

Abstract
Objective
Coagulation initiation by tissue factor (TF) is regulated by cellular inhibitors, cell surface availability of procoagulant phosphatidylserine (PS) and thiol-disulfide exchange. How these mechanisms contribute to keeping TF in a non-coagulant state and to generating prothrombotic TF remains incompletely understood.
Approach and Results
Here we study activation of TF in primary macrophages by a combination of pharmacological, genetic and biochemical approaches. We demonstrate that primed macrophages effectively control TF cell surface activity by receptor internalization. Following cell injury, ATP signals through the purinergic receptor P2rx7 induce release of TF+ microvesicles (MV). TF cell surface availability for release onto MV is regulated by the GTPase arf6 associated with integrin α4β1. Furthermore, MV proteome analysis identifies activation of Gαi2 as a participating factor in the release of MV with prothrombotic activity in flowing blood. ATP not only prevents TF and PS internalization, but furthermore induces TF conversion to a conformation with high affinity for its ligand, FVIIa. Although inhibition of dynamin-dependent internalization also exposes outer membrane procoagulant PS, the resulting TF+ MV distinctly lack protein disulfide isomerase and high affinity TF and fail to produce fibrin strands typical for MV generated by thrombo-inflammatory P2rx7 activation.
Conclusions
These data show that procoagulant phospholipid exposure is not sufficient and that TF affinity maturation is required to generate prothrombotic MV from a variety of cell types. These findings are significant for understanding TF-initiated thrombosis and should be considered in designing functional MV-based diagnostic approaches.
Keywords: Extracellular vesicles, Thrombo-inflammation, Thiol-isomerases
Subject codes: Thrombosis, Inflammation
Introduction
Tissue factor (TF) initiates extrinsic coagulation by serving as the cell surface receptor for coagulation factor VIIa (FVIIa). In cells of the myelo-monocytic lineage, which are central to the crosstalk of inflammation and coagulation, TF remains predominantly in a non-coagulant, encrypted form. Secondary signals can activate TF procoagulant function and thus cause thrombotic complications in disease1. Relevant injury and danger signals in this context are degradation of TF pathway inhibitor (TFPI) by neutrophil proteases2, complement activation3–5, protein disulfide isomerase (PDI) release by injured cells6, 7, and triggers of neutrophil extracellular traps incorporating TF8–11.
Additionally, extracellular ATP generated by tissue injury and inflammation activates the purinergic receptor P2xr7 that stimulates TF activity on smooth muscle cells, macrophages12, 13, and human dendritic cells14. Primarily recognized for promoting the release of the pro-inflammatory cytokine IL-1β, P2rx7 signaling also induces cell surface exposure of phosphatidylserine (PS) and the release of highly procoagulant TF on microvesicles (MV). Thiol-disulfide exchange reactions depending on the thioredoxin (TRX)–TRX reductase system are crucial for both intracellular caspase 1 activation and extracellular reductive changes inducing TF activation and MV release13. Thus, P2rx7-signaling is a central mechanistic link in the coupling of inflammation and coagulation.
Thiol-disulfide exchange and PDI have been implicated in regulating TF procoagulant activity3, 6, 12, 15–17. However, PS exposure is also influenced by PDI12, 18 and concomitantly induced by stimuli that promote TF activation1, 16, 19, 20; thus it remains controversial whether TF structural changes induced by PDI are relevant for enhancing TF procoagulant activity or whether PS exposure is sufficient21–23. Alternative conformations of TF are indicated by measurements of affinity for its ligand, FVIIa. For example, TF-FVIIa signaling requires FVIIa concentrations of 5–10 nM, whereas TF-dependent FX activation can be efficient with FVIIa at pM concentrations, suggesting that the procoagulant pool of cell surface TF has higher affinity for FVIIa15, 24. TF with high affinity for FVIIa has been demonstrated in cell binding studies on highly procoagulant monocytes and other cells25, 26. However, experimental approaches have so far not clearly dissected functional contributions of TF affinity versus procoagulant phospholipids in the context of physiologically relevant membranes.
While the functional crosstalk of TF with integrins in cell signaling and cancer progression is well established27, 28, we report here that the arf6 recycling pathway29 that controls integrin α4β1 function in macrophages30 also regulates TF surface availability. Thus, we found that ATP stimulation of P2rx7 uncouples the TF-integrin α4β1 complex from arf6 control, enabling TF incorporation into MV. Intervening at distinct points in these TF trafficking pathways, we have been able to dissect prothrombotic effects caused by procoagulant phospholipid exposure on MV from those of enhanced TF affinity for FVIIa required for prothrombotic activity in flowing blood.
Material and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Internalization controls TF cell surface activity
Cell surface TF activity in interferon γ- and LPS-primed, adherent macrophages is very low in the absence of secondary stimuli13. We followed antibody-tagged cell surface TF in primed macrophages carrying knocked-in human TF (TFKI macrophages)31 and found that constitutive internalization of cell surface TF was prevented by the dynamin inhibitor Dynasore32 (Figure 1A and Figure IA in the online-only Data Supplement). Dynamin controls both clathrin- and raft-dependent internalization33. Dynasore treatment increased cell surface phosphatidylserine (PS) exposure and TF activity (Figure 1B). Antibody blockade of TF prior to stimulation showed that Dynasore and ATP similarly stimulated the activity of TF that was already present on the cell surface. Dynasore also caused the release of MV with TF activity (Figure 1C). FACS analysis confirmed that Dynasore treatment released MV that carried TF and procoagulant PS and had scatter properties similar to MV from ATP-stimulated cells (Figure 1D). Release of TF antigen and activity by both agonists was prevented by acute blockade of lipid raft domains with the cholesterol-chelator filipin (Figure IB in the online-only Data Supplement), indicating mobilization of a similar lipid raft-localized pool of cell surface TF.
Figure 1. Inhibition of TF internalization promotes TF+ MV release.
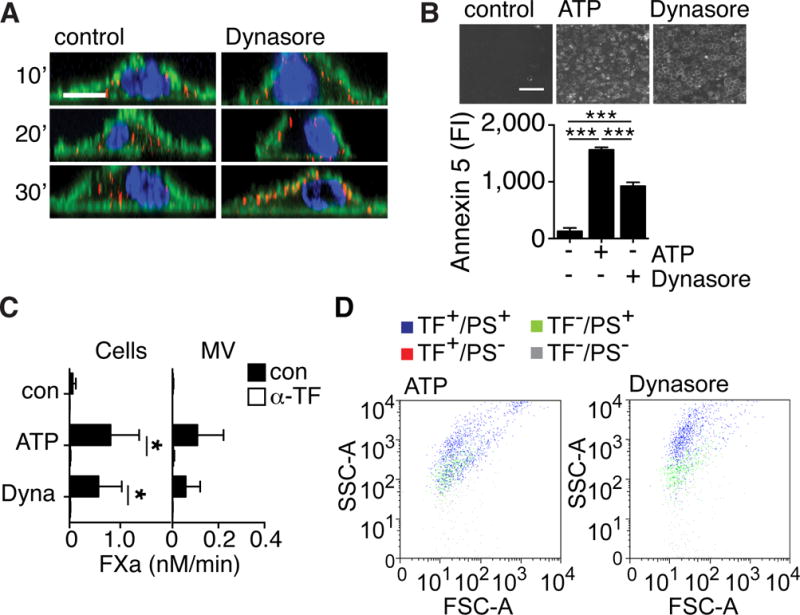
A, Cell surface TF was stained with anti-TF-Alexa 647 conjugates (red) for 10 minutes and followed in assay buffer for the indicated times without (control, left panels), or with Dynasore (right panels) stimulation. Fixed cells were counterstained for F-actin with phalloidin-Alexa488 (green) and nuclei with Hoechst (blue). Images were taken on a Zeiss LSM 710 with a 63x Plan-Apochromat NA 1.4 WD 190 mm oil emersion objective and processed using Image Browser Software; scale bar = 10 μm. B, PS exposure on control, ATP- and Dynasore-stimulated cells assessed by annexin 5-FITC staining. Images were taken on Nikon Eclipse fluorescent microscope with a 10x Plan NA 0.25 WD 105mm objective and analyzed using ImageJ software; scale bar = 500 μm; ***p<0.001, ANOVA (Tukey’s), n=3. C, TF FXa generation activity in the presence of 0.5 nM FVIIa on cells (left panel) and 2 nM FVIIa on MV (right panel) following ATP and Dynasore (Dyna) stimulation for 30 minutes. Cell-surface TF was blocked with anti-TF antibody (α-TF) prior to stimulation; *p<0.05 paired t-test, n=3. D, FACS analysis of MV released from cells activated with ATP or Dynasore following staining with anti-TF and lactadherin-FITC for detection of PS.
