SEC (Fig. 3B). We also analyzed SEC assembly upon IP of HA-AFF1. Like in the SEC/AFF4 complex, Cdk9 incorporation into the SEC/AFF1 was repressed. However, we did not visualize a difference in ELL2 incorporation between wild type or cyclin T1 KO cells. Incorporation of ELL2 was similar in wild type or cyclin T1 KO cells (Fig. S2).
Figure 3.
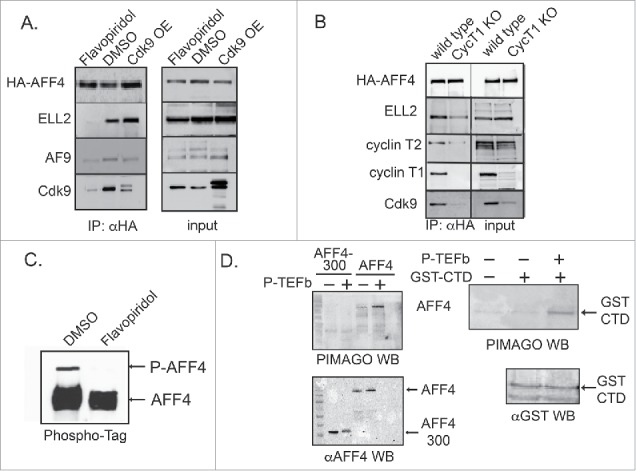
P-TEFb modulates SEC protein assembly. (A) Cdk9 kinase activity is required for the assembly of SEC Cells overexpressing HA-AFF4 were treated with Flavopiridol and subjected to immuno-precipitation with αHA IgG, followed by western blot analysis with the indicated antibodies to detect endogenous SEC and P-TEFb protein subunits. Western blot for input represent 10% of total lysate. (B) KO of cyclin T1 expression disrupts SEC/AFF4 assembly. Wild-type HEK, or cells that are depleted of cyclin T1 (cyclin T1 KO) were transfected with HA-AFF4 and subjected to immuno-precipitation with αHA IgG, followed by western blot with the indicated antibodies for detecting SEC and P-TEFb protein subunits. Western blot analysis for input represents 10% of total cell lysate. (C) AFF4 is phosphorylated by P-TEFb in cells. Phos-tag analysis of HA-AFF4 purified from control cells or cells treated with Flavopiridol. Cells were lysed 48 hours post transfection and subjected to SDS-PAGE analysis and western blot on Phos-tag acrylamide SDS-PAGE (Wako) as described. (D) AFF4 is phosphorylated by P-TEFb in cells. HA-AFF4 full-length and HA-AFF4–300 were expressed in cells, and 48 hours later were harvested and lysed. Cell lysate was subjected to immuno-precipitation with αHA and precipitated proteins were extensively washed before used as substrates for in-vitro kinase assay in the presence or absence of recombinant P-TEFb (Millipore). Recombinant GST-CTD was purified from bacteria and used as a positive control for P-TEFb phosphorylation. Proteins were analyzed on SDS-PAGE and western blot analysis using the “pIMAGO®-biotin Phosphoprotein Detection Kit” for the detection of protein phosphorylation.
To further explore the mechanisms by which P-TEFb modulates SEC function, we tested the ability of Cdk9 to phosphorylate AFF4 in-vitro and in cells. Previous results report that AFF1 is phosphorylated by cdk9, and that kinase activity of P-TEFb is enhanced by AFF1, leading to stimulation of transcription elongation and chromatin remodeling.56,57 We thus used Phos-tag analysis to detect AFF4 phopshorylation by P-TEFb in cells. Treating cells with Flavopiridol inhibited AFF4 phosphorylation by Cdk9 (Fig. 3C). In-vitro kinase assays complemented our analysis. HA-AFF4–300 or HA-AFF4-full-length were IP from cells by αHA antibody, and incubated with recombinant P-TEFb (Millipore), confirming that only full-length AFF4 is phosphorylated by P-TEFb (Fig. 3D).
P-TEFb modulates AFF1-AFF4 protein dimerization
Previous reports show that AFF1 and AFF4 dimerize.53 However, no functional significance was reported for protein dimerization. We initially confirmed AFF1 and AFF4 dimerization by IP experiments from cells that overexpress Flag-AFF1 and HA-AFF4 (Fig. 4A). To test the role of P-TEFb in AFF1/4 protein dimerization, IP experiments were performed in wild-type and in cyclin T1 KO cells (Fig. 4B). KO of cyclin T1 abolished AFF1/AFF4 dimerization, implying a role for P-TEFb in protein dimerization (Fig. 4B). However, upon treating cells with Flavopiridol, a Cdk9 kinase inhibitor, no effects on AFF1/4 protein dimerization were detected, suggesting that an active Cdk9 kinase of P-TEFb is not a required for AFF1/AFF4protein dimerization (Fig. 4C).
Figure 4.
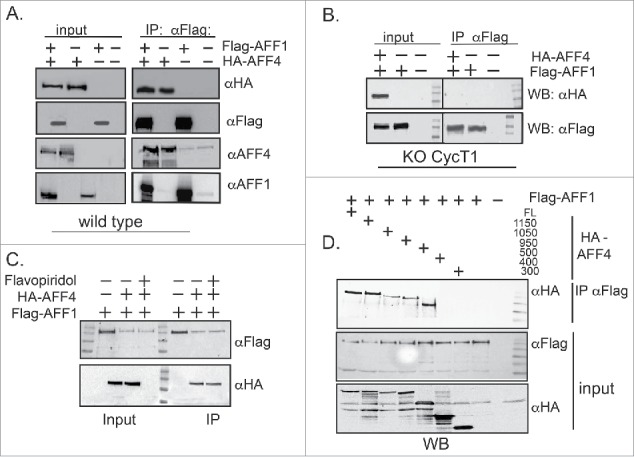
P-TEFb modulates AFF1/AFF4 dimerization. (A) AFF1–AFF4 protein dimerization HA-AFF4 and Flag-AFF1 were co-expressed in HEK293T. 48 hours post transfection, cells were lysed and subjected to immuno-precipitation with αHA IgG. IP reactions were analyzed by western blot with the indicated IgG to confirm protein association. Input represents 10% of total cell lysate. Both immuno-precipitated and input samples were also analyzed with AFF1 or AFF4 IgG, to confirm that proteins detected on the gel are indeed AFF1/4. (B) P-TEFb modulates AFF1/AFF4 protein dimerization. Cyclin T1 KO HEK cells were transfected with either HA-AFF4, Flag-AFF1, or both proteins. Cell lysate was subjected to αFlag IP followed by western blot with αHA IgG. (C) Kinase active Cdk9 is not required for AFF1 /AFF4 protein dimerization. Flavopiridol-treated cells (50 mM) were co-transfected with Flag-AFF1 and HA-AFF4, and subjected to IP with αFlag IgG. Immuno-precipitated samples were analyzed by western blot with αHA to confirm the association between AFF1 and AFF4. Input samples represent 10% of total cell lysate and were also analyzed by western blot with both αFlag and αHA IgG. (D) Domain mapping of AFF4 regions that mediate dimerization with AFF1. Flag-AFF1 was co-expressed with the indicated HA-AFF4 - C- terminal truncated mutants and subjected to IP with αFlag IgG. IP samples were analyzed with αHA to confirm association of AFF1 and AFF4 mutant proteins. Input represents 10% of total cell lysate and was also analyzed by WB with both αFlag and αHA IgG.
