

Abstract
The rate of water exchange between the inner-sphere of a paramagnetic ion and bulk water is an important parameter in determining the magnitude of the chemical exchange saturation transfer signal from paramagnetic CEST agents (paraCEST). This is governed by various geometric, steric, and ligand field factors created by macrocyclic ligands surrounding the paramagnetic metal ion. Our previous on-bead combinatorial studies of di-peptoid-europium(III)-DOTA-tetraamide complexes revealed that negatively charged groups in the immediate vicinity of the metal center strongly enhances the CEST signal. Here we report a solid phase synthesis and on-bead imaging of 76 new DOTA derivatives that are developed by coupling with a single residue onto each of the three arms of a DOTA-tetraamide scaffold attached to resin beads. This single residue predominantly carries negatively charged groups blended with various physico-chemical characteristics. We found that non-bulky negatively charged groups are best suited at the immediate vicinity of the metal ion, while positive, bulky and halogen containing moieties suppress the CEST signal.
Keywords: Combinatorial, CEST-MRI, DOTA
Graphical abstract
The rate of water exchange in chemical exchange saturation transfer (CEST) agents is governed by various geometric, steric, and ligand field factors surrounding the paramagnetic metal ion. Here we reporta solid phase synthesis and on-bead imaging of 76 new DOTA derivatives that are developed by modifying the three arms of a DOTA-tetraamide scaffold attached to beads. We found non-bulky negatively charged groups support the CEST signal, while positive, bulky and halogen containing moieties suppress the CEST signal.
1. Introduction
Combinatorial high throughput strategies are an important tool in modern day drug discovery, but have received little attention in identifying efficient diagnostic probes. Previously, we reported the synthesis and imaging of an on-bead combinatorial library of 80 di-peptoid-europium(III)-DOTA-tetraamide complexes.1 This new technique allows us to modify a DOTA-tetraamide scaffold rapidly and economically and acquire the image of an entire library at once to compare the effects of all chemical modifications. In our previous study, we used CEST-MRI as the model system and were able to study various physico-chemical factors that alter CEST image contrast and found that having negatively charged groups at immediate vicinity of the metal ion is favorable for CEST imaging (Figure 1a). Here we report the development of a much more focused library designed to further elucidate how negatively charged groups in combination with different physicochemical parameters as well as other functional groups at immediate vicinity of the metal center affect the CEST signal (Figure 1b).
Figure 1.
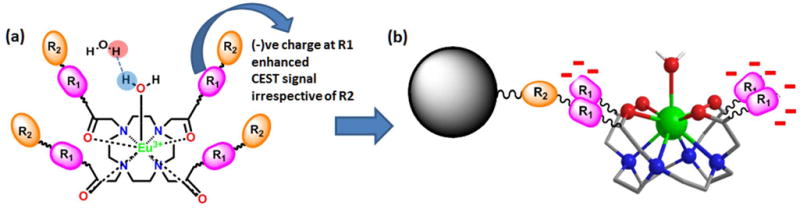
(a) Schematic of a europium(III) DOTA-tetraamide complex showing its interactions with inner-sphere and outer-sphere water molecules. Negatively charged groups on R1 were previously shown to increase CEST sensitivity. (b) The new library design includes negatively charged moieties combined with other physico-chemical characteristics again at R1.
Magnetic resonance imaging (MRI) is one of the main imaging modalities used in the clinic that generates image contrast based on differences in water (proton) densities and relaxation rates in different types of tissues.2 The differences in relaxation times (T1 & T2) of protons of different tissues or pools are generally small and thus, to enhance tissue contrast, exogenous agents are being used to alter the relaxation times.3 The most widely used exogenous contrast agents are complexes of paramagnetic gadolinium (III) that act by reducing the relaxation time of water protons.4 Other approaches for introducing image contrast are gaining in popularity, especially methods based on chemical exchange saturation transfer (CEST).5 CEST techniques offer some advantages over relaxation-based imaging agents including, i) the ability to switch the contrast “on” or “off” by application of a suitable radiofrequency (RF) pulse, and ii) the exquisite sensitivity of the CEST signal to the rates of water or proton exchange between a ligand or metal ion exchange site and bulk water. These features make this CEST-based methods attractive for developing biologically responsive sensors.6 The one remaining disadvantage is sensitivity.