We previously showed that the release of TF+ MV carrying γ-actin and PDI following P2rx7 activation occurred from filopodia13. Dynasore-treatment did not promote the formation of filopodia (Figure IC in the online-only Data Supplement), and, consistently, Dynasore-induced MV lacked surface actin characteristic of ATP-induced MV and carried markedly reduced levels of PDI relative to TF and integrin β1 (Figure 2A,B). We have previously shown that caspase-1 promotes TF trafficking and release on thrombo-inflammatory MV carrying proteins with free thiol groups. We therefore hypothesized that MV thiol-proteome analysis might uncover additional components of cellular events required for the generation of TF+ MV. The macrophage cell line RAW 264.7 facilitated scale up of MV protein recovery for mass spectrometry. Using this method, we identified Gαi2, 14-3-3ε, and 14-3-3β/α in the indicated thiol-labeled positions as constituents of ATP-induced MV (Figure IIA in the online-only Data Supplement). These proteins were of interest, since activation of Gαi2 is known to promote secretion34–37 and 14-3-3ε and β/α are inhibitors of the Gαi2-specific regulator of G protein signaling 16 (RGS16)38.
Figure 2. P2xr7 activation specifically incorporates Gαi2signaling components into TF+ MV.
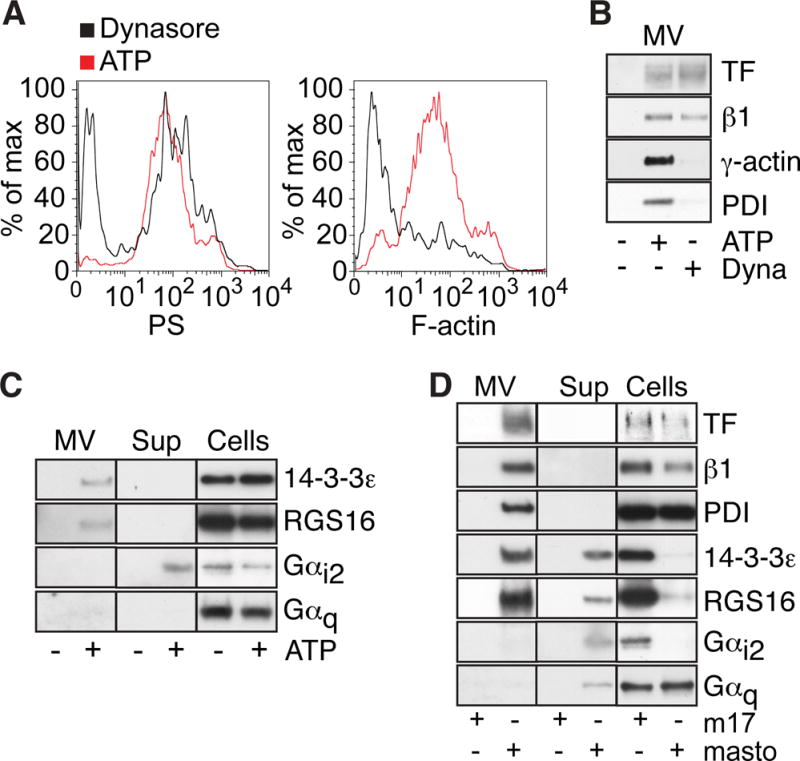
A, FACS detection of PS (lactadherin-FITC) and F-actin (phalloidin-Alexa 633) on MV released from ATP and Dynasore-treated cells. B, Representative Western blots of TF, integrin β1, γ-actin and PDI on MV from ATP and Dynasore-stimulated cells. C, Representative Western blots of 14-3-3ε, RGS16, Gαi2, and Gαq, in MV, MV-free supernatant (Sup) and cells following macrophage stimulation with or without ATP. D, Western blots of proteins characteristic for thrombo-inflammatory MV in macrophages stimulated with mastoparan or the inactive control peptide m17.
Western blotting furthermore showed that primary macrophages expressed only low levels of 14-3-3β/α. 14-3-3ε as well as the predicted partner RGS16 were detected in the MV fraction of ATP-stimulated macrophage (Figure 2C), but these proteins were absent from Dynasore-induced MV (Figure IIB in the online-only Data Supplement). Gαi2, but not Gαq was primarily detected in the MV-free cell supernatant fraction from ATP-stimulated primary macrophages (Figure 2C), suggesting that TF release was associated with the activation of selected G proteins by P2rx7 stimulation. Inhibition with pertussis toxin was not a feasible approach to demonstrate functional involvement of Gαi2 signaling in TF release, since this treatment is known to interfere with LPS priming and TF induction39. We therefore evaluated the contributions of Gαi2 activation to TF+ MV release by stimulating macrophages with mastoparan, a direct activator of Gαi240, 41. Similar to ATP-stimulation, mastoparan treatment led to release of Gαi2 into the MV-free extracellular space (Fig. 2D), and FACS analysis showed that mastoparan generated large quantities of TF and PS-positive MV (Figure IIC, D in the online-only Data Supplement). Mastoparan, but not the inactive control peptide m17, induced MV release of TF, integrin β1, PDI and γ-actin as well as 14-3-3ε and RGS16 (Figure 2D). These data provide evidence that Gαi2 is a downstream component of P2xr7 signaling and suggest a possible role for Gαi2 in a push reaction for release of MV, once TF and integrins associate with PDI- and actin-containing microdomains.
Integrin-associated arf6 controls TF availability on the cell surface
We next analyzed the released proteome to better understand the differences between MV generated by ATP stimulation versus dynamin blockade. Coomassie brilliant blue-stained gels showed a protein that was preferentially released into the supernatant of ATP-stimulated cells (Figure IIIA in the online-only Data Supplement). Mass spectrometry identified this protein as the small rho GTPase arf6. Western blotting confirmed that cellular levels of arf6 were markedly decreased after ATP stimulation and that arf6 was selectively released into the supernatant of ATP-stimulated cells (Figure 3A). Strikingly, arf6 was also depleted from Dynasore-stimulated cells, but predominantly recovered in the MV fraction, suggesting that transport of TF and integrins were mediated by arf6 and that P2rx7 activation induced dissociation of arf6 during MV release. Consistently, paxillin, an integrin adaptor protein that recruits arf6 regulators42, was found at similar levels in the MV fraction of ATP- as well as Dynasore-stimulated cells (Figure 3A).