We further mapped the domains of AFF1 and AFF4 that mediate protein dimerization by WB analysis (Fig. 4D). To do so, HA-AFF4 truncated proteins were generated based on the published structure of AFF4 and co-expressed in cells with Flag-AFF1.45,58 Our results show that AFF4-C-terminal truncation mutants up to AFF4–500 still associated with AFF1, while shorter AFF4 protein truncated mutants could not. We analyzed these C-terminal AFF4 truncated mutants for their ability to activate HIV transcription in the presence or absence of Tat (Fig. S1A+B). Interestingly, all AFF4 protein mutants supported HIV transcription when Tat is expressed. However, in the absence of Tat, only full-length (1162 amino residues) and to a lesser extent AFF4–1157, support HIV transcription, when Tat is not expressed. These results indicate that for activation of HIV transcription, a full-length AFF4 is required, and may imply it needs to dimerize with AFF1.
AFF4 and AFF1 occupancy on the HIV promoter
Our results show that in comparison with AFF1, AFF4 efficiently activates early HIV transcription when Tat is not expressed (Fig. 1). This may suggest that different SEC configurations enhance early HIV gene transcription to different extents. To elucidate the molecular mechanisms that dictate the distinction between AFF1 and AFF4 activation, we monitored their occupancy on the HIV promoter by ChIP-qPCR in cells that equally overexpress tagged HA-AFF1 and HA-AFF4 (Fig. 5). WB analysis confirmed similar AFF1 and AFF1 protein expression levels in wild type or cyclin T1 KO cells. ChIP experiments were performed with αHA-IgG to confirm that both proteins will be similarly immuno-precipitated. Control IgG confirmed specific IP of proteins. qPCR analysis was performed with two sets of primers, corresponding to either core HIV promoter (TSS primers), or to luciferase-coding gene (TAR-Luc) (Fig. 5). Our results show that AFF4 is recruited to the HIV promoter more efficiently than AFF1 (Fig. 5B+C). This is shown for both HIV core promoter (TSS primer) and for gene-coding sequences (TAR-Luc). Upon KO of cyclin T1 expression, the occupancy of AFF1 and AFF4 to the HIV promoter was significantly diminished. Depletion of cyclin T1 expression primarily affected AFF4 occupancy, compared with that of AFF1 (Fig. 5B). Cells were also treated with Flavopiridol to monitor the requirement of the Cdk9 kinase activity to AFF1 or AFF4 recruitment to the HIV promoter. In Flavopiridol treated cells, AFF1 and AFF4 occupancy on core promoter sites at the HIV promoter were reduced, implying a regulatory role for P-TEFb in recruiting SEC to the promoter (Fig. 5D).
Figure 5.
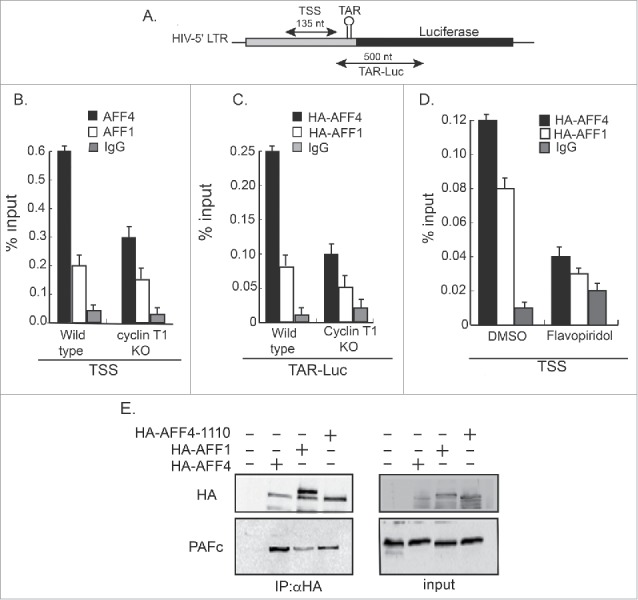
Recruitment of AFF1/4 proteins to chromatin on the HIV promoter. Wild-type, cyclin T1 KO, or Flavopiridol treated cells expressing the HIV-LTR-Luc plasmid (HEK-LTR-Luc) and either HA-AFF1, or HA-AFF4 were subjected to ChIP analysis using αHA or control IgG. Signals were obtained by qPCR using two sets of primers positioned either around transcription starting sites (TSS; B) or on the TAR and N-terminal luciferase coding gene (LTR-Luc:C). The recruitment of HA-AFF4 and HA-AFF4-to the HIV promoter was also performed upon treating cells with Flavopiridol (D). Panel (A) presents primers positions on the HIV promoter and coding region that were used for qPCR. Data is presented as percentage of input (20%). Protein inputs for both HA-AFF1 and HA-AFF4 were monitored by WB, confirming similar protein expression levels in cells that were used for the ChIP analysis. (E) Differential association of AFF1 and AFF4 with PAFc. HEK-LTR-Luc cells were transfected with HA-AFF1, HA-AFF4, or HA-AFF4–1110. Cells were harvested 48 hours post transfection and subjected to IP with αHA IgG. Western blot analysis was performed with the indicated antibodies to monitor protein association. 10% of cells lysate was used for input analysis.
To identify regions of AFF4 that control AFF4 recruitment to the HIV promoter, we used ChIP–qPCR and compared full-length AFF4 and AFF4–300 occupancy on the HIV promoter. We show that both AFF4-(1–300) and full-length AFF4 (1162 residue) similarly associate with the HIV promoter (Fig. S1C+D). Importantly, AFF4–300 (and more C-terminal AFF4 truncated mutants) did not activate HIV transcription in the absence of Tat, implying that this inability of AFF4–300 to activate HIV transcription is not caused due to differential occupancy of the protein to the viral promoter (Fig. S1 A+B).
To elucidate the molecular mechanisms that may lead to the differences between AFF1 and AFF4 to activate HIV gene transcription in the absence of Tat, we analyzed the association of AFF1 and AFF4 with P-TEFb and with PAFc that is reported to bridge the SEC to chromatin (Fig. 5E). Our analysis shows that AFF4 and AFF1 associate with PAFc. However, the association of AFF1 with PAFc is relatively weaker compared with AFF4. A shorter truncated mutant of AFF4, AFF4–1110, bound to PAFc equally well as the full length AFF4, suggesting that the C-terminus region of AFF4 is not involved in association of the SEC with chromatin (Fig. 5E).
