

Abstract
Chemotherapy-resistant human acute myeloid leukemia (AML) cells are thought to be enriched in quiescent immature leukemic stem cells (LSCs). To validate this hypothesis in vivo, we developed a clinically relevant chemotherapeutic approach treating patient-derived xenograft (PDX) with cytarabine. Cytarabine residual AML cells are enriched neither in immature, quiescent cells nor LSCs. Strikingly, cytarabine-resistant pre-existing and persisting cells displayed high levels of reactive oxygen species, showed increased mitochondrial mass, and retained active polarized mitochondria, consistent with a high oxidative phosphorylation (OXPHOS) status. Cytarabine residual cells exhibited increased fatty acid oxidation, upregulated CD36 expression and a HIGH OXPHOS gene signature predictive for treatment response in PDX and AML patients. HIGH OXPHOS but not LOW OXPHOS human AML cell lines were chemoresistant in vivo. Targeting mitochondrial protein synthesis, electron transfer, or fatty acid oxidation induced an energetic shift towards LOW OXPHOS and markedly enhanced anti-leukemic effects of cytarabine. Together, this study demonstrates that essential mitochondrial functions contribute to cytarabine resistance in AML and are a robust hallmark of cytarabine sensitivity and a promising therapeutic avenue to treat AML residual disease.
INTRODUCTION
Acute myelogenous leukemia (AML) is a heterogeneous disease characterized by a blockade in differentiation of hematopoietic stem cells and a clonal expansion of myeloid blasts in the bone marrow and peripheral blood. Standard “7+3” induction therapy, that combines a nucleoside analogue such as cytarabine (AraC) for 7 days with an anthracycline for 3 days, is highly effective in killing leukemic cells in AML. Despite a high rate of complete remission after these cytotoxic agents, the 5-year overall survival is very poor especially in patients over 60 years. Indeed, most patients relapse and only allogenic stem cell transplant is then curative (1,2). Relapses are caused by tumor regrowth initiated by chemoresistant leukemic cells (RLCs). Many hypotheses have been proposed to explain therapeutic resistance (drug efflux, detoxification enzymes, poor accessibility of the drug to the leukemic niche) (3,4), but none led to a complete understanding of the molecular mechanisms of AML resistance especially in vivo nor to new therapies, which would effectively eradicate RLCs.
It is also increasingly recognized that the causes of chemoresistance may reside in rare stem cell populations (5,6). Several laboratories have shown that the presence of high levels of leukemic stem cells (LSCs; CD34+CD38low/−CD123+ cells) at diagnosis correlates with adverse outcome in AML patients in terms of response to therapy and overall survival (7,8). These and other studies support the notion that chemoresistant cells represent leukemic stem cells (LSCs) (9,10), although this hypothesis has never been formally tested in vivo with clinically relevant doses. Recent research in our and other laboratories focusing on the phenotypic characterization of LSCs in highly immunodeficient NSG mice showed that LSCs are phenotypically heterogeneous in AML in vivo (11–14). Moreover, recent data suggested that LSCs are influenced by clonal genetic evolution, epigenetic alterations and their microenvironment, suggesting that they are themselves heterogeneous especially with regard to their chemoresistance capacities in vivo (15). AraC is used both in combination regimens for induction and as a single agent for post-remission therapy in AML patients. In cells, AraC is rapidly converted into AraC-triphosphate, which is incorporated into DNA strands during the S phase of the cell cycle inhibiting further DNA synthesis (16,17), thereby affecting preferentially rapidly dividing cells. Accordingly, RLCs are thought to be rare, quiescent and well adapted to hypoxic conditions (18–20).
Here, to exhaustively characterize the response of AML cells to AraC therapy, we treated 25 naive patient-derived xenograft (PDX) with a clinically relevant sub-lethal regimen of AraC, also used in previous studies (21,22). At the nadir of leukemic cell burden, in vivo AraC treatment has a strong cytoreductive effect mediated by death of both proliferating and quiescent AML cells. Surprisingly and as opposed to previous studies (9,10), this cytoreduction was not associated with any consistent changes in stem cell functions, such as CD34+CD38− phenotype, G0 status, stem cell gene markers/signature or frequency of LICs. Rather, we showed that AraC residual cells have mitochondrial-specific oxidative and bioenergetics features. Furthermore, we identified a specific HIGH OXPHOS gene signature in RLCs that is also predictive for treatment response in PDX. Accordingly, AML cells with a HIGH OXPHOS energetic phenotype are markedly less sensitive to AraC chemotherapy compared to LOW OXPHOS AML cells in NSG mice. Finally, modulation of mitochondrial OXPHOS status markedly affected the anti-leukemic effect of AraC in vitro and in vivo. Together, this study describes a novel model of chemotherapy resistance in AML that provides a better understanding of mechanisms underlying in vivo AraC resistance to new combinatorial therapies.
RESULTS
In vivo AraC treatment induces a significant reduction of tumor burden in AML-engrafted mice
To study the therapeutic response of primary human AML, we used our NSG-based PDX model for AML (14,23,24). Twenty-five primary AML patient specimens from two clinical sites were screened for their engraftment capacities in NSG mice and their genetic diversity (Table S1; Fig. S1A–D). Briefly, one to ten millions unsorted AML cells were injected into adult NSG mice after pre-conditioning with a sub-lethal treatment of busulfan one day prior injection (Fig. 1A). Engraftment efficiency was measured in peripheral blood or bone marrow aspirates by flow cytometry analysis of hCD45+CD33+CD44+ cells, starting at 8 weeks after xenotransplantation. Mice showing at least 50% of human AML engraftment were assigned to experimental groups to obtain balanced average engraftment levels in each cohort at initiation of therapy. Preliminary experiments were performed to determine the in vivo AraC regimen (3 or 5 consecutive daily treatments) and the optimum sub-lethal dose of AraC (10, 30, 60, 90, or 120 mg/kg/day) (Fig. S2A). Administration of 60mg/kg/day for 5 consecutive days was determined as the most efficient treatment (Fig. S2A–C) to observe a significant reduction in total cell tumor burden in bone marrow (BM) and spleen (SP) (Fig. S2D). Tumor reduction was not improved by treating mice for 7 days, or with higher doses and led increased mortality. Analogous to the patient response to chemotherapy, decrease in absolute white blood cell counts, hemoglobin, and platelets was observed in mice one week after treatment with the selected scheme (60mg/kg/day for 5 days) and resolved by 2 weeks after treatment. 60, 90, 120 mg/kg of AraC induced almost identical effects on blood parameters (Fig. S2E). Collectively, these data show that AraC is well tolerated in NSG mice at dose and schedule comparable to those administrated to AML patients (21), allowing us to study mechanisms involved in drug resistance.
Figure 1.
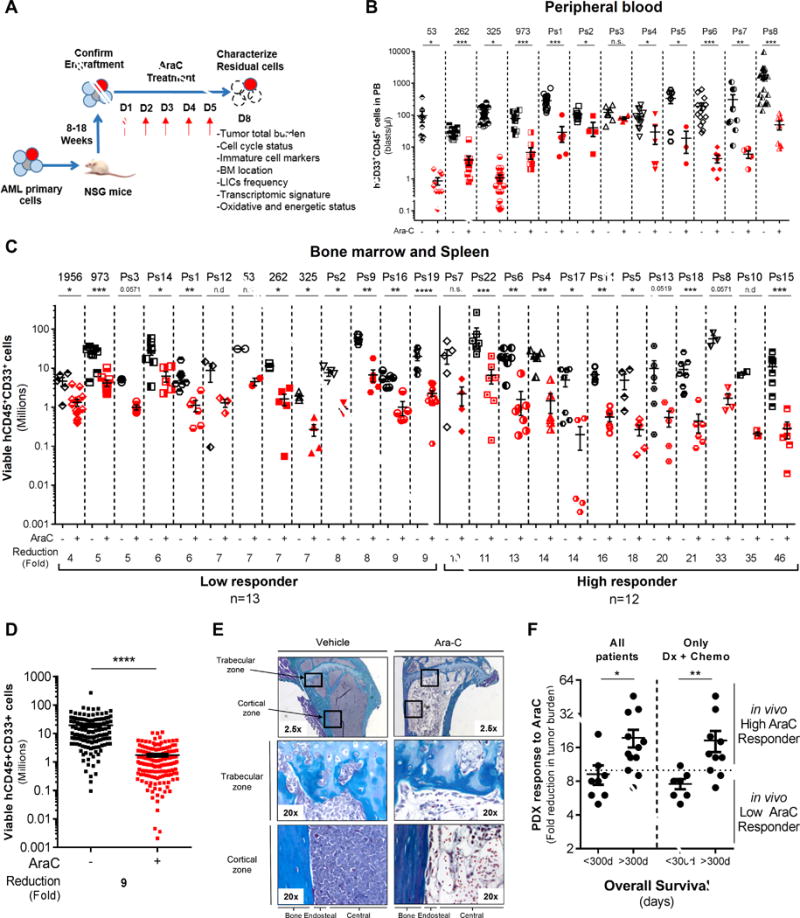
In vivo cytarabine (AraC) treatment induces a significant reduction of the total cell tumor burden in AML-engrafted mice. A, Schematic diagram of the chemotherapy regimen and schedule used to treat NSG-based PDX models with AraC. Peripheral blood engraftment was assessed between 8–18 weeks and mice were assigned to experimental groups of 4–10 mice with similar average engraftment per group. Mice were treated with vehicle (PBS) or 60mg/kg/d AraC given daily via IP injection for 5 days. Mice were sacrificed post-treatment at day 8 in order to characterize viable residual AML cells. B–C–D, Total number of human viable AML cells expressing CD45, CD33 and CD44 was analyzed and quantified using flow cytometry in AraC-treated AML-xenografted mice compared to PBS-treated AML-xenografted mice in peripheral blood (B; blasts per μL of mice blood) and in bone marrow and spleen (C; total cell tumor burden in Millions). Fold reduction of total cell tumor burden in AraC-treated mice compared with control-treated mice was calculated individually for each AML patient samples and in the entire PDX cohort. E, Goldner staining of bone marrow (tibia section at low/2.5x or high/20x magnification) shows engraftment and localization of AML cells at the cortical and trabecular region of the bone in vehicle (PBS)- and AraC-treated mice. F, Correlative analysis between the in vivo response to AraC using our PDX model and the overall survival of all matched AML patients or of those at diagnosis and that received intensive induction chemotherapy (Dx+Chemo). Graphs of mean ± sem. P values were determined by Mann-Whitney test. n.s., not significant. n.d., not determined. *, P≤0.05; **, P≤0.01; ***, P≤0.001.
These experiments demonstrated that the maximal response to AraC was achieved by day 8 after initiation of chemotherapy. Therefore, response to AraC treatment and various characteristics of resistant leukemic cells (thereafter, RLCs) were specifically monitored at D8 (3 days after the last administration of AraC; Fig. 1A). The total number of circulating human AML cells expressing the cell surface markers CD45 and CD33 in AraC-treated mice was determined by flow cytometry and compared to that of control, PBS-treated mice (Fig. 1B). At that time point, all AraC-treated mice had a significant reduction of human AML peripheral cells that were below the detection threshold (10 events/μL). Total cell tumor burden, e.g. total number of viable human AML blasts, was determined in murine BM and SP (Fig. 1C–D and Fig. S3A–B). Interestingly, the in vivo cytoreductive effect of this AraC regimen was significant in all patients but heterogeneous, ranging from 4- to 46-fold reduction. Based on this response in vivo, we defined low and high responders that exhibited a biological response below or above a 10-fold reduction, respectively (Fig. 1C–D; Fig. S3A–B). Additional histochemical analyses of tissue sections stained with Goldner’s trichrome or hematoxylin/eosin demonstrated a marked reduction of human blasts in both the trabecular and cortical zones of murine bone marrow and sternum following AraC treatment (Fig. 1E, Fig. S3C–D). Of note, we observed significant increase in adipocyte number after AraC treatment. These data demonstrate a strong cytotoxic effect of AraC on AML cells in this model, regardless of the intramedullary location of leukemic cells.