The CEST signal from Eu(III) complexes such as those described here is highly dependent on the rate of water molecule exchange between two pools of water protons (bulk water protons and the water molecule bound to metal center of the Eu(III) agent).4a,6a Previous studies have shown that prototropic exchange (exchange of protons only) occurs more slowly than water molecule exchange in EuDOTA-tetraamide complexes except at the extremes of very high and very low pH.7 Hence, we assume that the rate of water molecule exchange is the key factor in determining the intensity of the CEST signal in the complexes described herein. A general requirement for all CEST agents, paraCEST or diaCEST, is that chemical exchange must be slow enough to allow selective saturation of a proton or water molecule on the agent without saturation of the bulk water pool.5h, 7 Thus, a large frequency difference (Δω) between any two pools of protons is advantageous because chemical systems that undergo faster exchange (kex) between the two pools can be used while maintaining the basic CEST requirement, Δω ≥ kex. A large Δω, such as those seen for most paraCEST agents, has the added advantage of reducing off-resonance direct saturation of the bulk water signal thereby simplifying quantitative interpretation of the CEST signal.
Paramagnetic europium (III)-1, 4, 7, 10-tetraazacyclododecane-1, 4, 7, 10-tetraacetic acid (DOTA) tetraamide complexes have been widely reported as paraCEST agents. It has been reported that the tetra-amide derivatives with differing electronic, steric, and polar substituents on the four arms of DOTA can display widely variable water exchange rates and hence CEST contrast.5b,8 Consequently, CEST sensitivity can be improved substantially by modification of the side-chain chemical features in order to optimize the rate of water exchange in these complexes (= 2*pi*B1, namely, 1256 s−1 by applying for a “typical” safe saturation power B1 = 200 Hz for human use in clinical scanner).9 However; it is important to know how these various physico-chemical parameters alter the CEST signal when used in combination.
In the previous work, we synthesized 80 different derivatives of di-peptoid-europium(III)-DOTA-tetraamide complexes by attaching a cyclen derivative onto TentalGel resin beads using one arm of the macrocycle and modifying remaining three arms with two peptoid residues in a parallel synthesis approach.1 The entire library was imaged simultaneously to obtain a single CEST map that illustrated the sensitivity of the CEST signal to the identity of the two peptoid units on the side chains. The DOTA arms designed with amines with variable charges, polarity, hydrophobicity, and size provided a preliminary insight into how these physico-chemical parameters affect the water exchange mechanism. The observations from this study1 as well as our initial work8a,b led to the conclusion that europium(III)-DOTA-tetraamide complexes modified with a negatively charged group (almost independent of residue at the second position) closest to the europium(III) center have a constructive influence on the water exchange rate and hence the CEST signal (Figure 1a).
2. Results and discussion