AFF1 and AFF4 promote early steps of HIV transcription in the absence of Tat
To analyze the effects of SEC on early versus late elongation steps of HIV transcription, we monitored viral mRNA levels by qPCR, using primers located on either the promoter (short transcripts) or within luciferase gene body (long transcripts) (Fig. 6B+C). Over-expression of HA-AFF4 resulted in accumulation of short mRNA transcripts, corresponding to elevated transcription initiation (×15 fold). In the same conditions, AFF4 also enhanced the production of long transcripts, but to a lesser extent (×6 fold above basal levels). A similar pattern was also observed for AFF1. However, in this case, overall quantitated mRNA levels were lower than those detected for AFF4, both at early or late elongation steps of transcription. These results correspond with the lower activation seen for AFF1 (Fig. 1). KO of AFF1 or AFF4 expression abolished HIV transcription activation as detected by a decrease in accumulated of both long or short transcripts (Fig. 6C), corresponding to the decreased levels of HIV gene activation measured as luciferase readings in AFF1 or AFF4 KO cells (Fig. 1).
Figure 6.
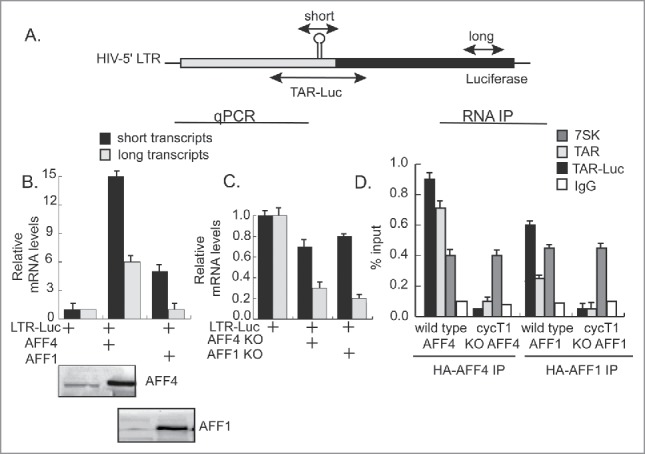
SEC promotes both initiation and elongation of transcription and is recruited to the HIV promoter via RNA. (A) Scheme of the HIV-Luc cassette and location of primers used for qPCR analysis and RNA IP. (B and C) In the absence of Tat, AFF1 and AFF4 promote initiation rather than elongation of transcription. RNA was isolated from cells over-expressing either HA-AFF1 or HA-AFF4 (panel B), or from cells where the corresponding protein expression was KO (C). Following cDNA synthesis, mRNA was amplified with the designated primers corresponding to either short (core promoter sites) or long transcripts, (luciferase coding genes). Results are shown as fold of mean enrichment relatively to data obtained in cells expressing LTR-Luciferase alone—set to 1. (D) P-TEFb modulates the association of SEC with HIV RNA. Cells expressing wild type or cyclin T1 KO cells were transfected with HIV LTR-Luciferase and subjected to RNA-immuno-precipitation (IP) with either αHA IgG, corresponding to HA-AFF1 and HA-AFF4. Non-immune human IgG served as control. RNA was extracted from immuno-precipitated and input samples (1%) and was then subjected to cDNA synthesis, which was further analyzed by qPCR using the indicated primers, located on either, short (core promoter sites) or TAR-Luc, located on both the TAR and N-terminal luciferase sequences. Primers that measured association to 7SK snRNA were also tested. qPCR reactions were performed in triplicates and presented as percentage of input over non-specific IgG.
AFF1 and AFF4 are recruited to the HIV promoter via RNA and associate with the 7SKsnRNA
SEC was previously reported to interact with P-TEFb and to facilitate Tat ability to extract P-TEFb from 7SKsnRNP, shifting P-TEFb equilibrium to TAR and enhancing transcription elongation.59,60 We tested the role of P-TEFb in the association of AFF1 and AFF4 with RNA by using RNA-IP in wild-type and cyclin T1 KO cells (Fig. 6D). We confirmed that both AFF1 and AFF4 associate with the 7SK snRNA. Furthermore, AFF4 associates more efficiently than AFF1 with the viral TAR—detected both on core promoter sites, as well as gene body sequences. Importantly, KO of cyclin T1 expression abolished association of AFF4 and AFF1 with RNA sequences, suggesting that P-TEFb is involved in controlling interactions of SEC with HIV RNA (Fig. 6D). KO of cyclin T1 expression had no effects on the association of AF4 proteins with the 7SK snRNA. We conclude that AF4 proteins associate onto the 7SK snRNA and the viral RNA and P-TEFb modulates this interaction onto the HIV RNA.
Discussion
The role of P-TEFb and SEC in stimulating transcription elongation has been well studied in recent years. SEC and its P-TEFb partner are recruited to gene promoters by activators that enhance the release of paused Pol II at promoter-proximal sites. The unique case of HIV is well-described, as it is heavily depended on the transcription function of SEC and P-TEFb. The Tat trans-activator acts as a master regulator that directly tethers the transcription elongation machinery to the viral promoter, promoting RNA Pol II release and subsequently transcription elongation. SEC increases the processivity of Pol II and overall enhance transcription elongation, while Cdk9 joins and phosphorylates the CTD of Pol II and pausing-inducing factors DSIF and NELF. Nevertheless, while the role of SEC in enhancing transcription elongation is well defined, its involvement in the early steps of transcription, where activators like Tat is not expressed, is less clear. Moreover, the mechanisms that govern SEC function are not fully understood.
Our current study uses HIV as a model for studying transcription control, and exploits genetic and pharmacologic tools to elucidate the function of SEC in enhancing both early steps of HIV transcription, when Tat is not expressed, as well as elongation of transcription when Tat and the elongation machinery are suited on the promoter. Early work on the control of transcription of HIV laid the foundation for our current knowledge of how RNA Pol II is released from promoter-proximal pausing sites. Herein, the expression of key players of the transcription elongation machinery, namely, P-TEFb/cyclin T1 and AFF1 or AFF4 of SEC was modulated, and the activation of HIV transcription was monitored in the presence or absence of Tat. Our results show that both AFF4, and to a lesser extent AFF1, efficiently activate HIV transcription in the absence of Tat (Fig. 1A). Significantly, when Tat is not expressed, AFF4 is more efficient in activating HIV transcription than AFF1. We find that this distinction between the two proteins stems from the differential occupancy of AFF1 versus AFF4 on the HIV promoter (Fig. 5). Furthermore, AFF4 association with PAFc is stronger than that of AFF1, providing an additional molecular explanation for its higher ability to activate HIV transcription in the absence of Tat (Fig. 5E).40
The expression of AFF1 or AFF4 was also depleted in Jurkat T cells that harbor integrated proviral HIV. We show that AFF1 or AFF4 KO has minor effects on HIV transcription in the presence of Tat (Fig. 2A), implying that these two proteins can complement each other for supporting Tat transactivation. However, when the expression of both subunits of SEC is depleted, Tat transactivation is completely diminished. (Fig. 2). This points to the key role of SEC in enhancing Tat-mediated transcription of HIV. In basal conditions when Tat is not expressed, KO of expression of AFF1, AFF4 or both subunits repress HIV transcription (Fig. 2A). This demonstrates that when Tat is not expressed, SEC contribution is also important.