We studied if the response to AraC in the PDX model (fold-reduction in BM and SP after AraC treatment) was correlated with clinical characteristics of the corresponding patients (Fig. S4). No correlation was observed between AraC response in vivo and either the specimen type (diagnosis versus relapse, S4A), gender (S4B), the clinical response to intensive “3+7” chemotherapy (S4D), the FAB classification (S4E), the expression of CD34 (S4F), the cytogenetic risk group (S4G), the FLT3-ITD allelic burden (S4I), or the mutational status (S4J). Strikingly, response to AraC in vivo was decreased in PDX from older patients (S4C) and correlated with FLT3-ITD mutation (S4H) and with a shorter overall survival (Fig. 1F). Finally, the in vitro cytotoxic response to AraC (as measured as IC50 by flow cytometry upon staining with Annexin V at 24hr or by Cell TiterGlo assay at 48hr) of cells from these AML patients did not correlate with their in vivo response to AraC in PDX (Fig. S4K). These results indicate that the analysis of this PDX-based preclinical model can predict patient clinical outcome and is a suitable model to study AraC response and residual disease in AML.
In vivo AraC treatment affects CD34+/−CD38+/− phenotypes in AML-engrafted mice
Previous reports showed that immature AML cells, that may represent LSCs or LICs, are enriched after chemotherapy. Such cells were initially described as CD34+CD38−, although LSCs have also been identified in more mature CD34+CD38+ populations in vivo (11,13,14). We evaluated the diversity of CD34+/−CD38+/− phenotypes after AraC treatment by analyzing the expression of these two cell surface markers in cells isolated from BM of AML PDX (as described in Fig. S5A; representative flow plot in Fig. S5B). We observed a significant change in phenotype after AraC treatment with an increase in CD34+CD38− population in only 3 out of 22 patients analyzed (Fig. 2A). Of note, four AML PDX showed a trend of decrease in CD34+CD38− population post-AraC (Ps12, Ps7, Ps18, Ps17; Fig2A). A significant increase in the percentage of CD34+CD38− cells after AraC treatment was observed in patients with the lowest level of this immature cell population, whereas patients with the highest basal level of CD34+CD38− cells exhibited a decrease of this sub-population (Fig. 2A–B). Analysis of the more mature CD34+CD38+ cells showed a variable response of this population of cells to AraC treatment that was not statistically different from that of control cells isolated from PBS-treated mice (325, Ps1, Ps2, Ps6 or Ps15; Fig. S5C–D). CD34 staining alone showed that CD34− cells displayed a variable response to AraC treatment (Ps2, 325 or Ps19, Ps17, Ps15, Ps22; Fig. S5E–F). Finally, we found that AraC decreased the absolute cell number of both CD34+CD38− (Fig. S6A–B) and CD34+CD38+ cells (Fig. S6C–D), indicating that AraC treatment affects both mature and immature AML cells in this model.
Figure 2.
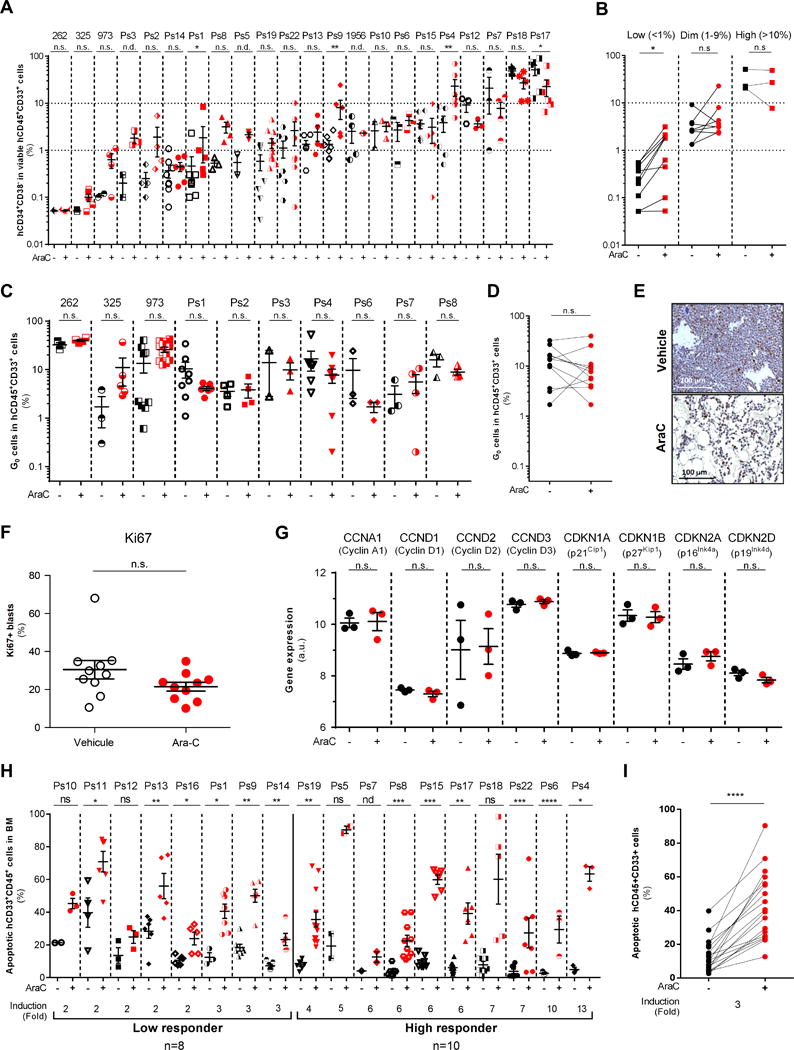
In vivo cytarabine (AraC) treatment does not induce any consistent changes in CD34+/−CD38+/− phenotypes nor in quiescent leukemic cells but increases apoptotic cell death in AraC-treated compared to PBS-treated AML-engrafted mice. A–B, Flow cytometry analyses of human viable (AnnexinV−/7-AAD−) CD45+CD33+ AML cells were performed to assess the percentage of CD34+CD38− cells in AraC-treated AML-xenografted mice compared to vehicle (PBS)-treated AML-xenografted mice (A) and stratified as following their basal level of CD34+CD38− phenotype (B). C–D, Hoechst/Pyronin Y-based flow cytometric assay was performed to measure cells in G0 in human viable (AnnexinV−/7-AAD−) CD45+CD33+ AML cells in AraC-treated AML-xenografted mice compared to vehicle (PBS)-treated AML-xenografted mice. E, Representative histological section showing Ki67 staining of the bone marrow. Scale bar, 100 μm. F, Ki67+ cycling cells quantification shows no difference between PBS-treated mice and AraC-treated mice. G, Expression level of genes involved into cell cycle checkpoint and G0-to-G1 transition in residual AML cells after AraC treatment compared to PBS. (H–I) Percent of human apoptotic CD45+CD33+ AML cells was assessed by AnnexinV/7AAD-based flow cytometric assay in AraC-treated AML-xenografted mice compared to vehicle (PBS)-treated AML-xenografted mice. Graphs of mean ± sem. P values were determined by Mann-Whitney test (A–C–F–G–H) or Wilcoxon matched-pairs signed rank test (B–D–I). n.s., not significant. n.d., not determined. *, P≤0.05; **, P≤0.01; ***, P≤0.001.
In vivo AraC treatment equally kills quiescent and proliferating AML cells
Next, we used flow cytometry after Hoechst/Pyronin Y staining (14,18,25) to assess the cell cycle status of bone marrow cells harvested from AML-xenografted mice after AraC. Peripheral blood mononuclear cells stained with Hoechst/Pyronin Y were used to define G0 quiescent cells (Fig. S7A). AML cells from 10 AML PDX specimens were analyzed at day 8. No enrichment of quiescent (G0) cells was detected in the residual human AML cell population after AraC- compared to PBS-treated AML PDX in 9 out of 10 patient samples (Fig. 2C–D). Moreover, we found no enhancement in quiescent G0 cells in the CD34+CD38− population after AraC treatment (Fig. S7B–C). Importantly, AraC treatment significantly decreased the absolute number of G0 cells in viable residual human CD45+CD33+ cells (Fig. S7D–E). These results differed from a previous study using a very high dose of AraC (1g/kg/d for 2 days) (9), and we next compared the two regimens. Consistent with published results, we observed an increase of cells in G0 only with 1g/kg AraC (Fig. S7F). However, the reduction of the total tumor burden with this high dose of AraC was only 4-fold, while it reached 33-fold with the 60mg/kg/day for 5 consecutive days regimen (Ps8; Fig. S7G), suggesting that the duration of treatment might be an important parameter of the AraC response efficacy. Importantly, most of the animals treated with this very high dose of AraC died within the first week of treatment, precluding a follow-up study of disease progression post-chemotherapy. The in vivo cell cycle profile of AraC-treated AML cells was confirmed upon staining with an antibody recognizing the cell cycle marker Ki67 that is expressed during all phases of the cell cycle (G1, S, G2) but not in quiescent G0 cells. As shown in BM sections prepared from representative control- or AraC- treated AML xenografted mice (Fig. 2E), no significant difference in the number of Ki67+ blasts was observed after AraC treatment in vivo (Fig. 2F). Moreover, analysis of the mRNA level of cell cycle-related genes did not show any change after AraC in residual human AML cells (n=3 patients analyzed in vivo; Fig. 2G). Taken together, these results indicated that human AML cells in mice were not highly proliferative and that quiescent G0 cells were not enriched after AraC chemotherapy although this treatment significantly reduced the total cell tumor burden in vivo. Accordingly, we showed that AraC strongly induced apoptotic cell death in all 18 patient samples analyzed in vivo (ranging from 2- to 13-fold induction; mean=3; Fig. 2H–I). This suggests that in vivo AraC equally kills quiescent and proliferating AML cells in vivo.
In vivo AraC treatment does not select LSCs but induces a specific gene signature of inflammatory and stress responses in RLCs
To determine whether LSCs (functionally defined hereafter as leukemia-initiating cells, LICs) were enriched in RLCs after AraC treatment, we performed a limiting dilution analysis on vehicle- and AraC-treated AML PDX from first recipient (Fig. S7H). Frequency of LSCs in each group was calculated using regression analysis (L-Calc software). We observed that AraC decreased the LSCs frequency in BM from PDX (Patient 1956) and induced no change in LSCs frequency in three other patient samples tested (Fig. 3A; BM, Table S2). Moreover, our analyses indicated that AraC did not change the LSCs frequency in SP from these four patient samples (SP, Table S2). These results indicate that some LSCs can be sensitive to AraC treatment in vivo but LSCs are typically similarly sensitive to AraC as bulk leukemic cells in some patients.
Figure 3.