These encouraging results led to the design of a more focused library to identify combinations of factors that could result in further increases in CEST sensitivity. Here, we employed the same rapid on-bead DOTA side chain modification synthesis strategy, mainly using negatively charged moieties built in the immediate vicinity of the central metal ion but varying the chemical properties of the moieties further away from the metal center (Figure 1b). The parental scaffold (Figure 2) for solid phase synthesis was initiated as previously described1 and shown in Supplementary Scheme 1. Briefly, with a small predetermined two 2-Methoxyethylamine peptoid linker formation further extended with β- alanine on TentaGel macro-beads (300 μm diameter), in order to keep the macrocycle away from the bead surface. The linker was coupled to 1, 4, 7, 10-tetraazacyclododecane-1, 4, 7-tris-tert-butyl acetate (DO3A tris-tert-butyl ester) which was then treated with TFA to remove the tert-butyl groups on the remaining three arms of DOTA. The carboxyl terminals were coupled with ethylene diamine to provide terminal amines to create the basic DOTA-tetraamide structure required for slow water exchange complexes and to provide terminal amines for adding additional chemical moieties for the library. With the goal of introducing structures having a combination of diverse physicochemical properties, the system was designed to include four types of well-established amide coupling reactions. 76 different moieties were added to the remaining three arms of DOTA using peptide (amino acid), anhydride, peptoid (amine), and carboxylic acid coupling reactions (Figure 2). Since different functional groups required different reaction conditions, unlike the previous study in which the residues were added via parallel synthesis, the reactions had to be conducted individually in reaction vessels. All of the reactions are insensitive to air and moisture and typically take place in high yield at each step. The groups chosen for the library provided a range of charge, polarity, and steric bulkiness around the europium (III) water exchange site (Figure 3). The parent compound shown in Figure 2 (insert) was synthesized using the same procedure reported previously.1 The resin beads containing this parent compound were then split equally among 76 reaction vessels (2 mL vessels, ~ 25 mg resin/ea.). Seventeen different amino acids (Figure 3) were added to the parent compound along with HBTU/HOBt/DIPEA (0.2 M/0.2 M/0.4 M) as standard peptide coupling conditions and left overnight on the shaker. Another group of 18 vessels were treated with 18 anhydrides (Figure 3) with 2M solutions each. Similarly, the third group of 13 vessels was reacted with carboxylic acids (Figure 3) overnight using DIC and HOBt in 3:4:6 in DMF. It is worth noting that the standard amide coupling using HBTU/HOBt also worked well with the most of the carboxylic acids but the yields were much improved for bulky moieties when using DIC/HOBt. Finally, the last set of 28 reaction vessels were coupled to amines using a two-step peptoid coupling condition (Figure 3). Bromoacetic acid (BAA) coupling brings the 2 carbon unit and the Br was replaced by primary amines. The completion of each reaction was individually checked via MALDI-TOF mass spectroscopy. The reactions showing partial or incomplete coupling, were reacted again using fresh reagents. Some compounds still failed to react completely with multiple rounds of coupling (4, 10), probably due to steric hindrance, but we opted to include them in the CEST screen as any compounds that produce a strong signal could be purified and analyzed to determine if the signal was due to our expected compound or an incomplete derivative. The on bead DOTA-tetraamide derivatives were then reacted with a 15-fold excess of EuCl3 overnight at pH 6.3.
Figure 2.
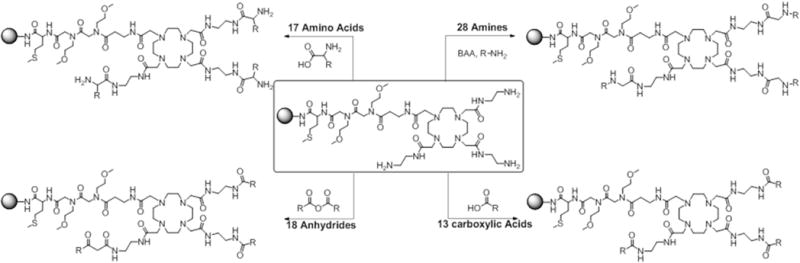
Reaction scheme of four types of coupling reactions used to modify parent DOTA complex.
Figure 3.
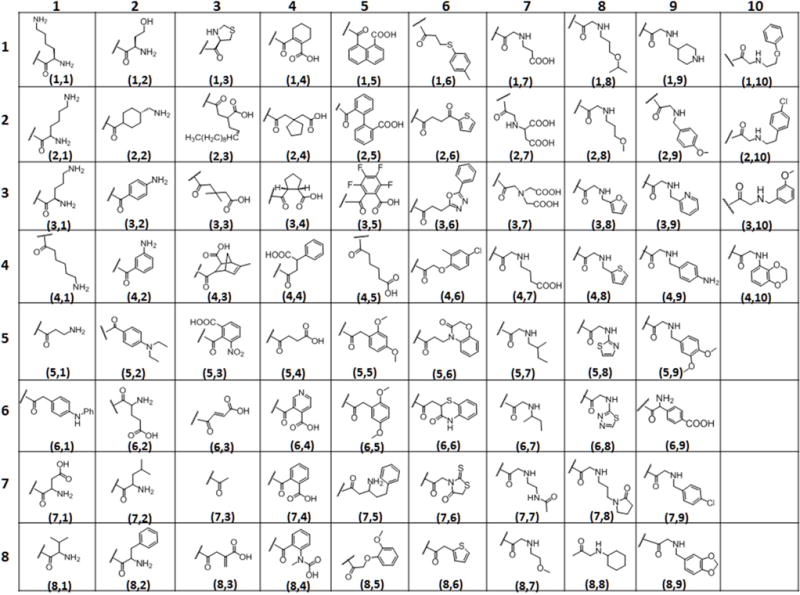
Table containing the subunits used to add physicochemical diversity to the library.