In agreement with our results, previous work defines the minor subset of SEC in promoting Tat-dependent HIV gene transcription. Depletion of SEC proteins, mainly AFF1, reduces latency reversal driven by JQ1, potentially by repressing transcription elongation, which is Tat dependent. In contrast, PMA or prostratin function via PKC-NFκB, thus less rely on SEC.52 Our work further extends this study and demonstrates that in conjugation with the requirement of SEC in transcription elongation (which is Tat-dependent), AFF1/4 enhance initial steps of HIV transcription, which are Tat-independent. Moreover, we show that in these Tat-free conditions, it is the recruitment of PAFc by AFF4 and (to a lesser extent) by AFF1, that drive transcription. We speculate that targeting these initial steps of HIV transcription might also allow the modulation of viral latency and promote HIV exist form this stage of transcription silencing.
Our results also confirm that in the absence of Tat, KO of cyclin T1 expression has only minor effects on the ability of AFF1 or AFF4 to support early HIV transcription activation (Fig. 1B). As expected, KO of cyclin T1 expression completely represses HIV Tat transactivation. These results suggest that P-TEFb is less required for early steps of transcription when Tat is not expressed, and it is SEC that plays a more major role in enhancing transcription in the absence of Tat (Fig. 2). Nevertheless, alternative P-TEFb configurations (Cdk9/CycT2a or T2b or CycK) may be recruited to the HIV promoter and support basal transcription steps in the absence of Tat despite KO of CycT1. Additionally, SEC by itself may bring P-TEFb to the viral promoter. Additional experiments will determine which of these P-TEFb configurations play a role in SEC gene activation and if Cdk9 is actually playing role in basal gene transcription, when Tat is absent. (Figs. 1B and 2).
Previous work from the Zhou laboratory also defined SEC as an essential factor for Tat transactivation and transcription elongation.52,53 Our study complements this work and confirms that SEC is also a key player in early steps of HIV transcription, and efficiently activates the HIV promoter without Tat. Their work also describes the potency of SEC/AFF1 in supporting HIV Tat transactivation relative to that of AFF4, and suggests that this property of AFF1 is due to its elevated affinity for Tat and P-TEFb relative to AFF4.53 Our study however demonstrates that AFF1 is less efficient in stimulating HIV transcription without Tat. In this setting, AFF4 is more potent in activating HIV transcription. Thus, the synergistic activation effects attributed for AFF1 on HIV Tat-mediated transcription, stem from its initial low ability to activate viral transcription when Tat is not expressed, compared with AFF4 (Fig. 5).
We demonstrate that P-TEFb plays a key role in modulating SEC function. P-TEFb mediates the recruitment of SEC to the HIV promoter, the dimerization of AFF4 and AFF1 scaffolds, the assembly of the SEC subunits, and the association of AFF1/4 with viral RNA (Figs. 3–6). KO of cyclin T1 represses AFF1/4 occupancy on the HIV promoter (Fig. 5), slightly modulates assembly of SEC subunits (Fig. 3), and dictates AFF1/4 dimerization (Fig. 4). Treating cells with Flavoliridol, exerts similar effects on AFF1/4 recruitment to the HIV promoter (Fig. 5) and on SEC assembly (Fig. 3). This is a first demonstration of a unique cyclin T1 function, which cyclin T2 cannot complement. The only additional example of such a distinction is the solely dependence of Tat transactivation in cyclin T1. However, KO of cyclin T1 only slightly represses AFF1, or AFF4-mediated activation of HIV gene transcription (Fig. 1B). In this case, alternative P-TEFb configurations might complement cyclin T1 depletion and supporting SEC functions. Upon Tat expression, P-TEFb regulatory role becomes highly critical, as KO of cyclin T1, which directly and uniquely binds Tat, completely abolishes Tat transactivation (Fig. 1B). Our work also confirms AFF1 and AFF4 dimerization,53 and demonstrates that P-TEFb modulates this step. Depletion of cyclin T1 expression disrupts AFF1/AFF4 dimerization (Fig. 4B). However, the kinase activity of Cdk9 is not required for protein dimerization, as Flavopiridol cannot inhibit protein dimerization (Fig. 4C). As Flavopiridol essentially halts all RNA Pol II transcription, and its specificity is questionable, other kinases may be tested for a possible regulatory role in SEC activity. Moreover, more specific Cdk9 inhibitors are currently being tested to confirm the requirement of Cdk9. We also show that AFF4, is a Cdk9 substrate verifying previous work that reported AF4 phosphorylation (56,57) (Fig. 3). Interestingly, we also defined the C-terminal region of AFF4 to mediate association with AFF1, as well as required for HIV transcription in the absent of Tat (Fig. 4; Fig. S1).
We conclude that P-TEFb phosphorylation of AFF1 and AFF4 modulates SEC assembly and recruitment to the viral promoter, presumably via PAFc binding. As Cdk9 kinase activity is not required for AFF1/4 dimerization, protein phosphorylation by Cdk9 is not involved in protein dimerization.
Finally, we demonstrate that SEC is recruited to chromatin on the HIV promoter via RNA (Fig. 6). AFF1 and AFF4 associate with RNA near TSS, and on coding regions. AFF4 associates with TAR and less efficiently with the abundant 7SK snRNA, (Fig. 6).60 While P-TEFb is not required for SEC association with 7SK snRNA, it is critical for RNA binding to TAR and core promoter regions. KO of cyclin T1 expression disrupts SEC interactions with TAR RNA, implying the regulatory role of P-TEFb in SEC functions (Fig. 6C). We are perusing additional experiments to validate the role of nascent RNA in enhancement of Pol II release and elongation of transcription.
Overall, this work highlights the key functions of SEC in controlling of both initiation and elongation of HIV gene transcription in the presence or absence of Tat. Moreover, within SEC, P-TEFb plays a key role in stimulating elongation of transcription through Tat, while also contributing to the early steps of transcription when Tat is not expressed. KO of AFF1 and AFF4, or cyclin T1 enabled us to dissect the contribution of each factor to the different steps of transcription. Importantly, P-TEFb modulates SEC functions. As HIV placed the foundations to our current knowledge of how Pol II pausing and release are regulated, we expect that this work will set way to the role of SEC/P-TEFb genome-wide, both when activators are expressed or stimuli are imposed and during a restating state of cells.
Experimental procedures
Cell lines
Human Embryonic 293T Kidney cells that express an integrated HIV-LTR-Luciferase (HEK-LTR-Luc) were maintained in Dulbecco's Modified Eagle's Medium (DMEM; Gibco), supplemented with 10% fetal bovine serum (Gibco), 2mg/mL L-glutamine, penicillin-streptomycin, and non-essential amino acids (Sigma, M7145). Similarly, Jurkat T cells stably expressing the integrated LTR-GFP and the LTR-Luciferase (Jurkat-LTR-Luc) were maintained in complete RPMI media. Cells were cultured at 37°C with 5% CO2.
Plasmids
HA-AFF4 and Flag-AFF1 expression plasmids were obtained from the Zhou laboratory (UC Berkeley). AF4 members were also sub-cloned into a lentiviral vector expressing an HA tag at their N-terminus. HA-Tat was kindly provided by the Peterlin laboratory, UCSF.
