

SUMMARY
Pancreatic beta-cell mass for appropriate blood glucose control is established during early postnatal life. Beta-cell proliferative capacity declines postnatally but the extrinsic cues and intracellular signals that cause this decline remain unknown. To obtain a high-resolution map of beta-cell transcriptome dynamics after birth, we generated single-cell RNA-seq data of beta-cells from multiple postnatal time points and ordered cells based on transcriptional similarity using a new analytical tool. This analysis captured signatures of immature, proliferative beta-cells and established high expression of amino acid metabolic, mitochondrial, and Srf/Jun/Fos transcription factor genes as their hallmark feature. Experimental validation revealed high metabolic activity in immature beta-cells and a role for reactive oxygen species and Srf/Jun/Fos transcription factors in driving postnatal beta-cell proliferation and mass expansion. Our work provides the first high-resolution molecular characterization of state changes in postnatal beta-cells and paves the way for the identification of novel therapeutic targets to stimulate beta-cell regeneration.
Graphical Abstract
INTRODUCTION
Pancreatic beta-cells maintain blood glucose homeostasis by secreting insulin in response to nutrients, such as glucose, amino acids, and lipids. Defects in beta-cell function and reduced beta-cell mass cause diabetes mellitus. The early postnatal period is important for establishing appropriate beta-cell mass as well as responsiveness to nutrient cues (Jermendy et al., 2011). During this period, beta-cell mass expands substantially in both mice and humans owing to a neonatal burst in beta-cell proliferation (Finegood et al., 1995; Gregg et al., 2012). This burst is followed by a sharp proliferative decline early postnatally and a more gradual decline during aging. The molecular pathways governing postnatal beta-cell growth have been under intense investigation in hopes of identifying therapeutic approaches for stimulating human beta-cell regeneration.
Studies have identified cyclin-dependent kinase 4 (Cdk4) and D-type cyclins as important regulators of postnatal beta-cell proliferation (Georgia and Bhushan, 2004; Kushner et al., 2005; Rane et al., 1999). Upstream of the basic cell cycle machinery, neonatal beta-cell proliferation is driven by Pdgf receptor-mediated signaling acting via the Ras/MAPK pathway (Chen et al., 2011) and calcineurin signaling through the transcription factor (TF) NFAT (Goodyer et al., 2012). Although several regulators of beta-cell proliferation have been identified, the upstream signals that cause cell cycle arrest of most beta-cells during early postnatal life remain unknown.
A major obstacle in defining the pathways and mechanisms that drive postnatal cell cycle arrest is the heterogeneity among individual beta-cells. Proliferative beta-cells are rare, and beta-cells may change their features asynchronously during early postnatal life. Hence, at a given time point, the beta-cell population may contain proliferative, quiescent, functionally mature, and immature beta-cells. This concept is supported by studies in adult mice showing heterogeneity of beta-cells with regard to their molecular features, proliferative capacity, and responsiveness to nutrient cues (Bader et al., 2016; Dorrell et al., 2016; Johnston et al., 2016).
Population-based gene expression profiling generates average measurements and masks the variation across individual cells, thus limiting insight into different cell states. By providing gene expression profiles of individual cells, single-cell RNA-seq can overcome this problem, as subpopulations of cells can be identified based on transcriptional similarity. In several contexts, this approach has revealed molecular profiles of distinct cell types not recognized at the population level (Macosko et al., 2015; Treutlein et al., 2014). Furthermore, in samples throughout a developmental time course, single-cell expression profiles can be used to order cells along a “pseudotemporal” developmental continuum; a method that has helped resolve cellular transitions (Bendall et al., 2014; Trapnell et al., 2014). However, this approach has not yet been applied to a maturation time course of a single cell type, where insight into cell state changes could be gained.
Here, we applied single-cell RNA-seq to reconstruct the postnatal developmental trajectory of pancreatic beta-cells. We isolated beta-cells at five different time points between birth and post-weaning and generated single-cell transcriptomes. We then developed a one-dimensional (1D) projection-based algorithm to construct a “pseudotemporal” trajectory of postnatal beta-cell development by ordering all profiled beta-cells based on transcriptional similarity. This analysis revealed remarkable changes in beta-cell metabolism during early postnatal life. We show that postnatal beta-cell development is associated with amino acid deprivation and decreasing production of mitochondrial reactive oxygen species (ROS), and demonstrate a role for amino acids and ROS in postnatal beta-cell proliferation and mass expansion.
RESULTS
Transcriptional Heterogeneity of Postnatal Beta-Cells
Pancreatic beta-cells acquire a fully differentiated phenotype after completion of a postnatal maturation process (Jermendy et al., 2011). To probe this process in vivo, we performed single-cell RNA-seq on sorted beta-cells from mIns1-H2B-mCherry mice (Benner et al., 2014) at postnatal day (P)1, P7, P14, P21, and P28 (Fig. 1A). As a control, population (bulk) cDNA libraries of the corresponding time points were also generated. To obtain reliable single-cell libraries, we applied several quality control criteria (see “Methods” and Fig. S1A,B). RNA-seq libraries from single cells and bulk samples were sequenced to an average depth of 4.3 million reads. Saturation analysis confirmed that this sequencing depth was sufficient to detect most genes represented in the single-cell libraries (Fig. S1C). On average, 6298 genes per library were detected. Libraries that contained fewer than 1 million unique reads and for which more than 15% of fragments mapped to mitochondrial protein coding genes were excluded (Table S1). Based on these criteria, we retained data from 14 bulk samples and 387 single cells (84 cells from P1, 87 cells from P7, 88 cells from P14, 68 cells from P21, and 60 cells from P28). To minimize technical noise and artifacts such as batch effects, we applied “Surrogate Variable Analysis” for sequencing experiments (SVA-seq) (Leek, 2014).
Figure 1. Single-cell RNA-sequencing of beta-cells during postnatal development.

A. Experimental overview. B. Correlation between average transcript levels of all genes detected in single cells and bulk cells. For single cells, gene expression was averaged across individual cells collected at different time points. Points are colored by postnatal (P) day collected (P1, red; P7, green; P14, blue; P21, orange; and P28, black). The same color-coding for groups was used across all figures. Pearson correlation coefficients r are given. C. Expression analysis of select genes showing variability in gene expression among single cells of the same age. Each point represents the gene expression level of an individual cell. Black bars represent the average of pooled single cells. D. Pairwise correlation of single-cell gene expression showing biological variation among single cells of the same age. The distribution of the pairwise Pearson’s correlation coefficients between single cells is plotted. E. Multidimensional scaling of single cell transcriptomes. The distance between any two cells reflects the similarity of their expression profiles. See also Figure S1 and Table S1.
To assess single-cell data quality, we compared the correlation between average transcript profiles of single cells and bulk cells of the same age. At all ages, the average profiles of single cells correlated highly with the bulk cell profile (r= 0.83-0.87; Fig. 1B). We then compared expression patterns of select genes. Average expression levels of Ucn3 and Mafb, two genes known to be regulated during postnatal beta-cell development (Blum et al., 2012; van der Meulen et al., 2012), showed temporal regulation similar to bulk experiments (Fig. 1C). Notably, Ucn3 and Mafb exhibited high variability in cell-to-cell gene expression, whereas the housekeeping gene Calm1 did not (Fig. 1C). This implies that the observed transcriptional heterogeneity reflects true biology and is not a technical artifact. At an individual cell level, Ucn3 and Mafb expression were negatively correlated (Fig. S1D), suggesting that declining expression of Mafb is accompanied by increasing expression of Ucn3 across individual cells. Globally, the genome-wide correlation of transcript expression between single cells at each time point showed only moderate correlation (r=0.3-0.7; Fig. 1D), indicating considerable gene expression heterogeneity between age-matched cells. To visualize the degree of similarity among all beta-cells from different time points, we employed multidimensional scaling analysis. The analysis showed progression of most cells along a single trajectory, implying a continuous beta-cell maturation process. While the majority of beta-cells grouped together by age collected, there was no clear separation between stages and cells from one age often crossed into the transcriptional space of other ages (Fig. 1E). This shows that a significant fraction of beta-cells from late postnatal time points bears higher similarity with cells from earlier than from age-matched time points and vice versa.
Reconstructing a Trajectory for Beta-Cell Maturation
Given the heterogeneity of beta-cells at each stage, we reasoned that ordering beta-cells by transcriptional similarity rather than time of collection could provide insight into the transcriptional dynamics associated with beta-cell maturation not captured when evaluating bulk gene expression data across the time course. With the recent growth in single-cell transcriptome data, numerous methods have been reported to computationally order individual cells according to the gradual transition of their transcriptomes (Campbell et al., 2015; Ji and Ji, 2016; Reid and Wernisch, 2016; Trapnell et al., 2014). The process of ordering cells in silico is called pseudotime reconstruction because it places individual cells on a virtual time axis along which the cells are presumed to travel as they differentiate or mature. We postulated that such de novo predicted developmental path could expose previously unrecognized transcriptional dynamics of postnatal beta-cell maturation. To construct a pseudotemporal time course of maturing beta-cells, we adapted a previously developed 1D-principle component analysis (PCA) method (Zagar et al., 2011). The method establishes a pseudotemporal trajectory by ordering single-cell profiles based on transcriptional similarity along a linear ruler, allowing for exploration of gene patterns over the reconstructed developmental trajectory of continuously placed cells. We observed minimal branching in our data (see Suppl. Information), suggesting adequacy of a linear trajectory for our data.
To construct a pseudotemporal trajectory, we considered the most variant genes ranked by median absolute deviation (MAD) (Table S2A). Genes in the upper quartile of the MAD distribution (n=4313) were used to place cells along a 1D trajectory that represents each cell’s likely placement along a continuum of beta-cell maturation (Fig. 2A). To assess the performance of our 1D-PCA-based cell ordering method, we conducted comparisons to other ordering methods, namely Monocle (Trapnell et al., 2014), TSCAN (Ji and Ji, 2016), Embeddr (Campbell et al., 2015), and DeLorean (Reid and Wernisch, 2016). The analysis revealed higher predicted accuracy of revealing the correct order of cells and smoother transition of gene expression through the cells with 1D-PCA (Suppl. Information). As further validation, we calculated similarity of the cell ordering with DeLorean and 1D-PCA and found similar placement of cells obtained with the two methods (Suppl. Information).
Figure 2. 1D-PCA orders beta-cells by maturation progression.

A. Workflow to generate a 1D-PCA-based pseudotemporal trajectory. Raw gene expression data was normalized by SVA-seq and processed using Median Absolute Deviation (MAD) to obtain the most variant genes. Cells were placed along a 1D trajectory that represents each cell’s likely placement along a continuum of maturation. B. Pseudotime ordering of single cells using 1D-PCA. Each data point represents a single cell colored by age collected. Median placement of cells from each age is marked on the pseudotime axis (black arrow). C. Samples were assigned to five groups based on time collected (left) or pseudotime ordering (right, pseudo-binned time). Each pseudo-binned time-ordered group contained an equal number of cells to that of the corresponding time-ordered point. D. Expression profiles of selected genes ordered by time collected (top panels), pseudo-binned time (middle panels), or pseudotime (bottom panels). See also Figure S2; S3 and Table S2, S3.
