

Abstract
Bacteria of the Burkholderia cepacia complex (Bcc) can cause devastating pulmonary infections in cystic fibrosis (CF) patients, yet the precise mechanisms underlying inflammation, recurrent exacerbations and transition from chronic stages to acute infection and septicemia are not known. Bcc bacteria are generally believed to have a predominant extracellular biofilm life style in infected CF lungs, similar to Pseudomonas aeruginosa, but this has been challenged by clinical observations which show Bcc bacteria predominantly in macrophages. More recently, Bcc bacteria have emerged in nosocomial infections of patients hospitalized for reasons unrelated to CF. Research has abundantly shown that Bcc bacteria can survive and replicate in mammalian cells in vitro, yet the importance of an intracellular life style during infection in humans is unknown. Here we studied the contribution of innate immune cell types to fatal pro-inflammatory infection caused by B. cenocepacia using zebrafish larvae. In strong contrast to the usual protective role for macrophages against microbes, our results show that these phagocytes significantly worsen disease outcome. We provide new insight that macrophages are critical for multiplication of B. cenocepacia in the host and for development of a fatal, pro-inflammatory response that partially depends on Il1-signalling. In contrast, neutrophils did not significantly contribute to disease outcome. In subcutaneous infections that are dominated by neutrophil-driven phagocytosis, the absence of a functional NADPH oxidase complex resulted in a small but measurably higher increase in bacterial growth suggesting the oxidative burst helps limit bacterial multiplication; however, neutrophils were unable to clear the bacteria. We suggest that paradigm-changing approaches are needed for development of novel antimicrobials to efficiently disarm intracellular bacteria of this group of highly persistent, opportunistic pathogens.
Author summary
Burkholderia cenocepacia is a bacterial pathogen that increases morbidity and mortality in cystic fibrosis patients, but can also cause serious hospital-acquired infections. In this work we demonstrate that macrophages, but not neutrophils, critically contribute to the development of a rapidly fatal inflammatory infection caused by B. cenocepacia. In contrast to findings with other important human pathogens, including Mycobacterium and Salmonella, Bcc bacteria cannot replicate in zebrafish embryos depleted of macrophages, identifying macrophages as a critical site for bacterial replication in vivo. Our study sheds light on the contribution of infected macrophages to infection outcome in an animal model, and contributes a paradigm changing concept of the infection mechanism of this class of bacterial pathogens.
Introduction
Bacteria belonging to the Burkholderia cepacia complex (Bcc) are ubiquitously found in the natural environment, specifically the rhizosphere of plants, and were first identified as the cause of onion rot; however, they are important opportunistic pathogens of individuals with cystic fibrosis (CF) and chronic granulomatous disease (CGD) [1]. More recently, they have emerged in nosocomial infections of both immunocompromised and immunocompetent patients hospitalized for reasons unrelated to CF or CGD; these infections have been correlated with contaminated surfaces and central venous access [2–4], but not much is known about reasons for the pathological effects. Multiple resistance to most clinically used antibiotics complicates treatment strategies [1,2].
In CF patients, pulmonary disease is the most important cause of morbidity and mortality. Vicious cycles of pulmonary obstruction, chronic microbial infections, and ineffective airway inflammation result in progressive deterioration of lung function [5]. The disease is characterized by massive neutrophil infiltration and production of pro-inflammatory cytokines, including IL-1β, TNFα and the neutrophil chemo attractant CXCL8 (IL8). Infections with Bcc bacteria aggravate clinical outcome with recurrent exacerbations and an increased pro-inflammatory state, and often result in fatal necrotizing pneumonia and septicemia known as Cepacia Syndrome. Neutrophils are fundamental in fighting infections, however, dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR) has been shown to result in an impairment in the ability of neutrophils to kill microbes and ineffective resolution of inflammation, which are considered as major causes of many of the pathological consequences seen in CF [6]. In B. cenocepacia-colonized CF patients neutrophil activation has been suggested to play an important role in the observed clinical deterioration [7]. In addition, human neutrophils have been shown to be able to reduce B. cenocepacia numbers in a reactive oxygen species (ROS)-dependent manner in vitro [8], contributing the increased sensitivity of CGD patients towards Bcc bacteria to a dysfunctional NADPH oxidase and the inability to produce an oxidative burst. Currently, no information is available about a role for neutrophils during nosocomial non-CF infections.
It has been demonstrated extensively using mammalian cell culture models that B. cenocepacia can build an intracellular niche and evade host immune killing [reviewed in 9–11]. In macrophages, B. cenocepacia has been shown to interfere with the phagolysosomal maturation process, membrane trafficking and the autophagy machinery [11–13]. Macrophages from individuals with CF or CGD have been shown to exhibit enhanced susceptibility towards B. cenocepacia infection, which has been attributed to reduced acidification and phagolysosomal fusion, defective autophagy and lower autophagic flux, respectively [12–14]. B. cenocepacia has also been detected in macrophages of experimentally infected rodents and zebrafish larvae [15–17]. While in vitro, macrophages stimulated with LPS or infected with B. cenocepacia display a significantly increased cytokine production [18–20], the direct contribution of Bcc-infected macrophages to pro-inflammatory state and disease outcome in vivo is not known.
Here we use a zebrafish (Danio rerio) infection model, taking advantage of the high similarity of its immune system to that of humans [21], and the excellent possibilities of intravital imaging of transparent zebrafish embryos that have contributed to better understanding microbe/host phagocyte interactions [22,23]. We demonstrate that macrophages are critical for proliferation of B. cenocepacia in zebrafish embryos. Moreover, we show that infection with B. cenocepacia leads to fatal inflammation requiring Il1-dependent signalling, and that Bcc bacteria can escape neutrophil defences. We discuss the implications of our findings in the context of development of therapeutic strategies to control Bcc infections.
Results
Proliferation of B. cenocepacia in the host and ensuing fatal infection critically depend on macrophages
Upon intravenous (iv) injection in zebrafish embryos, bacteria belonging to the Bcc are phagocytosed mainly by macrophages, in which they can replicate [17]. To better understand the explicit role of macrophages during infection with Bcc bacteria we used two strategies to deplete macrophages from zebrafish embryos; knockdown of pu.1 [24], a transcription factor involved in early myeloid progenitor formation, and chemical ablation.
B. cenocepacia K56-2, J2315 or B. cepacia CEP509, epidemic strains that we have previously shown to cause rapidly fatal systemic infection in zebrafish embryos [17], surprisingly showed a significant reduction in host mortality after depleting embryos of macrophages by pu.1 knockdown (Fig 1A, S1A Fig, S1C Fig and S1E Fig). Reduced mortality correlated with reduced bacterial burden in pu.1 knockdown compared to control embryos (Fig 1B, S1B Fig and S1D Fig). Intravital imaging of iv injected embryos, which express mCherry in macrophages (Tg(mpeg1:mCherry-F), referred to as mpeg1:mCherry, or Tg(mpeg:Gal4/uas-mCherry-NTR), referred to as mpeg1/umn) also showed that pu.1 knockdown embryos contained fewer bacteria at 24 hours post infection (hpi) than control embryos (Fig 1C and 1D and S2 Fig), demonstrating that macrophages are critical for bacterial replication. The applied concentration of pu.1 morpholino resulted in specific depletion of macrophages (Fig 1D compared to Fig 1C, 30 mpi), but not neutrophils (S1F Fig), although the effect was temporary as newly formed mCherry-positive macrophages appeared during infection (Fig 1D, 24 hpi and S2 Fig). In control embryos, the bacteria were rapidly seen associated with macrophages as shown previously [17], and the mCherry-positive signal was found to colocalise with bacteria up to 16 hpi; however, the mCherry positive signal had disappeared completely in the heavily infected control embryos, suggesting macrophages disappeared during the later stages of acute infection (Fig 1C and S2 Fig, and see below). The data suggest that an intramacrophage replication stage is critical for an increase in bacterial burden and for progression to rapidly fatal infection.
Fig 1. Macrophages are critical for virulence of B. cenocepacia.

(A,B) Embryo survival (average inoculum 17 CFU, representative experiment) (A) and bacterial burden (total of 3 experiments) over time (B) of control (black) and pu.1 knockdown embryos (red) injected iv with B. cenocepacia K56-2. (C,D) Representative fluorescence overlay images of an mpeg1:mCherry control and mpeg1:mCherry pu.1 knockdown embryo at 30 min and 24 h after injection with ~40 CFU B. cenocepacia K56-2 (blue). See also S2 Fig. (C) mCherry-positive macrophages (red) colocalise with K56-2 at 30 mpi, and are no longer detected at 24 hpi (insets show magnification). (D) mCherry-positive macrophages are absent in knockdown embryos at 30 mpi and start to re-appear at 24 hpi (insets show magnification). Scale bars, 100 μm. (E,F) Embryo survival (average inoculum 28 CFU, representative experiment) (E) and corresponding bacterial burden (n = 10 per group per time point). (F) of mpeg1/umn+ embryos, untreated or treated with 5mM Mtz or 0.2% DMSO, and mpeg1/umn−embryos treated with 5mM Mtz iv injected with B. cenocepacia K56-2. (B,F) Geometric means with each data point representing an individual embryo. Dead embryos marked as black open circles (not recorded for 5 embryos in (B), and in (F)). (A, B, E, F) * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001; **** p ≤ 0.0001. ns: not significant. See materials and methods for statistical tests. See also S1 Fig and S3 Fig.
Next, we adopted a chemically-inducible targeted cell ablation strategy based on the metronidazole/nitroreductase (Mtz/NTR) system [25]. Treatment of mpeg1/umn+ zebrafish embryos, which express the mCherry-NTR fusion protein in macrophages (S3A Fig), with the prodrug Mtz resulted in efficient specific ablation of fluorescent macrophages (S3B Fig and S3C Fig). Although DMSO- and Mtz-treated non-fluorescent mpeg1/umn- embryos (see materials and methods) showed slightly reduced bacterial load and mortality compared to non-treated infected control embryos (Fig 1E and 1F), Mtz-treated mpeg1/umn+ embryos lived significantly longer after iv injection of B. cenocepacia K56-2, with up to 60% of the embryos still alive at 5 days post infection (Fig 1E). Strikingly, no significant multiplication of B. cenocepacia was seen in macrophage-depleted embryos up to 24 hpi, in contrast to the infected control embryos (Fig 1F). The dependence on macrophages for replication was confirmed by real time imaging of infected DMSO- and Mtz-treated mpeg1/umn+ larvae (S4 Fig). Together, these data strengthen the pu.1 knockdown results, and demonstrate that macrophages are critical for multiplication of iv injected B. cenocepacia K56-2, J2315 and B. cepacia CEP509 and for the development of fatal infection in zebrafish embryos.
Macrophages clear less virulent Bcc strains inefficiently
Next, we determined the effect of macrophage depletion on the fate of three Bcc isolates that we have shown earlier to cause persistent infection in zebrafish embryos; iv-injected bacteria of these strains are able to survive and replicate in macrophages, yet they are unable to induce a pro-inflammatory response and disseminate from infected cells [17]. Pu.1 knockdown had no significant effect on host survival after infection with these strains (Fig 2A, S1G Fig and S1I Fig). For B. stabilis LMG14294 the absence of macrophages resulted in slightly but not significantly lower bacterial numbers at 48 hpi (Fig 2B), showing that, in agreement with the results described above for B. cenocepacia, these bacteria do not replicate efficiently in the absence of macrophages. B. cenocepacia J415 was cleared in 50% of the control, but not in pu.1 knockdown embryos at 48 hpi (S1H Fig), showing that macrophages have some protective role for the host against this strain. CFU counts were also higher at 48 hpi for B. vietnamiensis after pu.1 knockdown compared to control embryos (S1J Fig). Thus, although macrophages can reduce bacterial burden, they are unable to completely clear these less virulent Bcc strains, emphasizing the persistent character of this group of bacteria [17].
Fig 2. Macrophages clear less virulent Bcc strains inefficiently.

(A,B) Embryo survival (average inoculum 23 CFU, representative experiment) (A) and bacterial burden (total of 2 experiments) over time (B) of control (black) and pu.1 knockdown embryos (red) iv injected with B. stabilis LMG14294. Geometric means with each data point representing an individual embryo. * p ≤ 0.05, *** p ≤ 0.001; ns: not significant. See materials and methods for statistical tests. See also S1 Fig.