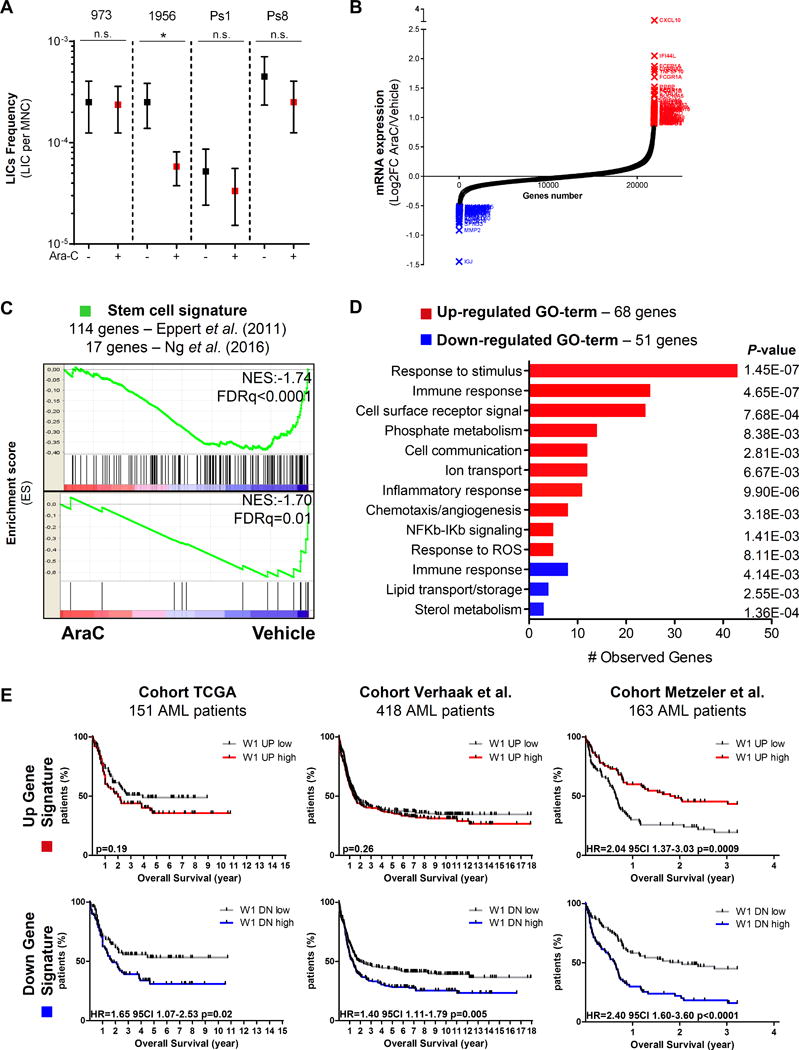
In vivo cytarabine (AraC) treatment does not enrich in leukemia-initiating stem cells (LICs) in AML-engrafted mice but uncovers a specific gene signature of immune/inflammatory stress response in AraC-resistant cells from AML-engrafted mice. A, The frequency of LICs from 4 different PDX was calculated for each treatment (vehicle/PBS versus AraC) group using regression analysis (L-Calc software). Graph of mean ± range. *P≤0.05. Mann-Whitney test was performed. B, Up- (red) and down- (blue) regulated gene signatures were generated from transcriptomes of human residual AML cells purified from AraC-treated AML-xenografted mice or vehicle (PBS)-treated AML-xenografted mice. C, Gene Set Enrichment Analysis (GSEA) of stem cell signatures functionally identified by Eppert et al. (13) or Ng et al. (28) was performed using transcriptomes of human residual AML cells purified from AraC-treated (red) compared to PBS/control-treated (blue) AML-xenografted mice. Kolmogorov-Smirnov statistical test was performed. D, Gene Ontology (GO) classification of up- (red) or down- (blue) regulated genes was identified in residual AML cells from AraC-treated compared to vehicle (PBS)-treated AML-xenografted mice by Genomatics software analysis. Fisher’s exact test was performed. E, Prognostic correlation of the up- (red, 68 genes) or down- (blue, 51 genes) regulated gene expression signature was performed in three independent AML cohorts of a total of 732 patients. Kaplan-Meier survival curve, number of patients and P values from log-rank tests are displayed.
To study the gene expression signature of RLCs in vivo, we performed a full genome transcriptomic analysis on viable human AML blasts purified from BM of mice treated with PBS or AraC in 3 different PDX specimens (Fig. 3B; Table S3). First, we performed a gene set enrichment analysis (GSEA) with two functionally identified stem cell signatures (13,26). Expression of these two stem cell signatures negatively correlated with that of residual AraC-resistant AML blasts in vivo (Fig. 3C; NES=−1.74, FDRq<0.0001 and NES=−1.70, FDRq=0.01, respectively), indicating that LSCs, as defined by these specific gene sets, were not enriched in vivo in viable RLCs at D8. Furthermore, computational analyses of these transcriptomic data identified two subsets of down- and up-regulated genes (51 and 68 genes, respectively; Fig. 3D) in viable residual post-AraC versus post-PBS human AML blasts (Table S3). These sets of genes were enriched in genes associated with immune and inflammatory responses (CXCL10, CXCL11, CXCL7, CCL23), ROS response (EPX, SESN3, GATM, HGF, HBB) as well as lipid and sterol metabolism (STARD4, STARD6, SLC27A6, SPNS3, PLA2G10, PRG2, HPGDS and DHRCR24, CYP51A1, INSIG1; Fig. 3B, Fig. 3D and Table S3). Finally, we studied the prognostic correlation of these up- or down-regulated gene signatures in transcriptomic datasets from three independently published cohorts of AML patients (The Cancer Genome Atlas, AML cohort; GSE12417; GSE14468) (27–29). We found that our up-regulated gene signature had no significant prognostic value in two out of three independent cohorts tested, while AML patients with a signature enriched in our down-regulated gene signature had a significant decrease in overall survival in all three cohorts (p=0.02, 0.005, 0.0001, respectively; Fig. 3E and Table S3). This suggests that the genes down-regulated by AraC treatment contribute to global chemoresistance mechanism in AML patients.
In vivo RLCs have elevated ROS levels and active mitochondria after AraC treatment
Based on the literature (30,31) and on our transcriptomic signature of RLCs enriched in inflammatory, stress, and ROS responses, we hypothesized that in vivo AraC chemotherapy could induce redox alteration and ROS production in RLCs. Pimonidazole (PIMO) reacts with free sulfhydryl groups, such as reduced cysteine residues on proteins to form adducts, that can then be detected by antibody staining (32). Thus, PIMO adduct formation can be used as a stable readout of the intracellular redox state in vivo. We injected PIMO in AraC- and PBS- treated AML mice xenografted in BM. RLCs showed higher intracellular PIMO staining than control AML cells in both trabecular and cortical zones of murine femurs (Fig. 4A, representative histochemical slides; Fig. 4B, quantification). AraC induced a significant reduction of human AML cells in BM of murine femur (Fig. 4C) as previously shown (Fig. 1 and Fig. S3D–E). These data suggest that AraC treatment leads to an enrichment/survival of RLCs in vivo regardless of their localization throughout the femur BM. Consistent with our gene expression profiling showing an increase in ROS responsive genes, DCF staining by flow cytometry showed a higher total ROS content in RLCs compared to PBS treated cells in all 15 different PDX analyzed (Fig. 4D–E). Accordingly, GSEA of a ROS signature generated by Houstis et al. (33) identified a positive correlation and an enrichment in ROS markers in RLCs following AraC in vivo (NES=1.54, FDRq=0.03; Fig. 4F).
Figure 4.
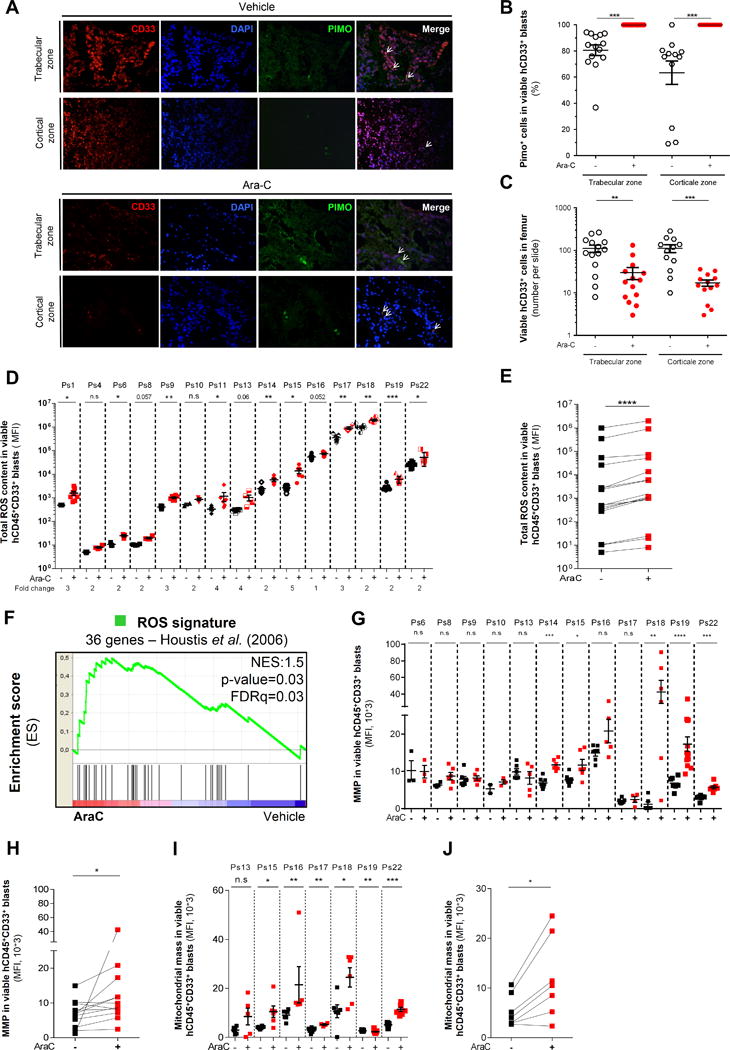
In vivo residual AML cells have an increased oxidative metabolism with higher ROS content and retained active mitochondria after cytarabine (AraC) treatment. A, Representative histological section of bone marrow showing intracellular pimonidazole staining of viable CD33+ AML cells in trabecular and cortical zone of the femur from mice treated with vehicle (PBS) and AraC (60mg/kg/d; 5 days). B, Quantification of pimonidazole positive cells showing altered intracellular redox state in viable CD33+ cells after AraC treatment in vivo. C, Number of viable CD33+ AML cells in femur in the regard of different histological (trabecular versus cortical) section in both PBS-treated and AraC-treated AML-xenografted mice. D–E, Intracellular ROS levels were assessed using DCF-DA probe and analyzed by flow cytometry in human viable (AnnexinV−/7-AAD−) CD45+CD33+ AML cells from AraC- and vehicle (PBS)-treated AML-xenografted mice. F, Gene Set Enrichment Analysis (GSEA) of ROS signature generated by Houstis et al. (2006) was performed from transcriptomes of human residual AML cells purified from AraC-treated (red) compared to vehicle (PBS)-treated (blue) AML-xenografted mice. Kolmogorov-Smirnov statistical test was performed. G–H, Active mitochondrial membrane potential was assessed in human viable (AnnexinV−/7-AAD−) CD45+CD33+ AML cells from AraC-treated and vehicle (PBS)-treated AML-xenografted mice by flow cytometry using the cell permeant fluorescent TMRE probe. I–J, Mitochondrial mass was measured in human viable (AnnexinV−/7-AAD−) CD45+CD33+ AML cells from AraC- and vehicle (PBS)-treated AML-xenografted mice by flow cytometry using MitoTracker® Green (MTG) probe. Graphs of mean ± sem. P values were determined by Mann-Whitney test (B–C–D–G–I) or Wilcoxon matched-pairs signed rank test (E–H–J). n.s., not significant. *, P≤0.05; **, P≤0.01; ***, P≤0.001.
As mitochondria are one of the major sources of intracellular ROS, we next assessed mitochondrial activity by measuring mitochondrial membrane potential in RLCs using tetramethylrhodamine ethyl ester (TMRE), a cell permeant lipophilic cationic fluorescent dye that concentrates in mitochondria in proportion to the transmembrane potential. Interestingly, mitochondrial membrane potential (MMP), as judged by TMRE labeling, was increased following AraC treatment in 7 PDX out of 12 tested (Fig. 4G–H), suggesting that RLCs retained active polarized mitochondria with no loss of MMP. Staining with MitoTracker Green, which passively diffuses across the plasma membrane and accumulates in mitochondria regardless of mitochondrial membrane potential, was higher in RLCs from 6 out of 7 patients treated with AraC compared with PBS-treated counterparts (Fig. 4I–J), suggesting an increase in mitochondrial mass and activity, underscoring the important role of mitochondrial metabolism that supports residual disease and AraC resistance in AML.