The projection of beta-cells along the 1D-PCA-based trajectory revealed a median placement of samples from each stage that agreed with the temporal order of the maturation time course from P1 to P28 (Fig. 2B). To further validate the time ordering method, we projected published RNA-seq data from single beta-cells of 3-month-old mice (Xin et al., 2016) onto our pseudotemporal trajectory and found that the median of these cells correctly projected at the end of the pseudotime spectrum (see Suppl. Information). Thus, our analysis generated a refined order of discrete beta-cell states independent of, but consistent with, prior knowledge.
Notably, single cells from an individual stage spanned a large spectrum of the pseudotemporal developmental scale, indicating significant transcriptional heterogeneity of beta-cells at each time point. To allow for comparison of gene expression trajectories between pseudotemporally ordered cells and collection time point averages, we created five “pseudo-time points” (= “pseudo-binned” cells) by selecting an equal number of cells for the pseudo-binned point as collected at a given time point. For example, 84 cells were collected at P1 and 87 cells at P7. Hence, we considered the first 84 cells along the pseudotemporal trajectory (Fig. 2B) as “pseudo-P1” and the next 87 cells as “pseudo-P7” (Fig. 2C). Illustrating the transcriptional heterogeneity of beta-cells at each stage, the pseudo-binning revealed beta-cells from multiple postnatal stages contributing to each pseudo-binned time point (Fig. S2A).
To validate that the pseudotemporal trajectory accurately captures transcriptional changes of postnatal beta-cell development, we analyzed expression profiles of genes known to be regulated during beta-cell maturation in our averaged collection time-specific, pseudo-binned, and pseudotime-ordered single-cell RNA-seq data. Consistent with previous bulk transcriptome data (Artner et al., 2010; Blum et al., 2012; van der Meulen et al., 2012), expression of Mafb decreased in both our collection time-based bulk data between P1 and P28 and the reconstructed pseudotemporal single-cell-based trajectories, while expression of Ucn3 and the regulator of insulin secretion G6pc2 increased (Fig. 2D; Fig. S2B). These results indicate that the primary phenotypic landmarks of beta-cell maturation were correctly reconstructed by our pseudotemporal ordering method.
Ordering of Cells Reveals Novel Maturation-Associated Changes in Gene Expression
We reasoned that the pseudotemporal ordering of cells could reveal transcriptional characteristics of rare cells within the beta-cell population, such as proliferating beta-cells. Although numbers of proliferating beta-cells are known to decline postnatally (Teta et al., 2005), mRNA levels of the proliferation markers Ki67, Cdk4, Rfc2, and Mcm3 showed little change in bulk samples between P1 and P28 (Fig. 2D; Fig. S2B), likely owing to the small contribution of these cells to the overall transcriptional profile of the bulk samples. By contrast, in the pseudotemporally-ordered time course, these proliferation genes exhibited declining expression during beta-cell maturation (Fig. 2D; Fig. S2B), suggesting that our pseudotemporal cell ordering method can resolve transcriptional features of immature proliferative beta-cells. In addition to proliferation genes, we observed decreasing expression of the mitochondrial respiratory chain component, Ndufv1, as well as the amino acid transporter Slc7a2 and amino acid sensing and mTOR signaling regulator Lamtor5 (Fig. 2D; Fig. S2B). These expression changes were not observed in bulk sample averages, indicating that our time ordering method can reveal novel molecular features associated with beta-cell maturation. To more globally assess to which extent gene expression patterns differ between collection-time- and pseudotime-ordered cells, we compared gene expression profiles from collection time averages to pseudo-binned averages (Fig. 2C). Analysis of increasing and decreasing genes in the transitions between two consecutive points confirmed that numerous patterns could be identified only by considering pseudotime-ordered cells (Fig. S3; Table S3A,B). Together, these findings provide evidence that 1D-PCA-based ordering of single beta-cells accurately reflects the transcriptional dynamics of beta-cell maturation and allows for de novo discovery of gene expression changes not revealed by population averages.
Metabolic Pathways Are Regulated during the Reconstructed Maturation Time Course
To identify groups of genes significantly regulated in the pseudotemporal trajectory, we examined gene sets, including MSigDB annotated pathways as well as clusters of transcriptionally correlated genes found in our data (de novo gene sets) (Fig. 3A). Briefly, continuous Gene Set Enrichment Analysis (GSEA) was performed with the aim of testing whether the genes in each set show coordinated increasing or decreasing expression during the pseudotemporal trajectory of postnatal beta-cell development. Gene sets showing a significant positive or negative correlation with the pseudotime coordinates, representing increasing and decreasing groups of genes, respectively, were selected (FDR < 0.25, Table S4). Analysis of annotated pathways revealed negative correlation with genes involved in cell cycle control and proliferation with the pseudotemporal time course (Fig. 3B; Table S4A), confirming the ability of our time ordering method to resolve transcriptomes of immature, proliferative beta-cells. In addition, there was overrepresentation of Gene Ontology (GO) categories associated with metabolic pathways, such as amino acid metabolism and mitochondrial respiration, as well as enrichment of hypoxia and reactive oxygen species (ROS) pathway GO-annotated gene sets. Combined, this suggests that beta-cells undergo fundamental changes in metabolism during early postnatal development.
Figure 3. Postnatal beta-cell development is associated with expression changes of genes regulating amino acid uptake and metabolism, mitochondrial respiration, and ROS production.

A. Schematic of workflow. To identify genes regulated during pseudotime, continuous GSEA was performed on annotated gene sets and de novo gene sets obtained by hierarchical clustering. B. Molecular pathways regulated in pseudotime from annotated gene sets. C. Heatmap showing average transcript expression of all genes within de novo gene sets showing significant correlation with pseudotime ordering (C1-C9). Number of genes in each set is shown on the right. D. Heatmaps for each de novo gene set showing expression of selected genes involved in cell proliferation (red), insulin secretion (green), regulation of ROS (black), mitochondrial function (orange), immediate early genes (purple), and amino acid metabolism (blue) with pseudotime. See also Figure S4 and Table S4.
In addition to analyzing a priori known gene sets, we also sought to capture molecular features distinguishing immature and mature beta-cells that might be poorly represented by annotated pathways. For this, we applied a clustering method to perform de novo gene set discovery (Fan et al., 2016). Hierarchical clustering was first applied to all genes expressed (RPKM > 1 in at least 2 cells; n=13899). In total, 78 groups of genes were found and were scored for significant correlation with pseudotime coordinates, using the same approach as for annotated pathways (Table S4B). From this, we identified nine clusters (C1-C9) that showed significant correlation with the reconstructed trajectory of beta-cell maturation (Fig. 3C; Table S4B). Each of the nine clusters contained genes that increased and decreased during the pseudotime course (Fig. S4). Cluster C1 contained predominantly upregulated genes and was enriched for regulators of insulin synthesis and secretion, such as Ins1/2, G6pc2, Iapp, and Ucn3 (Fig. 3C,D; Table S4C). Consistent with the observation that proliferative beta-cells can be resolved with our time ordering method (Fig. 2D), clusters C2 and C3 were enriched for genes encoding proteins involved in cell cycle control, comprising DNA replication, mitotic spindle assembly, and mitotic checkpoint proteins in cluster C2 and proteins with functions in the p53 and MAPK pathways in cluster C3 (Fig. 3D; Table S4C). Cluster C4 was enriched for immediate early genes (i.e. Fos, Jun, Atf3, Srf), which are regulators of cell growth signals (Mina et al., 2015). This cluster also contained the known regulator of postnatal beta-cell proliferation Pdgfa (Chen et al., 2011). A striking observation was the downregulation of numerous genes encoding proteins regulating mitochondrial function and ROS in cluster C5, including mitochondrial transporters (Slc25a3, Slc25a39), respiratory chain components (Ndufa5, Cox6a1, Uqcrb), and enzymes for ROS clearance and protection from oxidative damage (i.e. Prdx2, Sod1, Gpx4) (Fig. 3D; Table S4C). These findings suggest that mitochondrial respiration and ROS levels are highly regulated during postnatal beta-cell development. We further observed concordant regulation of multiple genes involved in amino acid metabolism in cluster C6 (Fig. 3D; Table S4C), namely a progressive decrease in the expression of the transmembrane amino acid transporters Slc7a2 and Slc38a5 as well as Gls and Glud1, which convert glutamine into glutamate and α-ketogluterate (αKG), respectively. Combined, these findings show that beta-cell maturation is associated with fundamental changes in the expression of genes associated with amino acid uptake and metabolism, as well as mitochondrial respiration and ROS production.
Amino Acid Availability Regulates Beta-Cell Proliferation
Amino acids promote cell proliferation by providing building blocks for protein and nucleotide synthesis. Based on the observed decrease in expression of multiple amino acid transporter and metabolism genes during maturation (Fig. 4A) and their co-variation with proliferation genes at the single-cell level (Fig. 4B), we hypothesized that amino acid deprivation could contribute to the postnatal decline in beta-cell proliferation. First, to assess whether amino acid availability to beta-cells changes during the early postnatal period, we measured levels of various amino acids in plasma from mice at P1 and P28. Plasma levels of most proteinogenic amino acids decreased between P1 and P28 (Fig. 4C), indicating environmental changes in beta-cell amino acid availability. Next, to determine whether amino acid and nucleotide availability is limiting for beta-cell proliferation, we supplemented cultures of islets from mice at P28 with nucleotides or individual amino acids. Addition of serine or tyrosine, or nucleotides, significantly increased proliferation rates of beta-cells at P28 (Fig. 4D); an effect that was not observed for non-beta islet cells (Fig. 4E). The results show that amino acid supplementation can, at least partially, restore beta-cell proliferation at a time point when proliferative rates have already declined. Accordingly, supplementation of serine, tyrosine or nucleotides increased expression of several proliferation genes, which are down-regulated in the pseudotemporal maturation time course (Fig. 4F). Notably, glutamine supplementation failed to enhance beta-cell proliferation (Fig. 4D), which could be because glutamine uptake rates were significantly lower in islets from mice at P28 compared to P1 (Fig. 4G). Combined, these findings suggest that changes in plasma amino acid levels as well as beta-cell amino acid uptake and metabolism could cause an amino acid-deprived state that contributes to the postnatal decline in beta-cell proliferation (Fig. 4H).
Figure 4. Amino acid supplementation increases beta-cell proliferation.

A. Amino acid transporter and metabolism genes downregulated with pseudotime with Pearson correlation coefficients. B. Heatmap showing Pearson correlation of gene expression in all 387 beta-cells comparing proliferation genes with genes encoding amino acid transporters and metabolizing enzymes downregulated with pseudotime. Proliferation genes are depicted in rows and amino acid transporters and metabolizing enzymes in columns. C. Plasma concentration of individual amino acids in mice at P1 (blue) and P28 (red). Data shown as mean ± SEM (n= 4 mice per group). D, E. Percentage of EdU+ beta-cells (D) and non-beta-cells (E) in islets from mice at P28 supplemented with nucleotides or amino acids. F. Quantitative RT-PCR analysis of proliferation genes after supplementation with serine, tyrosine or nucleotides. mRNA levels in control islets were set as 1. G. Glutamine uptake in mice at P1 (blue) and P28 (red). H. Schematic summarizing the observed gene expression changes during beta-cell maturation and effects on proliferation. In E-G, data are shown as mean ± SEM of three independent experiments. TCA, tricarboxylic acid cycle; Gls, glutaminase; Slc, solute carrier. * P < 0.05, ** P < 0.01, *** P < 0.001.
