

Hypoxia reduced the firing rates of cultured neurons. Depending on hypoxic depth and duration, activity recovered during hypoxia and upon return to normoxia. Recovery (partial) during hypoxia was associated with restored baseline connections rather than newly formed ones. Predominantly, baseline connections with most active postsynaptic electrodes recovered, supporting the notion of effective activity homeostasis. This compensatory mechanism remained effective during ~20 h of hypoxia. Beyond 20 h of compensation, loss of activity and connectivity became irreversible.
Keywords: stroke, energy depletion, in vitro model, synaptic failure, recovery, activity homeostasis
Abstract
In the core of a brain infarct, loss of neuronal function is followed by neuronal death within minutes. In an area surrounding the core (penumbra), some perfusion remains. Here, neurons initially remain structurally intact, but massive synaptic failure strongly reduces neural activity. Activity in the penumbra may eventually recover or further deteriorate toward massive cell death. Besides activity recovery, return of brain functioning requires restoration of connectivity. However, low activity has been shown to initiate compensatory mechanisms that affect network connectivity. We investigated the effect of transient hypoxia and compensatory mechanisms on activity and functional connectivity using cultured cortical networks on multielectrode arrays. Networks were exposed to hypoxia of controlled depth (10–90% of normoxia) and duration (6–48 h). First, we determined how hypoxic depth and duration govern activity recovery. Then, we investigated connectivity changes during and after hypoxic incidents, mild enough for activity to recover. Shortly after hypoxia onset, activity and connectivity decreased. Following 4–6 h of ongoing hypoxia, we observed partial recovery. Only if the hypoxic burden was limited did connectivity show further recovery upon return to normoxia. Partial recovery during hypoxia was dominated by restored baseline connections, rather than newly formed ones. Baseline strengths of surviving (persisting or recovered) and lost connections did not differ nor did baseline activity at their “presynaptic” electrodes. However, “postsynaptic” electrodes of surviving connections were significantly more active during baseline than those of lost connections. This implies that recovery during hypoxia reflects an effective mechanism to restore network activity, which does not necessarily conserve prehypoxia connectivity.
NEW & NOTEWORTHY Hypoxia reduced the firing rates of cultured neurons. Depending on hypoxic depth and duration, activity recovered during hypoxia and upon return to normoxia. Recovery (partial) during hypoxia was associated with restored baseline connections rather than newly formed ones. Predominantly, baseline connections with most active postsynaptic electrodes recovered, supporting the notion of effective activity homeostasis. This compensatory mechanism remained effective during ~20 h of hypoxia. Beyond 20 h of compensation, loss of activity and connectivity became irreversible.
every year, 13 million people suffer an ischemic stroke globally, which is lethal in 30% of all patients; another one-third is left permanently disabled (Mackay and Mensah 2004). The only effective treatment to improve outcome is acute recanalization by intravenous thrombolysis (Grond et al. 1998; Wardlaw et al. 1997) or intra-arterial thrombectomy (Goyal et al. 2016; Rodrigues et al. 2016). Treatments to promote recovery of ischemic cerebral damage are lacking. Moreover, when secondary damage of brain tissue leads to additional neurological impairment, which occurs in approximately one-third of patients during the first days after the infarct, there is no therapy available (Roger et al. 2011). Occlusion of a brain artery typically results in an infarct core, with loss of neuronal functioning followed by irreversible brain damage within minutes, surrounded by a penumbral region. In the penumbra, neuronal function is severely compromised, although damage is initially reversible. During the first days, the penumbra may further deteriorate or recover. The nature of the dynamic processes leading to either of these scenarios is only partly understood.
In an in vitro model of the penumbra, we observed that even under conditions with very little remaining oxygen (10% of normoxia), neurons survived for >12 h, although their activity was severely diminished by synaptic failure (le Feber et al. 2016). If oxygen were resupplied within 12 h, then activity could be restored and functional connectivity almost completely recovered, with ~90% similarity to baseline connectivity (le Feber et al. 2016). This observation is in agreement with the notion that ATP depletion leads to synaptic failure but initially, does not structurally affect neurons or synapses (Hofmeijer and van Putten 2012).