A single CEST image of the entire library was collected including the parent compound with chelated Eu(III), the parent compound lacking the metal ion, beads with europium salts only, and a few duplicates from the library [Figure 3. Compounds (5, 4), (7, 3), (1, 6) & (4, 8) where the designation refers to (row, column)]. A 384-well micro-test TM clear falcon plate was resized to 12 × 24 (288 wells) to fit in the 63 mm diameter quadrature coil of a Varian 9.4T NMR system. The beads with all derivatives and controls were then transferred to the inner 83 wells of a 288-well plate to register a single image. All the wells with beads were then filled with water and in order to make the sample homogenous; the remaining empty wells were also filled completely with water. The plate was then placed in the bore of a 9.4 T Varian scanner in a volume coil, and a single CEST image was acquired after applying a 10 μT frequency-selective pre-saturation pulse for 5s followed by a spin-echo sequence (Figure 4). Although a B1 of 10 μT is not necessarily the optimal power level for every Eu(III) complex in the library, this value was chosen from among a series of trial B1 values to provide the best visual difference in CEST intensities among the 76 compound library. Similarly, the length of the presaturation pulse (5 s) was chosen from among a series of trials using variable presaturation times of 3–7 s. The CEST image (Mo-Ms) was generated from the resultant water intensities in images obtained with the pre-saturation pulse set to −50 ppm (Mo, off control) versus a second image with the pre-saturation pulse set to +50 ppm (Ms, on). Given that each of the complexes studied in this library have a nearly identical coordination sphere surrounding the Eu(III) ion, the chemical shift of the exchanging bound water peak did not vary significantly over the entire library. Furthermore, any impact of variations in effective B1 across the samples was small compared to the linewidth of the exchanging CEST peaks. Hence, corrections for B1 or B0 variability were not necessary. We tabulated on bead CEST spectra for each of the 76 compounds of the entire library as well (Supplementary Figure S7).
Figure 4.

CEST images of the 76 compound library of europium(III)-DOTA-tetraamide complexes attached to resin beads. The color scale (on left) represents a percent change in bulk water signal intensity (1-Ms/Mo) after a presaturation pulse at +50 ppm (Ms) or −50 ppm (Mo). The last column contains experimental controls and repeats of one of each type of subunit reaction to show reproducibility.
The resulting image (Figure 4) showed multiple bright wells compared to the well containing the parent compound, an effect not seen in the previous study. Upon quantifying the signal intensities of each well, 21 of the 76 compounds showed a stronger CEST signal than the parent compound. In particular, compounds (3,3), (6,4), (6,3), (6,2) showed significant improvement over the parent compound. Of those 21 compounds, 52% carried a negative charge while only 19% carried a positive charge and one of them was in zwitter ionic form. It is worth noting that most of the positively charged compounds like amino acids with free amine groups shown in columns 1 and 2 (Figure 3 and Figure 4) proved ineffective in enhancing CEST, while the compounds containing small substituents like isopropyl (8,1) and isobutyl (7,2) with primary amine group showed some improvement over those containing two amine groups [(1,1); (2,1) and (3,1)] and long chain amines like (4,1) and (5,1). Among the aromatics, an amine group in the meta-position (4,2) displayed modestly improved CEST. One compound having a combined amine and carboxyl groups (zwitterion) (6,2) also showed improved CEST signals over the parent compound and another example of zwitter ion (7,1) had a CEST signal comparable to parent compound.
Further investigations of the negatively charged groups (mainly through free carboxylic groups on the side chains) revealed that 54% of those compounds displayed significantly improved CEST signals. (Figure 4, column 3, 4 and 5). These results were compatible with the data obtained in our previous study.1 All aliphatic acids and alicyclic showed moderate to good CEST. Compounds (5, 4) and (4,5) with succinate and adipate groups, respectively, exhibited similar CEST intensities while a few aromatic carboxylates [(6, 4), (7, 4)] displayed surprisingly strong CEST signals. In comparison, those derivatives containing larger aromatic carboxylates [(1, 5), (2, 5), and (3, 5)] displayed very weak CEST signals. This illustrates that negative charge on the extended side-chains is important for improving CEST sensitivity until the hydrophobic character of those groups begins to counteract those positive effects.
One of the main purposes of this focused library was to blend a variety of physico-chemical properties that might reveal an optimal structure. The derivatives synthesized from carboxylic acids and peptoid couplings did not produce efficient CEST agents, as the carboxylic group was used for amide bond formation. Most of these side chains had aromatic rings with different substitutions. This observation clearly suggests that the inefficiency of aromatic hydrophobic regions to regulate water exchange. Minimum CEST signals from peptoid derivatives could also be attributed to the presence of secondary amine (positive charge) along with the various substituents as in case of amino acids containing terminal primary amines. There were few derivatives that reached ~20% CEST enhancement [Figure 4, magenta color, (8, 6), (8, 7), (7, 7), (6, 8), (5, 8), (4, 8), (3, 8)]. Interestingly, strong CEST signals were observed when the DO3A scaffold was functionalized with simple acetyl groups as well [compound (7,3)].