Mitochondrial ROS Promotes Beta-Cell Proliferation
In addition to amino acid metabolic genes, numerous mitochondrial genes exhibited a significant decrease in expression during pseudotemporal maturation (Fig. 3D). Therefore, we postulated that beta-cell mitochondrial membrane potential might decrease postnatally. Indeed, islets at P28 exhibited lower mitochondrial membrane potential than at P1 (Fig. 5A; Fig. S5A). The mitochondrial to nuclear DNA ratio was similar in P1 and P28 islets (Fig. S5B), indicating that the overall number of mitochondria does not change postnatally. These results suggest that the early postnatal period is associated with a decline in mitochondrial respiration.
Figure 5. Mitochondrial ROS production promotes beta-cell proliferation.

A. FACS analysis of TMRM fluorescence intensity. Blue: postnatal day (P) 1 without FCCP; Green: P1 with FCCP; Red: P28 without FCCP; Purple: P28 with FCCP. Mitochondrial TMRE/MitoTracker Green uptake ratio is shown on the right. B. Schematic of pathways regulating ROS clearance by antioxidant enzymes. C. Downregulated genes with pseudotime involved in ROS regulation with their Pearson correlation coefficient. D. FACS analysis of MitosoxRed fluorescence intensity at P1 (blue) and P28 (red). Mitochondrial MitosoxRed/MitoTracker Green ratios are shown on the right. E. Schematic of alleles in RIP-Cre;mCAT;R26YFP mice (mCAT mice). Red triangles indicate loxP sites. F, G. Representative immunofluorescence staining for insulin (green), BrdU (red), and DAPI (blue) (F) and quantification of the percentage of beta-cells expressing BrdU (G) in 6-week-old control (RIP-Cre) and mCAT mice. White arrows indicate Ins+Ki67+ cells in (F). H. Quantification of the beta-cell area relative to total pancreatic area in 6-week-old mCAT and control mice. Data shown as mean ± SEM of three independent experiments (A,D) or three mice per group (G,H). Scale bar= 20 μm. SOD, superoxide dismutase; GR, glutamate receptor; GSH, glutathione; GSSG, glutathione disulfide; GPX, glutathione peroxidase; CAT, catalase; TRX, thioredoxin; PRX, peroxidase. * P < 0.05, ** P < 0.01. See also Figure S5.
Mitochondrial activity is an important source of ROS production. The abundance of ROS is determined by the balance between ROS production and ROS clearance through multiple antioxidant enzymes (Fig. 5B). Genes encoding antioxidant enzymes decreased during the pseudotemporal beta-cell maturation time course (Fig. 5C). To determine how the combination of decreased mitochondrial membrane potential and reduced expression of ROS eliminating enzymes affects overall beta-cell mitochondrial ROS abundance, we measured mitochondrial superoxide levels in islets from P1 and P28 mice. Despite reduced expression of antioxidant enzymes, mitochondrial superoxide levels were significantly lower at P28 (Fig. 5D).
ROS can enhance cell proliferation, but highly elevated ROS levels can also induce G2/M cell cycle arrest and reduce cell proliferation (Boonstra and Post, 2004). To determine how ROS affects beta-cell proliferation, we utilized a genetic model to stably overexpress the radical scavenger catalase specifically in beta-cell mitochondria. We generated mice carrying the RIP-Cre transgene, Cre recombinase-inducible human catalase (mCAT) inserted in the ubiquitously active GAPDH locus, and a conditional YFP reporter gene targeted to the Rosa-26 locus (R26YFP) (hereinafter called mCAT mice) (Fig. 5E). The insertion of mCAT in the GAPDH locus did not affect glucose homeostasis, as determined by glucose tolerance testing (Fig. S5C). Immunofluorescence analysis of YFP in pancreata from mCAT mice at 6 weeks revealed recombination in ~90% of beta-cells (Fig. S5D). Quantitative RT-PCR confirmed expression of human CAT mRNA in islets from mCAT mice (Fig. S5E). By staining islets with MitosoxRed, we further confirmed that mCAT mice exhibit lower levels of ROS than RIP-Cre control mice (Fig. S5F). Analysis of BrdU incorporation and Ki67 staining revealed a significant reduction in the percentage of proliferating beta-cells in mCAT mice compared to controls (Fig. 5F,G; Fig. S5G). Accordingly, total beta-cell mass in mCAT mice was significantly reduced (Fig. 5H). mCAT expression did not affect beta-cell identity and did not lead to conversion of beta-cells into other islet cell types (Glucagon+GFP+ cells = 1.3% in mCAT mice vs. 0.91% in control mice; no somatostatin+GFP+ cells were observed) (Fig. S5H). Beta-cell apoptosis and glucose-stimulated insulin secretion (GSIS) in islets were similar in mCAT and control mice (Fig. S5I,J). These results identify a specific role for mitochondrial ROS in promoting beta-cell proliferation and establishment of normal beta-cell mass.
Nutrient-Responsive Transcription Factors Mediate Maturation-Associated Gene Expression Changes
Having identified roles for amino acid availability and ROS in postnatal beta-cell proliferation, we next sought to identify the TFs that mediate maturation-associated gene expression changes and regulate beta-cell proliferation. First, to identify the most highly regulated genes during beta-cell maturation, we generated a list of genes positively and negatively correlating with the pseudotemporal trajectory (P <0.01; Fig. 6A; Table S5A). These criteria were met by 54 genes that increased and 3279 genes that decreased in expression during pseudotime. Second, to determine which TFs regulate these genes, we performed cis-regulatory analysis, focusing on enhancers identified by the presence of distal H3K27ac ChIP-seq signals in mouse islets, and annotated TF binding motifs at these enhancers (Fig. 6A). This analysis revealed binding site enrichment for TFs of the ETS and basic leucine zipper (bZIP) families at enhancers of both up- and down-regulated genes during beta-cell maturation (Fig. 6B,C; Table S5B). In addition, zinc finger (SP1 and Klf TFs), helix turn helix (Rfx TFs), and CCAAT motifs were among the twenty most highly enriched motifs at enhancer regions of downregulated genes (Fig. 6C; Table S5B). Binding sites for the previously identified regulators of beta-cell maturation, Neurod1 (Gu et al., 2010) and Mafa (Aguayo-Mazzucato et al., 2011), were also enriched at enhancers of downregulated genes (Table S5B); however, fewer target sequences contained Neurod1 and Mafa binding sites when compared to ETS, bZIP, and zinc finger motifs.
Figure 6. Transcription factors and target genes regulated during postnatal beta-cell development.

A. Workflow to identify TFs driving gene expression changes during pseudotime. Cis-regulatory analysis was performed on the most significant genes positively and negatively correlating with pseudotime (P <0.01) followed by annotation of TF binding motifs at their enhancers. B, C. Top TF binding motifs for up-regulated genes (B) and down-regulated genes (C) during pseudotime. Enrichment P values are shown. D. The top 2 up-regulated (red) and top 30 down-regulated (blue) TFs regulated with pseudotime with their Pearson correlation coefficient. E. Network of interactomic connections of TFs regulated during pseudotime with other pseudotemporally regulated genes. Green nodes represent TFs while light blue nodes represent pseudotemporally regulated genes. The size of the nodes is adjusted proportionally to the correlation coefficient of the gene expression level with pseudotime coordinates. Genes in red are associated with cell proliferation. See also Figure S6 and Table S5; S6.
To identify candidate TFs that could mediate the postnatal changes in beta-cell gene expression, we ordered TFs based on positive and negative correlation of their expression profiles with the pseudotemporal trajectory. This analysis revealed 202 significantly downregulated and two upregulated TFs (P < 0.01). Consistent with the motif analysis, the top thirty downregulated TFs comprised multiple members of the ETS (Etv1, Fev), bZIP (Atf3, Fosb, Fos, Jun, Maff, Junb, Atf4), and zinc finger (Klf10, Egr1, Klf4, Zfp868, Zfp655, Zbtb10, Egr2) families (Fig. 6D).
These ETS, bZIP and zinc finger TFs are core constituents of a TF network regulated in response to cellular stress (Espinosa-Diez et al., 2015). ROS abundance, Ras/MAPK and mTOR signaling, which we found to be regulated during postnatal beta-cell development (Fig. 3B; Fig. 5D; Table S4A), are potent inducers of Srf, Atf3, Atf4, Fosb, Fosl2, Fos, and Jun (Espinosa-Diez et al., 2015). By forming both homo- and heterodimers, these TFs regulate the expression of anti-oxidant genes and cellular responses to nutrient state, including cell proliferation. To understand the role of these TFs in postnatal beta-cell gene regulation, we performed network analyses using the STRING database to reveal connections of the TFs with other genes regulated during the pseudotemporal beta-cell maturation trajectory. We then applied a network propagation algorithm that prioritizes genes in the network based on the strength of their connections to a starting set of genes (Mulas et al., 2013). Using the TFs in the network as a starting set (Table S6A), the interest propagation algorithm retrieved an additional set of genes mostly related to cell proliferation (P = 0.0001, Fisher test) and mRNA processing (P = 1.85*10-7) as their most connected neighbors (Fig. 6E). Proliferation genes with high connectivity to the TFs Atf3/Atf4, Jun/JunB, Fos/Fosb, Egr1, and Srf included the regulator of postnatal beta-cell expansion Cdk4 (Rane et al., 1999), several components of the pre-replication complex (Mcm2/3/4/5), and Gsk3b, an important signaling hub in the regulation of beta-cell replication (Liu et al., 2010). When the most relevant connections of genes related to oxidative phosphorylation were searched through the network (Table S6B), we retrieved the TFs Jun and Fos (Fig. S6). Thus, our motif and network propagation analyses lend support to the model that changes in metabolic activity regulate postnatal beta-cell proliferation through nutrient-responsive TFs of the Jun/Fos family.
Srf Regulates Proliferation Genes in Primary Islets
Based on this model, expression levels of oxidative phosphorylation genes, Jun/Fos family TFs and proliferation genes should exhibit positive correlation at the single-cell level. To test this prediction, we selected the cell proliferation, oxidative phosphorylation, and TF genes most highly regulated in the pseudotemporal trajectory and calculated pairwise correlation coefficients considering all 387 beta-cells. The analysis revealed significant co-variation (P < 0.001) of both oxidative phosphorylation and TF genes with proliferation genes (Fig. 7A).
Figure 7. Srf regulates proliferation genes in beta-cells.

A. Heatmap showing Pearson correlation of gene expression profiles in all 387 beta-cells comparing proliferation genes with top pseudotemporally-regulated oxidative phosphorylation genes and TFs. Proliferation genes are depicted in red, oxidative phosphorylation genes in orange and TFs in black. B. Overview of RNA-seq analysis of lentiviral Srf overexpression in islets at P28. C. GSEA plots showing enrichment of proliferation genes regulated during pseudotime (left) and genes down-regulated during pseudotime (right) as an effect of Srf overexpression. RNA-seq data are from three independent transduction experiments. D. RPKM values of TFs Fos, Junb and Egr1 (top panel) and proliferation genes Mki67, Pcna and Ccne1 (bottom panel) in RNA-seq data from control and Srf-overexpressing islets. Data shown as mean ± SEM. E. Summary of metabolic regulators and effector TFs driving early neonatal beta-cell proliferation as revealed by reconstructing a pseudotemporal time course of beta-cell maturation, experimental validation, and prior literature. ** P < 0.01, *** P < 0.001. See also Figure S7 and Table S7.