Acute infection correlates with systemic phagocyte death
In contrast to macrophages, neutrophils do not efficiently phagocytose Bcc bacteria injected in the circulation [17]. Here, we analysed their behaviour during later stages after iv injection in mpx:GFP embryos. After uptake and replication of B. cenocepacia in macrophages, patrolling neutrophils often completely stretched around these infected cells without apparent success in eliminating them (S5A Fig). From this time point, recruited neutrophils were seen to degranulate close to infected cells which might contribute to the release of intracellular bacteria, although the mechanism for bacterial release or exit from infected macrophages is not clear (Fig 3A). The infection progressed, as described earlier [17] with the formation of infection sites throughout the body, defined as areas of local bacterial dissemination, repeated cycles of intracellular replication, and tissue damage. Here, we show that these infection sites are characterized by massive infiltration of both macrophages and neutrophils (Fig 3B). During acute infection caused by B. cenocepacia K56-2, GFP-positive neutrophils (Fig 3C) and mCherry-positive macrophages (Figs 1C and 3D and S5D Fig) rapidly disappeared from the whole embryo body, including the infection sites. In contrast, an increase in neutrophil numbers was observed in a percentage of the embryos during persistent infection with B. stabilis, and recruited neutrophils were only occasionally seen to accumulate near infected macrophages (Fig 3C, S5B Fig and S5C Fig).
Fig 3. Acute, but not persistent infection results in systemic phagocyte death.

(A) Sudan black staining of an mpx:GFP embryo (24 hpi), injected with ~45 CFU B. cenocepacia K56-2. Bright field, fluorescence and merged images showing recruited neutrophils (green) that release granules (stained by Sudan black as black deposit, white arrow) close to an infected cell containing red fluorescent bacteria. Arrow head, individual bacteria. Scale bars, 50 μm. (B) Image of the trunk region of an mpx:GFP; mpeg1:mCherry embryo 24 h post iv injection in the blood island with B. cenocepacia K56-2 (Turquoise), showing neutrophils (green) and macrophages (red) infiltrated in an infection site with multiple infected cells. BF, Bright field image, showing tissue damage. Scale bar, 50 μm. (C) Mean neutrophil numbers in non-infected control and mpx:GFP embryos injected at 50 hpf with B. cenocepacia K56-2 or B. stabilis LMG14294. See also S5B Fig and S5C Fig. (D) Mpeg1:mCherry embryos showing reduced macrophage numbers (red) at 24 hpi in B. cenocepacia K56-2-infected (~45 CFU) compared to non-infected control embryos. Scale bars, 0.5 mm. See S5D Fig for quantification. (E) Mean relative mpx and mpeg1 gene expression level (qRT-PCR) in embryos injected with on average 234 CFU of B. cenocepacia K56-2 (red bars) or 123 CFU of B. stabilis LMG14294 (pink bars) each normalised to a PBS-injected control group at each time point and analysed using Anova (error bars, SEM). Two independent experiments. Asterisks below each bar indicate significance compared to the PBS control at each time point, significance between groups per time point is indicated with a horizontal line. (F) Non-infected and B. cenocepacia K56-2 (~50 CFU, Turquoise indicated in red for better visualization) infected embryos at 24hpi with the cell-impermeable dye Sytox Green. Arrows, dead cells due to DMSO injection. Arrow heads, bacterial clusters. Scale bars, 100 μm. See also S5F Fig. (C). Each data point represents an individual embryo. (C,E) * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001; ns: non-significant. See materials and methods for statistical tests. See also S5 Fig.
Global expression analysis showed that the neutrophil-specific mpx and macrophage-specific mpeg1 genes were rapidly downregulated in embryos injected with K56-2, but not with B. stabilis (Fig 3E). The observed disappearance of fluorescent phagocytes during infection with B. cenocepacia K56-2, however, was not simply due to reduced expression levels of the mpx:GFP and mpeg1:mCherry transgenes; labelling of the leukocyte-specific protein L-plastin or with the macrophage-specific dye Neutral Red [26] also revealed drastically reduced phagocyte numbers in infected embryos, although infected macrophages still stained positive for L-plastin (S5E Fig). Importantly, staining with the cell impermeable dye SYTOX Green showed that K56-2-infected embryos were loaded with extracellular DNA, in contrast to few SYTOX-positive cells at the site of injection in non-infected embryos (Fig 3F). Systemic lysis of macrophages was confirmed in infected mpeg1/umn+ embryos, which show strong expression of mCherry in macrophages allowing visualization of their destruction as mCherry-positive debris (S4 Fig (24–48 hpi; DMSO-treated) and S5F Fig). Thus, excessive inflammation correlates with systemic neutrophil and macrophage death although infected macrophages stay intact.
Neutrophils constitute a major source of ROS during inflammation. To analyse whether the production of ROS contributes to disease outcome, we performed knockdown experiments of the gp91phox (or cybb) subunit of NADPH oxidase [27]. Knockdown of gp91phox did not significantly affect embryo survival and bacterial burden in embryos iv injected with B. cenocepacia K56-2, J2315 or B. stabilis LMG14294 (Fig 4A), suggesting ROS-mediated responses do not effectively contribute to host defence, or host-damaging inflammation.
Fig 4. Neutrophils do not contribute significantly to infection.

(A) Embryo survival (left; representative experiment) and bacterial burden (total of 2 experiments) over time (right panel, geometric mean) of control (black) and gp91 knockdown embryos (red) iv injected with B. stabilis. Each data point represents an individual embryo. (B) Embryo survival of mpx/umn+ embryos, untreated or treated with 10 mM Mtz or 0.2% DMSO, and mpx/umn−embryos treated with 10 mM Mtz iv injected with ~ 50 CFU B. cenocepacia K56-2. (C) Embryo survival (average inoculum 50 CFU, representative experiment) of control (black) and csf3R knockdown embryos (red) injected iv with B. cenocepacia K56-2. * p ≤ 0.05; ** p ≤ 0.01; ns: non-significant. See materials and methods for statistical tests.
To study whether neutrophils themselves contributed to the observed tissue damage and host mortality we depleted embryos from neutrophils in analogy to the chemical ablation of macrophages described above. We obtained fish lines expressing the mCherry-NTR fusion protein in neutrophils and pre-incubated mpx/umn+ larvae with Mtz or DMSO. No difference was observed after iv injection with B. cenocepacia K56-2 in non-treated, DMSO-treated, or Mtz-treated mpx/umn- larvae compared to Mtz-treated mpx/umn+ larvae (Fig 4B), showing that the absence of neutrophils does not significantly change disease outcome. In agreement with these results, knockdown of csf3R, which results in specific neutrophil ablation [28], did not affect embryo survival rates (Fig 4C). The finding that depletion of neutrophils did not have a net effect on host survival emphasizes the host-destructive role of macrophages during acute infection caused by B. cenocepacia.
Neutrophils rapidly phagocytose, but are unable to eradicate, subcutaneously introduced Bcc
Injection of Bcc in the circulation of embryos is marked by macrophage-dominated phagocytosis [17] and our data now show that neutrophils do not significantly contribute to the fatal outcome of these infections. To better understand the role of neutrophils upon phagocytosis of bacteria we performed experiments based on the elegant work by Colucci-Guyon and colleagues who showed that neutrophils efficiently sweep up surface-associated but not fluid-borne bacteria [29]. Intravital imaging of mpx:GFP embryos subcutaneously injected with B. cenocepacia K56-2 or B. stabilis LMG14294 showed that neutrophils were rapidly attracted to live and heat-killed bacteria, but not to PBS, and infiltrated the infected area (S1 Movie and S2 Movie). Surprisingly, after about 1 h we observed that both B. cenocepacia K56-2 and B. stabilis-containing GFP-positive neutrophils rounded up and spontaneously ejected their cell contents into the extracellular space covering a large area (Fig 5, S2 Movie), although this was not evident with lower infection doses (S1 Movie). The released bacteria remained clustered and other neutrophils in the area showed reduced mobility directly after the event (S2 Movie).
Fig 5. Neutrophils efficiently phagocytose surface-associated Bcc bacteria.

Confocal stacks after subcutaneous infection with B. stabilis in mpx:GFP embryos (STD intensity projection, 2 μm x 19 steps; T = 35–37 in S2 Movie). Arrows, rounded neutrophils (green) with vacuoles full of bacteria (red) at 64 minutes post injection (mpi) eject their cell contents in the surroundings (arrow heads 65.5 mpi), leaving bacterial clusters and cell debris (arrow heads, 66 min). Lower panels, MAX intensity projection of three consecutive slices (2 μm) at 65.5 mpi showing ejected cellular contents (diffuse GFP signal). Scale bars, 50 μm.
Although the majority of subcutaneous bacteria were phagocytosed by recruited neutrophils (S1 Movie and S2 Movie), macrophages also contributed to bacterial uptake at the infection site, and bacterial clusters localised inside macrophages at later time points (Fig 6A and S6A Fig). Subcutaneous injection of B. cenocepacia K56-2 resulted in massive infiltration of neutrophils and macrophages, and was followed by visible tissue damage and an increase in bacterial burden with neutrophils persisting at the infection site in 100% of the infected embryos (Figs 6B and 7A, S6B Fig and S6C Fig). Subcutaneous injection of B. stabilis also resulted in an increase in bacterial burden, although reduced compared to B. cenocepacia (Fig 7B). In contrast to B. cenocepacia infection, only 13.2% (±6.85% SEM) of the embryos (3 independent experiments, n = 8, 12, and 26, respectively) showed persistent neutrophil infiltration at the infection site at 22 hpi, while neutrophils had moved away in the majority of embryos, suggesting that the host was able to resolve the inflammation resulting from infection with B. stabilis (Fig 7C, left panel).
Fig 6. Macrophages, but not neutrophils, contribute to increased bacterial burden and pro-inflammatory responses towards subcutaneously introduced B. cenocepacia.
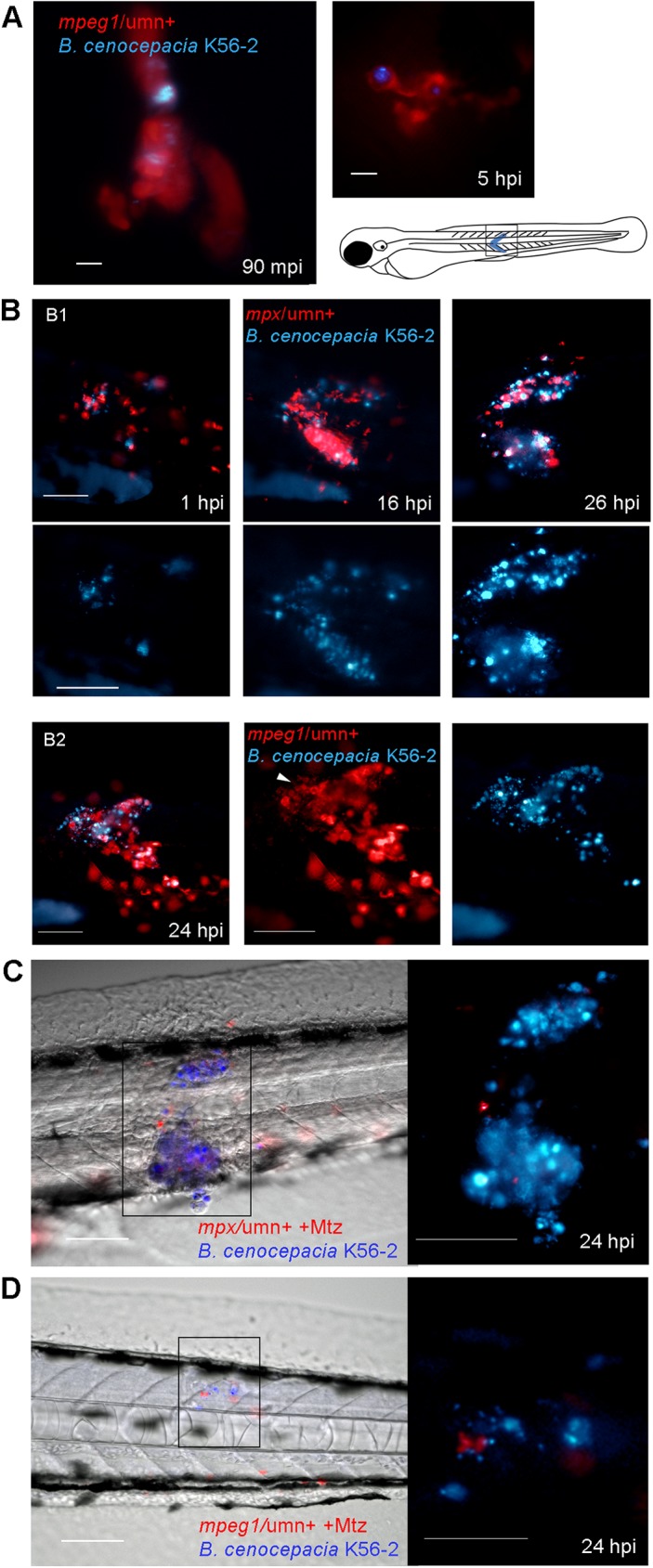
(A) Mpeg1/umn+ embryos were subcutaneously injected with B. cenocepacia K56-2 expressing Turquoise. Fluorescent overlay images were taken at 90 min and 5 h post infection, showing infected macrophages (red). Scale bars, 10 μm. (B) Images (red and blue overlay and below slightly enlarged individual fluorescence images with blue filter) of the indicated area (see drawing) of an mpx/umn+ (B1) and an mpeg1/umn+ (B2) embryo subcutaneously injected with B. cenocepacia K56-2 expressing Turquoise. B1 shows an embryo followed in time displaying neutrophil infiltration (red) and increase in bacterial burden (blue, see Fig 7A for quantification). B2 shows the image of an embryo with macrophage infiltration (red) and high bacterial burden at 24 hpi. Arrow head points at mCherry positive debris. See non-infected mpx/umn+ and mpeg1/umn+ control embryos in S6 Fig for comparison. Scale bars, 100 μm. (C) Image of the infected area of a representative mpx/umn+ embryo depleted of neutrophils with Mtz, and subcutaneously injected with B. cenocepacia K56-2 (Turquoise) at 24 hpi. Scale bar 100 μm, and 50 μm for inset. See also S6D Fig. (D) Image of a representative mpeg1/umn+ embryo depleted of macrophages with Mtz and subcutaneously injected with B. cenocepacia K56-2 (Turquoise) at 24 hpi. Scale bar 100 μm, and 50 μm for inset.