Figure 3. Availability of TF for incorporation into MV is regulated by arf6.
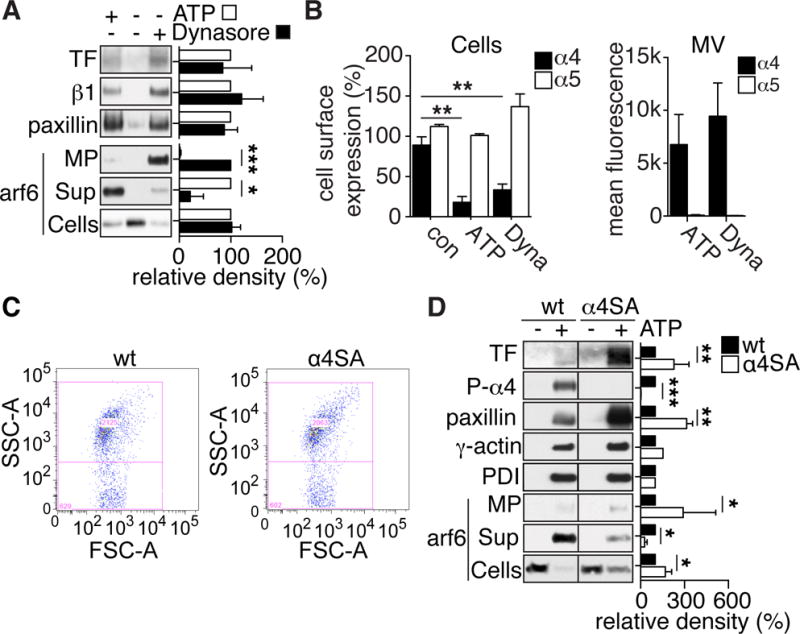
A, Representative Western blots of TF, integrin β1 and paxillin on MV, as well as of arf6 in MV, MV-free cell supernatant (Sup) and cells of control, ATP and Dynasore-stimulated macrophages. Bars present densitometric quantifications of Western blots from 3 independent experiments; *p<0.05, ***p<0.001, t-test. B, FACS detection of integrin α4 and α5 on the cell surface of macrophages after 30 minutes in assay buffer without (control), ATP or Dynasore (Dyna) stimulation; **p<0.01 ANOVA (Tukey’s), n=3. Right panel: FACS detection of integrin α4 and α5 on MVs released from ATP or Dynasore (Dyna)-stimulated macrophages, n=4. C, FACS detection of MV released from wild-type (wt) and α4SA macrophages stimulated with ATP for 30 minutes. Mean MV counts were indistinguishable between phenotypes (wt: 1611± 774, α4SA: 1601 ± 353, mean ± SD, n= 3). D, Western blots of TF, phospho-integrin α4, paxillin, γ-actin and PDI on MV and arf6 on MV, MV-free cell supernatant or cells of wild-type (wt) and α4SA ATP-stimulated macrophages. Bars represent densitometric quantifications of Western blots from 3 independent experiments; *p<0.05, **p<0.01, ***p<0.001, t-test.
Integrins α4β1 and α5β1 are known ligands for TF27 and are expressed by pro-inflammatory macrophages. FACS analysis showed that both integrin α subunits were equally expressed on the cell surface of quiescent macrophages, but upon ATP or Dynasore stimulation, integrin α4 levels decreased markedly, while integrin α5 surface levels remained unchanged (Figure 3B). Conversely, we detected integrin α4, but only very low levels of integrin α5, on released MV, suggesting that α4β1 traffics together with TF and represents the major integrin β1 heterodimer on TF+ MV.
We therefore studied integrin α4SA macrophages that express an integrin α4 mutant defective in phosphorylation-induced paxillin dissociation. Paxillin recruits negative regulators of arf6, leading to inactivation of arf6 in the receptor complex43. Of note, FACS staining showed similar integrin α5 expression by wild-type and α4SA macrophages (113% ± 10 % of wild-type; p=0.25, Wilcoxon rank-test, n=3), whereas integrin α4 levels were marginally increased in the mutant (145% ± 6.4 % of wild-type; p=0.13, Wilcoxon rank-test, n=4). Since arf6 is implicated in macrophage filopodia formation44 and cancer cell MV release45, we investigated whether integrin α4SA macrophages differed from wild-type macrophages in their ability to generate MV. Wild-type and integrin α4SA macrophages showed similar ATP-induced filopodia formation with visible TF staining (Figure IIIB in the online-only Data Supplement) and generated similar amounts of MV (Figure 3C), demonstrating that MV release was not measurably altered in α4SA macrophages.
ATP-induced MV released from wild-type and mutant macrophages carried similar high levels of integrin α4 and low levels of integrin α5 and showed higher TF procoagulant activity (Figure III C, D in the online-only Data Supplement). Consistent with the more stable interaction of integrin α4 with paxillin and arf6 in phosphorylation-deficient α4SA macrophages, both proteins were incorporated at increased levels into ATP-induced MV from mutant as compared to wild-type macrophages (Figure 3D). Conversely, lower levels of arf6 were released into the cell supernatant of ATP-stimulated α4SA versus wild-type macrophages. As expected, no phosphorylated integrin α4 was detectable in the α4SA MV. ATP-induced MV from α4SA macrophages carried more TF antigen, consistent with the increased TF activity. Thus, arf6 regulating the TF-associated integrin α4 influences cell surface levels of TF and determines availability of TF for incorporation into released MV.
Prothrombotic properties of MV carrying TF with high affinity for FVIIa
We next evaluated whether the release mechanism of TF was important for MV functional properties in flowing blood. We had previously shown that fibrin formation is induced dependent on TF following addition of ATP MV13. TF+/PS+ MV released from ATP- or Dynasore-stimulated cells were counted by FACS and equal amounts of MV were added to blood perfused over TF-negative macrophages (Figure 4A). Strikingly, only ATP-generated MV formed fibrin strands originating at the sites of localized particles and extending in the flow direction.
Figure 4. Prothrombotic properties of MV carrying TF with high affinity for FVIIa.