An overall summary of the chemical properties contributing to the strongest CEST signals are summarized in Figure 5. For example, negatively charged carboxylates generally yielded CEST signals in the 40–80% range. Within this group, negatively charges aliphatic were consistently better than negatively charged aromatics. More than half of the positively charged derivatives displayed less than a 20% CEST signal. Compounds containing primary and secondary amines tended to have weak CEST signals. None of positive charged moieties showed more than 40% CEST.
Figure 5.
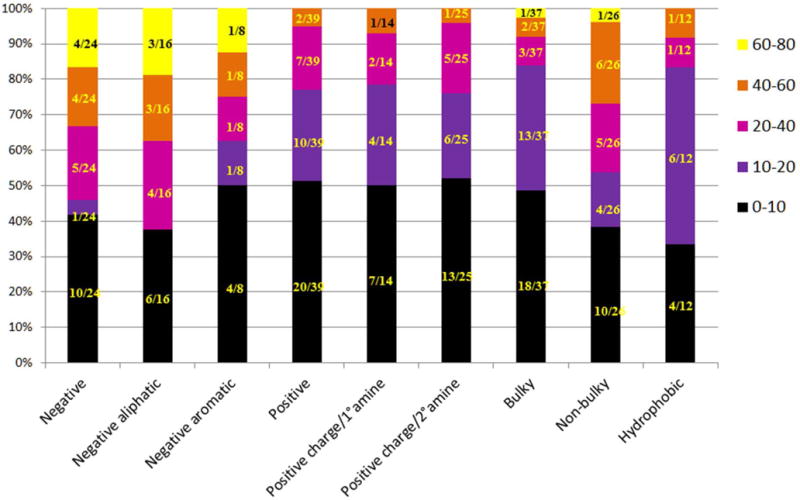
CEST intensity vs. physicochemical properties of modifications made. Color code is representative of the data presented in Figure 3 (the color scale shown in right side is equivalent to the color scale shown in Figure 3). The lighter colors represent a stronger CEST effect, while the darker colors represent a weaker CEST effect.
Another undesirable impact on the CEST signal was evident when bulkier hydrophobic groups were incorporated (hydrophobic bar). Overall, this comprehensive analysis shows that negatively charged compounds with smaller, less sterically crowded groups generally contribute to favorable CEST properties. This suggests that bulky and hydrophobic groups may hinder water access to the coordination sphere of the europium(III) ion where the single bound water molecule resides. One exception to general observations was the ortho-substituted (with a negatively charged group) aromatic ring structures that displayed an enhanced CEST signal. This may be an indication of electronic effects have a more substantial influence on CEST than steric effects.
3. Conclusion
In conclusion, we have performed simple and easy chemical modifications of the parent DOTA-tetraamide scaffold to bring about various changes that affect water molecule exchange rates or proton transfer rates as reflected in the CEST properties of imaging agents. Using this data as support, further modifications can be easily made using a similar combinatorial format testing even wider array of small sterically free compounds in an attempt to further improve the imaging sensitivity of CEST agents.
4. Experimental
4.1 General
1,4,7,10-tetraazacyclododecane-1,4,7-tris(tert-butyl acetate) [DO3A-tris(tert-butyl ester)] and N-Boc-1,2-diaminoethane were prepared using reported procedures and characterized by MALDI, NMR and HPLC methods.10 Tentagel macrobeads were purchased from Rapp Polymere (Germany). All the Fmoc protected amino acids and 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from EMD millipore (Billerica, MA). All primary amines, anhydrides, carboxylic acids, bromoacetic acid, N,N′-diisopropylcarbodiimide (DIC), N,N-diisopropylethylamine (DIPEA), piperidine, NMM, trifluoroacetic acid (TFA), and N,N-dimethylformamide (DMF) from Sigma-Aldrich (Milwaukee, WI, USA); Dichloromethane (DCM) and acetonitrile from Honeywell Burdick & Jackson (Morristown, NJ, USA). β-alanine tert-butylester hydrochloride (H-β-Ala-O-tert-bu.HCl) from EMD Biosciences (Gibbstown, NJ, USA). All of the chemical reagents and solvents from commercial sources were used without further purification. 5mL disposable reaction columns (Intavis AG) were used as reaction vessels for solid-phase synthesis. Syntheses of peptiods under microwave conditions were performed in a 1000 W microwave oven with 10% power.