Besides recovery after restoration of normoxia, network activity and functional connectivity showed partial recovery during hypoxia (~6–18 h after the onset of hypoxia). This probably reflects a compensatory mechanism aiming to maintain the total network activity within a certain (healthy) working range. Such so-called “homeostatic activity regulation” can be achieved by synaptic scaling: when the mean firing rate drops below a set point, excitatory synapses are upregulated (Turrigiano 2008), and inhibitory synapses are downregulated (Kilman et al. 2002). Initial local synaptic scaling has been shown to occur within 4–6 h, whereas global synaptic scaling takes place over a time period of up to 24–48 h (Turrigiano 2008). Synaptic scaling has an immediate effect on postsynaptic firing rates. Alternatively, activity homeostasis may also be achieved by growth of axons (Schmitz et al. 2009) and dendrites (Wong and Ghosh 2002) and the formation of spines and boutons (Florence et al. 1998). Such structural reorganization, however, takes place on longer time scales.
Whereas initial findings indicate the presence of activity homeostatic mechanisms, the exact mechanism and the effect on functional connectivity are unclear. Changes in functional connectivity may strongly influence brain functioning and cognition, as these largely depend on a network architecture with specific connections (Park and Friston 2013; Rychwalska 2013). In contrast, synaptic scaling is not necessarily restricted to recovery of the same synapses that constituted the prehypoxic connections.
To determine the effect of hypoxia and presumed compensatory mechanisms on functional connectivity, we investigated whether recovery of synaptic functioning during and after hypoxia is based on recovery of baseline connections or rather on formation of new ones (Fig. 1). Furthermore, we investigated which connections were preserved or recovered (further referred to as surviving connections) and which were lost. Finally, we examined whether baseline connectivity strength or baseline activity of the electrodes that constitute the connection was correlated with survival of connections.
Fig. 1.

Cartoon of activity and connectivity changes during and after hypoxia. Circles represent 6 imaginary neurons; colors, from white to yellow and red, indicate an increasing spiking frequency. A: initially, neurons had a mean baseline firing frequency and baseline functional connectivity (thicker arrows represent stronger connections). B: shortly after the onset of hypoxia, firing frequencies decreased, and functional connections became weaker or disappeared. C: then, under persisting hypoxic conditions, firing rates partially recovered. Furthermore, functional connectivity showed some recovery, some connections became stronger (black arrows), some baseline connections reappeared (blue arrow), and new connections appeared (green arrow). D: upon return to normoxia, activity and connectivity showed some further recovery. Posthypoxia connectivity partly consisted of baseline connections, but also, new connections were formed during hypoxia, whereas others were lost. We investigated to what extent new connections contributed to posthypoxia connectivity and whether survival of a baseline connection was associated to its baseline strength or to baseline activity of the neurons that span the connection.
METHODS
In Vitro Model of the Ischemic Penumbra
We developed an in vitro model of the ischemic penumbra, allowing study of individual neuronal functioning, synaptic connectivity, and consequent network functioning. It consists of a cultured cortical network on a multielectrode array (MEA) under controlled hypoxic conditions. We chose to restrict the available amount of oxygen, but not glucose, to enable direct manipulation of the available amount of ATP, without interrupting recordings or interfering with culture sterility. Manipulation of the available amount of glucose would challenge sterility, because our setup did not include a perfusion system.
The cultures contain a random mixture of all cortical cell types, including astrocytes, and self-organize into networks. Although such networks lack the typical cortical structure, network effects, such as activity-dependent neuronal survival (Ghosh et al. 1994) and synaptic scaling (Turrigiano 2012), are fully preserved. This makes the model perfectly suitable to study generic synaptic and neuronal functioning under varying conditions of hypoxia and network activity.
Cell Culture
Cortices were taken from newborn Wistar rats. The cells were mechanically dissociated by trituration and chemically by trypsin. Cells were plated on MEAs (Fig. 2). The centers were coated with polyethyleneimine, after which, a drop of growth medium was applied. The plating concentration was 1 million cells/ml, which resulted in a monolayer of cells with a density of ∼2,500 cells/mm2 after 2 days. Cultures were stored in an incubator with 5% CO2 to air mixture and near 100% humidity at 37°C. The culture medium was refreshed twice/wk, with (serum-free) R12 culturing medium (Romijn et al. 1984). Twenty-five cultures were used, which were kept in culture for 27 ± 11 days before experiments started. All cultures used in this study showed at least 30,000 action potentials in the first hour, recorded from at least 20 electrodes.