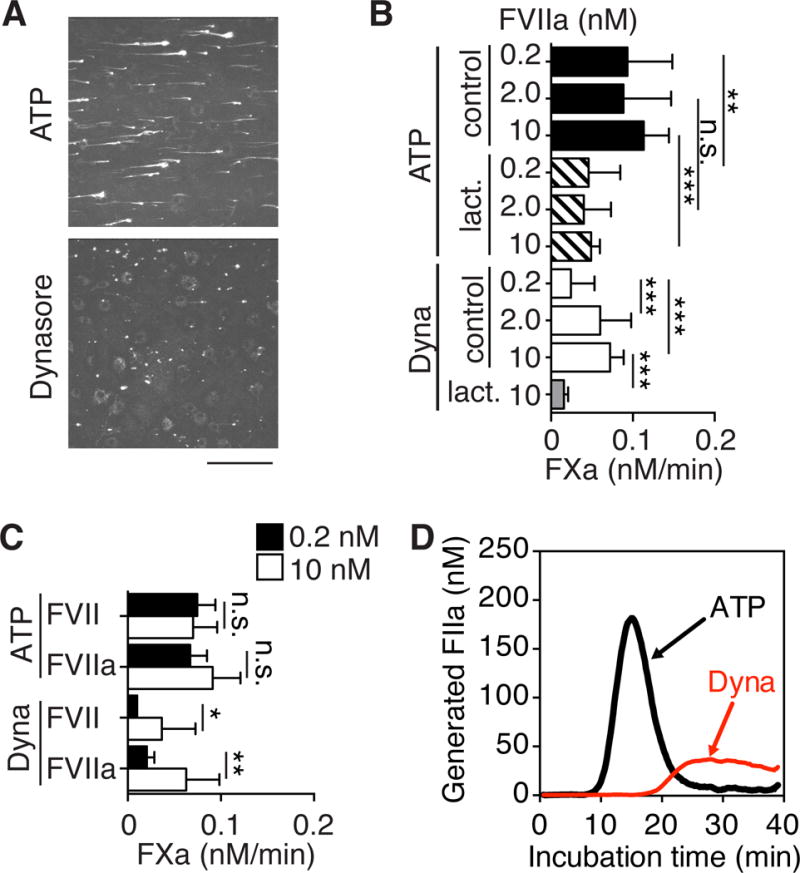
A, Whole C57BL/6J mouse blood containing equal amounts of TF+/PS+ MV derived from ATP- or Dynasore-stimulated TFKI macrophages as well as Alexa546-labeled anti-fibrin β-chain antibody was perfused over TFflox/LysM-cre macrophages for 2 min at the wall shear rate of 300 s−1. Following perfusion, stacks of confocal images (8-bit) were collected with a 2 μm spacing through the height of fluorescent objects using a Zeiss Axiovert 135M/LSM 410 microscope and Plan-Neofluar 40x/1.3 NA oil immersion objective. Confocal stacks were converted to 2-dimensional MAX projections. The figure shows a representative projection for each condition; scale bar =100 μm. B, FXa-generation with indicated concentrations of FVIIa and MV from ATP- or Dynasore-induced MV in the absence or presence of lactadherin (50 nM); **p<0.01, ***p<0.001, ANOVA, Tukey’s, n=3–10. C, FXa generation by ATP- or Dynasore-induced MV in the presence of 0.2 nM (closed bars) or 10 nM (open bars) FVII or FVIIa, *p<0.05, **p<0.01, t-test, n=3. D, Thrombin generation following addition of equal count of ATP- or Dynasore-generated MV to platelet-rich plasma. For quantification of thrombin generation see Figure IV C in the online-only Data Supplement.
MV are known to incorporate outer leaflet phosphatidylethanolamine (PE)46. Blocking PE with 1 μM Duramycin did not interfere with lactadherin binding to PS on MV released by either stimulus, but nevertheless completely abolished TF-dependent FXa generation as well as prothrombinase activity of both ATP- and Dynasore-induced MV (Figure IV A, B in the online-only Data Supplement). FXa generation by TF following activation by oxidation16 or thiol-modification19 is inefficiently inhibited by PS blockade with annexin 5. Similarly, the PS inhibitor lactadherin incompletely blocked FX activation on ATP generated MV independent of the concentrations of FVIIa used in the assay (Figure 4B). Unexpectedly, this experiment also indicated that ATP-generated MV carried TF that was saturated for maximal FX activation at much lower concentrations of FVIIa than seen with MV released following blockade of internalization with Dynasore. In addition, FX activation on Dynasore-induced MV was blocked with lactadherin, indicating that alternative procoagulant lipid composition is an unlikely cause for the enhanced prothrombotic activity of ATP-induced MV.
Thus, TF incorporated into PDI-containing MV exhibited high, subnanomolar affinity for its ligand FVIIa. ATP, but not Dynasore, MV carried TF that furthermore supported FX activation at lower concentrations of zymogen FVII (Figure 4C), consistent with the known similar affinity of activated and zymogen FVII for TF47. Thrombin generation assays in platelet-rich plasma also showed markedly decreased activity of Dynasore- as compared to ATP-induced MV (Figure 4D). Since MV generated by these agonists also differed in the composition of several proteins, we sought to identify a strategy that directly implicated TF in fibrin strand formation. We had previously shown in epithelial cells that anti-TF 10H10 preferentially inhibits and reacts with low affinity signaling pools of TF and only marginally interferes with cell surface FXa generation activity of TF15. Unexpectedly, anti-TF 10H10 significantly inhibited the activity of TF at low, but not high, FVIIa concentrations on released MV (Figure 5A). This effect was not prevented by the PDI inhibitor PACMA31 (Figure 5B). Anti-TF 10H10 had no effect on prothrombinase activity of ATP-induced MV (109 ± 13%, p=0.255, Wilcoxon rank-test, n=6), excluding indirect effects of the antibody on MV membrane lipid procoagulant properties. Furthermore, thiol labeling of MV was not altered by 10H10 treatment (Figure 5C), indicating that anti-TF 10H10 allosterically forces MV-localized TF to adopt a low affinity conformation for ligand binding or sterically interferes with FVIIa interactions required for high affinity binding on MV.
Figure 5. Affinity modulation of prothrombotic TF by anti-TF antibody 10H10.
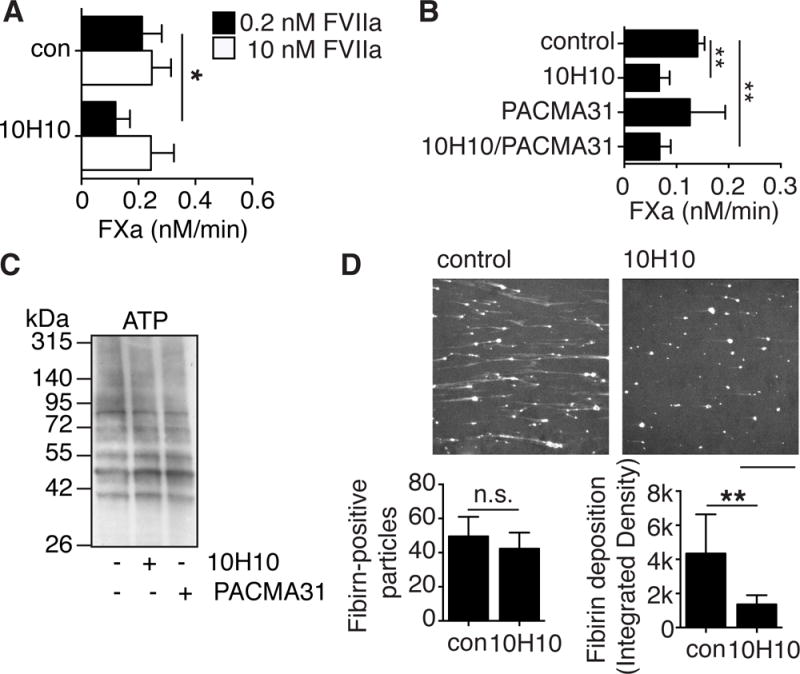
A, FXa generation by ATP-induced MV with 0.2 nM FVIIa (closed bars) and 10 nM FVIIa (open bars) in the presence of 10H10 (50 μg/ml); *p<0.05, paired t-test, n=3. B, FXa generation by ATP-induced MV with 0.2 nM FVIIa in the presence of 10H10 (50 μg/ml) and/or PACMA31 (10 μM); **p<0.01, ***p<0.001, ANOVA (Tukey’s), n=3. C, MV from ATP-stimulated macrophages were harvested and labeled with MPB in absence or presence of 10H10 (50 μg/ml) and/or PACMA31 (10 μM). D, Fibrin formation on TF-deficient macrophages analyzed as in Figure 4A, except that perfused blood contained MV from ATP-stimulated cells pre-incubated for 5 minutes on ice with either control IgG1 or anti-TF monoclonal antibody 10H10 (100 μg/ml); scale bar =100 μm. For statistical calculations, projections of three independent experiments, each including measurements in three preselected positions in the chamber, were analyzed with ImageJ. Number of fibrin-positive particles (p=0.30, t-test, n=3) (left panel), and total integrated density (mean grey value times surface area in pixels) of the fibrin-positive particles as measure for the amount of deposited fibrin on the surface (*p<0.05, t-test, n=3) (right panel).