Fig 7. The absence of Gp91 results in increased bacterial burden and neutrophil persistence during subcutaneous infections.

(A) Bacterial burden over time after subcutaneous injection of B. cenocepacia K56-2 in control (black circles) and gp91 knockdown embryos (open red circles). Average of two independent experiments. (B) Bacterial burden over time after subcutaneous injection of B. stabilis LMG14294 in control (black circles) and gp91 knockdown embryos (open red circles). Average of three independent experiments. (C) Fluorescent overlay images of the injected area of a representative mpx:GFP control MO and gp91 knockdown embryo (neutrophils in green) in time after subcutaneous injection with B. stabilis (red). Inset shows bacterial load at ~20 hpi. % at 22 and 21 hpi indicates percentage of control and gp91 knockdown embryos that show reduced neutrophil numbers (86.8%), and persistent neutrophil infiltration (71.3%), respectively, at the infection site. Scale bar, 100 μm. (A,B) * p ≤ 0.05; **** p ≤ 0.0001; ns: non-significant. See materials and methods for statistical tests.
To determine the individual contribution of neutrophils and macrophages during subcutaneous infections, mpx/umn+ and mpeg1/umn+ embryos depleted of neutrophils or macrophages, prior to infection using Mtz-mediated cell ablation, respectively, were subcutaneously injected with B. cenocepacia K56-2. The absence of neutrophils did not change bacterial multiplication (Fig 6C compared to Fig 6B). In subcutaneously injected Mtz-treated mpeg1/umn+ embryos, however, the absence of macrophages prevented bacterial multiplication (Fig 6D and S6D Fig). These data demonstrate that neutrophils are not able to control infection caused by subcutaneously injected B. cenocepacia K56-2, and that even upon neutrophil-dominated phagocytosis B. cenocepacia K56-2 is highly virulent through its interaction with macrophages.
Neutrophils of CGD patients are unable to mount an oxidative burst, and are less able to reduce Bcc numbers compared to those of healthy individuals [8,30]. Therefore, we analysed the effect of gp91 knockdown on subcutaneously injected B. cenocepacia K56-2 and B. stabilis LMG12494. Knockdown of gp91 resulted in a slight increase in bacterial burden compared to infected control embryos, although this was only significant for B. stabilis (Fig 7A, 7B and 7C). In addition, neutrophils were still massively recruited in 71.3% (±14.43% SEM) of the gp91 knockdown embryos infected with B. stabilis at 21 hpi (n = 8, 10, and 22 respectively, and Fig 7C) compared to 13.2% (as indicated above) for control embryos, showing that the absence of Gp91 reduces the host’s ability to resolve inflammation elicited by this strain.
Together the data show that even upon neutrophil-dominated phagocytosis, virulent B. cenocepacia K56-2 exerts its pro-inflammatory effects through macrophages. A functional ROS response helps the host to slow down multiplication of both B. cenocepacia and B. stabilis to some extent, and contributed to the resolution of inflammation during infection with B. stabilis, although no effect was seen with the virulent B. cenocepacia.
Positive and negative effects of Il1-signalling for the B. cenocepacia-infected host
Studies in mice have implicated Myd88, a key adaptor protein in innate immune signalling, in the pathological inflammation associated with B. cenocepacia infection [31]. In zebrafish, we found that the absence of Myd88 resulted in reduced mortality, with B. cenocepacia-infected myd88-/- mutant embryos living significantly longer than infected myd88+/+ embryos (S7A Fig), but no difference was observed in bacterial burden (S7B Fig). The reduced mortality found in myd88-/- mutants, however, was not consistently mimicked by morpholino knockdown of myd88, although no significant increase in mortality was found compared to control embryos (S7C Fig and S7D Fig). The data show that Myd88 is not critical for host defence, and suggest that Myd88-dependent and -independent signalling pathways contribute to fatal pro-inflammatory disease. The lack of a consistent host-protective effect of myd88 deficiency led us to focus on pro-inflammatory cytokines that might contribute to the fatal inflammation in B. cenocepacia-infected embryos.
To determine pro-inflammatory responses playing a role during acute and persistent infection of zebrafish embryos, we analysed global cxcl8 and il1b gene expression levels. In wildtype embryos, B. cenocepacia K56-2 induced a rapid and robust increase in il1b and cxcl8 expression, which remained high until at least 24 hpi (Fig 8A and 8B). In contrast, injection of B. stabilis resulted in significantly lower induction of expression of these pro-inflammatory cytokine genes, in agreement with the persistent character of the infection. To study the contribution of macrophages to the global increase in pro-inflammatory gene expression, we analysed infection-induced changes in il1b and cxcl8 expression in embryos that were depleted of macrophages using Mtz-mediated cell ablation. Although no significant difference was found in infection-induced cxcl8 expression at 4 and 7 hpi, the induction of il1b expression was significantly lower in B. cenocepacia K56-2 infected embryos that were depleted of macrophages compared to infected DMSO-treated control embryos (Fig 8C and 8D). Since il1b and cxcl8 expression were not affected by Mtz or solvent (DMSO) treatment in K56-2 or PBS injected wildtype embryos (S8A Fig), nor in PBS-injected macrophage-depleted mpeg1/umn+ embryos (Fig 8C and 8D), these results show that zebrafish macrophages contribute significantly to the infection-induced increase in global il1b expression.
Fig 8. B. cenocepacia K56-2 induces robust pro-inflammatory Il1b expression that is dependent on macrophages.

(A,B) Mean relative il1b (A) and cxcl8 (B) gene expression levels (qRT-PCR) in embryos injected with on average 250 CFU B. cenocepacia K56-2 (red bars) or on average 111 CFU B. stabilis LMG14294 (pink bars), normalized to a PBS-injected control group at each time point. Error bars represent mean with SEM of three biological replicates. Asterisks above each bar indicate significance compared to the PBS control at each time point, significance between groups per time point is indicated with a horizontal line. (C,D) mpeg1/umn+ embryos were pre-treated at 34 hpf for 15 h with DMSO or 5 mM Mtz. Randomized groups were injected with either PBS or with B. cenocepacia K56-2 (on average 150 CFU). Graphs show mean relative il1b (C) and cxcl8 (D) gene expression levels (qRT-PCR) normalized to the PBS-injected DMSO-treated group at each time point. Error bars represent mean with SEM of two biological replicates. See also S8 Fig. (E) Confocal stack images (green/red overlay (left panels) and red channel (right panels)) of il1b:GFP/mpeg1:mCherry embryos 8 h post iv injection with PBS, or B. cenocepacia K56-2 (DS-Red). Due to strong fluorescence of GFP in epithelial cells in the trunk and head region, images were taken over the yolk sac valley. Scale bars 10 μm. (F) Embryo survival (average inoculum 44 CFU, representative experiment) of control (black) and il1b knockdown embryos (red) injected iv with B. cenocepacia K56-2. (G,H) Embryo survival (G) and bacterial burden over time (H) of control (black) and Anakinra-treated embryos (red) injected iv with B. cenocepacia K56-2 (average inoculum 107 CFU for both groups). Representative experiment. (A-D, F-H) * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001; ns: non-significant.
The involvement of macrophages in the induction of il1b expression was further confirmed using an il1b-GFP reporter fish line that expresses membrane-targeted GFP from the il1b promoter sequence [32]. Intravenous injection of il1b:GFP/mpeg1:mCherry embryos with B. cenocepacia K56-2 resulted in expression of GFP in macrophages within 2 hours, with strong expression from 4 hpi (Fig 8E), confirming that macrophages significantly contribute to the global increase in il1b expression levels.
We then knocked down the expression of il1b, which resulted in enhanced mortality (Fig 8F), suggesting that Il1b signalling contributes to host defence. However, treatment of larvae with Anakinra, an antagonist of the Il1 receptor, resulted in a small but significant increase in survival of B. cenocepacia K56-2 infected embryos (Fig 8G), although bacterial multiplication (Fig 8H) and infection-induced expression of il1b and cxcl8 (S8B Fig), were not affected by Anakinra treatment. These apparently contrasting results suggest that Il1b contributes to host-protective and detrimental responses, while the balance of Il1-receptor mediated signalling leans towards fatal pro-inflammatory responses.
Discussion
Macrophages have been suggested to be important for Bcc virulence and invasiveness in infected CF patients since it was shown for the first time in the late 90’s that Bcc bacteria can survive in macrophages in vitro [33]. Bcc bacteria are strong biofilm producers and it is generally assumed that, as seen with Pseudomonas aeruginosa [34], they exist and persist as biofilm communities in the lungs of infected CF patients. Recent clinical evidence has challenged this belief showing a strong association of Bcc bacteria with cells in lung tissue samples taken from transplant and deceased CF patients [35,36]. This was highlighted in the recent study by Schwab and colleagues who found B. cenocepacia mainly in macrophages and not in biofilm-like structures in CF lung tissue [36]. In vitro, CF and CGD macrophages are more permissive to infection with B. cenocepacia than their healthy counterparts; however, these bacteria have also been shown to replicate in macrophages derived from healthy individuals, sometimes even to high numbers as exemplified for human monocyte-derived macrophages [11,13]. Of note, B. cenocepacia has been detected in macrophages present in deep skin infections of dogs that were receiving cyclosporine [37]. Thus, macrophages may contribute to pathogenicity caused by this group of bacteria not only in CF, but also in nosocomial infections of both immune competent and immune suppressed individuals [2–4,38]; the contribution of (infected) macrophages to pathogenesis in vivo is, however, still unexplored. Here we provide in vivo evidence that zebrafish macrophages are critical for multiplication of B. cenocepacia in the host, and subsequent induction of pro-inflammatory fatal infection (Fig 9). A key role for intramacrophage stages in pathogenicity has important consequences for the design of novel antimicrobial therapies.
Fig 9. Schematic showing the role of macrophages during acute infection by B. cenocepacia K56-2 in zebrafish embryos.

Macrophages are the major phagocytosing cells of iv injected bacteria (1) for which they provide a critical replication niche (3). In their absence, K56-2 does not replicate (bottom panel, 1). Intracellular bacteria induce a rapid and robust increase in pro-inflammatory cytokine expression (2). Il1 signalling contributes to fatal pro-inflammatory responses (4), characterized by massive neutrophil and macrophage infiltration (5) neutrophil degranulation (6), bacterial dissemination from infected cells (7), systemic phagocyte death (8), and tissue damage (9).
Many intracellular bacterial pathogens exploit macrophages for their virulence, however, the increased virulence of for instance Mycobacterium marinum [39], Staphylococcus aureus [40], and Salmonella Typhimurium [41] in macrophage-depleted zebrafish larvae shows that these phagocytes still have a beneficial role for the host. In sharp contrast, we show that genetic and chemical ablation of macrophages prior to infection considerably attenuated the virulence of Bcc strains that cause rapidly fatal infection in the macrophage-proficient host. Strikingly, in contrast to for instance S. Typhimurium [41], none of the analysed Bcc strains could replicate extracellularly in the absence of macrophages. This not only reveals the critical role of macrophages in proliferation of these bacteria and ensuing pro-inflammatory disease in the host, it also suggests that specific conditions prevented extracellular replication of the analysed strains upon introduction in the macrophage-depleted host. Of note, we have shown earlier that at later stages of infection B. cenocepacia re-enters the circulation and can replicate extracellularly [17], suggesting changes in host- or bacterial factors during intracellular or acute stages of infection support further extracellular bacterial proliferation. Our current research is focused on better understanding the inability of these strains to replicate extracellularly upon their delivery into the host, and whether this is a general feature of bacteria belonging to the Bcc.