High OXPHOS AML cell lines are more resistant to AraC chemotherapy and RLCs exhibit a HIGH OXPHOS gene signature in vivo
As suggested by our previous data and recently proposed by other groups in solid tumors (34–36), we hypothesized that RLCs responsible for relapse would have mitochondria-driven energetic features modulating their response to AraC in the context of the in vivo niche. To analyze whether the bioenergetic status of AML cells might be involved in AraC chemoresistance, we characterized mitochondrial ATP production and oxygen consumption rates (OCR) of six diverse human AML cell lines (molecular and mutational characteristics in Table S1) and defined their bioenergetic status as LOW (lowest mitochondrial ATP production and OCR: U937, KG1, KG1a) versus HIGH (highest mitochondrial ATP production and OCR: HL60, MV4-11, MOLM14) oxidative phosphorylation activity (LOW OXPHOS versus HIGH OXPHOS; Fig. S8A). Additionally, mitochondrial ROS content, mitochondrial membrane potential and mass, and sensitivity to galactose of four human AML cell lines were analyzed to further functionally define LOW and HIGH OXPHOS status and capacity (Fig. S8B–D). LOW and HIGH OXPHOS AML cell lines were next injected into adult NSG mice and treated with AraC (30 mg/kg/d) for 5 days starting 10 days after transplantation. LOW OXPHOS AML cells were sensitive to AraC in vivo as demonstrated by a significant increase in the overall survival of the AraC- compared to PBS-treated mice cohorts (Fig. 5A). In contrast, AML cells with HIGH OXPHOS were less sensitive to AraC chemotherapy in vivo compared to LOW OXPHOS AML cells (Fig. 5A). Furthermore, we observed a significant reduction of total cell tumor burden and a significant increase in apoptotic cell death in LOW OXPHOS AML U937 cells but no change in total cell tumor burden, apoptosis and loss of MMP in HIGH OXPHOS AML MOLM14 cells (Fig. 5B–D). Most notably, AraC residual cells have a significant increase in PIMO staining, total and mitochondrial ROS content as well as active MMP in LOW OXPHOS AML cells whereas these changes were less pronounced in HIGH OXPHOS AML cells (Fig. 5E–H, respectively).
Figure 5.
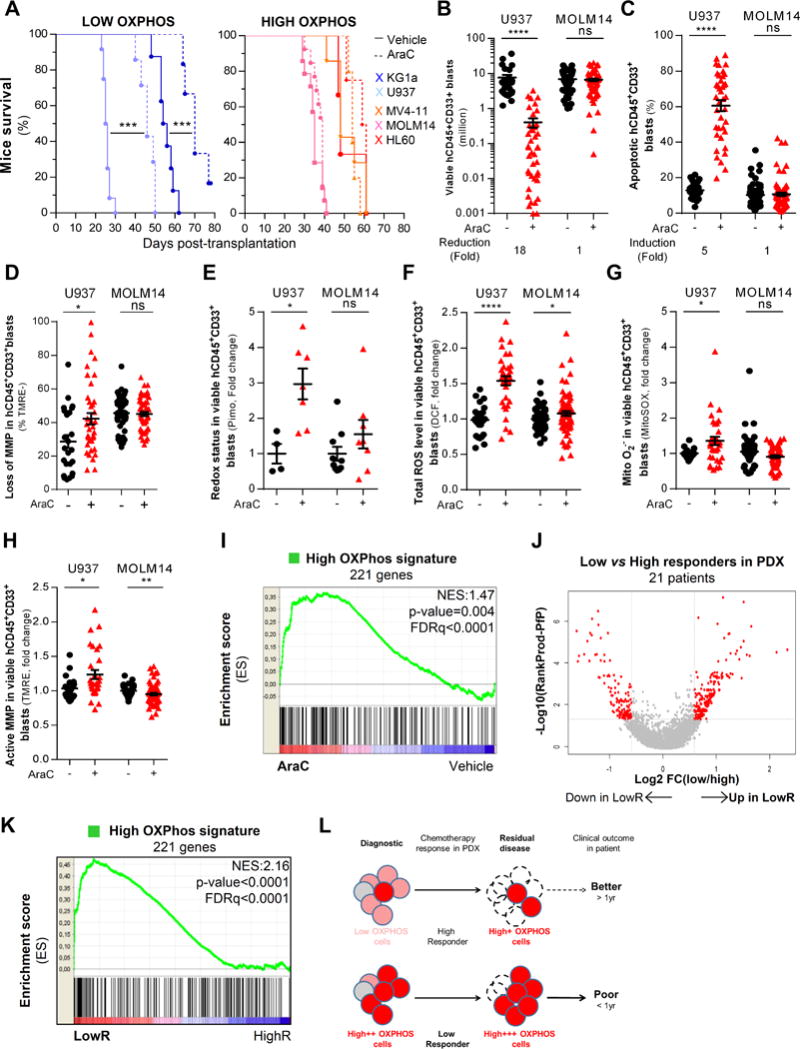
AML cells with high oxidative phosphorylation activity are more resistant to cytarabine (AraC) chemotherapy in vivo and residual human AML cells after AraC treatment in AML-engrafted mice exhibit a HIGH OXPHOS gene signature in vivo. A, Kaplan-Meier curves of mice survival were established for HIGH and LOW OXPHOS AML cell lines engrafted in NSG mice and treated with AraC (30mg/kg/day) or PBS during 5 days. Significance was determined by log-rank tests. B, Total cell tumor burden of human viable CD45+CD33+ AML cells was analyzed and quantified in PBS- and AraC-treated AML mice xenografted wit HIGH (MOLM14) and LOW (U937) OXPHOS AML cell lines using flow cytometry. C, Percent of human apoptotic CD45+CD33+ AML cells was analyzed using AnnexinV/7AAD-based flow cytometric assay in PBS- and AraC-treated MOLM14- or U937-xenografted mice. D, Loss of mitochondrial membrane potential was assessed with fluorescent TMRE probe using flow cytometry in human CD45+CD33+ AML cells in PBS- and AraC-treated MOLM14- or U937-xenografted mice. E, Intracellular redox status was assessed using pimonidazole probe and analyzed by flow cytometry in viable human AML cells from MOLM14- or U937-xenografted mice after PBS and AraC treatment. F, Intracellular total ROS levels were assessed using DCF-DA probe and analyzed by flow cytometry in viable human AML cells from MOLM14- or U937-xenografted mice after PBS and AraC treatment. G, Mitochondrial superoxide production was measured by MitoSOX probe using flow cytometry in viable human AML cells from MOLM14- or U937-xenografted mice after PBS and AraC treatment. H, Active mitochondrial membrane potential was assessed by flow cytometry using the fluorescent TMRE probe in viable human AML cells from MOLM14- or U937-xenografted mice after PBS and AraC treatment. I, Gene set enrichment analysis of the HIGH OXPHOS gene signature in viable human residual AML cells from AraC- and PBS-treated AML-xenografted mice. J, Volcano plot of most differentially expressed (230 up- and 151 down- regulated) genes identified in transcriptomes of AML patients that are Low compared to High Responders to AraC in PDX models. The adjusted p-values based on –log10 were plotted against the log2 ratio of gene expression level for all genes. K, Gene set enrichment analysis of the HIGH OXPHOS gene signature in transcriptomes of AML patients that are Low (red) compared to High (blue) responder in PDX models. L, Schematic diagram of the chemotherapy response, residual disease and their clinical outcome of HIGH OXPHOS versus LOW OXPHOS AML patients. Graphs of mean ± sem. Kolmogorov-Smirnov statistical test was performed. Mann-Whitney test was performed (A-K) n.s., not significant. *, P≤0.05; **, P≤0.01; ***, P≤0.001.
Finally, as the electron transport chain (ETC) complex I inhibitor metformin induces a well-described energetic shift from HIGH OXPHOS to LOW OXPHOS (so-called Pasteur effect, Fig. S9A compared to Fig. S8A), we performed a comparative transcriptomic analysis of HIGH (HL60, MOLM14) versus LOW (U937, KG1a) OXPHOS AML cell lines untreated (Table S4; Fig. S9B) or treated with metformin (Table S4). First, we showed that up-regulated genes in untreated HIGH OXPHOS compared to untreated LOW OXPHOS cell lines were involved into biological pathways such as ribosome, RNA/DNA metabolism, pyrimidine metabolism (Fig. S9C) and were specifically encoded for mitochondrial proteins involved in the TCA cycle, electron transport chain, ubiquinone Q10 biosynthesis, fatty acid metabolism and apoptotic pathways (Table S4; Fig. S9D). Using transcriptomes from metformin-treated AML cell lines, we further identified a HIGH OXPHOS gene signature of 221 transcripts (Fig. S9E; Table S4) with an enrichment of genes involved in metabolic processes (including ATP, nucleoside and glycerolipid metabolism), responses to stress, subcellular organization and cell cycle (Fig. S9F). Strikingly, this HIGH OXPHOS gene signature was also significantly enriched in the transcriptomes of RLCs from 3 AML PDX (NES=1.47; FDRq<0.0001; Fig. 5I).
In vivo chemotherapy spares pre-existing and persisting AML cells with a HIGH OXPHOS signature
Next, we performed a transcriptomic analysis of 21 AML primary samples used in this study (before xenotransplantation, Table S1) comparing low (n=10 patients) versus high (n=11 patients) responders to AraC in vivo in NSG mice (LowR vs. HighR; Fig. 5J) and identified a signature of 230 up- and 151 down- regulated genes in LowR compared to HighR (Table S5). Unexpectedly, we found that HIGH OXPHOS gene signature was also enriched in the transcriptomes of LowR in vivo (Fig. 5K). Altogether, these data strongly suggest that a HIGH OXPHOS phenotype may not only pre-exist before xenotransplantation and escape of chemotherapy but may also persist and be amplified by AraC treatment in vivo.
To test this hypothesis, we compared the ex vivo chemosensitivity of untreated AML cells and RLCs post-PDX. We observed a significant 5-to-10-fold increase in EC50 of AraC in RLCs compared to post-PBS treated mice when assayed immediately after sorting (plain symbol) and also after a subsequent 2-week culture (empty symbol; Fig. S10A). Consistent with these results, post-AraC sorted RLCs had also an increased EC50 of idarubicin compared to PBS-matched sorted AML cells from 2 cell lines and 2 patients (MOLM14, U937, Ps19, Ps22; Fig. S10B). Furthermore, we also purified high and low ROS fractions of AML cells (MOLM14, U937) in vitro and in vivo using DCF-stained cells and FACS-based sorting (Fig. S10C) and shown that high ROS cells exhibited a significantly higher EC50 of AraC compared to Low ROS cells in both PBS- and RLCs in vivo and in vitro (Fig. S10D).
Next, we determined whether RLCs have high ROS content and increased active mitochondria in vivo during (D3), immediately after the last dose of AraC treatment (D5) and after the standard D8 time-point in NSG mice. In addition to showing that AraC-induced apoptotic cell death and decreased total cell tumor burden occurred as early as D3 (Fig. S10E–F), we also observed the appearance of RLCs with an increased ROS content, mitochondrial mass and membrane potential starting at D3 (Fig. S10G–I). Of note, in this time course study, RLCs overexpressed CD36, CD44 and not CD123 (Fig. S10J–L). Selection for HIGH OXPHOS cells occurred without genetic selection as the major founder mutations are present at diagnosis in PDX throughout the same time-course (Fig. S10M–N). All of these data confirmed that in vivo chemotherapy spares pre-existing and persisting AML cells with a HIGH OXPHOS signature and CD36+CD44+ phenotype and enriches them in RLCs (Fig. 5L).