Remarkably, ATP-induced MV pre-incubated with 10H10 produced markedly reduced fibrin strand formation in flowing blood (Figure 5D). Quantification of fibrin-positive particles showed that similar numbers of control and 10H10-preincubated MV localized to the cell surface and initiated limited fibrin formation (Figure 5D). However, overall fibrin deposition was significantly decreased in the presence of 10H10. These data provided an independent line of evidence that the conformation of TF associated with high affinity ligand binding, rather than the MV phospholipid composition, was primarily responsible for efficient procoagulant fibrin strand formation under flow.
P2X7 receptor stimulation of smooth muscle and cancer cells induces the release of MV carrying TF with high affinity for FVIIa
In order to determine whether P2rx7-induced release of high affinity TF is unique for macrophages, we further analyzed MV release and procoagulant activity from other cell types expressing P2X7 receptors and TF. We have demonstrated previously that smooth muscle cells activate and release TF in response to ATP-induced P2rx7 activation12. As seen with macrophage-derived MV, FX activation was maximal at subnanomolar concentrations of FVIIa, consistent with the presence of TF with high affinity for FVIIa (Figure 6A).
Figure 6. MV carrying TF with high affinity for FVIIa are released from various cell types.
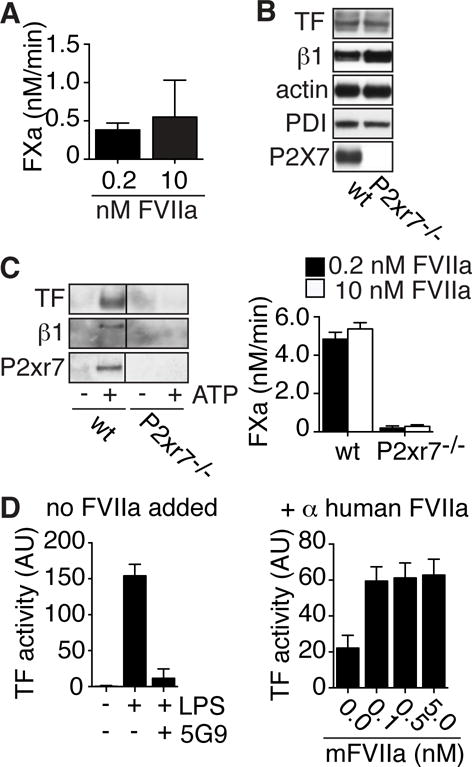
A, FXa generation by MV released from ATP-treated smooth muscle cells in the presence of 0.2 or 10 nM FVIIa. B, Western blot detection of TF, integrin β1, actin, PDI and P2xr7 in wt or P2rx7-deficient breast cancer cells from PyMT mice. C, Western blot detection of TF, integrin β1, and P2xr7 and TF activity in the presence of 0.2 nM (closed bars) or 10 nM (open bars) FVIIa on MV from wt and P2rx7-deficient cells stimulated for 30 minutes with 5 mM ATP. D, TF FXa generation activity on MV isolated from LPS stimulated blood. Note that in the left panel no FVIIa was added to the reaction and TF-dependence was confirmed with anti-human TF 5G9 (50 μg/ml). Mouse FVIIa (mFVIIa) was added to MV preparation in which human FVIIa was neutralized with specific monoclonal antibodies in the right panel.
We furthermore isolated breast cancer cells from the polyoma middle T (PyMT) model of spontaneous breast tumor development48. Cells isolated from P2rx7-deficient tumors lacked the P2rx7 protein based on Western blotting, but expressed levels of TF, integrin β1, actin and PDI similar to wt controls (Figure 6B). As expected, wt but not P2xr7−/− cells responded to ATP stimulation with the release of MV that, as previously seen with smooth muscle cells, carried the P2X7 receptor along with integrin β1 and TF. Importantly, FXa generation on these MV was maximal with 0.2 nM FVIIa (Figure 6C), demonstrating that injury signaling by the P2X7 receptor in a variety of cell types produces MV carrying TF with high affinity for its ligand FVIIa.
TF on human monocyte-derived MV has high affinity for FVIIa
In order to extent these findings to other pathophysiologically relevant injury signals activating TF, we studied TF function on blood monocyte-derived MV. TF expressed by blood monocytes is known to be rendered fully procoagulant dependent on PDI in the context of activation of the complement pathway3 and complement-dependent monocyte TF activation plays a pivotal role in venous thrombus development in mice5. In addition, LPS stimulation of human whole blood causes a PDI-dependent appearance of MV procoagulant activity5. In order to confirm that TF release from human cells was also associated with affinity maturation, we isolated MV from LPS-stimulated whole blood. We found that isolated MV activated FX efficiently in a TF-dependent manner without the addition of FVIIa (Figure 6D); thus MV TF apparently bound FVIIa with high affinity even in anticoagulated blood. We then used human FVIIa-specific monoclonal antibodies to dissociate the TF-FVIIa complex on isolated MV and obtained an estimate of TF affinity for FVIIa binding by using mouse FVIIa that shows no species incompatibility with human TF49. Murine FVIIa as low as 0.1 nM produced maximal FXa generation, confirming that the PDI-dependent release of MV from human cells also yields TF with high affinity for FVIIa (Figure 6D).
Discussion
The present study provides new insights into the cellular regulation of TF procoagulant activity. We have demonstrated that primary macrophages effectively control TF cell surface activity by dynamin-mediated receptor internalization. Inhibition of dynamin not only prevents TF uptake, but also promotes the release of TF+ MV. Characterization of the released soluble and MV proteomes led us to identify a genetically validated pathway in which integrin α4 regulates TF cell surface availability in an arf6-dependent manner. Following P2xr7 activation, Gαi2 signaling is furthermore implicated in a “push” for trafficking of TF onto MV with a distinct protein composition.
Arf6 has emerged as a master regulator of cell migration and adhesion by modulating intracellular trafficking of integrins29. Inactivation of arf6 prevents receptor uptake and alters recycling routes, ultimately leading to increased cell surface presence of integrins50–52. The coordinated incorporation of TF and integrin α4β1 into MV released from filopodia indicates a close association and common trafficking pathway of these receptors. Accordingly, functional inactivation of integrin α4-associated arf6 in α4SA macrophages results in increased availability of surface TF for incorporation into MV.