Fig. 2.

Experimental setup, protocol, and data analysis. A: dissociated cortical neurons were plated on a multielectrode array (top) and kept in culture for 27 ± 10 days before experiments started. Middle: close up of cultured cells and several electrodes. Bottom: example of recorded activity. B: after a general assessment of a culture, in which we determined whether it was active enough for experiments, and 2–3 electrodes that were able to induce network responses when stimulated electrically, cultures were subject to the outlined protocol. Six hours of normoxia were followed by 48 h of hypoxia (50, 70, or 90% normoxia) or normoxia (control recordings) and finally, at least 6 h of normoxia. During the entire experiment, cultures were stimulated at the selected electrodes during 1, 10- to 15-min period/2 h. Between stimulation periods, we recorded spontaneous activity. In a stimulation period, the electrodes were stimulated with biphasic square pulses, 200 μs/phase and 12–32 μA amplitude at a rate of 1 pulse/5 or 10 s. C: in response to stimulation at a certain electrode, all action potentials at the other electrodes were summed in 5 ms bins in the interval 0–150 ms. The top shows an example. The late phase (15–150 ms latencies) completely depended on synaptic signal propagation, and the area under the curve (bottom) was interpreted as a measure for synaptic functioning. D: example of recorded activity in periods of stimulation (left) and spontaneous activity (right). Tics indicate action potentials recorded at the indicated electrodes; the bottom curve shows summed activity in 1 s bins. Tics at electrode 60 indicate electrical stimulation. E: functional connectivity was inferred from conditional firing probabilities (CFP). CFPa,b was calculated as the probability that electrode b fired and action potential at t = τ, given that electrode a fired at t = 0. A typical example of CFPa,b is depicted.
In parallel to the cultures on MEAs, we cultured cells on polyethyleneimine-coated glass coverslips in 24-well plates (under normoxic control conditions) to quantify the ratio between the number of neurons and the number of astrocytes. These cultures were plated at the same density as cultures on MEAs. They were fixed in 4% paraformaldehyde [10 min, room temperature (RT)] after 19, 20, 21, 25, or 26 days in vitro (n = 2 for each day) and after permeabilization (0.2% Triton X-100, 5 min, RT) and blocking with 1% BSA (30 min, RT), incubated with primary antibodies: mouse anti-microtubule-associated protein 2 (MAP2; M9942, 1:200; Sigma-Aldrich, St. Louis, MO) and goat anti-glial fibrillary acidic protein (GFAP; SAB250046, 1:200; Sigma-Aldrich) overnight at 4°C. Secondary antibodies—donkey anti mouse-CF555 (SAB4600060, 1:500; Sigma-Aldrich) and donkey anti goat-488 (SAB4600032, 1:500; Sigma-Aldrich)—were incubated for 3 h at RT. Cultures were mounted in Mowiol, and each culture was photographed at three or four randomly chosen locations using a Nikon Eclipse 50i fluorescence microscope.
All procedures involving animals were conducted according to Dutch and European laws and guidelines and approved by the Dutch Animal Use Committee.
Data Recording and Induction of Hypoxia
We used MEAs and measurement setup (MEA1060-BC preamplifier and STG1002 stimulus generator), manufactured by Multi Channel Systems (Reutlingen, Germany; Fig. 2A). The MEAs had 60 electrodes that were 30 μm in diameter, spaced 200 μm apart. During measurements, MEAs were placed under a Plexiglas hood, and a humidified gas mixture of air and N2, completed with 5% CO2, was blown over the setup at a rate of 4.7 l/min. Mixtures of air and N2 could be delivered at any ratio and were computer controlled by mass flow controllers (Vögtlin Instruments, Aesch BL, Switzerland). Normoxic conditions were realized by setting the flow controllers to 100% air; hypoxic mixtures contained 10, 50, 70, or 90% air and 90, 50, 30, or 10 N2. We will refer to these conditions as 10, 50, 70, and 90% of normoxia, respectively. Cultures were sealed with a membrane permeable to O2 and CO2 but not to water. The temperature at the bottom of the MEA was kept at 36°C. Recordings were obtained at a sample rate of 16 kHz per channel using a custom LabVIEW program (National Instruments, Austin, TX), driving an NI PCI-6071E analog-to-digital convertor board. All analog signals were band-pass filtered (0.1–6 kHz) before sampling. Spikes were detected whenever the signal crossed a threshold of 5.5 times the (root mean square) noise level. Timestamps, channel numbers, and 6 ms wave shapes of each detected event were stored. Artifact detection was performed offline using an algorithm adapted from Wagenaar et al. (2005).