RLCs exhibit increased mitochondrial respiration and targeting HIGH OXPHOS enhances AraC chemotherapy efficacy in AML
Overall, our results strongly support the idea that mitochondrial OXPHOS activities greatly influence cytotoxicity of AraC. Interestingly, we showed that RLCs have increased mitochondrial oxygen consumption in vitro and in vivo (Fig. 6A) and an increased concentration of TCA cycle intermediates in vitro (Fig. 6B) compared to PBS-treated cells. Accordingly, we next sought to manipulate the mitochondrial energetic status toward HIGH OXPHOS or LOW OXPHOS in order to induce resistance or sensitivity to AraC, respectively. Thus, we first cultured LOW OXPHOS AML cells (U937) in presence of medium containing galactose instead of glucose as the sole sugar source, a treatment well documented to shift energetic metabolism from glycolysis to mitochondrial OXPHOS (12,37–39) (Fig. S11A). As expected, we observed an energetic shift toward HIGH OXPHOS with increased mitochondrial oxygen and pyruvate consumption in the presence of galactose and even higher values in combination with AraC (Fig. S11B), concomitant with decreased glucose consumption and lactate production (Fig. S11C), leading to a higher ATP production by mitochondria in cells grown in galactose medium (Fig. S11D–E). In addition, the shift to a HIGH OXPHOS phenotype blocked AraC-induced reduction of cell density and viability (Fig. S11F–G) as well as AraC-induced apoptotic cell death, in a dose- and time- dependent manner (Fig. S11H).
Figure 6.
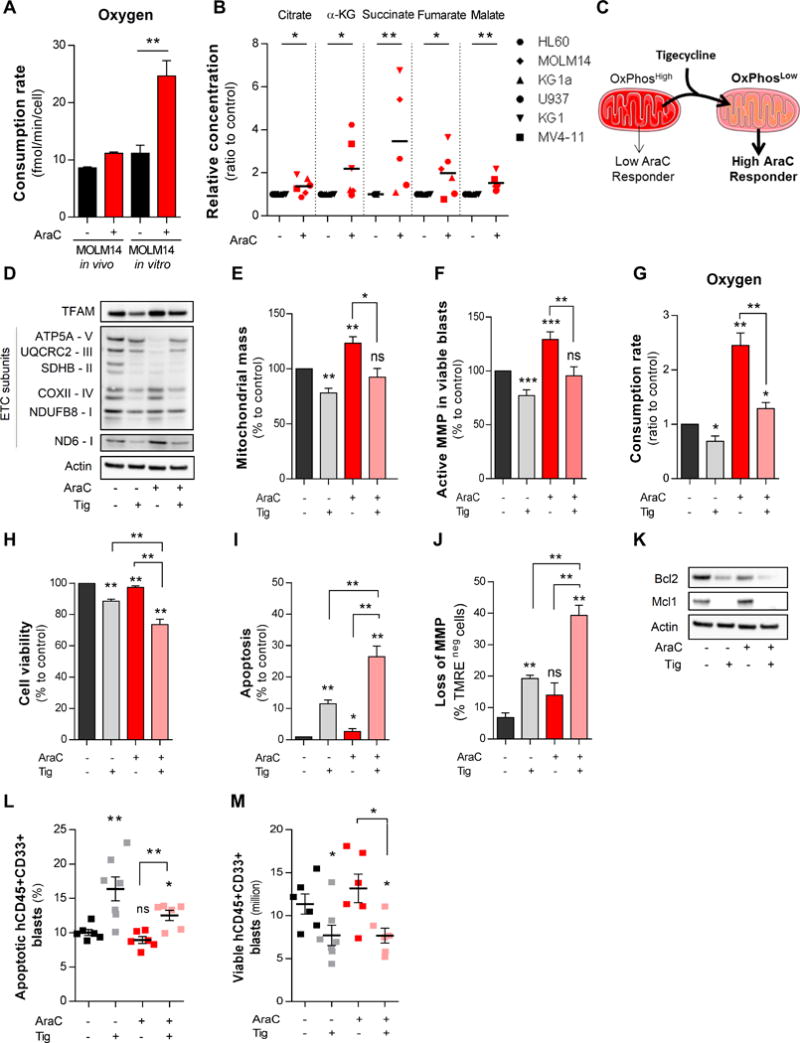
Cytarabine (AraC) residual AML cells increase mitochondrial respiration and targeting HIGH OXPHOS with tigecycline (TIG) enhances AraC chemotherapy efficacy in AML. A, Rates of oxygen consumption in HIGH OXPHOS MOLM14 cells were measured after PBS and AraC treatment both in vitro and in vivo using oxygraph with Clark electrode and Seahorse. B, Intracellular TCA cycle metabolites were quantified in PBS- or AraC-treated AML cell lines in vitro by ion chromatography coupled to mass spectrometry (IC-MS). C, Schematic diagram of TIG-induced effects on shifting HIGH OXPHOS towards LOW OXPHOS state that leads to a significant abrogation of AraC resistance in HIGH OXPHOS MOLM14 cells. D, Protein expression of mitochondrial transcription factor (mtTFAM) and oxidative phosphorylation (OXPHOS) complexes was assessed by western blot after 24h treatment with AraC (2μM) and TIG (50μM) in MOLM14 cells in vitro. E, Mitochondrial mass and F, active mitochondrial membrane potential were assessed by flow cytometry using the fluorescent MitoTracker® Green (MTG) and TMRE probes, respectively, in MOLM14 cells after PBS, TIG, AraC or TIG+AraC treatment. For each parameter the values were normalized to PBS-treated samples. G, Oxygen consumption rate in MOLM14 cells after PBS, TIG, AraC or TIG+AraC treatment. H, Cell viability after 24h of treatment were determined with trypan blue count. I, Percent of human apoptotic cells was measured in MOLM14 cells after PBS, TIG, AraC or TIG+AraC treatment using AnnexinV/7AAD-based flow cytometric assay. J, Loss of mitochondrial membrane potential was assessed in MOLM14 cells after PBS, TIG, AraC or TIG+AraC treatment by flow cytometry with TMRE probe. K, Protein expression of anti-apoptotic proteins (MCL1 and BCL2) was quantified in MOLM14 cells after PBS, TIG, AraC or TIG+AraC treatment by western blot analysis. L, Percentage of human apoptotic CD45+CD33+ AML cells was measured in MOLM14-xenografted mice after PBS, TIG, AraC or TIG+AraC treatment using AnnexinV/7AAD-based flow cytometry. M, Total cell tumor burden of viable human CD45+CD33+ AML cells was analyzed and quantified in MOLM14-xenografted mice after PBS, TIG, AraC or TIG+AraC treatment using flow cytometry. Graphs of mean ± sem. P values were determined by Mann-Whitney test. n.s., not significant. *, P≤0.05; **, P≤0.01; ***, P≤0.001.
Next, we determined whether pharmacological manipulation of HIGH OXPHOS cells toward a LOW OXPHOS phenotype would enhance cytotoxic effects of AraC in vitro and in vivo. In HIGH OXPHOS AML (MOLM14) cells, we tested pharmacological agents well known to inhibit OXPHOS activities through the inhibition of mitochondrial protein synthesis (Tigecycline, TIG (40); Ethidium Bromide, (36,41)), Electron Transport Chain complex I (ETCI; Phenformin or Metformin (42); Rotenone) or Electron Transport Chain complex III (ETCIII; antimycin A or atovaquone (43)). As expected and shown by Bozhena Jhas et al. (44), TIG-treated HIGH OXPHOS MOLM14 cells (Fig. 6C) exhibited a significantly decreased level of mitochondrial protein translation of ETCI and ETCIV (Fig. 6D), decreased mitochondrial mass and membrane potential (Fig. 6E–F) as well as oxygen consumption rate (Fig. 6G). Furthermore, TIG-treated MOLM14 cells were more sensitive to AraC than vehicle-treated non-energetically shifted MOLM14 cells through a stronger induction of apoptosis and a reduction of cell viability in vitro (Fig. 6H–K) and in vivo (Fig. 6L–M). Similar results were obtained after long-term ethidium bromide treatment used to generate mtDNA-depleted Rhozero (ρ°) cells (Fig. S12A). As compared to parental MOLM14 cells, ρ° MOLM14 cells had lower protein levels of ETCI and ETCIV complexes (Fig. S12B), which induced a LOW OXPHOS phenotype (Fig. S12C–F) and exhibited a 5-fold lower EC50 of AraC (Fig. S12G). Finally, treatments with inhibitors of ETCI (phenformin, Fig. S13A–K; metformin, Fig. S14A–I; rotenone, Fig. S15A–I) or of ETCIII (antimycin A, Fig. S16A–I; atovaquone, Fig. S17A–I) in HIGH OXPHOS MOLM14 cells yielded similar results.
High OXPHOS phenotype of RLCs is dependent on mitochondrial oxidation of fatty acids and a high CD36 signature in AML
As shown in Fig. 6A–B, AraC induced a 2.5-fold increase in OCR and increased TCA cycle intermediate concentrations. This OCR enhancement was associated with only a slight increase in pyruvate and glucose consumption and lactate production without any change in glutamine consumption in RLCs (Fig. 7A), suggesting a potential increase in mitochondrial uptake and utilization of other oxidizable substrates such as amino acids, acetate, ketones or fatty acids, which may support mitochondrial OXPHOS and respiration in the presence of AraC. Indeed, we observed an enrichment of genes involved in FA metabolism in transcriptomes of RLCs from AML PDX (Fig. 7B). Amongst the most differentially expressed genes involved in FA and lipid metabolism, we have identified FA translocase/receptor CD36 in RLCs from AML PDX (Fig. 7C) that is also markedly (21- to 60- fold) upregulated in AML cells either in bulk or LSCs from AML patients of two independent AML patient transcriptomic databases, analyzing a total of 1458 patients, as compared to normal HSCs (Fig. S18A). We confirmed that CD36 protein expression at the cell surface was also increased in viable RLCs in vitro and in vivo in 2 AML cell lines and 3 primary AML specimens (Fig. 7D). Moreover, AML patients in the TCGA cohort expressing the highest level of CD36 transcript had a poorer outcome (Fig. S18B). From those patients with the highest CD36 expression, we have generated a specific gene set (called High CD36 signature; Table S6) and performed a GSEA showing that this gene set is enriched in transcriptomes of our AML patients used in this study presenting the lower response to AraC in vivo when assayed in NSG mice (NES=1.45, FDR=0.04; bottom part of Fig. 7E) and in the transcriptomes of RLCs from AML PDX (NES=3.29, FDR<0.0001; top part of Fig. 7E). Interestingly, transcriptomes of AML patients with the higher CD36 expression are enriched in our HIGH OXPHOS gene signature (Fig. 7F) and in published gene signatures of OXPHOS, ROS, FA metabolism and FA oxidation (Fig. S18C–D) that have an adverse prognostic impact (Fig. S18E).
Figure 7.