Cytoskeleton remodeling contributes to other MV release pathways53 and in cancer cells, deregulated adhesive properties and increased arf6 activity appear to play a major role in generating MV45. Our results in untransformed macrophages indicate that arf6 rather serves to control release of TF onto MV. Inactivation and cellular secretion of arf6 through injury signals seems to be required to divert integrin α4β1 and TF trafficking towards sites of MV release. Although LPS-primed macrophages express lysophosphatic acid (LPA) receptors, which couple to Gαi subunits and are implicated in P2xr7-induced MV generation in osteoclasts54, pharmacological inhibitors of LPA receptors had no measurable effect on procoagulant MV release from macrophages (data not shown). Nevertheless, direct activation of Gαi2 with mastoparan produced MV with a composition that was remarkably similar to MV generated by P2xr7 signaling, implicating Gαi2 in this thrombo-inflammatory response of macrophages.
ATP-stimulation culminates in the release of TF on MV that incorporate PDI. TF released on these MV undergoes maturation that enables binding of its ligand FVIIa with subnanomolar affinity. Prior biochemical data showed that reduction or mutational elimination of the allosteric TF Cys186-Cys209 disulfide bond reduces the affinity of TF for its ligand FVIIa. Supraphysiological concentrations of FVIIa, however, are sufficient in some, but not all cell models to overcome the markedly diminished specific activity of reduced TF in FXa generation assays15, 17, 21, 23, 55. Determination of the distance between the reduced Cys186 and Cys209 residues with molecular rulers showed a close proximity compatible not only with ligand-induced fit, but also with oxidation-mediated formation of a disulfide bond56. The determined redox potential of the Cys186-Cys209 allosteric disulfide56 furthermore predicts that TRX primarily acts as a TF reductase, as experimentally shown57, whereas PDI would act as an extracellular oxidase of TF. Consistently, we find that only PDI containing MV carry TF with high affinity for FVIIa.
The functional importance of TF maturation to a high affinity conformation is supported by the analysis of isolated MV in thrombin generation assays and when added to flowing whole blood. MV generated by Dynasore treatment carried procoagulant PS and PE and had activity equivalent to ATP-generated MV in a prothrombinase assay. Although Dynasore-induced MV localized to cells and initiated local fibrin deposition in flowing blood, they failed to propagate fibrin strand formation typical for MV generated by stimulation with ATP13. Anti-TF 10H10 preferentially interacts with signaling, low affinity TF and minimally inhibits FXa generation by cell surface TF15. While anti-TF 10H10 was also non-inhibitory at high FVIIa concentrations with TF on MV, the antibody specifically interfered with high affinity MV TF activity and markedly inhibited the prothrombotic fibrin strand formation by MV in flowing blood.
The relevance of affinity maturation for TF prothrombotic MV activity in blood is somewhat counterintuitive, since TF activity was normal at plasma FVII concentration in the static FXa generation. Prior studies have shown that FVII and FVIIa bind with equal, ~5 nM affinity to lipid-free TF captured by the 10H10 antibody47. As shown by the elegant study of Sen et al.58, in vitro relipidation procedures used to achieve maximal TF activity not only increase affinity of TF for FVIIa, but remarkably cause a more than 2 log enhancement in the association rate of FVIIa relative to low affinity TF. The finding that TF affinity is primarily driven by the on rate rather than the off rate has implications for flow systems as opposed to static FXa generation assay with prolonged incubation times. We posit that such a rate enhancement for complex formation between FVIIa and TF becomes highly relevant for thrombogenic responses under flow not only in vitro but also under pathophysiological conditions in vivo. In addition, our data show that both ATP injury signal-induced MV as well as complement-dependent generation of MV in LPS-stimulated human whole blood carry TF with high affinity for FVIIa, indicating broad pathophysiological relevance of TF affinity maturation in thrombo-inflammatory diseases. TF+ MV have emerged as potential biomarkers to identify patients with hypercoaguable states that would benefit from thrombosis prophylaxis in various diseases. Measuring levels of circulating TF by antigen-based assays has yielded inconsistent results, since these assays do not necessarily discriminate between encrypted and decrypted TF59. The assessment of additional MV components, for instance PDI, may enhance the clinical prediction of TF antigen-based assays for evaluation of a patient’s risk for thrombosis. MV TF activity assays60, 61 may be optimized for increased sensitivity towards the prothrombotic form of TF with high affinity for its protease ligand, FVIIa. Thus, the novel insights into cellular mechanisms of MV generation and the heterogeneity of procoagulant TF+ MV presented here provides new perspectives for the evaluation of MV diagnostic strategies.
Supplementary Material
Highlights.
TF cell surface availability is controlled by integrin α4β1- and arf6-regulated trafficking
MV generated by pharmacological interruption of TF-integrin internalization differ in protein composition and function from MV released by P2rx7 cell injury signaling
Maturation of TF to a high affinity state is a key determinant for the prothrombotic activity of TF+ MV in blood.
Acknowledgments
We greatly appreciate the excellent technical assistance of Cynthia Biazak, Pablito Tejada, and Jennifer Royce.
Sources of funding
This study was supported by NIH grants P01-HL031950 (W.R., Z.M.R., M.H.G.), UM1-HL120877 and HL60742 (W.R.), R01-HL117722 (Z.M.R); the Humboldt Foundation of Germany (W.R.) and a fellowship of the Deutsche Forschungsgemeinschaft and NovoNordisk (A.S.R.).