Despite a significant reduction in bacterial multiplication in embryos experimentally depleted of macrophages, at later time points bacterial burden increased. This was more evident in pu.1 knockdown embryos than in embryos that were depleted of macrophages using Mtz-mediated cell ablation. It is not completely clear where B. cenocepacia starts to replicate at later time points in embryos depleted of macrophages but we believe that this is either in newly developed macrophages or, with Mtz, in macrophages that have survived treatment due to partial silencing of expression of the mpeg:Gal4/uas:mCherry-NTR transgenes. Further experiments are needed to determine whether persisting bacteria can enter and replicate in non-phagocytic cells in this model.
The pro-inflammatory state during B. cenocepacia infection was characterized by massive neutrophil and macrophage recruitment, neutrophil degranulation at the site of infection and tissue damage, culminating in fatal infection. The observed systemic phagocyte clearance might contribute to the pathological effects seen in zebrafish larvae. However, embryos that were experimentally depleted of neutrophils at the time of bacterial injection died as fast as infected neutrophil-proficient embryos. Thus the net contribution of neutrophils to disease outcome, whether host-protective or harmful, was undetectable, and emphasizes the pathological effect of the interaction between B. cenocepacia and macrophages.
In agreement with the finding that neutrophils need a surface for efficient phagocytosis [29], we show that neutrophils were massively recruited to and readily phagocytosed B. cenocepacia K56-2 and B. stabilis delivered by subcutaneous injection in zebrafish embryos, although macrophages were also seen to take up bacteria. As observed after iv delivery, B. cenocepacia caused pro-inflammatory disease with sustained infiltration of neutrophils and macrophages, and bacterial burden strongly increased, whereas B. stabilis was less virulent and most recruited neutrophils had left the infection site by 24 hpi. Experimental depletion of neutrophils prior to subcutaneous injection of B. cenocepacia did not have a major effect on bacterial replication and acute disease progression. In sharp contrast, the absence of macrophages prevented multiplication of B. cenocepacia and tissue damage. Thus, also with neutrophil-dominated phagocytosis and tissue localization of the bacteria, macrophages were critical for acute disease progression, as seen after intravenous delivery.
It has been reported that neutrophils which are unable to produce ROS through defects in the NADPH oxidase complex, as is the case in CGD patients, showed reduced killing of ingested Bcc, with the efficiency of killing depending on the bacterial strain [8]. CGD neutrophils were also unable to protect mice against Bcc challenge [42,43]. We did not find a significant effect of gp91 knockdown on virulence prior to intravenous injection of B. cenocepacia or B. stabilis, in agreement with our finding that neutrophils do not contribute measurably to disease outcome. However, subcutaneous delivery revealed that B. stabilis numbers increased slightly more in gp91 knockdown embryos compared to controls, indicating that in zebrafish embryos ROS plays some restrictive role against this strain. For B. cenocepacia, bacterial numbers in gp91 knockdown embryos were not significantly increased compared to those in control embryos, although we cannot exclude at this point that the dominant role of macrophages in virulence of B. cenocepacia masked a ROS-dependent effect.
While subcutaneously delivered B. cenocepacia showed sustained neutrophil recruitment at later time points even in the presence of functional NADPH oxidase, in most B. stabilis-infected embryos recruited neutrophils had left the infection site at later time points in a ROS-dependent manner, suggesting ROS play a role in the resolution of inflammation. The inability to efficiently resolve inflammation is reminiscent of neutrophil dominated abscesses found in the lungs of rats lacking ROS [43], which was contributed to Bcc-induced necrosis of neutrophils lacking a functional NADPH oxidase [44]. Starting around 1 hpi, neutrophils that had taken up B. stabilis or B. cenocepacia rounded up and expelled their cell contents into the environment as described for NETosis [45]. NETosis, a more recently described immune defence mechanism, depends on the production of ROS by NADPH oxidase [46]. Our results suggest that ROS-dependent release of NETs might be a host-protective mechanism to control B. stabilis infection. Together, our data demonstrate that macrophages, and not neutrophils, critically contribute to the detrimental effects during acute infections caused by intravenously and subcutaneously introduced B. cenocepacia.
Detection of bacterial ligands through Toll-like receptors (TLR) is essential for initiation of downstream immune signalling, principally through the key adaptor protein MYD88. Flagellin has been suggested as a key ligand in TLR5-mediated induction of the immune response to infection with Bcc bacteria [47,48]. However, recently it has also been described that glycosylation of B. cenocepacia flagellin reduces immune responses [49], and it remains essential to identify other bacterial ligands and host factors that are involved in the induction of this high, deregulated immune response and the inability to resolve the inflammation. In contrast to other pathogens, including Edwardsiella tarda and S. Typhimurium that have been shown to be more virulent in a zebrafish myd88-/- mutant [41], we found that B. cenocepacia-infected myd88-/- mutants lived longer than wildtype embryos. Although these findings are in agreement with results of Ventura et al [31] who showed that immunocompromised mice lacking MyD88 were protected against fatal challenge with B. cenocepacia, we were unable to reproducibly mimic these findings in myd88 knockdown experiments. Despite this inconsistency, however, the increased virulence in the absence of Myd88 described for other pathogens [41] was not observed, showing Myd88 does not have a measurable host-protective role against Bcc. Differences in experimental conditions and fish lines may contribute a difference in the balance between Myd88-dependent and -independent pathways, making it impossible to unmask a host detrimental role for Myd88 under certain conditions. Alternatively, Myd88-dependent host-protective and host detrimental effects may cancel each other out, and further research is needed to clearly define a role for Myd88 in acute fatal infection caused by B. cenocepacia in zebrafish.
Secretion of the pro-inflammatory cytokine IL1β has been shown to be strongly induced upon infection of macrophages and epithelial cells with B. cenocepacia. Activation of the pyrin inflammasome in monocytes and THP-1 cells by intracellular B. cenocepacia has been shown to be important for processing and secretion of IL1β in a type VI-secretion dependent manner [20]. We found that intracellular B. cenocepacia caused a rapid and robust increase in pro-inflammatory cytokine gene expression in zebrafish embryos, which contrasted to the low increase induced by B. stabilis, in agreement with the acute and persistent character of the infection, respectively. During acute infection, the induction of il1b, but not cxcl8 expression, was significantly lower in the absence of macrophages indicating an important role for macrophages in the global infection-induced il1b expression in zebrafish embryos. Knockdown of il1b enhanced host mortality, showing that Il1b is important for host defence against B. cenocepacia. However, treatment with the IL-1 receptor antagonist Anakinra, which has been approved in humans for the treatment of pro-inflammatory effects of rheumatoid arthritis, resulted in increased resistance of zebrafish larvae towards B. cenocepacia. Our results suggest that while Il1b contributes to pro-inflammatory responses that can either be host-protective or detrimental to the host, its absence is disadvantageous. The finding that Il1R signalling, which generally has a protective role in host defence, is detrimental for embryos infected with B. cenocepacia, was also reported in a mouse model of melioidosis, where IL-1r1-/- mice were more resistant than wildtype mice to B. pseudomallei infection [50]. A possible mechanism for the enhanced resistance to B. cenocepacia K56-2 by antagonising the Il1R receptor could be through a role for Anakinra in modulation of autophagy [51]. B. cenocepacia has been shown to reduce the expression of autophagy related genes [12] and to block autophagosome completion [13]. We are currently investigating a possible role for Anakinra in the restoration of autophagy in zebrafish macrophages infected with B. cenocepacia.
To date the molecular mechanisms that are at the basis of exacerbation, decline of lung function and often fatal septicemia in CF patients caused by Bcc bacteria are not known. Equally, it is unknown how these opportunists cause disease in both immune compromised and immune competent individuals. Based on our results, and those from others, we suggest that the intracellular stages of B. cenocepacia and the ensuing inflammatory response are essential therapeutic targets for the development of new therapies to combat these infections. We propose that zebrafish embryos are a valuable additional model to help advance our understanding of the interaction of Bcc bacteria with macrophages and neutrophils in CF and non-CF conditions in the context of an innate immune system. The importance of our findings is highlighted by the recent findings that several important extracellular opportunistic pathogens, including Staphylococcus aureus and Pseudomonas aeruginosa which cause the large majority of nosocomial infections, have only recently been reported to be able to survive, and replicate in host cells [52–54]. Thus, increased knowledge on the precise infection mechanisms of such opportunistic pathogens is crucial for the design of more effective antimicrobial therapies to disarm intracellular bacteria.
Materials and methods
Ethics statement
Zebrafish (Danio rerio) were kept and handled in compliance with the guidelines of the European Union for handling laboratory animals (http://ec.europa.eu/environment/chemicals/lab_animals/home_en.htm). Studies performed at VBMI are approved by the Direction Départementale de la Protection des Populations (DDPP) du Gard (ID 30-189-4) and the Comité d'Ethique pour l'Expérimentation Animale Languedoc-Roussillon (CEEA-LR-12186). Culture of myd88 mutant zebrafish was approved by the local animal welfare committee (DIerexperimentencommissie, DEC) of the University of Leiden (protocol 12232) and adhered to the 2010/63/EU directive. Infection-experiments were terminated before the larvae reached the free feeding stage and did not classify as animal experiments according to the 2010/63/EU Directive.
Zebrafish
Care and maintenance of zebrafish was as described previously [17]. Zebrafish lines used in this study are summarized in Table 1.
Table 1. Zebrafish lines used in this study.
| Name | Description | Reference |
|---|---|---|
| AB | Wildtype | - |
| AB “Golden” | Pigment mutation (slc24a5) | [55] |
| Tg(mpx:eGFPi114) | Neutrophil marker–GFP, referred to as mpx:GFP | [56] |
| Tg(mpeg1:mCherryF) ump2Tg | Macrophage marker with membrane localised mCherry-F, referred to as mpeg1:mCherry | [32] |
| Tg(fms:Gal4-VP16il86//UAS-E1b:nfsB-mCherryi149) | Fish line expressing a fusion protein of nitroreductase and mCherry under control of the Gal4 regulated UAS sequence in macrophages | [57] |
| Tg(mpeg1:Gal4-VP16/ UAS-E1b:kaedes1999t) | Line expressing the photo convertible protein Kaede under control of the Gal4-regulated UAS sequence in macrophages | [58] |
| myd88hu3568 | Mutant line with truncated MyD88, referred to as myd88-/- | [41] |
| Tg(mpeg1:Gal4-VP16-il86/; UAS-E1b:nfsB-mCherryi149) | Offspring from a cross between Tg(fms:Gal4; UAS-E1b:nfsB-mCherryi149) and Tg(mpeg1:Gal4-VP16 /UAS-E1b:kaedes1999t) fish, referred to as mpeg1/umn | [57, 58, this study] |
| Tg(mpx:Gal4-VP16/ UAS-E1b:nfsB-mCherryi149) | Offspring selected from a cross between Tg(mpx:Gal4.VP16)i222;Tg(UAS:Kaede)s1999t (59) and Tg(mpeg1:Gal4-VP16- il86/; UAS-E1b:nfsB-mCherryi149), referred to as mpx/umn | [57–59, this study] |
| Tg(il1b:GFP) | Fish expressing farnesylated GFP (membrane-targeted GFP; GFP-F) under the control of the il1b promoter, named il1b:GFP | [32] |
Bacterial strains and plasmids
Bcc strains used in this study are: B. cenocepacia K56-2/LMG18836 [60], B. cenocepacia J2315/LMG16656 and J415/LMG16654 [61], B. stabilis/LMG14294 [62], B. vietnamiensis FC441/LMG18836 [63], and B. cepacia CEP509/LMG18821 [63]. The mTurquoise2 gene (referred to as Turquoise) was amplified from pmTurquoise2-C1 [64] using primers TurquoiseNdeI-Fw (5’–GGAATTCCATATGGTGAGCAAGGGCGAGGAGC-3’; NdeI underlined) and TurquoiseXbaI-Rv (5’–GCTCTAGACTACTTGTACAGCTCGTCCATGCCGAG-3’; XbaI underlined), and cloned as an NdeI/XbaI fragment in pIN29 [17], replacing the DSRed gene, resulting in pIN289. Similarly, the eGFP gene was amplified from pEGFP-C3 (CLONTECH) using primers peGFP-Nde (5’–GGAATTCCATATGGTGAGCAAGGGCGAGGAG-3’; NdeI underlined) and peGFP-Xba (5’–GCTCTAGAATCACTTGTACAGCTCGTCCATGCCG-3’; XbaI underlined) and cloned as NdeI/XbaI fragment in pIN29, resulting in pIN301. The plasmids were transferred to B. cenocepacia by electroporation as described [17], and fluorescence verified. Bcc strains containing DsRed (pIN29 [17],Turquoise (pIN289, this study), or eGFP (pIN301, this study)) expression plasmids were grown in 5 ml of LB medium in the presence of 100 μg.ml-1 chloramphenicol at 37°C overnight.