We next sought to test whether these TFs could regulate proliferation genes in beta-cells. Previous studies have shown that Srf is an upstream activator of Fos, Fosb, Egr1, and Nr4a1 (Mina et al., 2015). We found that Srf itself and its downstream targets are downregulated during maturation (Fig. 6D), suggesting that Srf could be responsible for the regulation of a large portion of the maturation-dependent TFs. Moreover, Srf promotes recruitment of ETS TFs to DNA (Hassler and Richmond, 2001), and ETS TF motifs were highly enriched in genes regulated during maturation (Fig. 6B,C). To determine the role of Srf in beta-cell gene regulation, we overexpressed Srf via lentiviral delivery in primary mouse islets and performed RNA-seq (Fig. 7B). Quantitative RT-PCR analysis confirmed a significant increase of Srf mRNA (Fig. S7A). To determine whether proliferation genes are overrepresented among the genes regulated by Srf, we employed GSEA. Confirming a role for Srf in maintaining high expression of proliferative genes, proliferation-associated genes were expressed at significantly higher levels after Srf overexpression (Fig. 7C). Furthermore, mRNAs decreasing in expression during the pseudotemporal trajectory were enriched in Srf-overexpressing islets (Fig. 7C). Among the genes significantly induced by Srf were Fos, Junb and Egr1 (Fig. 7D; Table S7), demonstrating that Srf acts as an upstream regulator of these TFs also in islets. The induction of proliferation genes did not impair insulin secretion, as shown in GSIS assays of Srf overexpressing islets (Fig. S7B). These experiments identify Srf as a regulator of maturation-associated genes in beta-cells and suggest that declining Srf levels postnatally could contribute to decreasing beta-cell proliferation.
DISCUSSION
Here we have used single-cell transcriptomics to obtain a comprehensive view of transcriptional changes associated with mammalian postnatal beta-cell development. By quantifying gene expression in single beta-cells from different time points, we ordered cells along a continuous linear molecular trajectory to resolve the cellular heterogeneity of beta-cells. This analysis revealed hitherto unknown transcriptional dynamics associated with the declining proliferative capacity of beta-cells during postnatal development. Features we found to be associated with immature, proliferative beta-cells are high expression of regulators of amino acid metabolism and high ROS production as well as a network of nutrient-responsive TFs (Fig. 7E).
The analysis of single-cell RNA-seq data poses unique computational challenges that necessitate adaptation of existing workflows and development of new analytical strategies. Here we show that 1D-PCA-based ordering can be applied to single-cell data to accurately predict temporal dynamics of in vivo biology, as indicated by validation of known markers and higher accuracy in reconstructing the collection time of samples than achieved by other methods, including the unsupervised methods Monocle (Trapnell et al., 2014), TSCAN (Ji and Ji, 2016), and Embeddr (Campbell et al., 2015), and the supervised method DeLorean (Reid and Wernisch, 2016). PCA-based pseudotemporal ordering provides an intuitive representation with a single path traversing all cells and unlike minimum spanning tree-based methods, allows for the addition of new samples to the scale without changing the established path. Employing this feature, we show that previously published single beta-cell RNA-seq data from 3-month-old mice correctly projected onto the constructed pseudotemporal trajectory (see Suppl. Information). Projection of these external data onto our trajectory demonstrated that single beta-cells from 3-month-old mice exhibit a degree of heterogeneity comparable to very young mice. Our analysis implies that 1D-PCA-based ordering is well-suited for examining the trajectory of a single cell type along a continuous course of cell state changes.
Recently, markers unique to subpopulations of beta-cells have been identified and molecular characterization of these subpopulations has revealed gene expression differences (Bader et al., 2016; Dorrell et al., 2016). Consistent with these studies, we observed significant molecular heterogeneity among beta-cells at each analyzed time point. Dor and colleagues sorted replicating beta-cells based on a marker for S/G2/M phase and compared their transcriptome to beta-cells in G0/G1 (Klochendler et al., 2016). Some features enriched in replicating beta-cells, including high expression of components of the mitochondrial respiratory chain (Klochendler et al., 2016), were similar to the features of immature beta-cells identified by our study. However, there were also differences. For example, the TF genes Pdx1 and Nkx6.1 were expressed at lower levels in replicating beta-cells but not regulated in our trajectory of beta-cell maturation. The differences are not surprising because our cell ordering method does not group cells solely based on cell cycle characteristics, but associates a given cell with multiple, potentially independent aspects of transcriptional heterogeneity.
To place our data into the context of findings from recently published single-cell data of beta-cells from juvenile and adult humans (Wang et al., 2016) as well as young and aged mice (Xin et al., 2016), we compared gene signatures identified in these studies to genes regulated in our pseudotemporal trajectory. Wang et al. reported higher expression of alpha-cell lineage markers in juvenile compared to adult human beta-cells. We determined whether these alpha-cell signature genes decrease in expression during the pseudotemporal trajectory, but found no correlation (P = 0.07). This could indicate differences between rodent and human beta-cells, but could also be due to the specific time window covered in our study. We found that 34 genes with significantly lower expression in beta-cells from very old mice compared to 3-month-old mice (Xin et al., 2016) also decreased in expression in our maturation time course. Among these shared genes were the TFs Srf, Jun, Fos, Nr4a1, Fosl2, and Fosb, suggesting that expression of these TFs continues to decrease as beta-cells age. Previous studies have shown that Fos overexpression in islets stimulates beta-cell proliferation (Ray et al., 2016). Given our finding that Srf induces Fos and proliferation genes, decreasing Srf/Fos levels could be an important contributor to declining beta-cell proliferation rates early postnatally and during aging.
Two novel features revealed by the pseudotemporal ordering of beta-cells were declining expression of amino acid transporters and metabolizing enzymes as well as mitochondrial respiratory chain components during maturation. Our finding that mitochondrial gene expression decreases during beta-cell maturation appears, at first, to contradict a recent study reporting mRNA increases of oxidative phosphorylation and respiratory chain components when bulk islet samples of two- and six-week-old mice were compared (Yoshihara et al., 2016). The discrepancy could be explained by the different time window studied by Yoshihara and colleagues. Alternatively, it could be due to the increased ability to detect signatures of rare immature, proliferative beta-cells due to single-cell ordering in our study. The latter view is supported by the lack in regulation of cell cycle regulators and mitochondrial genes when we considered collection time averages of transcript levels (Fig. 2D; S2B). Thus, we propose that the here-identified subpopulation of beta-cells with high mitochondrial membrane potential represents a rare immature population with high proliferative capacity, whereas beta-cells that upregulate oxidative phosphorylation genes later postnatally represent cells with a mature insulin secretory response (Yoshihara et al., 2016).
We identified ROS as an important driver of early postnatal beta-cell proliferation and establishment of beta-cell mass. ROS has multiple roles in beta-cells. Discrete and transient increases in beta-cell ROS provide an important metabolic signal for GSIS (Supale et al., 2012). It is possible that decreasing levels ROS eliminating enzymes during maturation help enable the characteristic stimulus secretion-coupling of mature beta-cells. While transient increases in ROS provide an important signal for insulin secretion, oxidative stress caused by direct exposure to oxidants or glucotoxicity impairs beta-cell function (Supale et al., 2012). Obesity and insulin resistance are often associated with elevated plasma glucose levels, which through increased beta-cell mitochondrial metabolism could stimulate ROS production. In the insulin-resistant state, beta-cell proliferation is increased to help the organism adapt to higher insulin demand. An interesting question for future investigation is whether ROS drives beta-cell proliferation during metabolic adaptation.
Atf4, C/EBP and Ddit3 (Chop), which are downregulated during the maturation time course (Table S5A), are also downstream effectors of the ER stress pathway. Mild ER stress has been shown to promote beta-cell proliferation (Sharma et al., 2015), an observation that is consistent with the pro-proliferative gene regulatory network identified in this study. It has also been shown that reducing insulin expression promotes beta-cell proliferation (Szabat et al., 2016), suggesting that the observed increase in insulin expression during maturation could further contribute to the proliferative decline of beta-cells. However, different from Szabat et al. who observed reduced ER stress with low insulin expression, low insulin levels in our data were associated with high expression of ER stress markers. How ER stress-related signals are integrated in beta-cells to control proliferative responses clearly warrants further studies. The here-reported RNA-seq data sets provide a resource for further exploration of molecular signatures that define different beta-cell states. The ability to examine the connectivity between genes in unique beta-cell states will facilitate the discovery of targets for therapeutic intervention in diabetes.
STAR★Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Maike Sander M.D. (masander@ucsd.edu).
Experimental Model and Subject Details
Cell lines
HEK293T cell were maintained in medium A (DMEM containing 100 units/mL penicillin and 100 mg/mL streptomycin sulfate) supplemented with 10% fetal bovine serum (FBS).
Animals
Male and female mIns1-H2B-mCherry mice were used to sort beta-cells at P1, P7, P14, P21, and P28 (P1, n=15 mice; P7, n=14; P14, n=10; P21, n=4; P28, n=4). Male and female C57BL/6 mice at P1 and P28 were used to perform glutamine uptake experiment and mitochondrial function-related experiments. Male and female C57BL/6 mice at 4-6 weeks were used to perform amino acid supplementation experiment and lentiviral transduction experiment. C57BL/6.mCAT mice are kindly provided by Dr. Peter Rabinovitch. Beta-cell-specific mCAT overexpression was generated by crossing C57BL/6.mCAT mice with RIP-Cre mice and R26YFP mice. Studies were conducted in animals 6 weeks of age and included age- and sex-matched littermate control mice, which are RIP-Cre mice. To label proliferating beta-cells, 0.8mg/ml BrdU was supplied in the drinking water to 5-week-old mice for 7 days. All animal experiments were approved by the Institutional Animal Care and Use Committees of the University of California, San Diego. The numbers of animals studied per genotype are indicated within each experiment.
Mouse islet culture
Mouse islets were cultured in RPMI 1640 medium containing 10% FBS, 8.3 mM glucose, 2 mM glutamine, and 1% penicillin-streptomycin.
Method Details
Islet isolation and FACS sorting
Pancreata of mIns1-H2B-mCherry reporter mice at P1, P7, and P14 were dissected wholly without perfusion and digested with 1mg/ml Collagenase Type IV (Sigma). P21 and P28 pancreata were perfused through the common bile duct with 125 μg/ml Liberase TL (Roche). Islets were purified by density gradient centrifugation using Histopaque (Sigma), dissociated with Accumax (Life Technologies) and sorted by FACS on a FACSAria II (BD Biosciences). After excluding dead or damaged beta-cells and doublets, cells expressing mCherry were sorted (49.3%, 40.5%, 39.5%, 41.4%, and 40% of pre-gated cells were captured based on mCherry at P1, P7, P14, P21, and P28, respectively).