MALDI-TOF mass spectra were acquired on an Applied Biosystems Voyager-6115 mass spectrometer in positive reflector mode using alpha-Cyano-4-hydroxycinnamic acid as matrix.
4.2 On-bead bulk synthesis of parent compound (5). See supplementary information for synthesis scheme
Compound was synthesized on a solid phase using TentaGel macrobeads resin (Rapp polymer, with a 0.25 mmol/g loading, 300 μm diameters). Combinations of conventional protocols for Fmoc amino acids peptide and peptoid synthesis were used. The resin (100–200 mg for each synthesis) was placed in reaction vessels and swelled in N, N-dimethylformamide (DMF) for 1 hr. After solvent removal, the resin was treated with 0.2M Fmoc-Met-OH, 0.2M HBTU and 0.4M 4-methylmorpholine (NMM) in DMF (2.0 mL) at room temperature overnight. After washing with DMF (10 × 2.0 mL), Fmoc group was removed by 20% piperidine solution in DMF [2 × (2.0 mL × 10 min)] and the peptoid linker was inserted using a microwave (1000 W)-assisted synthesis protocol.11 Briefly, beads were treated with mixture of 2.0M bromoacetic acid (1.0 mL) and 3.2M N,N′-diisopropylcarbodiimide (DIC) (1.0 mL) followed by microwave (oven set at 10% power) 2 × 15 sec with a gentle shaking in between for 30 sec. After washing with DMF, beads were treated with 2.0M methoxyethylamine (2.0 mL), and again the coupling was performed in the microwave oven as described above. The procedure was repeated again to attach the second peptoid residue with DMF washing at every step. Fmoc-β-Ala-OH was then coupled using the peptide coupling condition described for the methionine coupling above. After washing with DMF and removal of the Fmoc group (20% piperidine), beads were treated again with bromoacetic acid/DIC mixture under microwave condition. Beads were then treated with 1.0M DO3A-tris(tert-butyl ester) solution (2.0 mL) in the microwave oven at 10 % power (3 × 15 s). After washing with DMF (10 × 2.0 mL) and dichloromethane (10 × 2.0 mL), tert-butyl groups were removed by treating with a 95% TFA, 2.5 % triisopropylsilane, and 2.5 % water mixture (3.0 mL) for 4 hrs at room temperature. Before the next addition, beads here were washed with DIPEA in DMF in order to neutralize acidic conditions. After washing with DCM (10 × 2.0 mL) and DMF (10 × 2.0 mL), beads were treated with N-Boc-1,2-diaminoethane in presence of HOBt (5.0eq) and DIC (5.5eq) as coupling reagents to give 5. Removal of the Boc group was performed again treating it with a 95% TFA, 2.5 % tri-isopropyl silane (TIS), and 2.5 % water mixture (3.0 mL) for 4 hrs at room temperature and then neutralized with DIPEA solution. The beads were thoroughly washed with DMF (10 × 2.0 mL).
4.3 Synthesis of library of 17 compounds using amino acids
The first 17 vessels containing beads with parent compounds were treated with 1mL solution of 0.2M amino acids, 0.2M HBTU, 0.2M HOBt, and 0.4M of DIPEA. The reactions were allowed to shake overnight. Fmoc protecting groups were removed with 1mL solution of 20% piperidine (2 times) for 10 minutes. Compounds having acid labile protecting groups (Boc protecting group) were further de-protected with 95% TFA solution with 2.5% of tri-isopropyl silane (TIS) and 2.5% of water. Acid treated beads were washed with DCM, DMF, and then neutralized with 10% piperidine for 10 minutes.
4.4 Synthesis of library of 18 compounds using anhydrides
2M solutions of different anhydrides were added to 18 reaction vessels containing beads with parent compound 5 and allowed overnight on the shaker. For some of the reactions for completion, DIPEA was also added in 1:1 (anhydride to DIPEA).
4.5 Synthesis of library of 13 compounds using carboxylic acids
The reactions were performed with 1mL solutions of 3.0eq organic acids in presence of 4.0eq of HOBt and 6.0eq of DIC in DMF overnight on the shaker.