Zebrafish lines and injection conditions
One to three nL of morpholinos (MO) (Genetools, Eugene, OR) dissolved in H2O with 0.05% Phenol Red (see S1 Table) was micro injected in the yolk at the 1–2 cell stage and eggs were incubated at 28°C in E3 medium (5mM NaCl, 0.17mM KCl, 0.33mM CaCl2, 0.33 mM MgSO4, 0.5x10-4% methylene blue). Methylene Blue was omitted when embryos were used for pixel count analysis to avoid strong autofluorescence of the yolk. Just prior to injection of bacteria the dechorionated embryos were anesthetized in 0.04% buffered 3-aminobenzoic acid ethyl ester methanesulfonate (tricaine, MS222) in E3. Embryos were micro injected in the blood island or caudal vein (30 hpf, 50 hpf), or subcutaneously (50 hpf). In knockdown experiments, no differences in virulence were found between embryos injected with nonspecific control MO compared to non-treated embryos (S1E Fig). MO-injected embryos were further compared to non-treated embryos (Control embryos). Embryos were manually dechorionated 2 hours before injection with Bcc bacteria. To prepare the inoculum, bacteria expressing fluorescent proteins were grown overnight in LB medium. Bacteria were collected by centrifugation at 3,000 g for 3 min and resuspended in PBS (Gibco) at the desired concentration with 0.05% phenol red solution (Sigma) for visualization of the injection. Subcutaneous injection was performed as described [29] with 0.1–0.5 nL of bacterial suspensions at an OD of 5. Iv injection was performed with 1–2 nL of bacterial suspension (50 CFU for survival assays and CFU counts, or 200 CFU for qRT PCR experiments) as described [17,65]. Embryos in each group were randomly attributed to survival assays after injection (exact numbers are indicated in survival graphs), or to CFU counts (n = 5 per experiment per indicated time point). Precise inoculum (T = 0) and bacterial burden, expressed as colony forming units (CFU), were determined by plating individually lysed embryos as described [65]. Briefly, after injection, embryos were rinsed in E3 medium, anesthetized in tricaine, and individually transferred to 1.5 ml Eppendorf tubes containing 45μl of 1x Trypsin-EDTA. Embryos were disrupted by pipetting (20–30 times) and 50 μL 2% Triton X-100 was added, mixed by flicking the tube, and incubated for 30 min at room temperature. To complete disruption the mixture was pipetted up and down 20–30 times. Depending on the number of bacteria, total lysate was plated on LB-agar plates with 100μg.ml-1 chloramphenicol or serial dilutions were made and 10μl of each dilution was deposited on a square LB-agar plate. Plates were incubated overnight at 37°C. The next day the number of colonies was determined. Graphs represent the individual number of CFU per embryo at the different time points. For treatment of embryos with Anakinra (Kineret), dechorionated embryos were incubated in 10 or 100 μM Anakinra in E3 for 4 hours prior to injection with bacteria. After injection, the treatment was continued with 10 μM Anakinra, and the medium was refreshed at 24 hpi.
Macrophage and neutrophil ablation
Offspring from a cross between Tg(fms:Gal4; UAS-E1b:nfsB-mCherryi149) and Tg(mpeg1:Gal4-VP16/UAS-E1b:kaedes1999t) fish (see Table 1) was selected for strong expression of mCherry in macrophages (mpeg/umn+), and raised to adulthood. Similarly, offspring was selected from a cross between Tg(mpx:Gal4.VP16)i222;Tg(UAS:Kaede)s1999t and Tg(mpeg1:Gal4-VP16- il86/; UAS-E1b:nfsB-mCherryi149), that specifically expressed mCherry in neutrophils, referred to as mpx/umn. Umn+ fish were crossed to AB, or heterozygote siblings were incrossed. For efficient ablation of macrophages and neutrophils, mpeg1/umn+ and mpx/umn+ embryos were preselected at 30 hpf for strong mCherry expression, and embryos with only few mCherry-positive cells were discarded. Non-fluorescent embryos from the same crosses were used as umn-negative (unm-) control. Metronidazole (Mtz; Sigma), freshly prepared in 0.2% DMSO, was used at 5 mM in E3. Fluorescent (umn+) and non-fluorescent (umn-) embryos were dechorionated and either left untreated (infection control), or incubated in 0.2%DMSO (DMSO effect) or 5mM Mtz (ablation and non-specific Mtz control, respectively) at 33 hpf for 15 h prior to iv injection of bacteria. The schedule for Mtz treatment and injection of bacterial suspensions in mpeg1/umn larvae, including controls, is shown in S3 Fig. Embryos were incubated at 28°C in the dark. Ablation was verified by fluorescence microscopy. Treatment of 15 h prior to infection, followed by an addition 24 h in Mtz, resulted in specific ablation of macrophages by the time of injection (see results). Mtz had no significant effect on log-phase growth of the bacteria in LB medium, and CFU counts were identical between DMSO and Mtz treatments. For complete ablation of neutrophils, 10 mM Mtz was used for pre-treatment. After injection 5 mM was applied; no difference in virulence was observed between pre-treatment with 5 or 10 mM.
Analysis of gene expression by qRT-PCR
At the indicated time points, 10 to 25 embryos were processed for RNA isolation, cDNA synthesis, and qRT-PCR analysis. The peptidylprolyl isomerase A-like (ppial) gene was used as a reference gene. For each condition, 10 to 25 embryos were transferred to 500μl of TRIzol, homogenized and stored at -80°C, as described [65]. RNA for each pool was extracted as described [66] and purified using the RNeasy MinElute Cleanup kit (Bio-Rad). Reverse transcription of each sample (500ng total RNA) was performed with the iScript cDNA synthesis kit (Bio-Rad) according to the manufacture. Quantitative RT-PCR was performed using the LightCycler 480 SYBR Green I Master mix (Roche), on a LightCycler 480. Each reaction was performed in a 10μl volume comprised of 2.5μl 10-fold diluted cDNA, 5μl of master mix and 10 pmol of each of the primers (S2 Table). Cycling parameters were: 95°C during 10 min to activate the polymerase, followed by 45 cycles of 95°C for 15 s and 60°C for 40 s. Fluorescence measurements were taken at the end of each cycle. Melting curve analysis was performed to verify that no primer dimers and non-specific product were amplified. Stability of the house keeping gene ppial was verified for each experiment. Results were analysed using the ΔΔCt method and represented as column bar graphs normalized to a PBS-injected control group at each time point, unless mentioned otherwise. Three biological control experiments were performed, each with two technical replicates, unless stated otherwise.
Cell death assay and Sudan black staining
Embryos were injected with 50–100 CFU red fluorescent B. cenocepacia K56-2, and 20 h later with 1 nL of the live cell impermeable nucleic acid stain SYTOX Green (1mM in DMSO, Invitrogen). As control embryos were injected with 1 nL of DMSO. Embryos were analysed between 30 min and 6 hours after injection of the dye by fluorescence microscopy. Sudan black staining was performed as described [67].
Antibody labelling
Zebrafish embryos were fixed in 4% Paraformaldehyde (PFA) in PBS with 0.4% Tween20 during 2 hours at room temperature or overnight at 4°C. Embryos were washed 4 times during 30 min with PBST (0.1% Tween 20 in PBS). Blocking was performed during 2 h at room temperature in 5% goat serum in PBST and embryos were incubated with 5% goat serum/PBST containing an antibody against the pan-leukocyte marker L-plastin (1:500 with gentle shaking at 4°C overnight [68]. Embryos were washed (3 x 30 min) with PBST at RT. Embryos were incubated with 2% goat serum/PBT containing secondary antibody anti-rabbit coupled with Alexa 350 (1:250) (Life Technologies). Embryos were rinsed twice with PBST and prepared for microscopy analysis.
Microscopy and fluorescent pixel quantification
Embryos were transferred to glass-bottom dishes (MatTek Corp., Ashland, MA) in E3 (inverted microscope) or embedded in 0.5% E3-agarose (confocal) containing 0.02% MS222. A Leica DM IRB inverted microscope (bright-field, differential interference contrast (DIC), and fluorescence imaging) coupled to a Coolsnap fx camera (Roper Scientific) was used. A Nikon AZ100 equipped for bright-field and fluorescence imaging, coupled with Coolsnap HQ2 (Roper Scientific) using MetaVue software was used to record full size embryos. Confocal microscopy was performed with an Olympus FV10i and images and movies were processed with Fluoview software and Image J. Images were processed further using Adobe Photoshop, and time-lapse videos made with image J (see specific details in movie legends). To quantify phagocytic cells, the fluorescent pixel quantification method was used as described [69]. Graphs depict macrophage and neutrophil numbers. BioImageXD [70] was used to obtain the image in S6A Fig.
Statistical analysis
Statistical analysis was performed using Prism 6 (GraphPad). Survival assays are represented in Kaplan-Meier graphs and analysed with a Log rank (Mantel-Cox) test. Representative experiments are shown with total numbers of embryos (n) indicated for each of the groups. In experiments determining CFU, and macrophage and neutrophil counts significance between multiple selected groups was determined using one-way Anova, with Sidak’s Multiple Comparisons test (the compared groups are indicated with significance level on each graph). The graphs show a summary of multiple biological replicates (see legends). CFU counts were log transformed and are presented in dot graphs showing the geometric mean per time point, with each embryo represented by a single dot. qRT-PCR data were log2-transformed, and significance of the log2-transformed data was analysed using one-way Anova, with Tukey’s Multiple Comparison Test at each time point by comparing treatments with each other and the PBS control (in case of multiple groups per time point). Columns indicate mean fold-change with SEM. Significance in relative fold-change is indicated for each treatment normalized to the corresponding PBS control with an asterisk above the column, or between two treatments with a connective line. Significance is indicated with: ns, non-significant, *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001. For some experiments, representative images are shown of at least 2 independent experiments, with a total of at least 20 embryos (Figs 6C, 6D and 8E).
Supporting information
Role of macrophages during infection with Bcc strains causing either persistent or acute infection. (A-D G-J) Embryo survival (A, C, G, and I) and bacterial burden (total of 2 experiments) over time (B, D, H, J) of control (black) and pu.1 knockdown embryos (red) injected iv with B. cenocepacia J2315 (A,B), B. cepacia CEP509 (C,D), B. cenocepacia J415 (G,H), and B. vietnamiensis FC441 (I,J), respectively. (E) Representative experiment (of at least three) showing embryo survival of control embryos (n = 47), pu.1 knockdown (n = 54) and nonspecific control MO (n = 52) embryos injected with B. cenocepacia K56-2 (average 53 CFU). (F) Representative fluorescence image at 24 hpi showing neutrophils (green) in an mpx:GFP pu.1 knockdown embryo injected with B. cenocepacia J2315 (red) (~50 CFU). Inset shows corresponding bright field image. Scale bar, 100 μm. (B, D, H, J) Geometric mean with each data point representing an individual embryo. Dead embryos are indicated as black open circles. (A-E and G-J) * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001; ns: non-significant. See materials and methods for statistical tests used.
(TIF)
Pu.1 knockdown prevents B. cenocepacia K56-2 multiplication. Fluorescence overlay images (red and green filters) of the indicated area (boxed area in embryo drawing) of two mpeg1:mCherry control embryos (left) and two mpeg1:mCherry pu.1 knockdown (right) imaged at 1, 3, 16, 20, 24, and 41 h after injection with ~50 CFU B. cenocepacia K56-2 harbouring pIN301 (green). The absence of macrophages prevents efficient bacterial replication. Bacteria in control embryos colocalise with macrophages (arrow heads). Fluorescent macrophages disappear from control embryos (>20 hpi). Appearance of macrophages in pu.1 knockdown embryos (white arrows).
(TIF)
Chemical ablation of macrophages using the NTR/Mtz system. (A) Schematic representation and treatment schedule of the chemical ablation strategy based on the nitroreductase (NTR)/ metronidazole (Mtz) system, shown for macrophage-specific ablation. See Materials and methods for details. (B) Representative fluorescence images of non-treated and Mtz-treated mpeg1/umn+ embryos, showing the efficacy of the Mtz treatment. Residual red fluorescence in treated embryos represented apoptotic cells. Scale bar, 0.5 mm. (C) Quantification of macrophage numbers in mpeg1/umn+ embryos (untreated or treated at 34 hpf with 5mM Mtz) at 0, 15 and 39 hours after treatment as indicated in (A). The efficacy of macrophage ablation by Mtz treatment was evaluated by pixel counting. The average macrophage numbers of two independent experiments (n = 6) are shown. ns, non-significant, ** p ≤ 0.01, **** p ≤ 0.0001. See material and methods for statistical analysis used.