Single-cell and bulk RNA-seq library preparation
Single sorted beta-cells were captured on medium-sized (10–17 μm cell diameter) microfluidic RNAseq chips (Fluidigm) using the FluidigmC1 system according to the Fluidigm protocol (PN 100-5950). For each C1 experiment, two bulk RNA controls (approximately 250 cells/sample) and a no-cell negative control were processed in parallel PCR tubes, using the same reagent mixes as used on chip. Multiplexed libraries were prepared using the Nextera XT DNA sample preparation kit (Illumina), and sequenced across 10 lanes of a HiSeq 2500 (Illumina) using 50-bp single-end sequencing.
RNA-seq data processing of single cell and bulk libraries
Single-end 50-bp reads were mapped to the UCSC mouse transcriptome (mm9) by STAR allowing for up to 10 mismatches (which is the default by STAR). Only the reads aligned uniquely to one genomic location were retained for subsequent analysis. Reads per kilobase of transcript per million fragments mapped (RPKM) expression levels of all genes were estimated by Cufflink using only the reads with exact matches. Libraries that contained fewer than 1 million reads or for which more than 15% of fragments mapped to mitochondrial reads were excluded. Single-cell samples with full values for number and fraction of aligned reads are provided in Table S1. Downstream analysis of RPKM values from both bulk and single cells RNA-Seq data sets was performed with custom scripts developed using the programming languages Python and R. Several software libraries from Orange and Bioconductor were adopted for data pre-processing, cell ordering and gene sets analysis. First, a moderated log-transformation was applied to both bulk and single-cell data sets. Specifically, the function log(dij + 1) was applied to the expression values, dij representing the RPKM estimate of the i-th gene in the j-th sample.
To remove unwanted variation, single-cell data was normalized with SVA-seq (Leek, 2014) using a set of “negative control” genes with low variation in the bulk data. Briefly, the top 5th percentile of expressed genes ranked by increasing values of Median Absolute Deviation (MAD) with high expression levels (average log-transformed RPKM > 5) were first selected as negative controls from the bulk data. The list was further filtered for genes with high expression levels in the single-cell data set (log-transformed RPKM > 5 in at least 2 cells). Genes used as negative controls, including known housekeeping genes, are listed in Table S2B.
The biological model was defined as the global average change in gene expression, with no explicit information on the stage of each cell provided to SVA-seq. The data corrected for unwanted variations was filtered by selecting the top quartile most variant genes (n=4313), ranked according to MAD, whose expression was considered for the inference of cell ordering models.
Saturation analysis
To identify the required sequencing depth, we subsampled raw data from bulk cell and individual single-cell libraries. To generate a single-cell ensemble data set, raw reads from all the single-cell RNA-seq libraries were bioinformatically pooled to mimic a bulk RNA-seq experiment. From these three data sets, saturation plots were generated by calculating the number of detected genes (RPKM > 0) as the number of reads sampled increased.
Cell ordering
To infer an ordered trajectory of single cells, we adapted a 1D-PCA-based unsupervised algorithm originally designed to build a differentiation scale, representing transcriptomic progression of bulk samples, as previously described (Mulas et al., 2012) and implemented in the Orange software. Briefly, we applied PCA to the pre-processed data matrix D= (dj,i) with dj,i representing the expression value of the i-th selected gene in the j-th cell. A real number p(dj ) was assigned to each cell j by projecting its expression profile to the first principal component:
| (1) |
wi being the elements of the first eigenvector of the covariance matrix DTD and i ranging from 1 to the total number of selected genes (m=4313). Projections p(dj ), j ranging from 1 to the total number of cells considered (n=387), were set as pseudotime coordinates and used to determine the order of cells, so that:
| (2) |
Ordering evaluation and comparison with other methods
The 1D pseudotime trajectory was compared with orderings obtained by applying other methods, using the same set of selected genes and all parameters set to default values, unless specified. To evaluate the accuracy of unsupervised algorithms, which do not use the sample collection time point to infer ordering, Pseudotemporal Ordering Score (POS) was used to count the number of cells ordered as expected from their true data collection time:
| (3) |
where Ti and Tj are sets of cells from time point i and j, p(x) and p(y) represents the pseudotime coordinates assigned to samples x and y, and δ equals to 0 or to (j – i)/D, if Ti = Tj or if Ti ≠ Tj, respectively. The constant D is computed to rescale POS values in the range [-1, 1].
To compare the 1D Pseudotime coordinates with orderings inferred with supervised methods, which use sample collection time to infer pseudotime coordinates, we relied on the “Roughness” of consecutively-placed cells (Reid and Wernisch, 2016). The Roughness score R was computed as a sum of distances of gene expression values between consecutive cells, from the beginning to the end of the trajectory, as ordered according to their pseudotime coordinates. Distance between a pair of consecutive cells j and j+1 was defined as the difference of their gene expression measurements dj+1 and dj:
| (4) |
dj,i being the expression value of the i-th selected gene in the j-th cell, as described above.
Ordering evaluation and comparison with other methods
The 1D pseudotemporal trajectory was compared with four additional methods, including unsupervised algorithms, namely TSCAN (Ji and Ji, 2016), Monocle (Trapnell et al., 2014), and Embeddr (Campbell et al., 2015), and a supervised method based on Gaussian processes that was recently added to the “DeLorean” R package, hereafter named as DeLorean (Reid and Wernisch, 2016).
The results of the pseudotemporal ordering score (POS) calculations used to evaluate the accuracy of unsupervised algorithms are reported below.
A confidence interval of the POS score of the PCA-based ordering was estimated with a bootstrap procedure, whereby random sets of cells were sampled with replacement by maintaining the same proportion of cells from the different stages. These samples were used as a training set, i.e. to determine weights wi, which were used to project the remaining samples, considered as a test set. A bootstrap sample contained about 63% of the cells in the original samples, whereas a test set was on average composed of the 37% of the cells. The procedure was iterated for 1000 times, obtaining an estimate of the POS statistic on the test sets. The 90% confidence interval is shown below and indicates a robust performance of the 1D-PCA as a pseudotime ordering method on our data set.
Evaluation of unsupervised methods on our single-cell data.
POS score obtained using TSCAN, Monocle and Embeddr. POS score obtained on the 1D Pseudotime Scale based on PCA and its corresponding 90% confidence interval [0.6015 - 0.7028].
| POS | |
|---|---|
| TSCAN | 0.17 |
| Monocle | 0.41 |
| Embeddr | 0.59 |
| 1D Pseudotime Scale | 0.65 |
As the sample collection time point is used by supervised methods to infer pseudotime coordinates, this information could not be used to evaluate the model generated by DeLorean with POS. For this reason, we evaluated the method by measuring the “Roughness” of gene expression values of consecutively-placed cells in the inferred trajectory. Using the top 20 genes selected with ANOVA, we obtained a Roughness value of 72.9 for DeLorean, while PCA-based ordering scored 72.5, indicating a slightly smoother transition of gene expression through the cells as projected with PCA. The same conclusion was obtained by applying different distance measures in place of Roughness to measure the total trajectory distance, namely Euclidean distance, cosine and correlation-based distances (data not shown). A null distribution was obtained computing the R score on sets of randomly ordered cells. After 1000 iterations, R scores obtained with DeLorean and 1D Pseudotime Scale were compared to the null distribution and p-values estimated as cumulative probabilities for each predicted path. Both 1D Pseudotime Scale and DeLorean orderings indicated a significant accuracy in reconstructing a smooth transition of transcriptomic values (P < 0.001 for both the approaches), with the PCA score being placed as more extreme compared to the left tail of the null distribution (see figure below). A measure of the similarity of the two orderings, as implemented in the TSCAN package and described in (Ji and Ji, 2016), indicated a similar placement of cells obtained with the two methods (similarity = 0.72).
Null distribution of Roughness obtained with random ordering.

Red and green arrows depict the Roughness values obtained by the 1D Pseudotime Scale and DeLorean, respectively.
Assessment of branching trajectories
The Wishbone method (Setty et al., 2016) was applied to explore bifurcating developmental trajectories, with the first three non-trivial diffusion components used to define a branched trajectory. As a starting point of the trajectory is required by the tool, we selected the first cell as projected by 1D-PCA. As shown with the dimensionality reduction method tSNE (t-distributed stochastic neighbor embedding), a branch was observed using Wishbone, with a limited number of cells deviating from the main trajectory. Similar results were obtained by choosing the cell with the lowest value of Insulin (Ins2) as a starting point, assuming that insulin levels increase during postnatal maturation, as well as by using different diffusion components (data not shown). By analyzing patterns of interest, we confirmed an increase in Ins2 and a decreasing pattern in Atf3 and Srf expression. As shown in the figure below, both branches identified by Wishbone showed these patterns, with slightly different dynamics in the two.
Cell association to different branches detected by Wishbone.
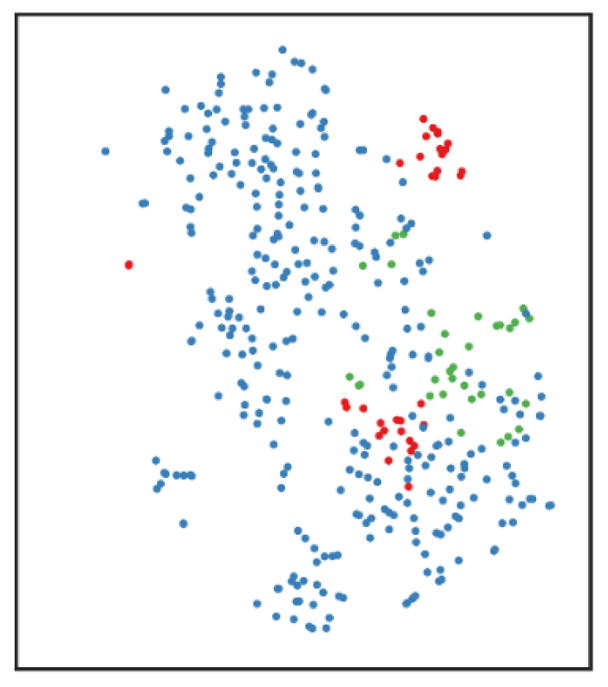
tSNE map: cells belonging to the main trajectory are depicted in blue, cells deviating into two different branches are shown in green and red.
Expression values of Ins2, Atf3 and Srf scaled in a (0-1) range and displayed across the cell trajectory identified by Wishbone.

Following a bifurcation point, two cell trajectories belonging to different branches are depicted with dotted and dashed lines.
Comparison to external data sets
Bioinformatic comparison with published gene signatures from mouse and human studies was performed using Gene Set Enrichment Analysis (GSEA). A signature of alpha-cell signature genes found to be highly expressed in beta-cells from juvenile human donors (Wang et al., 2016) was analyzed for correlation with pseudotime coordinates, using GSEA as described in detail in the section “Pseudotime analysis of gene sets”. The same approach was used for a signature of differentially expressed genes between beta-cells from 3-month-old and 26-month-old mice (Xin et al., 2016). Data from Xin et al. (Xin et al., 2016), including single-cell samples from 3-month-old mice (P90), was normalized with SVA-seq to allow for comparison of samples from different laboratories, with the same set of negative control genes used for our single-cell data. The PCA model described in Equation 1 was used to project expression values of genes selected to infer the model, corresponding to the top quartile most variant genes in our single-cell data set (n=4313). The 1D Pseudotime Scale including projections of these additional samples is shown below. A quantitative measure of the heterogeneity of single cells was computed as the range R spanned of pseudotime coordinates for both P28 (R=90.1) and P90 (R=91.5).