All experiments contained an initial normoxic period of 2–6 h (further referred to as baseline), followed by a hypoxic period of 48 h and return to normoxia of at least 6 h (Fig. 2B).
Data Analysis
Array-wide firing rate.
For the quantification of the amount of activity, we determined array-wide firing rate (AWFR) as the summed number of action potentials of all electrodes in 1 h time bins. To compare experiments, all curves were normalized to the average baseline AWFR and aligned at the onset and cessation of the hypoxic period. Changes in AWFR could arise from changes in neuronal excitability or synaptic efficacies.
Area under the curve of the synaptically mediated phase of stimulus responses.
To quantify excitatory synaptic efficacy, we stimulated cultures through one of the electrodes (biphasic current pulses, 200 μs/phase, 12–32 μA). We discriminated two phases in the poststimulus time histograms (PSTHs): the early phase (1–15 ms latencies) and the synaptically mediated phase (15–150 ms latencies). Whereas the early phase contained action potentials that were directly induced by the stimulation current and thus resistant to synaptic blockade, it may also include fast [AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)-mediated], synaptically induced action potentials (Suresh et al. 2016). Conversely, the synaptically mediated phase has been shown to disappear upon excitatory synaptic blockade (Fedorovich et al. 2017; Marom and Shahaf 2002; Wagenaar et al. 2004) and thus to depend on excitatory synaptic transmission. We calculated the area under the curve of synaptically mediated phase of stimulus responses (Asyn) as the area under the curve of the PSTH at 15–150 ms latencies (Fig. 2C). We did not further analyze the early phase of stimulus responses because of its mixed nature. Before the beginning of each experiment, we selected two to three electrodes that showed a clear network response in this latency range. Interpulse intervals were chosen between 5 and 10 s, such that subsequent stimulus responses showed no visible decline. The selected electrodes were stimulated 30 or 40 times in pseudorandom order at the beginning of every 2 h of recording, and average PSTHs were determined per electrode per 2 h (Fig. 2B).
Functional connectivity.
Spontaneous activity (Fig. 2D), recorded between the stimulation periods, was used to infer functional connectivity. Long-term recordings were divided into data blocks of 10,000 spikes each. In each data block, we calculated conditional firing probabilities for all possible electrode pairs (a,b), as explained in detail in le Feber et al. (2007). In short, given that electrode a recorded an action potential at time (t) = 0, we calculated the probability that electrode b recorded an action potential shortly thereafter, at t = latency (τ; 0 < τ < 500 ms). Curves that clearly deviated from flat [see le Feber et al. (2007) for exact criteria] indicated that two electrodes were functionally connected. In this case, the vast majority of all curves was bell shaped, similar to a (truncated) Gaussian distribution, and the height of the (main) peak was interpreted as the strength of a connection (M; Fig. 2E). Otherwise, M was set to 0. The total number of connections in each data block was determined as the number of pairs with strength M > 0.
In case of synaptic connections, electrode a would be on the presynaptic side of the connection and electrode b at the postsynaptic side. However, functional connections may also arise if two neurons share a common input. In that case, we cannot say that either electrode is at the presynaptic side. Our analysis does not discriminate between these possibilities [see le Feber et al. (2009)], and all functional connections are interpreted as if activity recorded at electrode a may (or may not) initiate a response, recorded at electrode b. For readability, we will refer to electrode a as the initiating electrode (the “presynaptic” electrode) and to electrode b as the responding (the “postsynaptic”) electrode.