(TIF)
Mtz-mediated ablation of macrophages prevents B. cenocepacia K56-2 multiplication. Fluorescence overlay images (red and green filter) of two DMSO-treated (left panels) and two Mtz-treated (right panels) mpeg1/umn+ embryos imaged at 1, 3, 24, and 48 h after iv injection with ~50 CFU B. cenocepacia K56-2 harbouring pIN301 (green). Bacterial replication in control embryos resulted in macrophage cell death, seen as mCherry positive debris (>24 hpi) (see also S5F Fig). At 28 and 48 hpi individual fluorescence images (green filter) are shown below the overlay images. Drawing indicates imaged area, marking the embryo sac extension (green) prone to autofluorescence. Arrow head indicates mCherry positive macrophages in apoptosis.
(TIF)
Behaviour and fate of neutrophils and macrophages during acute and persistent infection. (A). Confocal stack images of a time series, first image ~20 hours after infection of an mpx:GFP embryo with B. cenocepacia K56-2. Patrolling neutrophil inspects heavily infected macrophage (white arrow). Individual bacteria (arrow head), possibly released from infected cell nearby (open arrow), are being moved around by the neutrophil. The last image shows the same area 90 minutes later with the infected macrophage still intact. (B,C) Mpx:GFP embryos (50 hpf) were injected with B. stabilis LMG14294. (B) Representative images of control and B. stabilis infected mpx:GFP embryos with increased neutrophil numbers. Neutrophils were more dispersed in B. stabilis-infected compared to non-infected control embryos, where most neutrophils were resting in the caudal hematopoietic tissue. (C) The number of neutrophils in infected and non-infected control embryos was determined by pixel counting at different time points after injection. Each data point represents one embryo. The graph represents one of the three experiments represented in Fig 3C, but includes an additional time point at 5 days post infection, which was not determined in the other 2 experiments. A percentage of embryos injected with B. stabilis contained more neutrophils than control embryos (see also (B)). *, p ≤ 0.05. (D) Mpeg1:mCherry embryos were injected with B. cenocepacia K56-2 (~45 CFU) and the number of macrophages was evaluated by pixel counting at 0 and 24 hpi (30 and 54 hpf, respectively). The results are related to the corresponding experiment shown in Fig 3D. Each data point represents one embryo. Significance was determined using a one-way Anova with Sidak’s Multiple Comparisons test. **** p ≤ 0.0001. Two independent experiments (n = 5). (E) Image showing B.cenocepacia K56-2 (red) in an L-plastin labelled macrophage (blue) at 24 hpi. Scale bar, 50 μm. Inset, magnification red/ blue filters, scale bar 10 μm. (F) Images (bright field, green/red overlay, and detailed image with red/green filter), of a non-infected mpeg1/umn+ embryo (left panels) and mpeg1/umn+ embryo (right panels) iv infected with B. cenocepacia K56-2 (green, arrows) at 24 hpi. The images are similarly treated to enhance the red fluorescence to visualize the mCherry positive debris. The close up shows individual macrophages (left panel, red), and red fluorescent debris (arrow heads). Related to Fig 3F.
(TIF)
Macrophages, but not neutrophils, contribute to increased bacterial burden and pro-inflammatory responses towards subcutaneous B. cenocepacia. (A) mpeg/umn+ embryo infected subcutaneously at 2 dpf with B. cenocepacia K56-2 (green). Confocal stack (59 slices, 1 μm) at 24 hpi showing bacteria in macrophages. A close up of the indicated area is shown in 3D, with volume rendering for GFP signal and surface rendering of mCherry signal without (middle panel) and with 50% surface transparency (right panel). Scale bar 50 μm. (B) Fluorescence images showing non-infected mpx/umn+ and mpeg1/umn+ control embryos at 3 dpf, the time point that resembles 24 h post subcutaneous injection in Fig 6. (C) Image overlay (bright field, red and blue filters) of the tail region of an mpx/umn+ embryo after subcutaneous injection with B. cenocepacia K56-2 expressing Turquoise, presenting tissue damage by 24 hpi. Scale bar, 100 μm. (D) Image overlay (bright field, red, blue and green filters) of the tail region of an Mtz-treated mpeg1/umn+; mpx:GFP embryo 24 h after subcutaneous injection with B. cenocepacia K56-2 expressing Turquoise. Scale bar, 100 μm.
(TIF)
(A) Embryo survival (average inoculum 35 CFU, representative experiment) (B) and bacterial burden (average of 2 experiments) over time of wildtype control (myd88+/+) and mutant (myd88-/-) embryos iv injected with B. cenocepacia K56-2. (C, D) Embryo survival (average inoculum 39 (C) and 75 (D) CFU) of control (black) and myd88 knockdown embryos (red) iv injected with B. cenocepacia K56-2.
(TIF)
DMSO, Mtz and Anakinra do not affect il1b and cxcl8 expression levels. (A) mpx:GFP embryos were either non-treated or pre-treated at 34 hpf for 15 h with DMSO or 5 mM Mtz. Randomized groups were injected with PBS or B. cenocepacia K56-2 (on average 270 CFU). Graphs show mean relative il1b and cxcl8 gene expression levels, normalized to the PBS-injected non-treated control group at 4 hpi. Error bars represent mean with SEM of two biological replicates. (B) Control and Anakinra-treated mpx:GFP embryos were iv injected with PBS or B. cenocepacia K56-2 (average 120 CFU). Graphs show mean relative il1b and cxcl8 gene expression levels, normalized to the PBS-injected non-treated control group at each time point. Error bars represent mean with SEM of two biological replicates. ns = non-significant.
(TIF)
Morpholinos used in this study.
(DOCX)
Primers used for qRT-PCR experiments.
(DOCX)
Phagocytosis of surface-associated B. cenocepacia by neutrophils. Live imaging of an mpx:GFP embryo, injected subcutaneously at 50 hpf with DSRed expressing B. cenocepacia, every 90 seconds from 20 mins pi, for the duration of 75 mins. A time lapse of 50 confocal images with maximal intensity projection is shown (19 steps x 2 μm) at a rate of 2 frames per sec. Bacteria are rapidly phagocytosed by neutrophils, and remain visible in individual small vacuoles. Bacteria are also phagocytosed by GFP-negative macrophages, seen as individual bacterial clusters that are not colocalising with GFP-positive neutrophils.
(AVI)
Phagocytosis of surface-associated B. stabilis by neutrophils induces cell rupture. Live imaging of an mpx:GFP embryo, injected subcutaneously over a somite at 50 hpf with DSRed expressing B. stabilis, every 90 seconds from 10 minutes pi. Total duration 128 mins representing the period between 20 mpi and 148 mpi. A time lapse of 85 confocal images with standard deviation projection is shown (19 steps x 2 μm) at a speed of 2 frames per sec. Bacteria are rapidly phagocytosed by neutrophils, and are directed into large growing vacuoles (See Fig 5). Between 64–67 mpi two neutrophils full of bacteria round up (arrows) and eject their cell contents into the extracellular space (arrow heads appear), see also Fig 5. After the event, the bacteria remain clustered, and mobility of surrounding neutrophils is dramatically reduced, before they start moving again. Other neutrophils (indicated with arrows and consecutive numbers 3, 4 etc) undergo the same phenomenon, although sometimes the collapse is less explosive. The neutrophil indicated with number 4 seems to expel part of its contents including the phagocytosed bacteria, but the neutrophil takes up the contents again and after 35 mins, effectively “bursts” (indicated with arrow 4).
(AVI)
Acknowledgments
We greatly acknowledge J. Goedhart for providing pmTurquoise2-C1, G. Lieschke for mpeg1:Gal4/uas-kaede fish, G. Lutfalla for mpeg1:mCherry-F and il1b reporter fish, P. Martin and Y. Feng for L-plastin antibodies, and S. Renshaw for mpx:GFP, mpx;Gal4;uas-Kaede and fms:Gal4;umn fish. We thank Y. Tasrini for constructing pIN301.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was supported by l'Institut National de la Santé et de la Recherche Médicale INSERM and Université de Montpellier. ACV was recipient of a ‘Chercheur d’avenir’ award from La Région Languedoc Roussillion, and JM from a doctoral grant from the French Ministry of Higher Education and Research and a ”Bourse de stage d’Excellence "Eole" du Réseau franco-néerlandais”. MBG was supported by “Vaincre la Mucoviscidose”. JRF was recipient of a doctoral grant from Fundação Ciência e Tecnologia (FCT), Portugal. MV and AHM were supported by the Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Ministry of Education (SSM06010), Culture and Science. Work in the groups of ACV and AHM was also supported by the Marie-Curie Initial Training Network FishForPharma (PITN-GA-2011-289209). LZ was Marie-Curie fellow in the FishForPharma network. MCG was recipient of the French Ministry of Higher Education and Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Drevinek P, Mahenthiralingam E. Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clin Microbiol Infect. 2010; 16(7):821–30. doi: 10.1111/j.1469-0691.2010.03237.x [DOI] [PubMed] [Google Scholar]
- 2.Medina-Pascual M, Valdezate S, Villalón P, Garrido N, Rubio V, Saéz-Nieto J. Identification, molecular characterisation and antimicrobial susceptibility of genomovars of the Burkholderia cepacia complex in Spain. Eur J Clin Microbiol Infect Dis. 2012;31:3385–96. doi: 10.1007/s10096-012-1707-6 [DOI] [PubMed] [Google Scholar]
- 3.Katsiari M, Roussou Z, Tryfinopoulou K, Vatopoulos A, Platsouka E, Maguina A. Burkholderia cenocepacia bacteremia without respiratory colonization in an adult intensive care unit: epidemiological and molecular investigation of an outbreak. Hippokratia. 2012;16(4):317–23. [PMC free article] [PubMed] [Google Scholar]
- 4.Bressler A, Kaye K, LiPuma J, Alexander B, Moore C, Reller L, et al. Risk factors for Burkholderia cepacia complex bacteremia among intensive care unit patients without Cystic Fibrosis: A case-control study. Infect Control Hosp Epidemiol. 2007;28(8):951–8. doi: 10.1086/519177 [DOI] [PubMed] [Google Scholar]
- 5.Hartl D, Gaggar A, Bruscia E, Hector A, Marcos V, Jung A, et al. Innate immunity in cystic fibrosis lung disease. J Cyst Fibros. 2012; 11(5):363–82. doi: 10.1016/j.jcf.2012.07.003 [DOI] [PubMed] [Google Scholar]
- 6.Pohl K, Hayes E, Keenan J, Henry M, Meleady P, Molloy K, et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood. 2014; 129: 999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes JE, Stewart J, Barclay GR, Govan JR. Priming of neutrophil respiratory burst activity by lipopolysaccharide from Burkholderia cepacia. Infect Immun. 1997;65(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speert DP, Bond M, Woodman RC, Curnutte JT. Infection with Pseudomonas cepacia in chronic granulomatous disease: role of nonoxidative killing by neutrophils in host defense. J Infect Dis. 1994;170(6):1524–31. [DOI] [PubMed] [Google Scholar]
- 9.Saldías MS, Valvano MA. Interactions of Burkholderia cenocepacia and other Burkholderia cepacia complex bacteria with epithelial and phagocytic cells. Microbiology. 2009;155(9):2809–17. [DOI] [PubMed] [Google Scholar]
- 10.Ganesan S, Sajjan US. Host Evasion by Burkholderia cenocepacia. Front Cell Infect Microbiol. 2011;1:25 doi: 10.3389/fcimb.2011.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valvano MA. Intracellular survival of Burkholderia cepacia complex in phagocytic cells. Can J Microbiol. 2015;61(9):607–15. doi: 10.1139/cjm-2015-0316 [DOI] [PubMed] [Google Scholar]
- 12.Abdulrahman BA, Khweek AA, Akhter A, Caution K, Kotrange S, Abdelaziz DA, et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy. 2011;7:1359–70. doi: 10.4161/auto.7.11.17660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al-Khodor S, Marshall-Batty K, Nair V, Ding L, Greenberg DE, Fraser IDC. Burkholderia cenocepacia J2315 escapes to the cytosol and actively subverts autophagy in human macrophages. Cell Microbiol. 2014;16:378–95. doi: 10.1111/cmi.12223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamothe J, Valvano MA. Burkholderia cenocepacia-induced delay of acidification and phagolysosomal fusion in cystic fibrosis transmembrane conductance regulator (CFTR)-defective macrophages. Microbiology. 2008;154:3825–34. doi: 10.1099/mic.0.2008/023200-0 [DOI] [PubMed] [Google Scholar]
- 15.Chiu CH, Ostry A, Speert DP. Invasion of murine respiratory epithelial cells in vivo by Burkholderia cepacia. J Med Microbiol. 2001;50(7):594–601. doi: 10.1099/0022-1317-50-7-594 [DOI] [PubMed] [Google Scholar]
- 16.Sajjan U, Thanassoulis G, Cherapanov V, Lu A, Sjolin C, Steer B, et al. Enhanced susceptibility to pulmonary infection with Burkholderia cepacia in Cftr—/- mice. Infect Immun. 2001;69(8):5138–50. doi: 10.1128/IAI.69.8.5138-5150.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vergunst AC, Meijer AH, Renshaw SA, O’Callaghan D. Burkholderia cenocepacia creates an intramacrophage replication niche in zebrafish embryos, followed by bacterial dissemination and establishment of systemic infection. Infect Immun. 2010;78(4):1495–508. doi: 10.1128/IAI.00743-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruscia EM, Zhang P-X, Ferreira E, Caputo C, Emerson JW, Tuck D, et al. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator-/- mice. Am J Respir Cell Mol Biol. 2009;40(3):295–304. doi: 10.1165/rcmb.2008-0170OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kopp BT, Abdulrahman BA, Khweek AA, Kumar SB, Akhter A, Montione R, et al. Exaggerated inflammatory responses mediated by Burkholderia cenocepacia in human macrophages derived from Cystic fibrosis patients. Biochem Biophys Res Commun. 2012;424(2):221–7. doi: 10.1016/j.bbrc.2012.06.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gavrilin MA, Abdelaziz DHA, Mostafa M, Abdulrahman BA, Grandhi J, Akhter A, et al. Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. J Immunol. 2012;188(7):3469–77. doi: 10.4049/jimmunol.1102272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Vaart M, Spaink HP, Meijer AH. Pathogen recognition and activation of the innate immune response in zebrafish. Adv Hematol. 2012;159807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature. 2014;505:218–22. doi: 10.1038/nature12799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mostowy S, Boucontet L, Mazon Moya MJ, Sirianni A, Boudinot P, Hollinshead M, et al. The zebrafish as a new model for the In vivo study of Shigella flexneri interaction with phagocytes and bacterial autophagy. PLoS Pathog. 2013;9(9):e1003588 doi: 10.1371/journal.ppat.1003588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su F, Juarez MA, Cooke CL, Lapointe L, Shavit JA, Yamaoka JS, et al. Differential regulation of primitive myelopoiesis in the zebrafish by Spi-1/Pu.1 and C/ebp1. Zebrafish. 2007;4(3):187–99. doi: 10.1089/zeb.2007.0505 [DOI] [PubMed] [Google Scholar]