Abbreviations
- FVII
coagulation factor VII
- FX
coagulation factor X
- MV
microvesicles
- PDI
protein disulfide isomerase
- PS
phosphatidylserine
- TF
tissue factor
Footnotes
Disclosures
none
References
- 1.Langer F, Ruf W. Synergies of phosphatidylserine and protein disulfide isomerase in tissue factor activation. Thromb Haemost. 2014;111:590–597. doi: 10.1160/TH13-09-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Massberg S, Grahl L, von Bruehl ML, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16:887–896. doi: 10.1038/nm.2184. [DOI] [PubMed] [Google Scholar]
- 3.Langer F, Spath B, Fischer C, Stolz M, Ayuk FA, Kroger N, Bokemeyer C, Ruf W. Rapid activation of monocyte tissue factor by antithymocyte globulin is dependent on complement and protein disulfide isomerase. Blood. 2013;121:2324–2335. doi: 10.1182/blood-2012-10-460493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silasi-Mansat R, Zhu H, Popescu NI, Peer G, Sfyroera G, Magotti P, Ivanciu L, Lupu C, Mollnes TE, Taylor FB, Kinasewitz G, Lambris JD, Lupu F. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood. 2010;116:1002–1010. doi: 10.1182/blood-2010-02-269746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subramaniam S, Jurk K, Hobohm L, Jackel S, Saffarzadeh M, Schwierczek K, Wenzel P, Langer F, Reinhardt C, Ruf W. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood. 2017;129:2291–2302. doi: 10.1182/blood-2016-11-749879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reinhardt C, von Bruhl ML, Manukyan D, Grahl L, Lorenz M, Altmann B, Dlugai S, Hess S, Konrad I, Orschiedt L, Mackman N, Ruddock L, Massberg S, Engelmann B. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest. 2008;118:1110–1122. doi: 10.1172/JCI32376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho J, Furie BC, Coughlin SR, Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest. 2008;118:1123–1131. doi: 10.1172/JCI34134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880–15885. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kambas K, Chrysanthopoulou A, Vassilopoulos D, Apostolidou E, Skendros P, Girod A, Arelaki S, Froudarakis M, Nakopoulou L, Giatromanolaki A, Sidiropoulos P, Koffa M, Boumpas DT, Ritis K, Mitroulis I. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann Rheum Dis. 2014;73:1854–1863. doi: 10.1136/annrheumdis-2013-203430. [DOI] [PubMed] [Google Scholar]
- 10.von Bruhl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, Tsironidou V, Giatromanolaki A, Skendros P, Konstantinides S, Ritis K. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J. 2015;36:1405–1414. doi: 10.1093/eurheartj/ehv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furlan-Freguia C, Marchese P, Gruber A, Ruggeri ZM, Ruf W. P2X7 receptor signaling contributes to tissue factor-dependent thrombosis in mice. J Clin Invest. 2011;121:2932–2944. doi: 10.1172/JCI46129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothmeier AS, Marchese P, Petrich BG, Furlan-Freguia C, Ginsberg MH, Ruggeri ZM, Ruf W. Caspase-1-mediated pathway promotes generation of thromboinflammatory microparticles. J Clin Invest. 2015;125:1471–1484. doi: 10.1172/JCI79329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baroni M, Pizzirani C, Pinotti M, Ferrari D, Adinolfi E, Calzavarini S, Caruso P, Bernardi F, Di Virgilio F. Stimulation of P2 (P2X7) receptors in human dendritic cells induces the release of tissue factor-bearing microparticles. FASEB J. 2007;21:1926–1933. doi: 10.1096/fj.06-7238com. [DOI] [PubMed] [Google Scholar]
- 15.Ahamed J, Versteeg HH, Kerver M, Chen VM, Mueller BM, Hogg PJ, Ruf W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci USA. 2006;103:13932–13937. doi: 10.1073/pnas.0606411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen VM, Ahamed J, Versteeg HH, Berndt MC, Ruf W, Hogg PJ. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochemistry. 2006;45:12020–12028. doi: 10.1021/bi061271a. [DOI] [PubMed] [Google Scholar]
- 17.Versteeg HH, Ruf W. Tissue factor coagulant function is enhanced by protein-disulfide isomerase independent of oxidoreductase activity. J Biol Chem. 2007;282:25416–25424. doi: 10.1074/jbc.M702410200. [DOI] [PubMed] [Google Scholar]
- 18.Popescu NI, Lupu C, Lupu F. Extracellular protein disulfide isomerase regulates coagulation on endothelial cells through modulation of phosphatidylserine exposure. Blood. 2010;116:993–1001. doi: 10.1182/blood-2009-10-249607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le D, Rapaport S, Rao LVM. Studies of the mechanism for enhanced cell surface factor VIIa\tissue factor activation of factor X on fibroblast monolayers after their exposure to N-ethylmaleimide. Thromb Haemost. 1994;72:848–855. [PubMed] [Google Scholar]
- 20.Bach RR, Moldow CF. Mechanism of tissue factor activation on HL-60 cells. Blood. 1997;89:3270–3276. [PubMed] [Google Scholar]
- 21.Kothari H, Nayak RC, Rao LV, Pendurthi UR. Cystine186-cystine 209 disulfide bond is not essential for the procoagulant activity of tissue factor or for its de-encryption. Blood. 2010;115:4273–4283. doi: 10.1182/blood-2009-09-241356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kothari H, Rao LVM, Pendurthi U. Cys186-Cys209 disulfide-mutated tissue factor does not equal cryptic tissue factor: no impairment in decryption of disulfide mutated tissue factor. Blood. 2010;116:502–503. [Google Scholar]
- 23.Ruf W, Versteeg HH. Tissue factor mutated at the allosteric Cys186-Cys209 disulfide bond is severely impaired in decrypted procoagulant activity. Blood. 2010;116:500–501. doi: 10.1182/blood-2010-04-281287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petersen LC, Albrektsen T, Hjorto GM, Kjalke M, Bjorn SE, Sorensen BB. Factor VIIa/tissue factor-dependent gene regulation and pro-coagulant activity: effect of factor VIIa concentration. Thromb Haemost. 2007;98:909–911. [PubMed] [Google Scholar]
- 25.Broze GJ. Binding of human factor VII and VIIa to monocytes. J Clin Invest. 1982;70:526–535. doi: 10.1172/JCI110644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fair DS, MacDonald MJ. Cooperative interaction between factor VII and cell surface-expressed tissue factor. J Biol Chem. 1987;262:11692–11698. [PubMed] [Google Scholar]
- 27.Dorfleutner A, Hintermann E, Tarui T, Takada Y, Ruf W. Crosstalk of integrin α3β1 and tissue factor in cell migration. Mol Biol Cell. 2004;15:4416–4425. doi: 10.1091/mbc.E03-09-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Versteeg HH, Schaffner F, Kerver M, Petersen HH, Ahamed J, Felding-Habermann B, Takada Y, Mueller BM, Ruf W. Inhibition of tissue factor signaling suppresses tumor growth. Blood. 2008;111:190–199. doi: 10.1182/blood-2007-07-101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D’Souza-Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol. 2006;7:347–358. doi: 10.1038/nrm1910. [DOI] [PubMed] [Google Scholar]
- 30.Feral CC, Neels JG, Kummer C, Slepak M, Olefsky JM, Ginsberg MH. Blockade of alpha4 integrin signaling ameliorates the metabolic consequences of high-fat diet-induced obesity. Diabetes. 2008;57:1842–1851. doi: 10.2337/db07-1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Snyder LA, Rudnick KA, Tawadros R, Volk A, Tam SH, Anderson GM, Bugelski PJ, Yang J. Expression of human tissue factor under the control of the mouse tissue factor promoter mediates normal hemostasis in knock-in mice. J Thromb Haemost. 2008;6:306–314. doi: 10.1111/j.1538-7836.2008.02833.x. [DOI] [PubMed] [Google Scholar]