4.6 Synthesis of library of 28 compounds using amines
Finally, last 28 compounds were synthesized using amines, via two-step peptoid reactions of acylation followed by a substitution reaction with the desired amines. Compounds with protecting group were treated with 95% TFA solution with 2.5% of tri-isopropyl silane (TIS) and 2.5% of water solution to remove their acid labile protecting group. Acid treated beads were washed with DCM, DMF, then neutralized with 10% piperidine for 10 minutes, and finally washed with water. Beads that did not require acid treatment were washed with DCM, DMF, and then water.
4.7 Europium chelation of the library
Each vessel was allowed to have the resin equilibrate in water for 2 hours before metal complexation with 0.2M EuCl3 overnight at a pH of 6.3. Beads were then washed 10 times each with water and stored at 4°C.
4.8 Cleavage and mass analysis
Microwave assisted cyanogen bromide cleavage was used to remove the compounds from the resin for mass analysis. 10–20 beads from each compound were placed in a 4.0 mL glass vial and 50μL each of cleavage solution (95% H2O, 2% HCl, and 3% CNBr) and water were added to the vials. Vials (5–10 at a time) were placed in the center of the microwave around a vial with 3.0 mL H2O and microwaved 3 times for 35 seconds at 10% power. Compounds were dried and dissolved in a 1:1 acetonitrile/water mixture for mass analysis.
4.9 CEST-MR imaging of the library.1
In vitro imaging was performed on a Varian 9.4 T animal system using a 63 mm diameter quadrature volume coil. A 384 well microtest TM clear falcon plate was resized to 12 × 24 (288 wells) to fit in the 63 mm diameter quadrature coil. The resin samples (~3/4 of the well full) were placed with surrounding wells filled with water at the gradient isocenter with the temperature maintained at 20°C using warmed air and a thermocouple. A single coronal plane (64 × 64 × 2 mm) passing through the bottom portion of each sample well was selected for imaging. Image-based CEST spectra were acquired with the fast spin-echo sequence (TR/TE = 69.8 ms/8.4 ms, echo train 8, averages 2, 64 × 64 × 1 pixel matrix) were used to measure the CEST frequency (Δω) for each well. Images were then acquired using the fast spin-echo sequence (TR/TE = 69.8 ms/8.4 ms, echo train 8, averages 2, 64 × 64 × 1 pixel matrix) with saturation at both +50 ppm (“on”) and −50 ppm (“off”). A 5 s long, 10 μT saturation pulse was used for every TR. A CEST image was created by subtracting the two images (off-on). Spatial susceptibility effects within the samples were minimized using two methods. First, the 8 × 11 sample wells were surrounded by a two-well thick boarder of pure water. Second, after the first set of images was taken, the sample was rotated by 180° and imaged again. The two CEST images at 0° and 180° were then averaged. These methods assured that the CEST variations from well to well were due to changes in chemical configuration and not susceptibility effects.
Supplementary Material
Acknowledgments
We would like to acknowledge the staff members of the Advanced Imaging Research Center at UT-Southwestern Medical Center. This work was supported by National Institutes of Health (NIH) grants R21EB015602, R01CA115531, P41EB015908 and, in part, by the Harold C. Simmons Cancer Center through an NCI Cancer Center Support Grant, 1P30-CA142543, and the Robert A. Welch Foundation (AT-584).
Footnotes
Supplementary data
Supplementary data contains list of reactants, CEST imaging protocol and MALDI TOF mass spectrometry analysis.