- 25.Curado S, Stainier DYR, Anderson RM. Nitroreductase-mediated cell/tissue ablation in zebrafish: a spatially and temporally controlled ablation method with applications in developmental and regeneration studies. Nat Protoc. 2008;3(6):948–54. doi: 10.1038/nprot.2008.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herbomel P, Thisse B, Thisse C. Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive process. Dev Biol. 2001;238(2):274–88. doi: 10.1006/dbio.2001.0393 [DOI] [PubMed] [Google Scholar]
- 27.Yang C-T, Cambier CJ, Davis JM, Hall CJ, Crosier PS, Ramakrishnan L. Neutrophils exert protection in the early tuberculous granuloma by oxidative killing of Mycobacteria phagocytosed from infected macrophages. Cell Host Microbe. 2012;12(3):301–12. doi: 10.1016/j.chom.2012.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palha N, Guivel-Benhassine F, Briolat V, Lutfalla G, Sourisseau M, Ellett F, et al. Real-time whole-body visualization of Chikungunya virus infection and host interferon response in zebrafish. PLoS Pathog. 2013;9(9):e1003619 doi: 10.1371/journal.ppat.1003619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colucci-Guyon E, Tinevez J-Y, Renshaw SA, Herbomel P. Strategies of professional phagocytes in vivo: unlike macrophages, neutrophils engulf only surface-associated microbes. J Cell Sci. 2011;124:3053–9. doi: 10.1242/jcs.082792 [DOI] [PubMed] [Google Scholar]
- 30.Greenberg DE, Marshall-Batty KR, Brinster LR, Zarember KA, Shaw PA, Mellbye BL, et al. Antisense phosphorodiamidate morpholino oligomers targeted to an essential gene inhibit Burkholderia cepacia complex. J Infect Dis. 2010;201(12):1822–30. doi: 10.1086/652807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ventura GMDC, Balloy V, Ramphal R, Khun H, Huerre M, Ryffel B, et al. Lack of MyD88 protects the immunodeficient host against fatal lung inflammation triggered by the opportunistic bacteria Burkholderia cenocepacia. J Immunol. 2009;183(1):670–6. doi: 10.4049/jimmunol.0801497 [DOI] [PubMed] [Google Scholar]
- 32.Nguyen-Chi M, Phan QT, Gonzalez C, Dubremetz J-F, Levraud J-P, Lutfalla G. Transient infection of the zebrafish notochord with E. coli induces chronic inflammation. Dis Model Mech; 2014;7(7):871–82. doi: 10.1242/dmm.014498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saini LS, Galsworthy SB, John MA, Valvano MA. Intracellular survival of Burkholderia cepacia complex isolates in the presence of macrophage cell activation. Microbiology. 1999;145:3465–75. doi: 10.1099/00221287-145-12-3465 [DOI] [PubMed] [Google Scholar]
- 34.Bjarnsholt T, Jensen PØ, Fiandaca MJ, Pedersen J, Hansen CR, Andersen CB, et al. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr Pulmonol. 2009;44:547–58. doi: 10.1002/ppul.21011 [DOI] [PubMed] [Google Scholar]
- 35.Sajjan U, Corey M, Humar A, Tullis E, Cutz E, Ackerley C, et al. Immunolocalisation of Burkholderia cepacia in the lungs of cystic fibrosis patients. J Med Microbiol. 2001;50(6):535–46. doi: 10.1099/0022-1317-50-6-535 [DOI] [PubMed] [Google Scholar]
- 36.Schwab U, Abdullah LH, Perlmutt OS, Albert D, Davis CW, Arnold RR, et al. Localization of Burkholderia cepacia complex bacteria in cystic fibrosis lungs and interactions with Pseudomonas aeruginosa in hypoxic mucus. Infect Immun. 2014;82(11):4729–45. doi: 10.1128/IAI.01876-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banovic F, Koch S, Robson D, Jacob M, Olivry T. Deep pyoderma caused by Burkholderia cepacia complex associated with ciclosporin administration in dogs: a case series. Vet Dermatol. 2015;26(4):287–e64. doi: 10.1111/vde.12210 [DOI] [PubMed] [Google Scholar]
- 38.Gautam V, Singhal L, Ray P. Burkholderia cepacia complex: beyond pseudomonas and acinetobacter. Indian J Med Microbiol. 2011;29(1):4–12. doi: 10.4103/0255-0857.76516 [DOI] [PubMed] [Google Scholar]
- 39.Clay H, Davis JM, Beery D, Huttenlocher A, Lyons SE, Ramakrishnan L. Dichotomous role of the macrophage in early Mycobacterium marinum infection of the zebrafish. Cell Host Microbe. 2007;2(1):29–39. doi: 10.1016/j.chom.2007.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prajsnar TK, Cunliffe VT, Foster SJ, Renshaw SA. A novel vertebrate model of Staphylococcus aureus infection reveals phagocyte-dependent resistance of zebrafish to non-host specialized pathogens. Cell Microbiol. 2008;10(11):2312–25. doi: 10.1111/j.1462-5822.2008.01213.x [DOI] [PubMed] [Google Scholar]
- 41.van der Vaart M, van Soest JJ, Spaink HP, Meijer AH. Functional analysis of a zebrafish myd88 mutant identifies key transcriptional components of the innate immune system. Dis Model Mech. 2013;6(3):841–54. doi: 10.1242/dmm.010843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dinauer MC, Gifford MA, Pech N, Li LL, Emshwiller P. Variable correction of host defense following gene transfer and bone marrow transplantation in murine X-linked chronic granulomatous disease. Blood. 2001;97(12):3738–45. [DOI] [PubMed] [Google Scholar]
- 43.Sousa SA, Ulrich M, Bragonzi A, Burke M, Worlitzsch D, Leitão JH, et al. Virulence of Burkholderia cepacia complex strains in gp91phox-/- mice. Cell Microbiol. 2007;9(12):2817–25. doi: 10.1111/j.1462-5822.2007.00998.x [DOI] [PubMed] [Google Scholar]
- 44.Bylund J, Campsall PA, Ma RC, Conway BD, Speert DP. Burkholderia cenocepacia induces neutrophil necrosis in chronic granulomatous disease. J Immunol. 2005;174:3562–9. [DOI] [PubMed] [Google Scholar]
- 45.Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol. 2012;198(5):773–83. doi: 10.1083/jcb.201203170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–41. doi: 10.1083/jcb.200606027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ventura GM, Le Goffic R, Balloy V, Plotkowski M-C, Chignard M, Si-Tahar M. TLR 5, but neither TLR2 nor TLR4, is involved in lung epithelial cell response to Burkholderia cenocepacia. FEMS Immunol Med Microbiol. 2008;54(1):37–44. doi: 10.1111/j.1574-695X.2008.00453.x [DOI] [PubMed] [Google Scholar]
- 48.Urban TA, Griffith A, Torok AM, Smolkin ME, Burns JL, Goldberg JB. Contribution of Burkholderia cenocepacia flagella to infectivity and inflammation. Infect Immun. 2004;72(9):5126–34. doi: 10.1128/IAI.72.9.5126-5134.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hanuszkiewicz A, Pittock P, Humphries F, Moll H, Rosales AR, Molinaro A, et al. Identification of the flagellin glycosylation system in Burkholderia cenocepacia and the contribution of glycosylated flagellin to evasion of human innate immune responses. J Biol Chem. 2014;289(27):19231–44. doi: 10.1074/jbc.M114.562603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ceballos-Olvera I, Sahoo M, Miller MA, del Barrio L, Re F. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1β is deleterious. PLoS Pathog. 2011;7(12); e1002452 doi: 10.1371/journal.ppat.1002452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci USA. 2014;111(9):3526–31. doi: 10.1073/pnas.1322831111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Angus AA, Lee AA, Augustin DK, Lee EJ, Evans DJ, Fleiszig SMJ. Pseudomonas aeruginosa induces membrane blebs in epithelial cells, which are utilized as a niche for intracellular replication and motility. Infect Immun. 2008;76(5):1992–2001. doi: 10.1128/IAI.01221-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lovewell RR, Patankar YR, Berwin B. Mechanisms of phagocytosis and host clearance of Pseudomonas aeruginosa. AJP Lung Cell Mol Physiol. 2014;306(7):L591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flannagan RS, Heit B, Heinrichs DE. Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination. Cell Microbiol. 2016;18:514–35. doi: 10.1111/cmi.12527 [DOI] [PubMed] [Google Scholar]
- 55.Lamason RL, Mohideen MPK, Mest JR, Wong AC, Norton HL, Aros MC, et al. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science. 2005;310:1782–6. doi: 10.1126/science.1116238 [DOI] [PubMed] [Google Scholar]
- 56.Renshaw SA, Loynes CA, Trushell DMO, Elworthy S, Ingham PW, Whyte MKB. A transgenic zebrafish model of neutrophilic inflammation. Blood. 2006;108(13):3976–8. doi: 10.1182/blood-2006-05-024075 [DOI] [PubMed] [Google Scholar]
- 57.Gray C, Loynes CA, Whyte MKB, Crossman DC, Renshaw SA, Chico TJA. Simultaneous intravital imaging of macrophage and neutrophil behaviour during inflammation using a novel transgenic zebrafish. Thromb Haemost. 2011;105(5):811–9. doi: 10.1160/TH10-08-0525 [DOI] [PubMed] [Google Scholar]
- 58.Ellett F, Pase L, Hayman JW, Andrianopoulos A, Lieschke GJ. Mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood. 2011;117(4):e49–56. doi: 10.1182/blood-2010-10-314120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ellett F, Elks PM, Robertson AL, Ogryzko NV, Renshaw SA. Defining the phenotype of neutrophils following reverse migration in zebrafish. J Leukocyte Biol. 2017;98(6):975–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Darling P, Chan M, Cox AD, Sokol PA. Siderophore production by Cystic Fibrosis Isolates of Burkholderia cepacia. Inf Immun. 1998;66(2):874–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Govan JRW, Brown PH, Maddison J, Doherty CJ, Nelson JW, Dodd M, et al. Evidence for transmission of Pseudomonas cepacia by social contact in cystic fibrosis. Lancet. 1993;342(8862):15–9. [DOI] [PubMed] [Google Scholar]
- 62.Revets H, Vandamme P, Van Zeebroeck A, De Boeck K, Struelens M, Verhaegen J, et al. Burkholderia (Pseudomonas) cepacia and cystic fibrosis: the epidemiology in Belgium. Acta Clin Belg. 1996;51(4):222–30 [DOI] [PubMed] [Google Scholar]
- 63.Mahenthiralingam E, Coenye T, Chung JW, Speert DP, Govan JR, Taylor P, et al. Diagnostically and experimentally useful panel of strains from the Burkholderia cepacia complex. J Clin Microbiol. 2000;38(2):910–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goedhart J, von Stetten D, Noirclerc-Savoye M, Lelimousin M, Joosen L, Hink MA, et al. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat Commun. 2012;3:751 doi: 10.1038/ncomms1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mesureur J, Vergunst AC. Zebrafish embryos as a model to study bacterial virulence. Methods Mol Biol. 2014;1197:41–66. doi: 10.1007/978-1-4939-1261-2_3 [DOI] [PubMed] [Google Scholar]
- 66.Cui C, Benard EL, Kanwal Z, Stockhammer OW, van der Vaart M, Zakrzewska A, et al. Infectious disease modeling and innate immune function in zebrafish embryos. Methods Cell Biol. 2011;105:273–308. doi: 10.1016/B978-0-12-381320-6.00012-6 [DOI] [PubMed] [Google Scholar]
- 67.Le Guyader D, Redd MJ, Colucci-guyon E, Murayama E, Kissa K, Briolat V, et al. Origins and unconventional behavior of neutrophils in developing zebrafish. Blood. 2008;111:132–41. doi: 10.1182/blood-2007-06-095398 [DOI] [PubMed] [Google Scholar]
- 68.Feng Y, Santoriello C, Mione M, Hurlstone A, Martin P. Live imaging of innate immune cell sensing of transformed cells in zebrafish larvae: parallels between tumor initiation and wound inflammation. PLoS Biol. 2010;8(12):e1000562 doi: 10.1371/journal.pbio.1000562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ellett F, Lieschke GJ. Computational quantification of fluorescent leukocyte numbers in zebrafish embryos. Methods Enzymol. 2012;506:425–35. doi: 10.1016/B978-0-12-391856-7.00046-9 [DOI] [PubMed] [Google Scholar]
- 70.Kankaanpää P, Paavolainen L, Tiitta L, Karjalainen M, Päivärinne J, Nieminen J, et al. BioImageXD: an open, general-purpose and high-throughput image-processing platform. Nature Methods. 2012;9(7):683–689. doi: 10.1038/nmeth.2047 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Role of macrophages during infection with Bcc strains causing either persistent or acute infection. (A-D G-J) Embryo survival (A, C, G, and I) and bacterial burden (total of 2 experiments) over time (B, D, H, J) of control (black) and pu.1 knockdown embryos (red) injected iv with B. cenocepacia J2315 (A,B), B. cepacia CEP509 (C,D), B. cenocepacia J415 (G,H), and B. vietnamiensis FC441 (I,J), respectively. (E) Representative experiment (of at least three) showing embryo survival of control embryos (n = 47), pu.1 knockdown (n = 54) and nonspecific control MO (n = 52) embryos injected with B. cenocepacia K56-2 (average 53 CFU). (F) Representative fluorescence image at 24 hpi showing neutrophils (green) in an mpx:GFP pu.1 knockdown embryo injected with B. cenocepacia J2315 (red) (~50 CFU). Inset shows corresponding bright field image. Scale bar, 100 μm. (B, D, H, J) Geometric mean with each data point representing an individual embryo. Dead embryos are indicated as black open circles. (A-E and G-J) * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001; ns: non-significant. See materials and methods for statistical tests used.