Transfections and luciferase reporter assays
HEK-LTR-Luc cells were co-transfected with the indicated expression plasmids using lipofectamine. 48 hours post transfection, cells were harvested and their luciferase activity was measured according to the manufacture manual (Promega). Luciferase readings were normalized to Renilla expression and are presented relative to the readings obtained in cells that expressed wild-type cells—set to 1. Similar protocol was performed to analyze transcription activation from other promoters—PGK-Luciferase or EF1α–DsRed. In this case, lentiviruses were used to transduce target cells at an MOI of 1. Luciferase readings were monitored as described, while DsRed expression levels were detected by FACS. Results are representative of the mean value of triplicate wells; error bars show ± SEM.
CRISPR mediated gene silencing
For KO of protein expression, Jurkat-LTR-Luc or HEK-LTR-Luc cells were infected with lentiviral vectors expressing the Cas9/sgRNA for KO cyclin T1; AFF1; or AFF4 (Addgene #49535). To KO, the expression of each protein, a mix of three different sgRNA oligos were used and cloned into the lentiviral vector. VSV-G pseudotyped lentiviruses were generated as described below and used for infection. Infected cells were further selected with media supplemented with 1–2 μg/mL of puromycin, and single clones were isolated by limiting dilution before expanded. KO of protein expression was validated by WB with specific antibodies. Two separate clones were further sequenced independently to confirm mutations in the specific reading frame of the gene. Genomic DNA from selected clones, where cyclin T1, AFF4, or AFF1, expression was depleted by CRISPR, was extracted and fragments around the sgRNA editing sites were amplified using Pfu-Taq polymerase. A-tails were added to the PCR fragments with Taq polymerase and the fragments were further cloned into pCDNA–TOPO before sent for sequence analysis.
Production of pseudotyped lentivirus and infection
HEK293T cells were transfected with packaging HIV expression plasmids, pGag-Pol; pRT; pTat; pREv; and the env VSV-G. In the case of generating KO cells, a lentivector expressing Cas9/sgRNA was used. For the production of a Tat-expressing virus (HIV-Tat-BFP), the HIV-LTR-Tat and the blue-florescence protein (BFP) was used as trans-vector. Cell supernatant containing virus was collected 48 hours post transfection, spinned down at 2000 rpm for 10 minutes and then filtered through a 0.45 u and aliquot for further experiments.
For measuring cell viability or percentage of infectivity by FACS analysis, cells were infected with the indicated VSV-G pseudotyped viruses, pHR-LTR-DsRed; HIV-Tat-BFP; and CMV-GFP.
Protein immune-precipitation antibodies and binding experiments
HEK-293T cells grown in 10 cm plate were transfected with 10 μg of the indicated AFF1 plasmid, using Lipofectamine (Invitrogen). 48 hours post transfection, cells were harvested and lysed with 500 μL lysis buffer (0.15% Triton; 20 mM Tris-Cl pH 7.6; 200 mM NaCl; 0.72 mM EDTA; 10% Glycerol; 1mM DTT, protease inhibitors cocktail (Sigma; 1:200 dilution)). Lysates were incubated on ice for 1 hour and then and centrifuged at 14000 rpm for 10 minutes at 4°C. Cleared supernatants were then incubated overnight with gentle rocking with 1 μg of α-HA antibody (Abcam ab-9110) or αFlag antibody (sigma). 50 μL was taken of the lysis for input analysis. Following, lysates were incubated with BSA-pre-blocked protein A-sepharose beads (Invitrogen) at 4°C for 2 hours with gentle rocking. Beads were extensively washed ×3 times with washing buffer (lysis buffer containing 0.05% Triton), and were then precipitated by centrifugation at 3000 rpm at 4°C for 5 minutes. IP proteins were heated at 95°C for 5 minutes in Laemmli sample buffer and resolved by SDS-PAGE, followed by WB using the indicated antibodies: αAFF4 (abcam; ab57077); αFlag (M2-Sigma; A2220); αAFF1 (A302–344A; Bethyl laboratories); αCyclin T1 (abcam; ab27963); αELL2 antibody (abcam; ab115027); αAF9 antibody (abcam; ab154492); αPAFc (rabbit; ab20662). Secondary antibodies conjugated to HRP were from Jackson immune-research laboratories; α-mouse (Jackson-115–035–062); α-rabbit (Jackson- 111–035–045); α-sheep 313–035–045.
In vitro kinase assays
10 cm plates of wild type or cycT1 KO HEK 293T cells were transfected with 10 μg of the indicated plasmids (either HA-AFF4 full length, or HA-AFF4 1–300) using Lipofectamine 2000 (Invitrogen). 2 μg of GFP were added to the transfection mixture for monitoring transfection efficiency. 48 hours post transfection cells were harvested and washed twice with PBS and lysed with 500 μL optimized IP buffer containing 0.15% Triton, 20 mM Tris-Cl pH 7.6, 200 mM NaCl, 0.72 mM EDTA, 10% Glycerol, 1mM DTT, protease inhibitors cocktail 1:50 (Sigma), NaVo4 0.5M, NaF1. Cells were harvested and incubated for 30 minutes on ice, following by centrifugation at 14000 rpm for 10 minutes at 4°C. Supernatant was then collected and proteins were IP with 1 μg of anti-HA (Abcam) antibody overnight at 4°C on a rocker. 50 μL before antibody was collected for input control. The next day 50 μL of BSA pre-blocked protein A beads were added and incubated for additional 2 hours at 4°C on a rocker. Beads were washed ×3 times with washing buffer, centrifuged for 3 minutes at 3000 rpm at 4°C. Beads samples were then divided into two aliquots and re-suspended with 40 μL kinase buffer A containing 200 μM ATP, with or without recombinant P-TEFb (50 ng; Millipore). Kinase reactions were incubated for 1 hour at 30°C and stopped by adding sample buffer and boiling at 98°C for 5 minutes. Protein phosphorylation was monitored by Phos-Tag acrylamide (Wako) analysis, which provides a phosphate affinity SDS-PAGE for mobility shift detection of phosphorylated proteins.
pIMAGO kinase analysis
In vitro kinase samples were boiled at 98°C for 5 minutes with sample buffer and loaded onto an SDS PAGE acrylamide gels. Gels were transferred to a PVDF membrane, which was pre activated with methanol. Membranes were blocked with ×1 blocking buffer for overnight at 4°C, and further incubated at room temperature for 1 hour with ×1 pIMAGO buffer mixed with pIMAGO reagents as described in the manufacturer protocol (pIMAGO®-biotin Phosphoprotein Detection Kit for WB HRP-based detection (ECL)). This kit is a universal phospho-protein detection technology that enables sensitive and specific recognition of phosphorylated protein residues. Membranes were washed ×3 with washing buffer and once with ×1 TBST for 5 minutes each. Avidin-HRP mixed in blocking buffer was added to the membrane and incubated for 1 hour, followed by ×3 washing steps, 5 minutes each with ×1 TBST. Membranes were analyzed using in Chemi DOC imager (Bio-Rad).