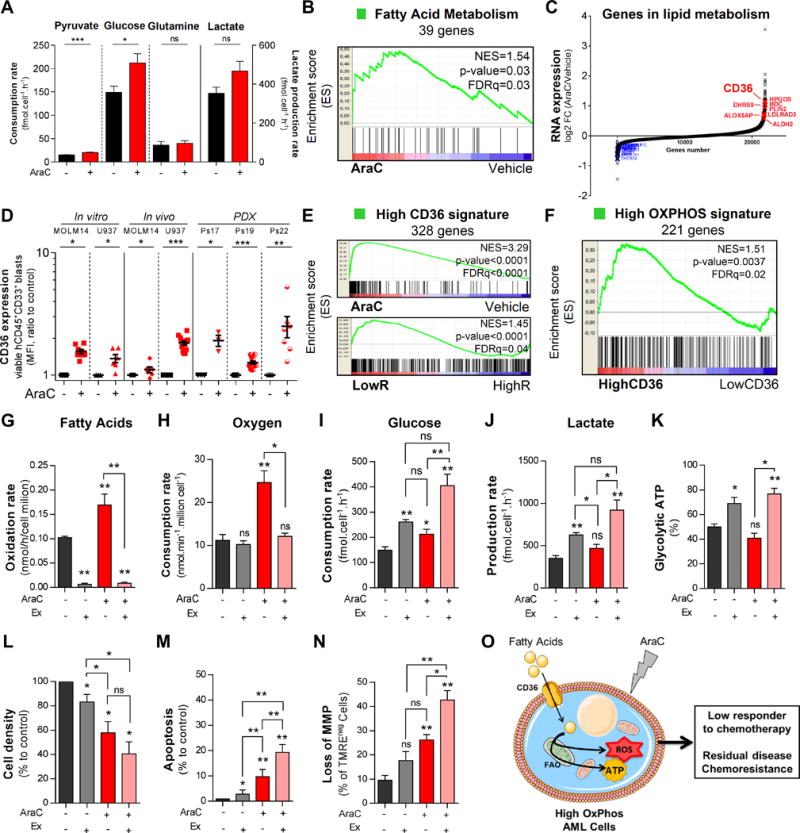
Cytarabine (AraC) residual cells increases mitochondrial fatty acid oxidation and targeting HIGH OXPHOS metabolism with Etomoxir enhances AraC chemotherapy efficacy in AML. A, Metabolomic profiling of extracellular metabolites was performed to quantify the rate of production and consumption of extracellular glucose, glutamine, pyruvate and lactate following 24h AraC (2μM) treatment in MOLM14 cells using 1D1H NMR spectra. B, Gene set enrichment analysis of fatty acid metabolism gene signature was performed with transcriptomes of viable human residual AML cells from AraC and PBS-treated AML-xenografted mice. C, Genes involved in lipid metabolism including CD36 are upregulated in AraC-residual AML cells compared to vehicle-treated AML cells in PDX. D, Cell surface expression of CD36 was analyzed and quantified in viable human CD45+CD33+ AML cells after PBS and AraC treatment from U937 and MOLM14 cell lines in vitro and in vivo, and in three different PDX using flow cytometry. E, Gene Set Enrichment Analysis of High CD36 gene signature was performed from transcriptomes of human residual AML cells purified from vehicle (PBS)- and AraC treated xenografted mice (top) and from transcriptomes of AML patients that are Low versus High Responder to AraC treatment in NSG mice (bottom). Kolmogorov-Smirnov statistical test was performed. F, Gene Set Enrichment Analysis of HIGH OXPHOS gene signature was performed from transcriptomes of AML patients that are the highest CD36 mRNA expression compared to those with the lowest expression in TCGA cohort. G, Fatty acid oxidation rate was evaluated 24h after treatment of MOLM14 cells with PBS, AraC, Etomoxir (Ex, 200μM) and AraC+Ex. This assay assessed the labeled 14CO2 after incubation with [1-14C] palmitate acid using scintillation counter and normalized to cell number. H, Oxygen Consumption Rate was measured by Clark electrode at 24h post-treatment of MOLM14 cells with PBS, AraC, Ex and AraC+Ex in vitro. H–J, Glucose consumption and lactate production rate in extracellular medium were evaluated using 1D 1H NMR spectra. K, Percentage of glycolytic ATP production were quantified in MOLM14 cells after PBS, AraC, Ex and AraC+Ex in vitro treatment by CellTiter-Glo® Assay kit using a plate reading spectrophotometer. L, Cell density was determined with trypan blue count after 24h treatment of MOLM14 cells with PBS, AraC, Ex and AraC+Ex in vitro. M, Percentage of apoptotic cells was measured after 24h treatment of MOLM14 cells with PBS, AraC, Ex and AraC+Ex using AnnexinV/7AAD-based flow cytometry. N, Loss of mitochondrial membrane potential was assessed following 24h treatment of MOLM14 cells with PBS, AraC, Ex and AraC+Ex by fluorescent TMRE probe staining using flow cytometry. O, Schematic diagram of mechanism-of-resistance of AraC based on fatty acids as key source for maintaining HIGH OXPHOS metabolism and support cell survival upon AraC treatment. Graphs of mean ± sem. P values were determined by Mann-Whitney test. n.s., not significant. *, P≤0.05; **, P≤0.01; ***, P≤0.001.
Accordingly, inhibition of FAO should be a promising chemosensitizing intervention in AML. Thus, we tested the effect of the FAO inhibitor Etomoxir, which inhibits carnitine O-palmitoyltransferase 1 (CPT1), the enzyme that catalyzes the rate-limiting step (FA shuttling through the inner mitochondrial membrane) of FAO. First we showed that, AraC alone increased FAO in MOLM14 cells (Fig. 7G), concomitant with OCR enhancement upon AraC treatment in vitro. Etomoxir inhibited FAO and OCR (p=0.0159, Fig. 7H) induced by AraC in MOLM14 cells, showing that these cells do indeed oxidize mostly FA upon AraC treatment. This treatment led to an energetic shift toward LOW OXPHOS metabolism (Fig. 7I–K), as described above with other mitochondrial and OXPHOS inhibitors, thereby potentiating of the cytotoxicity of AraC in MOLM14 cells (Fig. 7L–N). Altogether, these results suggest a new model of AraC resistance based on the role of the CD36-FAO-OXPHOS axis in AML (Fig. 7O, Table S7 and Fig. S18F).
DISCUSSION
Prognosis is very poor in AML patients due to frequent relapse caused by RLCs (1,2). However, molecular mechanisms of AML resistance are still largely unknown, especially in vivo, and new therapies, which effectively eradicate RLCs, are an urgent medical need. Previous reports hypothesized that RLCs in AML were enriched in LSCs with an immature cell phenotype and in G0 cells (9,10). However, these studies used a high dose of AraC, poorly tolerated, or did not formally assess LSC number by secondary transplantation. We have developed a clinically relevant and well-tolerated approach to deliver AraC in NSG mice engrafted with AML. This regimen reduced AML tumor burden in all mice and allows identification of high- and low- responders to AraC treatment in vivo. Importantly, this PDX model robustly correlates with clinical outcome of respective patients. AraC treatment in this pertinent PDX model induced apoptotic cell death of leukemic blasts. Furthermore, RLCs were not consistently enriched for immature cells, cells in G0 or LSCs as formally assessed by limiting dilution analysis (LDA) into secondary recipient mice, in agreement with a recent study (22). Compared to normal CD34+ cells, we further show in this study that most leukemic cells are not quiescent (G0/G1: 80–90% with 0.5–10% in G0) in PDX and AML patients, confirming previous studies by us and others in AML patients (14,18,25). However, AraC effectively kills leukemic cells, including CD34+ cells, suggesting that quiescence is not a crucial attribute necessary for AraC sensitivity in vivo. While we have used AraC as a single agent in our current studies to mimic AraC schedules and doses, most induction treatment regimens used in AML patients combine AraC with an anthracycline. However, the NSG mouse strain has a deficiency in the DNA repair enzyme, and thus, NSG mice tolerate DNA double strand breaks-inducing agents very poorly (21). Therefore, other mouse models will need to be employed to determine if anthracyclines spare LSCs or quiescent cells.
We have further shown that, as compared to chemosensitive cells, pre-existing and persisting RLCs were distinctly more resistant to drugs (e.g. AraC, IDA), exhibited an enriched HIGH OXPHOS gene signature, displayed high ROS levels with modified intracellular redox status, showed increased mitochondrial mass, and retained active, polarized, mitochondria, all features consistent with a HIGH OXPHOS status. Relevant in this regard, a previous publication also showed that AraC enhances mitochondrial activities including mitochondrial respiration and cytochrome c oxidase (45). We also demonstrated that AML cell lines with a HIGH OXPHOS status are resistant to AraC in vivo and that the HIGH OXPHOS gene signature is also enriched at diagnosis in transcriptomes of AML patients who had a lower response to AraC in NSG mice and a lower overall survival. Interestingly, the gene signature enriched in genes involved into mitochondrial functions strongly correlated with a negative prognosis index in TCGA, was also recently identified in the transcriptomes of AML cell lines less sensitive to AraC in vitro (46). We propose that, in the context of the in vivo BM niche, AraC resistance is likely determined by redox/metabolic/energetic status of AML cells, independent of their stem cell features. In agreement with previously published works (11,13–15), we can further propose that LSCs are not only heterogeneous in their cell surface immunophenotype, but also in their sensitivity to AraC in vivo. We and others have previously shown that ROS-dependent AMPK- or SUMO- signaling pathways are strongly involved in AML survival upon metabolic or genotoxic stresses, especially in hypoxic BM, but not in SP niches (47,48). Overall, our data and a growing body of evidence in the literature suggest that chemoresistance may be more strongly driven by cellular metabolism rather than by cellular quiescence or stage of maturity of CSCs.
Our results further indicate that mitochondrial and OXPHOS activities greatly influence the sensitivity and in vivo efficacy of chemotherapeutic agents. We observed that RLCs exhibited higher mitochondrial oxygen consumption that is dependent on increased TCA cycle activity and FAO, and not solely on glucose, pyruvate or glutamine consumption, and is sensitive to FAO inhibitors. A recent publication reported that adipose tissue serves as reservoir for LSCs in chronic myeloid leukemia (CML) and their respective crosstalk result in lipolysis to fuel FAO of CML cells (49). These authors further observed that in this type of myeloid leukemia, a distinct population of LSCs, which expresses CD36 and higher FAO with a quiescent and drug-resistant phenotype, are protected from chemotherapy by adipocytes (49). As CD36 is also known to have an adverse prognostic impact in AML patients (50), we found that RLCs overexpressed not only the CD36 gene and cell surface receptor but also genes involved in FA and lipid metabolism. Thus, as shown in carcinoma metastasis (51), cachexia (52) or CML (49), upregulation of CD36 might drive intracellular FA metabolism, increase FAO, maintains OXPHOS state and activity of RLCs by fueling long chain FA uptake from the extracellular BM matrix and medullar adipocytes, and protects cells from AraC. Adipocytes colonize the empty space of the BM post-AraC as observed in our histochemical analyses. Bidirectional interplay between BM adipocytes and leukemic blasts is crucial to support energy and cell survival in AML (53,54). Cachectic-like metabolic reprogramming might occur in the early time points of the post-chemotherapeutic residual disease processes in AML patients to support energy requirements for AML regrowth and relapse. Of note, a high expression of CD36 in AML patients at diagnosis is associated with a poor prognosis in TCGA and our HIGH OXPHOS gene signature is enriched in transcriptomes of AML patients who overexpressed CD36 in TCGA. Together, our data suggest that CD36 might be a robust biomarker of residual disease and of this specific metabolic state (Fig. S18F). However, prospective clinical studies will be needed to confirm these preclinical results and to examine the role of other FA transporters in AML given their molecular link with obesity and body mass index in this pathology (55).
Interestingly, Sriskanthadevan et al. (56) have shown that, while AML cells have increased mitochondrial mass and oxygen consumption compared to normal hematopoietic cells, they have a lower spare reserve capacity of their respiratory chain that renders them susceptible to oxidative or metabolic (but not genotoxic) stress. Here, we propose that metabolic flexibility as well as mitochondrial dependency are two crucial requirements of AraC resistance of AML cells in vivo. An OXPHOS-dependent energetic flexibility may be responsible for resistance to oncogene ablation or induced senescence, oxidative stress, radiation and chemotherapeutics (35,36,57–60), suggesting that resistance in cancer is associated with a shift toward a HIGH OXPHOS status. Of note, this model may explain several recent studies inducing chemosensitivity in AML cells through modulation of mitochondrial metabolism. For example, inhibition of mitochondrial fatty acid transport and β-oxidation (etomoxir or ST1326) (61–63) and inhibition of the mitochondrial protease ClpP, which interacts with respiratory chain proteins (64), alters chemosensitivity in vitro.