Projection of external data on the Pseudotime Scale.

Cells from 3-month-old mice (P90) and their corresponding median projection are depicted in pink.
Analysis of time-ordered and pseudo-binned time expression profiles
Genes with log-transformed RPKM > 1 in at least 2 cells, used in all subsequent analysis, were considered to construct time-ordered and pseudobinned-time expression profiles. The average of samples from each of the five time points was considered for each gene to obtain time-ordered profiles. Pseudotime profiles were constructed by assigning pseudotime-ordered cells to five bins, each of them with size equal to the number of cells collected at the corresponding time point.
Pseudotime analysis of gene sets
Hallmarks and curated gene sets from KEGG, REACTOME and BioCarta part of the MSigDB compendium (http://software.broadinstitute.org/gsea/msigdb/index.jsp) were used as annotated gene sets. Genes with log-transformed RPKM > 1 in at least 2 cells were clustered (Hierarchical clustering based on absolute values of Pearson correlation, Ward method) to obtain de novo gene sets. The Dynamic Tree Cutting method (R package ‘cutreeDynamic’ with minimum cluster size = 10, method = “hybrid”, deepSplit = 4) was used to obtain clusters. The GSEA tool (http://www.broad.mit.edu/gsea) for continuous phenotypes was used to identify annotated and de novo genes with expression profiles correlated with the pseudotime trajectory. GSEA was run with the vector of pseudotime coordinates p = p(d1), … , p(d387) set as the “continuous phenotype” and significant gene sets with coordinated increasing or decreasing activity were selected with corrected P value < 0.25.
Pseudotime analysis of individual genes
Genes with log-transformed RPKM > 1 in at least 2 cells were ranked based on their correlation with pseudotime coordinates. Significance of correlation values was assessed on a set of 1000 randomly permuted gene expression profiles.
Identification of enhancer regions and motif analysis
To identify enhancer regions, we used previously published H3K27ac ChIP-seq datasets (GSM1677162 and GSM1677164). ChIP-seq peak identification, quality control, and motif analysis were performed using HOMER. Briefly, genome enriched regions of H3K27ac were identified using the ‘findPeaks’ command in HOMER with settings of ‘–style histone’: 500 bp peaks with 3-fold enrichment and 0.01 FDR significance over local tags. To identify active enhancers of target genes, enhancer sites defined by ChIP-seq enrichment of H3K27ac were filtered by the following criteria: (1) regions were at least 3 kb away from annotated TSSs; (2) regions were within 200 kb from annotated target gene TSSs. For motif analysis, transcription factor motif finding was performed on ± 200 bp relative to the peak center defined by ChIP-seq analysis using HOMER. Peak sequences were compared to random genomic fragments of the same size and G/C content was normalized to identify motifs enriched in the ChIP-seq targeted sequence.
Gene correlation analysis
To identify co-variation of proliferation-related genes with other selected genes, we measured Pearson correlation for each pair of pseudotime-ordered gene expression profiles in the selected categories. Proliferation-related genes retrieved from previous annotation (Buettner et al., 2015) were ranked by correlation with pseudotime coordinates and the 30 most down-regulated were compared with genes involved in amino acid metabolism of interest, identified from the pseudotime analysis of individual genes. Similarly, the top 30 regulated among transcription factors (AnimalTFDB database, http://www.bioguo.org/AnimalTFDB/) and oxidative phosphorylation-related genes (Gene Ontology database, http://amigo.geneontology.org/amigo/term/GO:0006119) were compared to proliferation genes. For each comparison, statistical significance of the global correlation of each category with proliferation genes was assessed by referring the average correlation of genes in the two groups to a null distribution obtained with random sampling, as described in the Statistics section.
Network analysis
Links between the top 90th percentile of pseudotime-regulated genes (n = 1389) were retrieved through the STRING repository (version 10.0, http://string-db.org/). Only the most reliable protein associations were retained (combined confidence score > 0.7) and used to assign weights to each network link. Genes with annotation in the STRING database (n=1356) were prioritized using an interest propagation algorithm described previously (Mulas et al., 2013). Briefly, given a set of nodes of interest Gint, the method assigns ‘propagation scores’ to all the other nodes, with values proportional to their connectivity to Gint in the network. Transcription factors selected from the AnimalTFDB database (http://www.bioguo.org/AnimalTFDB/) and oxidative phosphorylation-related genes from the GO database (http://amigo.geneontology.org/amigo/term/GO:0006119) were used separately as initial nodes of interest and the top 90th percentile of the distribution of propagation scores obtained was used as a threshold to select the most relevant genes. Proliferation-related genes included in the network were retrieved from previous annotation (Buettner et al., 2015) and genes involved in mRNA processing were identified from their correspondent GO category (http://amigo.geneontology.org/amigo/term/GO:0006397).
Serum amino acid detection
Serum glutamine concentrations from P1 and P28 mice (n=4) were measured using a YSI 2950 enzymatic analyzer. To measure other amino acids, 5 μl serum was mixed by vortexing first with 200 μl methanol (50% v/v in water with 20 μM L-norvaline as internal standard) and second with 100 μl of chloroform before centrifugation for 10 min at 13,000 rpm. The upper (polar) phase was dried, derivatized, and analyzed. Amino acids in samples were quantified against varied amounts of standards run in parallel using MetaQuant.
Glutamine uptake measurements
Overnight recovered P1 and P28 islets were washed in PBS, and then plated (50 islets/time point in triplicate) into 96 well plates in 100 μl culture medium containing 2mM glutamine. After 24 hrs, supernatant and islets were collected separately. Wells without cells containing only media served as controls. Supernatants were centrifuged (10000 rpm for 10min, 4°C) and then stored at -80°C until analysis. Islets were lysed in RIPA buffer and protein concentrations for each well containing supernatant were measured. Glutamine concentrations in supernatants were measured using a YSI 2950 enzymatic analyzer. Glutamine uptake rates were calculated by subtracting experimental glutamine concentrations from control sample glutamine concentrations and expressed as pmol of glutamine per hour per microgram of cell protein. Three independent experiments were performed.
Measurement of beta-cell proliferation with amino acid or nucleotide supplementation
Isolated islets from 4 to 6-week-old C57BL6 mice were cultured overnight, and then supplied with fresh medium supplemented with 1 mM of proline, serine, lysine, tyrosine (Sigma), glutamine for a total of 3 mM (Life Technologies), or nucleotides (1X GS Media Supplement, Millipore). Islets were then cultured for an additional 48 hours with the thymidine analogue EdU, which was added to the medium for the last 24 hours. Cell proliferation was detected with Click-iT EdU Alexa Fluor 488 (Life Technologies) and rabbit Alexa-Fluor-647-conjugated insulin mAb (Cell Signaling Technology) using BD FACSCanto II, and analyzed by Flowjo 8.7. Three independent experiments were performed.
Mitochondrial membrane potential and mitochondrial ROS detection by FACS analysis
P1 and P28 islets were allowed to recover overnight, dissociated with trypsin-EDTA treatment for 5 min at 37 °C, and washed twice with Krebs solution containing 4 mM glucose. For detection of mitochondrial membrane potential, dissociated islet cells were incubated with 10 nM of the fluorescent probe TMRM (Life Technologies) and 200 nM MitoTracker Green (Life Technologies) for 1 h at 37°C in Krebs solution containing 4 mM glucose with or without 50 μM FCCP (Sigma). For detection of mitochondrial ROS, dissociated islet cells were incubated with 5 μM MitosoxRed (Life Technologies) and 200 nM MitoTracker Green (Life Technologies) for 1 hour at 37 °C in Krebs solution containing 4 mM glucose. Cells were washed with PBS once, scored by FACS using BD FACSCanto II, and analyzed by Flowjo 8.7. TMRM and MitosoxRed levels were normalized to MitoTracker Green. Three independent experiments were performed.
ΔΨm analysis
P1 and P28 islets were dispersed with accutase (Life Technology A1110501) for 10 min, plated on Greiner Cellview glass bottom 10 mm 4-compartment confocal dishes, and cultured overnight for recovery. The following day, dispersed cells were stained for 45 min with 15 nM TMRE (Life Technologies) and 200 nM MitoTracker Green in Krebs buffer under 4 mM glucose concentration after which they were washed twice with buffer containing 15 nM TMRE and 4 mM glucose. The cells were imaged on a Zeiss LSM880 confocal microscope. Then glucose was supplemented for a total of 16 mM glucose and cells were imaged at 30 min. Subsequently, 50 μM FCCP was added and cells were imaged at 10 min. The resulting images were quantified for fluorescence intensity in the red and green channels (TMRE and MitoTracker Green, respectively). Live cells were defined as having an at least 10% increase in the TMRE/MitoTracker Green ratio in 4 mM glucose compared with FCCP treatment. Relative change in ΔΨm was calculated by the fold change of TMRE/MitoTracker Green ratio in 16 mM glucose over 4 mM glucose.
Mitochondrial DNA quantification
To measure mitochondrial DNA copy number, total DNA from mCherry-sorted beta-cells from P1 and P28 mice was isolated using DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer’s instructions. Mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) content were determined by real-time PCR using specific primers for the mitochondrial cytochrome c oxidase subunit II (Cox2) gene and the nuclear gene Rsp18. The ratio of mtDNA to nDNA content was calculated for each time point. Experiments were performed four times.
Immunohistochemistry, beta-cell mass measurements, and TUNEL assay
Mouse pancreata were analyzed by immunostaining using the following primary antibodies: guinea pig anti-insulin (Dako), 1:1000; rabbit anti-Ki67 (Thermo Fisher Scientific), 1:200; rat anti-BrdU (Novus Biologicals), 1:250; goat-anti-GFP (Abcam), 1:1000; rabbit-anti-MafA (Bethyl Labs), 1:1000; rabbit-anti-Pdx1 (Abcam), 1:500; rabbit-anti-Nkx6.1 (LifeSpan BioSciences), 1:250; mouse-anti-glucagon (Sigma), 1:100; mouse-anti-somatostatin (BCBC), 1:2000. Primary antibodies were detected with donkey-raised secondary antibodies conjugated to Cy3 or Cy5, (Jackson ImmunoResearch), and nuclei were counterstained with DAPI (Sigma) at 0.1 μg/ml. Images were captured on a Zeiss Axio Observer Z1 microscope with an ApoTome module and processed with Zeiss AxioVision 4.8 software. For beta-cell mass measurements, images covering an entire pancreas section were tiled using a Zeiss Axio Oberver Z1 microscope with the Zeiss ApoTome module. The insulin+ and total pancreas areas were measured using ImageJ and beta-cell mass was calculated as follows: Insulin+ area/total pancreatic area. For examination of apoptosis, TUNEL analysis was performed using ApopTag Red In Situ Apoptosis Kit as specified by the manufacturer (Thermo Fisher Scientific).