Protein purification
pGEX GST-CTD plasmid was transformed into BL21 bacteria, and the fallowing day a single colony was picked and transferred into a 2 mL LB-Amp starter growth media, followed by growth in a 50 mL media for overnight growth. The growing culture then transferred into 200 mL LB+amp media and incubated, while shaking, until reached to 0.6–0.8 O.D. Protein expression was induced by adding 100 mM IPTG, and the culture was further grown for 3 hours. Bacterial culture was harvested by centrifugation at 5000 rpm for 10 minutes 4°C and the pellet was then re-suspended in 4 mL of EBC-DTT buffer [50 mM Tris-Cl (PH 8),120 mM NaCl, 0.5% NP-40, 5mM DTT and supplemented with protein inhibitors.1;50] Cells were lysed by adding 2 mg/mL lysozyme while placed on ice for 15 minutes. Next, cell pellet was sonicated for 30 seconds in a four-time cycle interval, while kept on ice. Following, sonicated cells were centrifuged for 15 minutes at 4000 rpm at 4°C. In a clean 15 mL tube, the supernatant was then diluted with 4 mL EBC-DTT buffer (supplemented with PI 1:50), and 500 μL of equilibrated GST beads were added for further 2-hour incubation (gentle shaking at 4°C). Beads were precipitated by centrifugation at 3000 rpm at 4°C, and further washed ×3 with EBC-DTT buffer. Elution was performed with buffer containing 50 Mm Tris pH 8, 120 mM NaCI, mM DTT, 10 mM glutathione added fresh for 2 hours on rocker at 4°C. Purified protein was collected liquated and stored in −80°C.
Chromatin immuno-precipitation analysis (ChIP)
Protein-DNA complexes were cross-linked with 1% formaldehyde for 10 minutes, washed twice with phosphate buffered saline (PBS). Cell cross-link was stopped by adding glycine (0.125 M; 5 minutes), followed by washing with PBS and then lysis in 0.5 mL lysis buffer (50 mM HEPES pH 7.5, 140 mM NaCl, 1% Triton ×100, 1 mM EDTA, 0,1% SDS and 1% protease inhibitor cocktail; 10 minutes on ice). Nuclear pellet DNA was fragmented by sonication with following settings: amplitude 40%, for 10 cycles 20 seconds on/40 seconds off )Sonics Vibra Cell(. Samples were centrifuged (15 minutes, 14000 rpm, 4°C) and the soluble chromatin fraction (50 μg) was collected and immune-precipitated overnight with an HA-IgG targeting HA-AFF1 or HA-AFF4 (5 μg). Precipitated DNA fragments were extracted with phenol–chloroform and amplified by real-time qPCR with the specific designated primers—short primers around core promoter sites; TAR-Luc that covered both TAR and terminal luciferase gene. All signals were normalized to the input DNA. For each reaction, signals generated by non-specific IgG in the control IP were performed.
RNA precipitation and analysis of long and short transcripts
HEK-293T wild type of cyclin T1 KO cells were grown on 10 cm plates and were transfected with 3 μg of HIV LTR-Luc. Cells were lysed with 800 μL of RNA-IP buffer (0.5% NP-40; 20 mM HEPES pH 7.8; 100 mM KCl; 0.2 mM EDTA) supplemented with RNAseA inhibitors (NEB MO314S) and protease inhibitors (Sigma), added fresh before use. Cell lysates were incubated on ice for 10 minutes and centrifuged at 5000 ×g for 5 minutes at 4°C. Supernatants were collected and then divided into two aliquots. The first was incubated overnight with gentle rocking with 1 μg of the HA-IgG antibody, and the second was incubated with a control non-immune anti-human IgG. Next, protein A-Sepharose beads were pre-blocked with BSA and yeast tRNA and then were added for additional 2 hours at 4°C. 50 μL of cell lysate was collected for input and stored at −80°C until RNA extraction. Beads were then extensively washed with washing buffer containing; 0.1% NP-40; 20 mM HEPES pH 7.8; 100 mM KCl; 0.2 mM EDTA; PtdIns cocktail and RNAse inhibitor. Washing was performed for 10 minutes with gentle rocking at 4°C, while Eppendorf tubes were replaced at least once to avoid unspecific RNA binding to the tubes. Following wash, beads were centrifuged 3000 rpm for 3 minutes. Beads were re-suspended in 100 μL of lysis buffer and extracted with 100 μL TriReagent (Sigma) followed by ethanol precipitation. Input samples were extracted using the same protocol. Total precipitate RNA concentrations were measured and 1 μg was used for the synthesis of cDNA, using cDNA high capacity kit (Applied Bio-system) in a total volume of 20 μL. RNA concentrations of inputs were measured and 1 μg was used for the synthesis of cDNA. Samples were further analyzed by real-time PCR analysis using KAPA Syber Green fast mix with the indicated primers. 2 μL of 1:10 and 1:100 dilution of input were used to generate a standard curve and determine the quality control of the reaction. 2 μL of undiluted IP samples were used as templates. Reactions were analyzed in triplicates and presented as percentage of input. Indicated primers targeted 7SK snRNA; TAR (short primers), TAR-Luc located on both TAR sequences and N-terminal luciferase sites, and primers located around luciferase coding genes (long). For analysis of mRNA levels, RNA purification and cDNA synthesis steps were directly performed in the indicated cells directly or after transfection of HA-AFF1 and AFF4. qPCR analysis was performed with the indicated long (coding regions) and short (TAR) primers to differentiate initiation versus elongation of transcription.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work is supported by grants to Ran Taube from the Israel Cancer Research Foundation (ICRF) and the Leukemia Research Foundation (LRF). Part of this work is also supported by the Israel Cancer Association (ICA), as well as funds from Ben-Gurion University of the Negev (BGU) Israel and the Faculty of Health Sciences at BGU.
Authors' contribution
A.K and S.K. performed all the described experiments and equally contributed to this work. A.K. and R.T. designed the scope of the study, analyzing data, and helped in writing and revising the manuscript. The authors thank other members of the Taube laboratory in reading the manuscript and sharing their input of the work.