Although there is a broad heterogeneity in oxidative and metabolic requirements and capacities as well as mitochondrial energetics in AML cells, targeting mitochondrial OXPHOS presents a promising therapeutic avenue in AML (this study; 42,56,65). Furthermore, an important translational result would be to show that treatment of primary AML cells with a drug inhibiting OXPHOS, delivered in combination with chemotherapy would increase chemosensitivity in vivo and to translate these results to clinical applications. Importantly, initial results suggest that inhibition of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase (first-in-class anti-mitochondrial TCA cycle agent, CPI-613; (66)) may have clinical activity in patients with high risk AML.
In summary, our data show that mechanisms of resistance of AML to AraC in vivo are not contingent on quiescent LSCs, but rather depend upon both the pre-existence of resistant clones and/or the capacity of cells to adapt their energetic metabolism towards a HIGH OXPHOS phenotype in response to AraC. At diagnosis, HIGH and LOW OXPHOS AML cells coexist in variable proportions, while immediately post-chemotherapy HIGH OXPHOS cells predominantly persist and survive, especially in poor responders to AraC in vivo. Our data thus show that mitochondrial OXPHOS is a crucial contributory factor in AML chemoresistance, and a HIGH OXPHOS signature and metabolism are for the first time identified as a key hallmark of chemoresistance in vivo. Importantly, our data strongly suggest that an induced shift of human AML cells from a HIGH- to a LOW- OXPHOS status would lead to chemosensitization.
METHODS
Primary cells
Primary AML patient specimens are from two clinical sites (UPENN, University of Pennsylvania, Philadelphia, USA and TUH, Toulouse University Hospital, Toulouse, France). Frozen samples were obtained from patients diagnosed with AML at UPENN in accordance with U.S. Common Rules and at TUH after signed informed consent in accordance with the Declaration of Helsinki, and stored at the HIMIP collection (BB-0033-00060). According to the French law, HIMIP biobank collections has been declared to the Ministry of Higher Education and Research (DC 2008-307 collection 1) and obtained a transfer agreement (AC 2008-129) after approbation by the “Comité de Protection des Personnes Sud-Ouest et Outremer II” (ethical committee). Clinical and biological annotations of the samples have been declared to the CNIL (Comité National Informatique et Libertés ie Data processing and Liberties National Committee). In the USA, AML samples were obtained from the Stem Cell and Xenograft Core Facility at the University of Pennsylvania School of Medicine. Samples were obtained from patients presenting with AML at the Hospital of the University of Pennsylvania with informed consent in accordance with institutional guidelines. Peripheral blood or bone marrow samples were frozen in fetal calf serum with 10% DMSO and stored in liquid nitrogen. The percentage of blasts was determined by flow cytometry and morphological characteristics before purification.
Mice xenograft model
Animals were used in accordance to a protocol reviewed and approved by the Institutional Animal Care and Use Committee of Région Midi-Pyrénées (France) or at the University of Pennsylvania (UPENN). NOD/LtSz-scid/IL-2Rγchainnull (NSG) mice were produced at the Genotoul Anexplo platform at Toulouse (France) using breeders obtained from Charles River Laboratory. At UPENN, mice were acquired from Jackson Laboratories and bred at UPENN. Mice were housed and human primary AML cells were transplanted as reported previously (14,23,24). Briefly, mice were housed in sterile conditions using HEPA filtered micro-isolators and fed with irradiated food and sterile water. Transplanted mice were treated with antibiotic (baytril) for the duration of the experiment. Mice (6–9 weeks old) were sublethally treated with busulfan (30 mg/kg/d) 24hr before injection of leukemic cells. Leukemia samples were thawed at room temperature, washed twice in PBS, and suspended in Hanks balanced salt solution at a final concentration of 1–10 million cells per 200 μL of Hanks balanced salt solution per mouse for tail vein injection. Daily monitoring of mice for symptoms of disease (ruffled coat, hunched back, weakness and reduced mobility) determined the time of killing for injected animals with signs of distress. If no signs of distress were seen, mice were initially analyzed for engraftment 8 weeks after injection except where otherwise noted.
Cytarabine treatment
8 to 18 weeks after AML cell transplantation and when mice were engrafted (tested by flow cytometry on peripheral blood or bone marrow aspirates), NSG mice were treated by daily intraperitoneal (IP) injection of 10, 30, 60, 90 or 120 mg/kg AraC for 5 days or 1 g/kg AraC for 2 days kindly provided by the Pharmacy of the TUH (Toulouse, France). For control, NSG mice were treated daily with IP injection of vehicle, PBS 1X. Mice were monitored for toxicity and provided nutritional supplements as needed.
Assessment of leukemic engraftment
Peripheral blood was obtained at the time of first AraC dose (day 0) and at the time of dissection (day 8) two days after the last dose of AraC to determine the fraction of human blasts using flow cytometry. NSG mice were humanely killed in accordance with European ethic protocols. Bone marrow (mixed from tibias and femurs) and spleen were dissected in a sterile environment and flushed in Hanks balanced salt solution with 1% FBS. MNCs from peripheral blood, bone marrow and spleen were labeled with FITC-conjugated anti-hCD3, PE-conjugated anti-hCD33, PerCP-Cy5.5-conjugated anti-mCD45.1, APC-conjugated anti-hCD45 and PeCy7-conjugated anti-hCD44 (all antibodies from Becton Dickinson, BD, except FITC-conjugated anti-hCD3 from Ozyme Biolegend) to determine the fraction of human blasts (hCD45+mCD45.1−hCD33+hCD44+ cells) using flow cytometry. Analyses were performed on a Life Science Research II (LSR II) flow cytometer with DIVA software (BD) or Cytoflex flow cytometer with CytoExpert software (Beckman Coulter). The number of AML cells/ul peripheral blood and number of AML cells in total cell tumor burden (in bone marrow and spleen) were determined by using CountBright beads (Invitrogen) using described protocol.
Cell lines and culture conditions
Human AML cell lines were maintained in minimum essential medium-α supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) in the presence of 100 units per ml of penicillin and 100 μg/ml of streptomycin, and were incubated at 37°C with 5% CO2. The cultured cells were split every 2–3 days and maintained in an exponential growth phase. All AML cell lines were purchased at DSMZ (Braunschweig, Germany) or ATCC (Manassas, VA, USA) and their liquid nitrogen stock were renewed every two years. These cell lines have been routinely tested for Mycoplasma contamination in the laboratory. U937 was obtained from the DSMZ in February 2012 and from the ATCC in January 2014. MV4-11 and HL-60 were obtained from the DSMZ in February 2012 and February 2016. KG1 was obtained from the DSMZ in February 2012 and from the ATCC in March 2013. KG1a was obtained from the DSMZ in February 2016. MOLM14 was obtained from Pr. Martin Carroll (University of Pennsylvania, Philadelphia, USA) in 2011 and from the DSMZ in June 2015. DSMZ and ATCC cell banks provides authenticated cell lines by cytochrome C oxidase I gene (COI) analysis and short tandem repeat (STR) profiling. Furthermore, the mutation status was also verified by targeted re-sequencing of a panel of 40 genes frequently mutated in AML as described in Supplementary methods. Clinical and mutational features of our AML cell lines are described in Supplementary Table S1.
Statistical analyses
We assessed the statistical analysis of the difference between 2 sets of data using non-parametric Mann-Whitney test one-way or two-way (GraphPad Prism, GraphPad). P values of less than 0.05 were considered to be significant (* P<0.05, ** P<0.01 and *** P<0.001).
For in vitro and in vivo analyses of cytarabine residual disease, see Supplementary Methods.
Supplementary Material
SIGNIFICANCE.
Cytarabine-resistant AML cells exhibit metabolic features and gene signatures consistent with a HIGH oxidative phosphorylation (OXPHOS) status. In these cells, targeting mitochondrial metabolism through CD36-FAO-OXPHOS axis induces an energetic shift towards LOW OXPHOS and strongly enhanced anti-leukemic effects of cytarabine, offering a promising avenue to design new therapeutic strategies and fight cytarabine resistance in AML.
Acknowledgments
We thank The Cancéropoles PACA and GSO, Sonia Zaghdoudi, Pierre-Luc Mouchel, all members of mice core facilities (UMS006, ANEXPLO, Inserm, Toulouse) for their support and technical assistance, and Prof. Véronique De Mas, Cécile Demur and Eric Delabesse for the management of the Biobank BRC-HIMIP (Biological Resources Centres-INSERM Midi-Pyrénées “Cytothèque des hémopathies malignes”) that is supported by CAPTOR (Cancer Pharmacology of Toulouse-Oncopole and Région). We thank the Platform of Experimental Histophathology of INSERM/UPS, the flow cytometry core facilities of U1043-CPTP, U1048-I2MC and U1037-CRCT for technical assistance, and Anne-Marie Benot, Muriel Serthelon and Stéphanie Nevouet for their daily help about the administrative and financial management of our Team RESISTAML. We also thank Jean Delpouy, his family and friends for his generous support. The authors also thank Drs. Florence Cabon and Laurent Le Cam for critical reading of the manuscript. This work is dedicated to Prof. Christophe Cazaux.
Grant Support
This work was also supported by grants from Association Laurette Fugain (RESISTAML; J-E.S.), Fondation ARC (SFI20121205478; J-E.S.), Région Midi-Pyrénées (J-E.S.), the program “Investissement d’Avenir” IMODI (J-E.S.; O.D.), the Laboratoire d’Excellence Toulouse Cancer (TOUCAN; contract ANR11-LABEX), the Programme Hospitalo-Universitaire en Cancérologie (CAPTOR; contract ANR11-PHUC0001), INCA (PLBIO 2012-105, METAML; J-E.S., C.R.), Plan Cancer 2014-BioSys (FLEXAML; J-E.S.), Fondation Toulouse Cancer Santé (RESISTAML; C.R., J-E.S.), the Association “La confrérie de la Fève” and the Association G.A.E.L. MetaToul (Metabolomics & Fluxomics Facitilies, Toulouse, France, www.metatoul.fr) is gratefully acknowledged for carrying out metabolome analysis. MetaToul and LEMM are part of the national infrastructure MetaboHUB-ANR-11-INBS-0010 (The French National infrastructure for metabolomics and fluxomics, www.metabohub.fr). MetaToul is supported by grants from the Région Midi-Pyrénées, the European Regional Development Fund, the SICOVAL, the Infrastructures en Biologie Sante et Agronomie (IBiSa, France), the Centre National de la Recherche Scientifique (CNRS) and the Institut National de la Recherche Agronomique (INRA). M.C. is supported by the Veterans Affairs Administration (1I01BX000918-01) and National Institute of Health (1R01CA149566-01A1). F.d.T. has a fellowship from the Fondation de France.
Footnotes
Disclosure of Potential Conflict of interest
The authors declare no conflict of interest.