(TIF)
Pu.1 knockdown prevents B. cenocepacia K56-2 multiplication. Fluorescence overlay images (red and green filters) of the indicated area (boxed area in embryo drawing) of two mpeg1:mCherry control embryos (left) and two mpeg1:mCherry pu.1 knockdown (right) imaged at 1, 3, 16, 20, 24, and 41 h after injection with ~50 CFU B. cenocepacia K56-2 harbouring pIN301 (green). The absence of macrophages prevents efficient bacterial replication. Bacteria in control embryos colocalise with macrophages (arrow heads). Fluorescent macrophages disappear from control embryos (>20 hpi). Appearance of macrophages in pu.1 knockdown embryos (white arrows).
(TIF)
Chemical ablation of macrophages using the NTR/Mtz system. (A) Schematic representation and treatment schedule of the chemical ablation strategy based on the nitroreductase (NTR)/ metronidazole (Mtz) system, shown for macrophage-specific ablation. See Materials and methods for details. (B) Representative fluorescence images of non-treated and Mtz-treated mpeg1/umn+ embryos, showing the efficacy of the Mtz treatment. Residual red fluorescence in treated embryos represented apoptotic cells. Scale bar, 0.5 mm. (C) Quantification of macrophage numbers in mpeg1/umn+ embryos (untreated or treated at 34 hpf with 5mM Mtz) at 0, 15 and 39 hours after treatment as indicated in (A). The efficacy of macrophage ablation by Mtz treatment was evaluated by pixel counting. The average macrophage numbers of two independent experiments (n = 6) are shown. ns, non-significant, ** p ≤ 0.01, **** p ≤ 0.0001. See material and methods for statistical analysis used.
(TIF)
Mtz-mediated ablation of macrophages prevents B. cenocepacia K56-2 multiplication. Fluorescence overlay images (red and green filter) of two DMSO-treated (left panels) and two Mtz-treated (right panels) mpeg1/umn+ embryos imaged at 1, 3, 24, and 48 h after iv injection with ~50 CFU B. cenocepacia K56-2 harbouring pIN301 (green). Bacterial replication in control embryos resulted in macrophage cell death, seen as mCherry positive debris (>24 hpi) (see also S5F Fig). At 28 and 48 hpi individual fluorescence images (green filter) are shown below the overlay images. Drawing indicates imaged area, marking the embryo sac extension (green) prone to autofluorescence. Arrow head indicates mCherry positive macrophages in apoptosis.
(TIF)
Behaviour and fate of neutrophils and macrophages during acute and persistent infection. (A). Confocal stack images of a time series, first image ~20 hours after infection of an mpx:GFP embryo with B. cenocepacia K56-2. Patrolling neutrophil inspects heavily infected macrophage (white arrow). Individual bacteria (arrow head), possibly released from infected cell nearby (open arrow), are being moved around by the neutrophil. The last image shows the same area 90 minutes later with the infected macrophage still intact. (B,C) Mpx:GFP embryos (50 hpf) were injected with B. stabilis LMG14294. (B) Representative images of control and B. stabilis infected mpx:GFP embryos with increased neutrophil numbers. Neutrophils were more dispersed in B. stabilis-infected compared to non-infected control embryos, where most neutrophils were resting in the caudal hematopoietic tissue. (C) The number of neutrophils in infected and non-infected control embryos was determined by pixel counting at different time points after injection. Each data point represents one embryo. The graph represents one of the three experiments represented in Fig 3C, but includes an additional time point at 5 days post infection, which was not determined in the other 2 experiments. A percentage of embryos injected with B. stabilis contained more neutrophils than control embryos (see also (B)). *, p ≤ 0.05. (D) Mpeg1:mCherry embryos were injected with B. cenocepacia K56-2 (~45 CFU) and the number of macrophages was evaluated by pixel counting at 0 and 24 hpi (30 and 54 hpf, respectively). The results are related to the corresponding experiment shown in Fig 3D. Each data point represents one embryo. Significance was determined using a one-way Anova with Sidak’s Multiple Comparisons test. **** p ≤ 0.0001. Two independent experiments (n = 5). (E) Image showing B.cenocepacia K56-2 (red) in an L-plastin labelled macrophage (blue) at 24 hpi. Scale bar, 50 μm. Inset, magnification red/ blue filters, scale bar 10 μm. (F) Images (bright field, green/red overlay, and detailed image with red/green filter), of a non-infected mpeg1/umn+ embryo (left panels) and mpeg1/umn+ embryo (right panels) iv infected with B. cenocepacia K56-2 (green, arrows) at 24 hpi. The images are similarly treated to enhance the red fluorescence to visualize the mCherry positive debris. The close up shows individual macrophages (left panel, red), and red fluorescent debris (arrow heads). Related to Fig 3F.
(TIF)
Macrophages, but not neutrophils, contribute to increased bacterial burden and pro-inflammatory responses towards subcutaneous B. cenocepacia. (A) mpeg/umn+ embryo infected subcutaneously at 2 dpf with B. cenocepacia K56-2 (green). Confocal stack (59 slices, 1 μm) at 24 hpi showing bacteria in macrophages. A close up of the indicated area is shown in 3D, with volume rendering for GFP signal and surface rendering of mCherry signal without (middle panel) and with 50% surface transparency (right panel). Scale bar 50 μm. (B) Fluorescence images showing non-infected mpx/umn+ and mpeg1/umn+ control embryos at 3 dpf, the time point that resembles 24 h post subcutaneous injection in Fig 6. (C) Image overlay (bright field, red and blue filters) of the tail region of an mpx/umn+ embryo after subcutaneous injection with B. cenocepacia K56-2 expressing Turquoise, presenting tissue damage by 24 hpi. Scale bar, 100 μm. (D) Image overlay (bright field, red, blue and green filters) of the tail region of an Mtz-treated mpeg1/umn+; mpx:GFP embryo 24 h after subcutaneous injection with B. cenocepacia K56-2 expressing Turquoise. Scale bar, 100 μm.
(TIF)
(A) Embryo survival (average inoculum 35 CFU, representative experiment) (B) and bacterial burden (average of 2 experiments) over time of wildtype control (myd88+/+) and mutant (myd88-/-) embryos iv injected with B. cenocepacia K56-2. (C, D) Embryo survival (average inoculum 39 (C) and 75 (D) CFU) of control (black) and myd88 knockdown embryos (red) iv injected with B. cenocepacia K56-2.
(TIF)
DMSO, Mtz and Anakinra do not affect il1b and cxcl8 expression levels. (A) mpx:GFP embryos were either non-treated or pre-treated at 34 hpf for 15 h with DMSO or 5 mM Mtz. Randomized groups were injected with PBS or B. cenocepacia K56-2 (on average 270 CFU). Graphs show mean relative il1b and cxcl8 gene expression levels, normalized to the PBS-injected non-treated control group at 4 hpi. Error bars represent mean with SEM of two biological replicates. (B) Control and Anakinra-treated mpx:GFP embryos were iv injected with PBS or B. cenocepacia K56-2 (average 120 CFU). Graphs show mean relative il1b and cxcl8 gene expression levels, normalized to the PBS-injected non-treated control group at each time point. Error bars represent mean with SEM of two biological replicates. ns = non-significant.
(TIF)
Morpholinos used in this study.
(DOCX)
Primers used for qRT-PCR experiments.
(DOCX)
Phagocytosis of surface-associated B. cenocepacia by neutrophils. Live imaging of an mpx:GFP embryo, injected subcutaneously at 50 hpf with DSRed expressing B. cenocepacia, every 90 seconds from 20 mins pi, for the duration of 75 mins. A time lapse of 50 confocal images with maximal intensity projection is shown (19 steps x 2 μm) at a rate of 2 frames per sec. Bacteria are rapidly phagocytosed by neutrophils, and remain visible in individual small vacuoles. Bacteria are also phagocytosed by GFP-negative macrophages, seen as individual bacterial clusters that are not colocalising with GFP-positive neutrophils.
(AVI)
Phagocytosis of surface-associated B. stabilis by neutrophils induces cell rupture. Live imaging of an mpx:GFP embryo, injected subcutaneously over a somite at 50 hpf with DSRed expressing B. stabilis, every 90 seconds from 10 minutes pi. Total duration 128 mins representing the period between 20 mpi and 148 mpi. A time lapse of 85 confocal images with standard deviation projection is shown (19 steps x 2 μm) at a speed of 2 frames per sec. Bacteria are rapidly phagocytosed by neutrophils, and are directed into large growing vacuoles (See Fig 5). Between 64–67 mpi two neutrophils full of bacteria round up (arrows) and eject their cell contents into the extracellular space (arrow heads appear), see also Fig 5. After the event, the bacteria remain clustered, and mobility of surrounding neutrophils is dramatically reduced, before they start moving again. Other neutrophils (indicated with arrows and consecutive numbers 3, 4 etc) undergo the same phenomenon, although sometimes the collapse is less explosive. The neutrophil indicated with number 4 seems to expel part of its contents including the phagocytosed bacteria, but the neutrophil takes up the contents again and after 35 mins, effectively “bursts” (indicated with arrow 4).
(AVI)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.