GSIS assays
Islets were allowed to recover overnight, washed and pre-incubated for 1 hour in Krebs solution containing 2.8 mM glucose. Afterwards, groups of 10 islets were transferred to a 96 well dish into solutions of 2.8 mM glucose or 16.8 mM glucose. After incubation for 1 hour, supernatant was collected and islets were lysed overnight in a 2% acid:80% ethanol solution. Insulin was then measured in supernatants and lysates using a mouse insulin ELISA kit (ALPCO). Secreted insulin was calculated as percentage of total insulin content.
Glucose tolerance tests
Mice were fasted for 6 hours after the onset of the light phase. Basal blood glucose was sampled at 0 min, and glucose administered by intraperitoneal injection at a dose of 1.5mg/kg of 10% glucose. Blood samples were taken at 20, 40, 60, 90, and 120 min after glucose administration.
Lentivirus production and transduction
GFP-tagged lentiviral plasmid (Origene PS100071) or GFP-tagged Srf lentiviral plasmid (Origene MR208120L2) was transfected with pCMV-R8.74 (Addgene 22036) and pMD2.G expression plasmid into HEK293T cells. Transfection was performed using PEIsolution (1 mg/ml) and lentiviral supernatants were collected at 48 hours and 72 hours after transfection. The lentivirus was further concentrated by ultracentrifugation at 4 °C. The titer ranged from 5x108 to 1x109 TU/ml.
Lentiviral transduction was carried out as follows: after isolation, islets were cultured overnight and treated with accutase for 10 min. 5x103 dispersed cells were seeded per well in a 96 v-bottom plate (Fisher Scientific, 12565481) and transduced with lentivirus at MOI 5-6 in the presence of 0.8 ng/ml polybrene. Single cells were re-aggregated by centrifugation at 365 g for 5 min, and medium was changed after overnight culture.
RNA-seq analysis of lentiviral transduced islets
Cells were collected 72 hours after transduction and RNA was extracted using RNeasy Micro Kit (Qiagen). Three biological replicates of RNA-seq libraries were generated with SMART-Seq® v4 Ultra® Low Input RNA Kit for Sequencing (Clontech) and Illumina Nextera XT DNA sample preparation kit (Illumina), multiplexed and sequenced on the HiSeq 4000 system (Illumina) using 50 bp single-end sequencing. On average, 25 million reads were generated from each library. Reads were mapped as described above by Cufflink. Differential gene expression in Srf-overexpressing and control samples was assessed by Cuffdiff. Gene sets for GSEA were defined as: i) genes up- or down-regulated along pseudotime (P < 0.05); ii) proliferation-related genes regulated during pseudotime (P < 0.05).
Quantitative PCR analysis
DNA or cDNA from islets were mixed with SYBR GreenERTM qPCR Supermix Universal (Thermo Fisher Scientific) according to manufacturer’s protocol. Reactions were performed in a 96-well format using Biorad PCR system. Relative mRNA levels were calculated using the comparative CT method and normalized to Calm1 mRNA. A complete list of primers and sequences can be found below.
| Primer | Name Sequence |
|---|---|
| ms-Calm1-F | GCTGCAGGATATGATCAACG |
| ms-Calm1-R | GCTGCAGGATATGATCAACG |
| ms-Srf-F | CTGACAGCAGTGGGGAAAC |
| ms-Srf-R | GCTGGGTGCTGTCTGGAT |
| ms-Cox2-F | ATAACCGAGTCGTTCTGCCAAT |
| ms-Cox2-R | TTTCAGAGCATTGGCCATAGAA |
| ms-Rsp18-F | TGTGTTAGGGGACTGGTGGACA |
| ms-Rsp18-R | CATCACCCACTTACCCCCAAAA |
| ms-Pim2-F | GAGGCCGAATACCGACTTG |
| ms-Pim2-R | CCGGGAGATTACTTTGATGG |
| ms-Pim3-F | ACATGGTGTGTGGGGACAT |
| ms-Pim3-R | ATAAGCTGCTGGCACTCTGG |
| ms-Sik1-F | GACGGAGAGCGTCTGATACC |
| ms-Sik1-R | GGTCCTCGCATTTTTCCTC |
| ms-Plk2-F | TGAAGGTGGGAGACTTTGGT |
| ms-Plk2-R | TGGGGTTCCACATATTGTTCT |
| ms-Apitd1-F | CCGCAGGAGTTCTCTCACC |
| ms-Apitd1-R | GAGACAGCCGACCGTGTAGT |
| hu-Catalase-F | TCATCAGGGATCCCATATTGTT |
| hu-Catalase-R | CCTTCAGATGTGTCTGAGGATTT |
Quantification and Statistical Analysis
Quantification
For beta-cell mass measurements, four to six sections, at least 100 μm apart, from each pancreas were tiled using a Zeiss Axio Oberver Z1 microscope with the Zeiss ApoTome module. The insulin+ and total pancreas areas were measured using ImageJ and beta-cell mass was calculated as follows: Insulin+area/total pancreatic area. For all quantifications of proliferation, apoptosis and markers, at least 500 beta-cells per mouse were examined.
Statistical Analysis
Experimental comparisons
All experiments were independently repeated at least three times. Results are shown as means ± SEM. Statistical analyses were conducted using Prism 5 software (GraphPad Software). Statistical comparisons between groups were analyzed for significance by an unpaired two-tailed Student’s t test or paired two-tailed Student’s t test. Glucose tolerance testing significance was determined by one-way ANOVA. Differences are considered significant at p < 0.05. The exact values of n, statistical measures (mean ± SEM) and statistical significance are reported in the figures and in the figure legends.
GSEA significance
Significance for GSEA results was assessed with 1000 permutations and FDR was used to correct for multiple testing. The exact thresholds used for FDR-based selection are specified in the Results.
Permutation-based significance
Random sampling and bootstrap approaches were used to obtain null distributions and confidence intervals, respectively, with the number of iteration set to 1000. Null distributions of different scores, including Roughness, fold change of consecutive time (and pseudo-) time points and gene correlations with pseudotime, were obtained by computing the scores on randomly ordered samples. Null distributions for average correlations of gene groups were obtained by randomly sampling sets of genes from the data, with sizes equal to the number of genes in each group under study. For each score tested, a P value was estimated as a cumulative probability from the corresponding null distribution. The confidence interval of the POS score for the PCA-based ordering was estimated with a bootstrap procedure, whereby random sets of cells were sampled with replacement by maintaining the same proportion of cells from the different stages.
Significance of proportions
Significance of overlaps between lists of genes resulting from the network propagation analyses with proliferation- or mRNA processing-related genes was assessed through a one-tailed Fisher Exact test, as implemented in the Python library scipy.stats.
Data and Software Availability
Data
The accession number for the RNA-seq data reported in this manuscript is NCBI GEO: GSE86479. The accession number for the H3K27ac ChIP-seq and input datasets is GSM1677162 and GSM1677164, respectively, and available in NCBI GEO: GSE68618
Software
Custom R and Python scripts are provided as Data S1.
Supplementary Material
Key Resources Tables.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea pig anti-Insulin | DAKO | A0564 RRID:AB_10013624 |
| Rabbit anti-Ki67 | Lab Vision Corp | RM-9106-S0 RRID:AB_149919 |
| Rat anti-BrdU | Novus Biologicals | NB500-169 RRID:AB_10002608 |
| Goat-anti-GFP | Abcam | ab13970 RRID:AB_300798 |
| Rabbit-anti-MafA | Bethyl Labs | A300-611A RRID:AB_2297116 |
| Rabbit-anti-Pdx1 | Abcam | AB47267 RRID:AB_777179 |
| Rabbit-anti-Nkx6.1 | LifeSpan BioSciences | LS-C143534 RRID:AB_10947571 |
| Mouse-anti-Glucagon | Sigma | G2654 AB_259852 |
| Mouse-anti-Somatostatin | BCBC | AB1985 RRID:AB_10014609 |
| Alexa-Fluor-647-conjugated insulin mAb | Cell Signaling Technology | 9008s |
| Chemical Reagents, Peptides, and Recombinant Proteins | ||
| Collagenase Type IV | Sigma | 639207 |
| Liberase TL | Roche | 05401020001 |
| Histopaque | Sigma | 10771 |
| Accumax | Life Technologies | AM105 |
| Accutase | Life Technologies | A1110501 |
| SYTOX® Blue Dead Cell Stain | Life Technologies | S34857 |
| TMRM | Life Technologies | T668 |
| MitoTracker Green | Life Technologies | M7514 |
| MitosoxRed | Life Technologies | M36008 |
| L-Proline | Sigma | P5607 |
| L-Serine | Sigma | S4311 |
| L-Lysine | Sigma | L5501 |
| L-Tysorine | Sigma | T8566 |
| FCCP | Thermo Fisher Scientific | NC0904863 |
| GS System GS Media Supplement | Millipore | GSS-1016-C |
| Critical Commercial Assays | ||
| C1 Single-Cell Auto Prep Reagent Kit for RNA-seq | Fluidigm | 100-6201 |
| SMARTer Ultra Low RNA kit for Illumina Sequencing | Clontech | 634833 |
| Click-iT EdU Alexa Fluor 488 imaging kit | Life Technologies | C10337 |
| iScript cDNA Synthesis Kit | BioRad | 1708891 |
| DNeasy Blood & Tissue Kit | QIAGEN | 69504 |
| RNeasy Micro Kit | QIAGEN | 74004 |
| LIVE/DEAD Viability/Cytotoxicity Kit for mammalian cells | Life Technologies | L3224 |
| ApopTag Red In Situ Apoptosis Kit | Thermo Fisher Scientific | S7165 |
| mouse insulin ELISA kit | Alpco | 80-INSHU-E10.1 |
| SMART-Seq® v4 Ultra® Low Input RNA Kit for Sequencing | Clontech | 634889 |
| Nextera XT DNA Sample Preparation Kit | Illumina | FC-131-1096 |
| Nextera XT DNA Sample Preparation Index Kit | Illumina | FC-131-1002 |
| Advantage® 2 PCR Kit | Clontech | 639207 |
| Deposited Data | ||
| RNAseq data | This manuscript | GEO:GSE86479 |
| H3K27ac_ChIP | GEO: GSE68618 | GSM1677162 |
| H3K27ac_Input | GEO: GSE68618 | GSM1677164 |
| Experimental Models: Cell Lines | ||
| HEK293T cells | ATCC | |
| Experimental Models: Organisms/Strains | ||
| Mouse: RIP-Cre | The Jackson Laboratory | 003573 |
| Mouse: R26YFP | The Jackson Laboratory | 006148 |
| Mouse: C57BL/6 | Charles River Laboratories | 027 |
| Mouse: mIns1-H2B-mCherry | Benner et al., 2014 | |
| Mouse: C57BL/6.mCAT | Dai et al., 2011 | |
| Oligonucleotides | ||
| Primers for quantitative PCR | This manuscript | See table in quantitative PCR analysis section |
| Recombinant DNA | ||
| pLenti-C-mGFP | Origene | PS100071 |
| pLenti-C-mGFP-Srf | Origene | MR208120L2 |
| pCMV-R8.74 | Addgene | 22036 |
| pMD2.G | Addgene | 12259 |
| Software and Algorithms | ||
| FlowJo 8.7 software | https://www.flowjo.com/solutions/flowjo | N/A |
| ImageJ software | https://imagej.nih.gov/ij/ | N/A |
| Prism 5 software (GraphPad Software) | https://www.graphpad.com/scientific-software/prism/ | N/A |
| STAR | https://github.com/alexdobin/STAR | N/A |
| Cufflink and Cuffdiff | http://cole-trapnell-lab.github.io/cufflinks/ | N/A |
| Orange | http://orange.biolab.si/, https://bitbucket.org/biolab/orange-differentiation | N/A |
| Bioconductor | https://www.bioconductor.org/ | N/A |
| GSEA | http://www.broad.mit.edu/gsea | N/A |
| HOMER | http://homer.ucsd.edu/homer/ | N/A |
| STRING | http://string-db.org/ | N/A |
| Monocle | Trapnell et al., 2014 | http://cole-trapnell-lab.github.io/monocle-release/ |
| TSCAN | Ji et al., 2016 | https://github.com/zji90/TSCAN |
| Embeddr | Campbell et al., 2015 | https://github.com/kieranrcampbell/embeddr |
| DeLorean | Reid et al., 2016 | https://cran.r-project.org/web/packages/DeLorean/index.html |
| 1D Pseudotime Scale | This manuscript | See scripts in Data S1 |
Acknowledgments
We thank P. Rabinovitch for mCAT mice. We acknowledge support of S. Naik at the UCSD Stem Cell Genomics Core for assistance with the fluidigm C1 system, the UCSD Human Embryonic Stem Cell Core for cell sorting, the UCSD IGM Genomic Center (supported by P30 DK064391) for library preparation and sequencing, and O. Zagnitko at the Sanford Burnham Prebys Medical Discovery Institute Cancer Metabolism Core for metabolite measurements. We are grateful to Y. Song, O. Botvinnik, and L. Jamal-Schafer for advice on single-cell RNA-seq and computational analysis. We also thank Nancy Rosenblatt for mouse husbandry and members of the Sander lab for discussions and comments on the manuscript. This work was supported by National Institutes of Health grants DK068471 and DK078803 to M.S., an Iacocca Family Foundation fellowship to C.Z, and a JDRF postdoctoral fellowship (3-PDF-2015-83-A-N) to W.J.