References
- 1.Napolitano R, Soesbe TC, De Leon-Rodriguez LM, Sherry AD, Udugamasooriya DG. J Am Chem Soc. 2011;133:13023–13030. doi: 10.1021/ja201123f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Damadian R. Science. 1971;171:1151–1153. doi: 10.1126/science.171.3976.1151. [DOI] [PubMed] [Google Scholar]
- 3.(a) Strijkers GJ, Mulder WJ, van Tilborg GA, Nicolay K. Anticancer Agents Med Chem. 2007;7:291–305. doi: 10.2174/187152007780618135. [DOI] [PubMed] [Google Scholar]; (b) Lauffer RE. Chem Rev. 1987:901–927. [Google Scholar]
- 4.(a) Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Chem Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]; (b) Aime S, Crich SG, Gianolio E, Giovenzana GB, Tei L, Terreno E. Coord Chem Rev. 2006;250:1562–1579. [Google Scholar]
- 5.(a) Woods M, Woessner DE, Sherry AD. Chem Soc Rev. 2006;35:500–511. doi: 10.1039/b509907m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Viswanathan S, Kovacs Z, Green KN, Ratnakar SJ, Sherry AD. Chem Rev. 2010;110:2960–3018. doi: 10.1021/cr900284a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang S, Merritt M, Woessner DE, Lenkinski RE, Sherry AD. Acc Chem Res. 2003;36:783–790. doi: 10.1021/ar020228m. [DOI] [PubMed] [Google Scholar]; (d) van Zijl PC, Yadav NN. Magn Reson Med. 2011;65:927–948. doi: 10.1002/mrm.22761. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Cakic N, Savic T, Stricker-Shaver J, Truffault V, Platas-Iglesias C, Mirkes C, Pohmann R, Scheffler K, Angelovski G. Chem Comm. 2016;52:9224–9227. doi: 10.1039/c6cc04011j. [DOI] [PubMed] [Google Scholar]; (f) Du K, Harris TD. J Am Chem Soc. 2016;138:7804–7807. doi: 10.1021/jacs.6b03060. [DOI] [PubMed] [Google Scholar]; (g) Hingorani DV, Montano LA, Randtke EA, Lee YS, Cárdenas-Rodríguez J, Pagel MD. Contrast Media Mol Imaging. 2016;11:130–138. doi: 10.1002/cmmi.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Li AX, Wojciechowski F, Suchy M, Jones CK, Hudson RHE, Menon RS, Bartha R. Magn Reson Med. 2008;59:374–381. doi: 10.1002/mrm.21482. [DOI] [PubMed] [Google Scholar]; (i) Olatunde AO, Dorazio SJ, Spernyak JA, Morrow JR. J Am Chem Soc. 2012;134:18503–18505. doi: 10.1021/ja307909x. [DOI] [PubMed] [Google Scholar]; (j) Pumphrey A, Yang Z, Ye S, Powell DK, Thalman S, Watt DS, Abdel-Latif A, Unrine J, Thompson K, Fornwalt B, Ferrauto G, Vandsburger M. NMR in Biomed. 2016;29:74–83. doi: 10.1002/nbm.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Sherry AD, Woods M. Annu Rev Biomed Eng. 2008;10:391–411. doi: 10.1146/annurev.bioeng.9.060906.151929. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang Y, Coman D, Ali MM, Hyder F. Contrast Media Mol Imaging. 2015;10:51–58. doi: 10.1002/cmmi.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woods M, Pasha A, Zhao P, Tircso G, Chowdhury S, Kiefer G, Woessner DE, Sherry AD. Dalton Trans. 2011;40:6759–6764. doi: 10.1039/c1dt10616c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Mani T, Opina AC, Zhao P, Evbuomwan OM, Milburn N, Tircso G, Kumas C, Sherry AD. J Biol Inorg Chem. 2014;19:161–171. doi: 10.1007/s00775-013-1031-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dixon WT, Ren J, Lubag AJ, Ratnakar J, Vinogradov E, Hancu I, Lenkinski RE, Sherry AD. Magn Reson Med. 2010;63:625–632. doi: 10.1002/mrm.22242. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Viswanathan S, Ratnakar SJ, Green KN, Kovacs Z, De Leon-Rodriguez LM, Sherry AD. Angew Chem Int Ed Engl. 2009;48:9330–9333. doi: 10.1002/anie.200904649. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mani T, Tircso G, Togao O, Zhao P, Soesbe TC, Takahashi M, Sherry AD. Contrast Media Mol Imaging. 2009;4:183–191. doi: 10.1002/cmmi.279. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ratnakar SJ, Woods M, Lubag AJ, Kovacs Z, Sherry AD. J Am Chem Soc. 2008;130:6–7. doi: 10.1021/ja076325y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Sherry AD, Wu Y. Curr Opin Chem Biol. 2013;17:167–174. doi: 10.1016/j.cbpa.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Woessner DE, Zhang S, Merritt ME, Sherry AD. Magn Reson Med. 2005;53:790–9. doi: 10.1002/mrm.20408. [DOI] [PubMed] [Google Scholar]
- 10.Breipohl G, Uhlmann E, Will DW. Patent. 1998;5817811 [Google Scholar]
- 11.Olivos HJ, Alluri PG, Reddy MM, Salony D, Kodadek T. Org Lett. 2002;4:4057–4059. doi: 10.1021/ol0267578. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.