To investigate the effect of hypoxia on functional connectivity strength, all strengths of individual connections were normalized to their average baseline value, as measured during the initial normoxic period of 2–6 h, averaged per culture and then averaged across cultures. This method yielded multiple connectivity matrices in the normoxic baseline phase. Every connection that was found in at least one of these baseline connectivity matrices was defined to be a baseline connection. All other connections that appeared during the experiment were considered new connections. We investigated the appearance of new connections during and after hypoxia and possible dependency on the hypoxic depth. Furthermore, we investigated which of the baseline connections disappeared during the experiment and which ones persisted in relation to their baseline strength and the baseline firing rate of both electrodes spanning the functional connection.
Determinants of Recovery
We investigated two factors that might determine which connections survive and which are lost: baseline strength of connections and baseline activity, as recorded from the two electrodes that span the functional connection (Fig. 1).
To determine whether the survival of connections was associated with their baseline strength, we compared the average baseline strength of the set of surviving connections (all baseline connections with strength M > 0 in the postbaseline data block) with that of the set of lost connections (all baseline connections with strength M = 0 in the postbaseline data block).
Alternatively, baseline activity (measured under normoxic conditions) of the two electrodes constituting a functional connection might be associated with survival. For all data blocks recorded during and after hypoxia, we determined the sets of surviving and lost connections. For both sets, we composed two lists of electrodes that included the following: 1) initiating or 2) responding in one or more connections. Then, we calculated the average baseline activity of initiating and responding electrodes in surviving and lost connections.
A lost connection did not change the sets of initiating and responding electrodes if both remained active in other connections. Therefore, we also calculated the average baseline activity of all initiating and responding electrodes, weighed by the number of functional connections that they constituted.
To eliminate the possible influence of large differences in activity between cultures, we calculated the ratio of baseline activity of electrodes associated by surviving connections to baseline activity of electrodes associated by lost connections. We calculated this ratio for initiating and responding electrodes in four phases: baseline (before the onset of hypoxia), hypox6–12 (6–12 h after the onset of hypoxia), hypox24–30 (24–30 h after the onset of hypoxia), and post hypox (0–6 h after return to normoxia). To visualize possible changes during and after hypoxia, all ratios were normalized to their baseline values. Statistical significance was evaluated using a paired t-test or two-way ANOVA, with P < 0.05 as the threshold for significance.
RESULTS
We applied 48 h of hypoxia at 90% of normoxia (n = 4 cultures), 70% (n = 4), 50% (n = 3), and 10% (n = 4). Control recordings of ~60 h at normoxia were obtained in six cultures. We combined these data with an earlier recorded data set (le Feber et al. 2016) that contained measurements before, during, and after hypoxic periods (at 10% of normoxia) of varying duration (6, 12, 24, or 48 h).
The lowering of the oxygen concentration to 10% of normoxia led to an increasingly pronounced drop in the AWFR (Fig. 3A). AWFR did not decrease any faster beyond 50% of normoxia (Fig. 3A). Figure 3B shows Asyn at these hypoxic depths.
Fig. 3.

Array-wide firing rate (AWFR), stimulus responses, number of functional connections, and average normalized connectivity strength before, during, and after hypoxic period of varying depth and duration. A–D: the effect of 48 h of hypoxia at varying depths. AWFR, summed activity of all electrodes; Asyn, area under the curve of synaptically mediated phase of stimulus responses (see Fig. 2C). Gray backgrounds indicate hypoxic periods. E and F: the effect of hypoxia (10% of normoxia) at varying durations (6, 12, 24, or 48 h). Solid parts of the curves indicate the hypoxic period; dashed parts indicate normoxia. All curves show median values; error bars reflect interquartile ranges (25–75% quartiles).
Figures 3, C–F, shows the number of functional connections and the average connection strength (normalized to baseline) in 48 h hypoxia experiments of varying depth (Fig. 3, C and D) and at 10% of normoxia in experiments of varying duration (Fig. 3, E and F). Both the number of connections as well as the average connection strength generally decreased during hypoxia. Superimposed on this general decline, most curves show some recovery in the period between 6 and 18 h, even during persisting hypoxia. If the hypoxic burden were mild enough (short or shallow), there was further recovery upon restoration of the oxygen supply. There was no significant correlation between this recovery and culture age (Pearson correlation coefficient: −0.13 < ρ < 0; P > 0.48). Additional measurements of 24 h at 10% of normoxia (n = 6; data not shown) revealed that although AWFR, stimulus responses, and functional connectivity partially recovered upon return to normoxia, this recovery gradually disappeared again between 5 and 24 h after return to normoxia in four (67%) cultures. Figure 4 summarizes the severity of induced changes in a color-coded space of hypoxic depth and duration.