Footnotes
AUTHOR CONTRIBUTIONS
C.Z., F.M., and M.S. designed the experiments and strategy for data analysis. C.Z., T.G., N.M., F.L., and W.J. performed experiments with input from M.S. and O.S.S. F.M., C.Z., Y.S., Y.T. performed data analysis with input from M.S. and G.W.Y. M.O.H. provided mice. C.Z., F.M., A.C.C., and M.S. wrote and edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguayo-Mazzucato C, Koh A, El Khattabi I, Li WC, Toschi E, Jermendy A, Juhl K, Mao K, Weir GC, Sharma A, et al. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia. 2011;54:583–593. doi: 10.1007/s00125-010-2026-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, Magnuson MA, Stein R. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes. 2010;59:2530–2539. doi: 10.2337/db10-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader E, Migliorini A, Gegg M, Moruzzi N, Gerdes J, Roscioni SS, Bakhti M, Brandl E, Irmler M, Beckers J, et al. Identification of proliferative and mature beta-cells in the islets of Langerhans. Nature. 2016;535:430–434. doi: 10.1038/nature18624. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Davis KL, Amir el AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, Pe’er D. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell. 2014;157:714–725. doi: 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner C, van der Meulen T, Caceres E, Tigyi K, Donaldson CJ, Huising MO. The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC Genomics. 2014;15:620–637. doi: 10.1186/1471-2164-15-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum B, Hrvatin SS, Schuetz C, Bonal C, Rezania A, Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol. 2012;30:261–264. doi: 10.1038/nbt.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonstra J, Post JA. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene. 2004;337:1–13. doi: 10.1016/j.gene.2004.04.032. [DOI] [PubMed] [Google Scholar]
- Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, Teichmann SA, Marioni JC, Stegle O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol. 2015;33:155–160. doi: 10.1038/nbt.3102. [DOI] [PubMed] [Google Scholar]
- Campbell K, Ponting CP, Webber C. Laplacian eigenmaps and principal curves for high resolution pseudotemporal ordering of single-cell RNA-seq profiles. 2015 doi: 10.1101/027219. BioRxiv. [DOI] [Google Scholar]
- Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, Schorle H, Sage J, Kim SK. PDGF signalling controls age-dependent proliferation in pancreatic beta-cells. Nature. 2011;478:349–355. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell C, Schug J, Canaday PS, Russ HA, Tarlow BD, Grompe MT, Horton T, Hebrok M, Streeter PR, Kaestner KH, et al. Human islets contain four distinct subtypes of beta cells. Nat Commun. 2016;7:11756–11765. doi: 10.1038/ncomms11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sanchez-Perez P, Cadenas S, Lamas S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015;6:183–197. doi: 10.1016/j.redox.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Salathia N, Liu R, Kaeser GE, Yung YC, Herman JL, Kaper F, Fan JB, Zhang K, Chun J, et al. Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat Methods. 2016;13:241–244. doi: 10.1038/nmeth.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finegood DT, Scaglia L, Bonner-Weir S. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes. 1995;44:249–256. doi: 10.2337/diab.44.3.249. [DOI] [PubMed] [Google Scholar]
- Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004;114:963–968. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyer WR, Gu X, Liu Y, Bottino R, Crabtree GR, Kim SK. Neonatal beta cell development in mice and humans is regulated by calcineurin/NFAT. Dev Cell. 2012;23:21–34. doi: 10.1016/j.devcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, Wright AJ, Atkinson MA, Rhodes CJ. Formation of a human beta-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab. 2012;97:3197–3206. doi: 10.1210/jc.2012-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Stein GH, Pan N, Goebbels S, Hornberg H, Nave KA, Herrera P, White P, Kaestner KH, Sussel L, et al. Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell Metab. 2010;11:298–310. doi: 10.1016/j.cmet.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassler M, Richmond TJ. The B-box dominates SAP-1-SRF interactions in the structure of the ternary complex. EMBO J. 2001;20:3018–3028. doi: 10.1093/emboj/20.12.3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermendy A, Toschi E, Aye T, Koh A, Aguayo-Mazzucato C, Sharma A, Weir GC, Sgroi D, Bonner-Weir S. Rat neonatal beta cells lack the specialised metabolic phenotype of mature beta cells. Diabetologia. 2011;54:594–604. doi: 10.1007/s00125-010-2036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Ji H. TSCAN: Pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res. 2016;44:e117. doi: 10.1093/nar/gkw430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston NR, Mitchell RK, Haythorne E, Pessoa MP, Semplici F, Ferrer J, Piemonti L, Marchetti P, Bugliani M, Bosco D, et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab. 2016;24:389–401. doi: 10.1016/j.cmet.2016.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klochendler A, Caspi I, Corem N, Moran M, Friedlich O, Elgavish S, Nevo Y, Helman A, Glaser B, Eden A, et al. The Genetic Program of Pancreatic beta-Cell Replication In Vivo. Diabetes. 2016;65:2081–2093. doi: 10.2337/db16-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. 2005;25:3752–3762. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT. svaseq: removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res. 2014;42:e161. doi: 10.1093/nar/gku864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Tanabe K, Baronnier D, Patel S, Woodgett J, Cras-Meneur C, Permutt MA. Conditional ablation of Gsk-3beta in islet beta cells results in expanded mass and resistance to fat feeding-induced diabetes in mice. Diabetologia. 2010;53:2600–2610. doi: 10.1007/s00125-010-1882-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mina M, Magi S, Jurman G, Itoh M, Kawaji H, Lassmann T, Arner E, Forrest AR, Carninci P, Hayashizaki Y, et al. Promoter-level expression clustering identifies time development of transcriptional regulatory cascades initiated by ErbB receptors in breast cancer cells. Sci Rep. 2015;5:11999–12012. doi: 10.1038/srep11999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulas F, del Fabbro E, Zupan B, Bellazzi R. Interest propagation for knowledge extraction and representation. Stud Health Technol Inform. 2013;186:182–186. [PubMed] [Google Scholar]
- Mulas F, Zagar L, Zupan B, Bellazzi R. Supporting regenerative medicine by integrative dimensionality reduction. Methods Inf Med. 2012;51:341–347. doi: 10.3414/ME11-02-0045. [DOI] [PubMed] [Google Scholar]
- Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- Ray JD, Kener KB, Bitner BF, Wright BJ, Ballard MS, Barrett EJ, Hill JT, Moss LG, Tessem JS. Nkx6.1-mediated insulin secretion and beta-cell proliferation is dependent on upregulation of c-Fos. FEBS Lett. 2016;590:1791–1803. doi: 10.1002/1873-3468.12208. [DOI] [PubMed] [Google Scholar]
- Reid JE, Wernisch L. Pseudotime estimation: deconfounding single cell time series. Bioinformatics. 2016;32:2973–2980. doi: 10.1093/bioinformatics/btw372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma RB, O’Donnell AC, Stamateris RE, Ha B, McCloskey KM, Reynolds PR, Arvan P, Alonso LC. Insulin demand regulates beta cell number via the unfolded protein response. J Clin Invest. 2015;125:3831–3846. doi: 10.1172/JCI79264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supale S, Li N, Brun T, Maechler P. Mitochondrial dysfunction in pancreatic beta cells. Trends Endocrinol Metab. 2012;23:477–487. doi: 10.1016/j.tem.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Szabat M, Page MM, Panzhinskiy E, Skovso S, Mojibian M, Fernandez-Tajes J, Bruin JE, Bround MJ, Lee JT, Xu EE, et al. Reduced Insulin Production Relieves Endoplasmic Reticulum Stress and Induces beta Cell Proliferation. Cell Metab. 2016;23:179–193. doi: 10.1016/j.cmet.2015.10.016. [DOI] [PubMed] [Google Scholar]
- Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–375. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen T, Xie R, Kelly OG, Vale WW, Sander M, Huising MO. Urocortin 3 marks mature human primary and embryonic stem cell-derived pancreatic alpha and beta cells. PLoS One. 2012;7:e52181. doi: 10.1371/journal.pone.0052181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Schug J, Won KJ, Liu C, Naji A, Avrahami D, Golson ML, Kaestner KH. Single-Cell Transcriptomics of the Human Endocrine Pancreas. Diabetes. 2016;65:3028–3038. doi: 10.2337/db16-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Y, Okamoto H, Kim J, Ni M, Adler C, Cavino K, Na E, Murphy AJ, Yancopoulos GD, Lin C, et al. Single-Cell RNAseq Reveals That Pancreatic beta-Cells From Very Old Male Mice Have a Young Gene Signature. Endocrinology. 2016;157:3431–3438. doi: 10.1210/en.2016-1235. [DOI] [PubMed] [Google Scholar]
- Yoshihara E, Wei Z, Lin CS, Fang S, Ahmadian M, Kida Y, Tseng T, Dai Y, Yu RT, Liddle C, et al. ERRgamma Is Required for the Metabolic Maturation of Therapeutically Functional Glucose-Responsive beta Cells. Cell Metab. 2016;23:622–634. doi: 10.1016/j.cmet.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagar L, Mulas F, Garagna S, Zuccotti M, Bellazzi R, Zupan B. Stage prediction of embryonic stem cell differentiation from genome-wide expression data. Bioinformatics. 2011;27:2546–2553. doi: 10.1093/bioinformatics/btr422. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.