- 32.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 33.McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- 34.Ferry X, Eichwald V, Daeffler L, Landry Y. Activation of betagamma subunits of G(i2) and G(i3) proteins by basic secretagogues induces exocytosis through phospholipase Cbeta and arachidonate release through phospholipase Cgamma in mast cells. J Immunol. 2001;167:4805–4813. doi: 10.4049/jimmunol.167.9.4805. [DOI] [PubMed] [Google Scholar]
- 35.Aridor M, Traub LM, Sagi-Eisenberg R. Exocytosis in mast cells by basic secretagogues: evidence for direct activation of GTP-binding proteins. J Cell Biol. 1990;111:909–917. doi: 10.1083/jcb.111.3.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shefler I, Seger R, Sagi-Eisenberg R. Gi-mediated activation of mitogen-activated protein kinase (MAPK) pathway by receptor mimetic basic secretagogues of connective tissue-type mast cells: bifurcation of arachidonic acid-induced release upstream of MAPK. J Pharmacol Exp Ther. 1999;289:1654–1661. [PubMed] [Google Scholar]
- 37.Bueb JL, Mousli M, Bronner C, Rouot B, Landry Y. Activation of Gi-like proteins, a receptor-independent effect of kinins in mast cells. Mol Pharmacol. 1990;38:816–822. [PubMed] [Google Scholar]
- 38.Abramow-Newerly M, Ming H, Chidiac P. Modulation of subfamily B/R4 RGS protein function by 14-3-3 proteins. Cell Signal. 2006;18:2209–2222. doi: 10.1016/j.cellsig.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 39.Fan H, Li P, Zingarelli B, Borg K, Halushka PV, Birnbaumer L, Cook JA. Heterotrimeric Galpha(i) proteins are regulated by lipopolysaccharide and are anti-inflammatory in endotoxemia and polymicrobial sepsis. Biochim Biophys Acta. 2011;1813:466–472. doi: 10.1016/j.bbamcr.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yokokawa N, Komatsu M, Takeda T, Aizawa T, Yamada T. Mastoparan, a wasp venom, stimulates insulin release by pancreatic islets through pertussis toxin sensitive GTP-binding protein. Biochem Biophys Res Commun. 1989;158:712–716. doi: 10.1016/0006-291x(89)92779-4. [DOI] [PubMed] [Google Scholar]
- 41.Ozaki Y, Matsumoto Y, Yatomi Y, Higashihara M, Kariya T, Kume S. Mastoparan, a wasp venom, activates platelets via pertussis toxin-sensitive GTP-binding proteins. Biochem Biophys Res Commun. 1990;170:779–785. doi: 10.1016/0006-291x(90)92159-w. [DOI] [PubMed] [Google Scholar]
- 42.Kondo A, Hashimoto S, Yano H, Nagayama K, Mazaki Y, Sabe H. A new paxillin-binding protein, PAG3/Papalpha/KIAA0400, bearing an ADP-ribosylation factor GTPase-activating protein activity, is involved in paxillin recruitment to focal adhesions and cell migration. Mol Biol Cell. 2000;11:1315–1327. doi: 10.1091/mbc.11.4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishiya N, Kiosses WB, Han J, Ginsberg MH. An alpha4 integrin-paxillin-Arf-GAP complex restricts Rac activation to the leading edge of migrating cells. Nat Cell Biol. 2005;7:343–352. doi: 10.1038/ncb1234. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Q, Calafat J, Janssen H, Greenberg S. ARF6 is required for growth factor- and rac-mediated membrane ruffling in macrophages at a stage distal to rac membrane targeting. Mol Cell Biol. 1999;19:8158–8168. doi: 10.1128/mcb.19.12.8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, D’Souza-Schorey C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol. 2009;19:1875–1885. doi: 10.1016/j.cub.2009.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Larson MC, Woodliff JE, Hillery CA, Kearl TJ, Zhao M. Phosphatidylethanolamine is externalized at the surface of microparticles. Biochim Biophys Acta. 2012;1821:1501–1507. doi: 10.1016/j.bbalip.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dickinson CD, Ruf W. Active site modification of factor VIIa affects interactions of the protease domain with tissue factor. J Biol Chem. 1997;272:19875–19879. doi: 10.1074/jbc.272.32.19875. [DOI] [PubMed] [Google Scholar]
- 48.Schaffner F, Versteeg HH, Schillert A, Yokota N, Petersen LC, Mueller BM, Ruf W. Cooperation of tissue factor cytoplasmic domain and PAR2 signaling in breast cancer development. Blood. 2010;116:6106–6113. doi: 10.1182/blood-2010-06-289314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petersen LC, Norby PL, Branner S, Sorensen BB, Elm T, Stennicke HR, Persson E, Bjorn SE. Characterization of recombinant murine factor VIIa and recombinant murine tissue factor: a human-murine species compatibility study. Thromb Res. 2005;116:75–85. doi: 10.1016/j.thromres.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 50.Eva R, Crisp S, Marland JR, Norman JC, Kanamarlapudi V, ffrench-Constant C, Fawcett JW. ARF6 directs axon transport and traffic of integrins and regulates axon growth in adult DRG neurons. J Neurosci. 2012;32:10352–10364. doi: 10.1523/JNEUROSCI.1409-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang Y, Joshi S, Xiang B, Kanaho Y, Li Z, Bouchard BA, Moncman CL, Whiteheart SW. Arf6 controls platelet spreading and clot retraction via integrin alphaIIbbeta3 trafficking. Blood. 2016;127:1459–1467. doi: 10.1182/blood-2015-05-648550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dunphy JL, Moravec R, Ly K, Lasell TK, Melancon P, Casanova JE. The Arf6 GEF GEP100/BRAG2 regulates cell adhesion by controlling endocytosis of beta1 integrins. Curr Biol. 2006;16:315–320. doi: 10.1016/j.cub.2005.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morel O, Jesel L, Freyssinet JM, Toti F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler Thromb Vasc Biol. 2011;31:15–26. doi: 10.1161/ATVBAHA.109.200956. [DOI] [PubMed] [Google Scholar]
- 54.Panupinthu N, Zhao L, Possmayer F, Ke HZ, Sims SM, Dixon SJ. P2X7 nucleotide receptors mediate blebbing in osteoblasts through a pathway involving lysophosphatidic acid. J Biol Chem. 2007;282:3403–3412. doi: 10.1074/jbc.M605620200. [DOI] [PubMed] [Google Scholar]
- 55.Rehemtulla A, Ruf W, Edgington TS. The integrity of the Cys186-Cys209 bond of the second disulfide loop of tissue factor is required for binding of factor VII. J Biol Chem. 1991;266:10294–10299. [PubMed] [Google Scholar]
- 56.Liang HP, Brophy TM, Hogg PJ. Redox properties of the tissue factor Cys186-Cys209 disulfide bond. Biochem J. 2011;437:455–460. doi: 10.1042/BJ20110718. [DOI] [PubMed] [Google Scholar]
- 57.Wang P, Wu Y, Li X, Ma X, Zhong L. Thioredoxin and thioredoxin reductase control tissue factor activity by thiol redox-dependent mechanism. J Biol Chem. 2013;288:3346–3358. doi: 10.1074/jbc.M112.418046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sen P, Neuenschwander PF, Pendurthi UR, Rao LV. Analysis of factor VIIa binding to relipidated tissue factor by surface plasmon resonance. Blood Coagul Fibrinolysis. 2010;21:376–379. doi: 10.1097/MBC.0b013e328333b084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hisada Y, Alexander W, Kasthuri R, Voorhees P, Mobarrez F, Taylor A, McNamara C, Wallen H, Witkowski M, Key NS, Rauch U, Mackman N. Measurement of microparticle tissue factor activity in clinical samples: A summary of two tissue factor-dependent FXa generation assays. Thromb Res. 2016;139:90–97. doi: 10.1016/j.thromres.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tesselaar ME, Romijn FP, Van DL I, Prins FA, Bertina RM, Osanto S. Microparticle-associated tissue factor activity: a link between cancer and thrombosis? J Thromb Haemost. 2007;5:520–527. doi: 10.1111/j.1538-7836.2007.02369.x. [DOI] [PubMed] [Google Scholar]
- 61.Tatsumi K, Antoniak S, Monroe DM, III, Khorana AA, Mackman N. Evaluation of a new commercial assay to measure microparticle tissue factor activity in plasma: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12:1932–1934. doi: 10.1111/jth.12718. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.