References
- [1].Jonkers I, Lis JT. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 2015; 16:167-177; PMID:25693130; https://doi.org/ 10.1038/nrm3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kwak H, Lis JT. Control of transcriptional elongation. Annu Rev Genet 2013; 47:483-508; PMID:24050178; https://doi.org/ 10.1146/annurev-genet-110711-155440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Smith E, Shilatifard A. Transcriptional elongation checkpoint control in development and disease. Genes Dev 2013; 27:1079-1088; PMID:23699407; https://doi.org/ 10.1101/gad.215137.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell 2006; 23:297-305; PMID:16885020; https://doi.org/ 10.1016/j.molcel.2006.06.014 [DOI] [PubMed] [Google Scholar]
- [5].Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 2012; 13:720-731; PMID:22986266; https://doi.org/ 10.1038/nrg3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Conaway JW, Conaway RC. Transcription elongation and human disease. Annu Rev Biochem 1999; 68:301-319; PMID:10872452; https://doi.org/ 10.1146/annurev.biochem.68.1.301 [DOI] [PubMed] [Google Scholar]
- [7].Izumi K, Nakato R, Zhang Z, Edmondson AC, Noon S, Dulik MC, Rajagopalan R, Venditti CP, Gripp K, Samanich J et al.. Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin. Nat Genet 2015; 47:338-344; PMID:25730767; https://doi.org/ 10.1038/ng.3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Natarajan M, Schiralli Lester GM, Lee C, Missra A, Wasserman GA, Steffen M, Gilmour DS, Henderson AJ. Negative elongation factor (NELF) coordinates RNA polymerase II pausing, premature termination, and chromatin remodeling to regulate HIV transcription. J Biol Chem 2013; 288:25995-26003; PMID:23884411; https://doi.org/ 10.1074/jbc.M113.496489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Core LJ, Waterfall JJ, Gilchrist DA, Fargo DC, Kwak H, Adelman K, Lis JT. Defining the status of RNA polymerase at promoters. Cell Rep 2012; 2:1025-1035; PMID:23062713; https://doi.org/ 10.1016/j.celrep.2012.08.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fromm G, Gilchrist DA, Adelman K. SnapShot: Transcription regulation: pausing. Cell 2013; 153:930–930 e931; PMID:23663787; https://doi.org/17994021 10.1016/j.cell.2013.04.011 [DOI] [PubMed] [Google Scholar]
- [11].Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, Adelman K. RNA polymerase is poised for activation across the genome. Nat Genet 2007; 39:1507-1511; PMID:17994021; https://doi.org/ 10.1038/ng.2007.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gilchrist DA, Nechaev S, Lee C, Ghosh SK, Collins JB, Li L, Gilmour DS, Adelman K. NELF-mediated stalling of Pol II can enhance gene expression by blocking promoter-proximal nucleosome assembly. Genes Dev 2008; 22:1921-1933; PMID:18628398; https://doi.org/ 10.1101/gad.1643208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fuda NJ, Ardehali MB, Lis JT. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 2009; 461:186-192; PMID:19741698; https://doi.org/ 10.1038/nature08449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998; 92:451-462; PMID:9491887; https://doi.org/ 10.1016/S0092-8674(00)80939-3 [DOI] [PubMed] [Google Scholar]
- [15].Li J, Liu Y, Rhee HS, Ghosh SK, Bai L, Pugh BF, Gilmour DS. Kinetic competition between elongation rate and binding of NELF controls promoter-proximal pausing. Mol Cell 2013; 50:711-722; PMID:23746353; https://doi.org/ 10.1016/j.molcel.2013.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem 1995; 270:12335-12338; PMID:7759473; https://doi.org/ 10.1074/jbc.270.21.12335 [DOI] [PubMed] [Google Scholar]
- [17].Yamaguchi Y, Wada T, Watanabe D, Takagi T, Hasegawa J, Handa H. Structure and function of the human transcription elongation factor DSIF. J Biol Chem 1999; 274:8085-8092; PMID:10075709; https://doi.org/ 10.1074/jbc.274.12.8085 [DOI] [PubMed] [Google Scholar]
- [18].Eick D, Geyer M. The RNA polymerase II carboxy-terminal domain (CTD) code. Chemical reviews 2013; 113:8456-8490; PMID:23952966; https://doi.org/ 10.1021/cr400071f [DOI] [PubMed] [Google Scholar]
- [19].Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol Cell Biol 2004; 24:787-795; PMID:14701750; https://doi.org/ 10.1128/MCB.24.2.787-795.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kim JB, Sharp PA. Positive transcription elongation factor B phosphorylates hSPT5 and RNA polymerase II carboxyl-terminal domain independently of cyclin-dependent kinase-activating kinase. J Biol Chem 2001; 276:12317-12323; PMID:11145967; https://doi.org/ 10.1074/jbc.M010908200 [DOI] [PubMed] [Google Scholar]
- [21].Lis JT, Mason P, Peng J, Price DH, Werner J. P-TEFb kinase recruitment and function at heat shock loci. Genes Dev 2000; 14:792-803; PMID:10766736. [PMC free article] [PubMed] [Google Scholar]
- [22].Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, Handa H. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell 2006; 21:227-237; PMID:16427012; https://doi.org/ 10.1016/j.molcel.2005.11.024 [DOI] [PubMed] [Google Scholar]
- [23].Michels AA, Fraldi A, Li Q, Adamson TE, Bonnet F, Nguyen VT, Sedore SC, Price JP, Price DH, Lania L et al.. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J 2004; 23:2608-2619; PMID:15201869; https://doi.org/ 10.1038/sj.emboj.7600275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nguyen VT, Kiss T, Michels AA, Bensaude O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 2001; 414:322-325; PMID:11713533; https://doi.org/ 10.1038/35104581 [DOI] [PubMed] [Google Scholar]
- [25].Yang Z, Zhu Q, Luo K, Zhou Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 2001; 414:317-322; PMID:11713532; https://doi.org/ 10.1038/35104575 [DOI] [PubMed] [Google Scholar]
- [26].Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell 2003; 12:971-982; PMID:14580347; https://doi.org/ 10.1016/S1097-2765(03)00388-5 [DOI] [PubMed] [Google Scholar]
- [27].Zhou Q, Li T, Price DH. RNA polymerase II elongation control. Annu Rev Biochem 2012; 81:119-143; PMID:22404626; https://doi.org/ 10.1146/annurev-biochem-052610-095910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sano M, Wang SC, Shirai M, Scaglia F, Xie M, Sakai S, Tanaka T, Kulkarni PA, Barger PM, Youker KA et al.. Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure. EMBO J 2004; 23:3559-3569; PMID:15297879; https://doi.org/ 10.1038/sj.emboj.7600351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Barboric M, Yik JH, Czudnochowski N, Yang Z, Chen R, Contreras X, Geyer M, Matija Peterlin B, Zhou Q. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res 2007; 35:2003-2012; PMID:17341462; https://doi.org/ 10.1093/nar/gkm063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bartholomeeusen K, Xiang Y, Fujinaga K, Peterlin BM. BET bromodomain inhibition activates transcription via a transient release of P-TEFb from 7SK snRNP. J Biol Chem 2012; 19:36609-36616; PMID:22952229; https://doi.org/23255218 10.1074/jbc.M112.410746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, Aull K, Li PC, Planelles V, Bradner JE et al.. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013; 12:452-462; PMID:23255218; https://doi.org/ 10.4161/cc.23309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].He N, Pezda AC, Zhou Q. Modulation of a P-TEFb functional equilibrium for the global control of cell growth and differentiation. Mol Cell Biol 2006; 26:7068-7076; PMID:16980611; https://doi.org/ 10.1128/MCB.00778-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Krueger BJ, Varzavand K, Cooper JJ, Price DH. The mechanism of release of P-TEFb and HEXIM1 from the 7SK snRNP by viral and cellular activators includes a conformational change in 7SK. PLoS One 2010; 5:e12335; PMID:20808803; https://doi.org/ 10.1371/journal.pone.0012335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Schulte A, Czudnochowski N, Barboric M, Schonichen A, Blazek D, Peterlin BM, Geyer M. Identification of a cyclin T-binding domain in Hexim1 and biochemical analysis of its binding competition with HIV-1 Tat. J Biol Chem 2005; 280:24968-24977; PMID:15855166; https://doi.org/ 10.1074/jbc.M501431200 [DOI] [PubMed] [Google Scholar]