References
- 1.Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood. 2005;106 doi: 10.1182/blood-2005-01-0178. [DOI] [PubMed] [Google Scholar]
- 2.Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29:487–94. doi: 10.1200/JCO.2010.30.1820. [DOI] [PubMed] [Google Scholar]
- 3.Ghiaur G, Wroblewski M, Loges S. Acute myelogenous leukemia and its microenvironment: A Molecular conversation. Semin Hematol. 2015;52:200–6. doi: 10.1053/j.seminhematol.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Pallis M, Turzanski J, Higashi Y, Russell N. P-glycoprotein in acute myeloid leukaemia: therapeutic implications of its association with both a multidrug-resistant and an apoptosis-resistant phenotype. Leuk Lymphoma. 2002;43:1221–8. doi: 10.1080/10428190290026277. [DOI] [PubMed] [Google Scholar]
- 5.Buzzai M, Licht JD. New molecular concepts and targets in acute myeloid leukemia. Curr Opin Hematol. 2008;15:82–7. doi: 10.1097/MOH.0b013e3282f3ded0. [DOI] [PubMed] [Google Scholar]
- 6.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–37. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Rhenen A, Feller N, Kelder A, Westra AH, Rombouts E, Zweegman S, et al. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin Cancer Res. 2005;11:6520–7. doi: 10.1158/1078-0432.CCR-05-0468. [DOI] [PubMed] [Google Scholar]
- 8.Vergez F, Green AS, Tamburini J, Sarry J-EE, Gaillard B, Cornillet-Lefebvre P, et al. High levels of CD34+CD38low/-CD123+ blasts are predictive of an adverse outcome in acute myeloid leukemia: a Groupe Ouest-Est des Leucemies Aigues et Maladies du Sang (GOELAMS) study. Haematologica. 2011;96:1792–8. doi: 10.3324/haematol.2011.047894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315–21. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 10.Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol. 2010;28:275–80. doi: 10.1038/nbt.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–75. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- 12.Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(−) fraction. Blood. 2010;115:1976–84. doi: 10.1182/blood-2009-02-206565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–93. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 14.Sarry JE, Murphy K, Perry R, Sanchez PV, Secreto A, Keefer C, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J Clin Invest. 2011;121:384–95. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kreso A, Dick JE. Evolution of the Cancer Stem Cell Model. Cell Stem Cell. 2014;14:275–91. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Kufe DW, Major PP, Egan EM, Beardsley GP. Correlation of cytotoxicity with incorporation of ara-C into DNA. J Biol Chem. 1980;255:8900–97. [PubMed] [Google Scholar]
- 17.Kufe DW, Munroe D, Herrick D, Egan E, Spriggs D. Effects of 1-beta-D-arabinofuranosylcytosine incorporation on eukaryotic DNA template function. Mol Pharmacol. 1984;26:128–34. [PubMed] [Google Scholar]
- 18.Guan Y, Gerhard B, Hogge DE. Detection, isolation, and stimulation of quiescent primitive leukemic progenitor cells from patients with acute myeloid leukemia (AML) Blood. 2003;101:3142–9. doi: 10.1182/blood-2002-10-3062. [DOI] [PubMed] [Google Scholar]
- 19.Raaijmakers MHGP. Niche contributions to oncogenesis: emerging concepts and implications for the hematopoietic system. Haematologica. 2011;96:1041–8. doi: 10.3324/haematol.2010.028035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 21.Wunderlich M, Mizukawa B, Chou FS, Sexton C, Shrestha M, Saunthararajah Y, et al. M. cells are differentially sensitive to chemotherapy treatment in a human xenograft model, Blood. Blood. 2013;121:e90–97. doi: 10.1182/blood-2012-10-464677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griessinger E, Anjos-Afonso F, Pizzitola I, Rouault-Pierre K, Vargaftig J, Taussig D, et al. A niche-like culture system allowing the maintenance of primary human acute myeloid leukemia-initiating cells: a new tool to decipher their chemoresistance and self-renewal mechanisms. Stem Cells Transl Med. 2014;3:520–9. doi: 10.5966/sctm.2013-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanchez PV, Perry RL, Sarry JE, Perl AE, Murphy K, Swider CR, et al. A robust xenotransplantation model for acute myeloid leukemia. Leukemia. 2009;23:2109–17. doi: 10.1038/leu.2009.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saland E, Boutzen H, Castellano R, Pouyet L, Griessinger E, Larrue C, et al. A robust and rapid xenograft model to assess efficacy of chemotherapeutic agents for human acute myeloid leukemia. Blood Cancer J. 2015;5:e297. doi: 10.1038/bcj.2015.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKenzie JL, Gan OI, Doedens M, Dick JE. Reversible cell surface expression of CD38 on CD34-positive human hematopoietic repopulating cells. Exp Hematol. 2007;35:1429–36. doi: 10.1016/j.exphem.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 26.Ng SWK, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540:433–7. doi: 10.1038/nature20598. [DOI] [PubMed] [Google Scholar]
- 27.Network TCGAR. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland M-C, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112:4193–201. doi: 10.1182/blood-2008-02-134411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verhaak RGW, Wouters BJ, Erpelinck CAJ, Abbas S, Beverloo HB, Lugthart S, et al. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009;94:131–4. doi: 10.3324/haematol.13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–3. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lagadinou EDD, Sach A, Callahan K, Rossi RMM, Neering SJJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–41. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varia MA, Calkins-Adams DP, Rinker LH, Kennedy AS, Novotny DB, Fowler WC, et al. Pimonidazole: a novel hypoxia marker for complementary study of tumor hypoxia and cell proliferation in cervical carcinoma. Gynecol Oncol. 1998;71:270–7. doi: 10.1006/gyno.1998.5163. [DOI] [PubMed] [Google Scholar]
- 33.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–8. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 34.Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. 2012;22:547–60. doi: 10.1016/j.ccr.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–32. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tan ASS, Baty JWW, Dong L-F, Bezawork-Geleta A, Endaya B, Goodwin J, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015;21:81–94. doi: 10.1016/j.cmet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 37.Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy Substrate Modulates Mitochondrial Structure and Oxidative Capacity in Cancer Cells. Cancer Res. 2004;64 doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- 38.Khan AUH, Rathore MG, Allende-Vega N, Vo D-N, Belkhala S, Orecchioni S, et al. Human Leukemic Cells performing Oxidative Phosphorylation (OXPHOS) Generate an Antioxidant Response Independently of Reactive Oxygen species (ROS) Production. EBioMedicine. 2016;3:43–53. doi: 10.1016/j.ebiom.2015.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, et al. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol. 2010;28:249. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Škrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell. 2011;20:674–88. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herst PM, Levine DM, Berridge MV. Mitochondrial gene knockout HL60ρ0 cells show preferential differentiation into monocytes/macrophages. Leuk Res. 2005;29:1163–70. doi: 10.1016/j.leukres.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 42.Scotland S, Saland E, Skuli N, de Toni F, Boutzen H, Micklow E, et al. Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia. 2013;27:2129–38. doi: 10.1038/leu.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-Demonacos M, Cappello AR, et al. No Title. Oncotarget. 2016;7:34084–99. doi: 10.18632/oncotarget.9122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jhas B, Sriskanthadevan S, Skrtic M, Sukhai MA, Voisin V, Jitkova Y, et al. Metabolic Adaptation to Chronic Inhibition of Mitochondrial Protein Synthesis in Acute Myeloid Leukemia Cells. PLoS One. 2013;8:e58367. doi: 10.1371/journal.pone.0058367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muus P, Van den Bogert C, De Vries H, Pennings A, Holtrop M, Haanen C. 1-beta-D-arabinofuranosylcytosine (Ara-C) enhances mitochondrial activities in human leukaemic cells. Br J Cancer. 1991;64:29–34. doi: 10.1038/bjc.1991.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan H, Wen L, Tan D, Xie P, Pang F, Zhou H, et al. Association of a cytarabine chemosensitivity related gene expression signature with survival in cytogenetically normal acute myeloid leukemia. Oncotarget. 2016;8:1529–40. doi: 10.18632/oncotarget.13650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bossis G, Sarry J-E, Kifagi C, Ristic M, Saland E, Vergez F, et al. The ROS/SUMO Axis Contributes to the Response of Acute Myeloid Leukemia Cells to Chemotherapeutic Drugs. Cell Rep. 2014;7:1815–23. doi: 10.1016/j.celrep.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 48.Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell. 2015;17:585–96. doi: 10.1016/j.stem.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adane Ye H, Khan B, Sullivan N, Minhajuddin T, Gasparetto MM, et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell. 2016 doi: 10.1016/j.stem.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perea G, Domingo A, Villamor N, Palacios C, Juncà J, Torres P, et al. Adverse prognostic impact of CD36 and CD2 expression in adult de novo acute myeloid leukemia patients. Leuk Res. 2005;29:1109–16. doi: 10.1016/j.leukres.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 51.Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS-O, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2016;541:41–5. doi: 10.1038/nature20791. [DOI] [PubMed] [Google Scholar]
- 52.Fukawa T, Yan-Jiang BC, Min-Wen JC, Jun-Hao ET, Huang D, Qian C-N, et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat Med. 2016;22:666–71. doi: 10.1038/nm.4093. [DOI] [PubMed] [Google Scholar]
- 53.Shafat MS, Oellerich T, Mohr S, Robinson SD, Edwards DR, Marlein CR, et al. Leukemic blasts program bone marrow adipocytes to generate a pro-tumoral microenvironment. Blood. 2017 doi: 10.1182/blood-2016-08-734798. [DOI] [PubMed] [Google Scholar]
- 54.Tabe Y, Yamamoto S, Saitoh K, Sekihara K, Monma N, Ikeo K, et al. Survival of acute monocytic leukemia cells is driven by fatty acid oxidation-mediated activation of AMPK in bone marrow adipocytes. Cancer Res. 2017 doi: 10.1158/0008-5472.CAN-16-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tavitian S, Denis A, Vergez F, Berard E, Sarry A, Huynh A, et al. Impact of obesity in favorable-risk AML patients receiving intensive chemotherapy. Am J Hematol. 2016;91:193–8. doi: 10.1002/ajh.24228. [DOI] [PubMed] [Google Scholar]
- 56.Sriskanthadevan S, Jeyaraju DV, Chung TE, Prabha S, Xu W, Skrtic M, et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood. 2015;125:2120–30. doi: 10.1182/blood-2014-08-594408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 498:109–12. doi: 10.1038/nature12154. [DOI] [PubMed] [Google Scholar]
- 58.Porporato PE, Payen VL, Pérez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 8:754–66. doi: 10.1016/j.celrep.2014.06.043. [DOI] [PubMed] [Google Scholar]
- 59.Vazquez F, Lim J-H, Chim H, Bhalla K, Girnun G, Pierce K, et al. GC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 23:287–301. doi: 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ippolito L, Marini A, Cavallini L, Morandi A, Pietrovito L, Pintus G, et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget. 2016;7:61890–904. doi: 10.18632/oncotarget.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120:142–56. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Estañ MC, Calviño E, Calvo S, Guillén-Guío B, Boyano-Adánez M del C, de Blas E, et al. Apoptotic Efficacy of Etomoxir in Human Acute Myeloid Leukemia Cells. Cooperation with Arsenic Trioxide and Glycolytic Inhibitors, and Regulation by Oxidative Stress and Protein Kinase Activities. PLoS One. 2014;9:e115250. doi: 10.1371/journal.pone.0115250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ricciardi MR, Mirabilii S, Allegretti M, Licchetta R, Calarco A, Torrisi MR, et al. Targeting the leukemia cell metabolism by the CPT1a inhibition: Functional preclinical effects in leukemias. Blood. 2015;126:1925–9. doi: 10.1182/blood-2014-12-617498. [DOI] [PubMed] [Google Scholar]
- 64.Cole A, Wang Z, Coyaud E, Voisin V, Gronda M, Jitkova Y, et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell. 27:864–76. doi: 10.1016/j.ccell.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Basak NP, Banerjee S. Mitochondrial dependency in progression of acute myeloid leukemia. Mitochondrion. 2015;21:41–8. doi: 10.1016/j.mito.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 66.Pardee TS, Miller LD, Pladna K, Isom S, Ellis LR, Berenzon D, et al. TCA Cycle Inhibition By Cpi-613 Increases Sensitivity to Chemotherapy in Older and Poor Risk Acute Myeloid Leukemia (AML) Blood. 2016;128 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.