- [35].Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 2005; 19:523-534; PMID:16109376; https://doi.org/ 10.1016/j.molcel.2005.06.027 [DOI] [PubMed] [Google Scholar]
- [36].Schroder S, Cho S, Zeng L, Zhang Q, Kaehlcke K, Mak L, Lau J, Bisgrove D, Schnolzer M, Verdin E et al.. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J Biol Chem 2012; 287:1090-1099; PMID:22084242; https://doi.org/ 10.1074/jbc.M111.282855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 2005; 19:535-545; PMID:16109377; https://doi.org/ 10.1016/j.molcel.2005.06.029 [DOI] [PubMed] [Google Scholar]
- [38].Takahashi H, Parmely TJ, Sato S, Tomomori-Sato C, Banks CA, Kong SE, Szutorisz H, Swanson SK, Martin-Brown S, Washburn MP et al.. Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell 2011; 146:92-104; PMID:21729782; https://doi.org/ 10.1016/j.cell.2011.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lu X, Zhu X, Li Y, Liu M, Yu B, Wang Y, Rao M, Yang H, Zhou K, Wang Y et al.. Multiple P-TEFbs cooperatively regulate the release of promoter-proximally paused RNA polymerase II. Nucleic Acids Res 2016; 44:6853-6867; PMID:27353326; https://doi.org/ 10.1093/nar/gkw571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].He N, Chan CK, Sobhian B, Chou S, Xue Y, Liu M, Alber T, Benkirane M, Zhou Q. Human polymerase-associated factor complex (PAFc) connects the super elongation complex (SEC) to RNA polymerase II on chromatin. Proc Natl Acad Sci U S A 2011; 108:E636-645; PMID:21873227; https://doi.org/ 10.1073/pnas.1107107108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kim J, Guermah M, Roeder RG. The human PAF1 complex acts in chromatin transcription elongation both independently and cooperatively with SII/TFIIS. Cell 2010; 140:491-503; PMID:20178742; https://doi.org/ 10.1016/j.cell.2009.12.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shi X, Finkelstein A, Wolf AJ, Wade PA, Burton ZF, Jaehning JA. Paf1p, an RNA polymerase II-associated factor in Saccharomyces cerevisiae, may have both positive and negative roles in transcription. Mol Cell Biol 1996; 16:669-676; PMID:8552095; https://doi.org/ 10.1128/MCB.16.2.669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Luo Z, Lin C, Shilatifard A. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol 2012; 13:543-547; PMID:22895430; https://doi.org/ 10.1038/nrm3417 [DOI] [PubMed] [Google Scholar]
- [44].Lin C, Garrett AS, De Kumar B, Smith ER, Gogol M, Seidel C, Krumlauf R, Shilatifard A. Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC). Genes Dev 2011; 25:1486-1498; PMID:21764852; https://doi.org/ 10.1101/gad.2059211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Schulze-Gahmen U, Lu H, Zhou Q, Alber T. AFF4 binding to Tat-P-TEFb indirectly stimulates TAR recognition of super elongation complexes at the HIV promoter. Elife (Cambridge) 2014; 3:e02375; PMID:23471103; https://doi.org/23471103 10.7554/eLife.00327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schulze-Gahmen U, Upton H, Birnberg A, Bao K, Chou S, Krogan NJ, Zhou Q, Alber T. The AFF4 scaffold binds human P-TEFb adjacent to HIV Tat. Elife 2013; 2:e00327; PMID:23471103; https://doi.org/ 10.7554/eLife.00327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Shilatifard A, Lane WS, Jackson KW, Conaway RC, Conaway JW. An RNA polymerase II elongation factor encoded by the human ELL gene. Science 1996; 271:1873-1876;PMID:8596958;https://doi.org/ 10.1126/science.271.5257.1873 [DOI] [PubMed] [Google Scholar]
- [48].Ott M, Geyer M, Zhou Q. The control of HIV transcription: keeping RNA polymerase II on track. Cell Host Microbe 2011; 10:426-435; PMID:22100159; https://doi.org/ 10.1016/j.chom.2011.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell 2010; 38:439-451;PMID:20471949; https://doi.org/ 10.1016/j.molcel.2010.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol Cell 2010; 38:428-438; PMID:20471948; https://doi.org/ 10.1016/j.molcel.2010.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Luo Z, Lin C, Guest E, Garrett AS, Mohaghegh N, Swanson S, Marshall S, Florens L, Washburn MP, Shilatifard A. The super elongation complex family of RNA polymerase II elongation factors: gene target specificity and transcriptional output. Mol Cell Biol 2012; 32:2608-17; PMID:22547686; https://doi.org/ 10.1128/MCB.00182-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li Z, Lu H, Zhou Q. A minor subset of super elongation complexes plays a predominant role in reversing HIV-1 latency. Mol Cell Biol 2016;36:1194-1205; PMID:26830226; https://doi.org/ 10.1128/MCB.00994-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lu H, Li Z, Zhang W, Schulze-Gahmen U, Xue Y, Zhou Q. Gene target specificity of the super elongation complex (SEC) family: how HIV-1 Tat employs selected SEC members to activate viral transcription. Nucleic Acids Res 2015; 43:5868-5879; PMID:26007649; https://doi.org/ 10.1093/nar/gkv541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 2010; 37:429-437; PMID:20159561; https://doi.org/ 10.1016/j.molcel.2010.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 2010; 17:198-212; PMID:20153263; https://doi.org/ 10.1016/j.ccr.2009.12.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet 2007; 16:92-106; PMID:17135274; https://doi.org/ 10.1093/hmg/ddl444 [DOI] [PubMed] [Google Scholar]
- [57].Esposito G, Cevenini A, Cuomo A, de Falco F, Sabbatino D, Pane F, Ruoppolo M, Salvatore F. Protein network study of human AF4 reveals its central role in RNA Pol II-mediated transcription and in phosphorylation-dependent regulatory mechanisms. Biochem J 2011; 438:121-131; PMID:21574958; https://doi.org/ 10.1042/BJ20101633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gu J, Babayeva ND, Suwa Y, Baranovskiy AG, Price DH, Tahirov TH. Crystal structure of HIV-1 Tat complexed with human P-TEFb and AFF4. Cell Cycle 2014; 13; PMID:24727379; https://doi.org/26171280 10.4161/cc.28756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lu H, Li Z, Xue Y, Schulze-Gahmen U, Johnson JR, Krogan NJ, Alber T, Zhou Q. AFF1 is a ubiquitous P-TEFb partner to enable Tat extraction of P-TEFb from 7SK snRNP and formation of SECs for HIV transactivation. Am J Blood Res 2015; 5(1):10–24; PMID:24367103; https://doi.org/26171280 10.1073/pnas.1318503111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Scholz B, Kowarz E, Rossler T, Ahmad K, Steinhilber D, Marschalek R. AF4 and AF4N protein complexes: recruitment of P-TEFb kinase, their interactome and potential functions. Am J Blood Res 2015; 5:10-24; PMID:26171280 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.