Fig. 4.
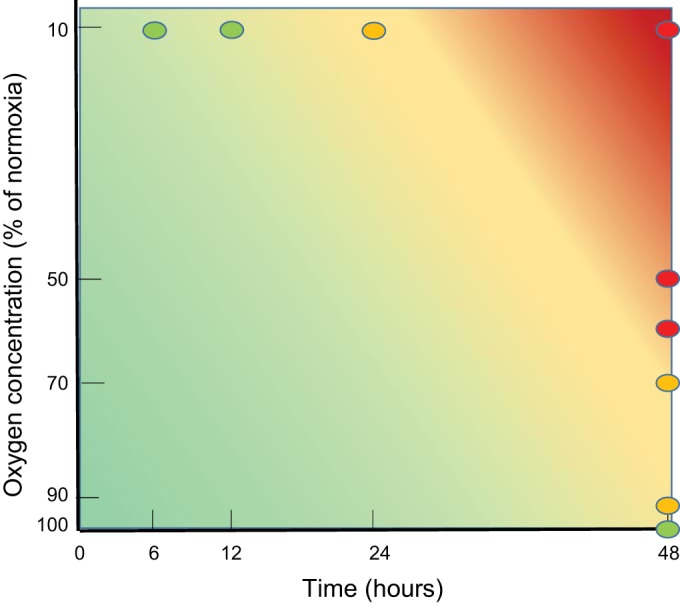
Summary of the effects of hypoxia of varying depth and duration. Changes in synaptic and neuronal functioning were reversible if the hypoxic period were sufficiently mild (short duration and/or high remaining oxygen fraction), indicated in green. More severe hypoxia or the increase of the duration of the hypoxic period (yellow) led to partially irreversible changes. Long periods of severe hypoxia induced irreversible damage, followed by massive cell death (red). Circles indicate experimentally determined effects; background represents interpolation.
Restoration of Functional Connectivity Depends on Restoration of Baseline Connections
In the vast majority of cultures, the formation of new connections (Fig. 5E) was limited compared with the number of surviving baseline connections (Fig. 5C).
Fig. 5.

Temporal development of the number of connections. A and B: under normoxic conditions, the set of connections slowly drifts. There was no difference between the baseline strengths of persisting and lost connections. The ratio of baseline strengths of persisting and lost connections during baseline does not differ from that during later phases. C and D: at 70% of normoxia, several connections are lost at approximately hour 10 (after 4 h of hypoxia), but they reappear at approximately hour 18 (after 12 h of hypoxia). The persisting or recovered connections tended to have higher baseline strength than lost ones, but this was not significant (2-way ANOVA, P > 0.21). Note differences in the vertical scale between A and C. E: the average number of new connections. Different colors indicate different hypoxic depths; the thick black line is the average. Until ~24 h after the onset of hypoxia, the number of new connections developed at a seemingly constant rate, independent of the hypoxic conditions. Beyond 30 h, the number of new connections decreased, particularly in experiments with more severe hypoxia. Error bars represent SE.
Baseline contained one to three data blocks of 2 h each. All connections that were found in at least one of the baseline data blocks were classified as baseline connections. The connections that were found in individual baseline data blocks fluctuated, and on average, only one-half of all baseline connections was found per data block of 2 h, resulting in two equally sized sets (surviving and lost) during baseline.
In normoxic control experiments (Fig. 5A), the set of surviving connections slowly decreased after baseline in favor of the set of lost connections.
Figure 5C shows the number of surviving and lost connections in the 48-h experiments at 70% of normoxia. During hypoxia, the number of surviving connections first dropped and then showed some recovery between 10 and 20 h (4–14 h after the onset of hypoxia), roughly the same period that showed recovery in the other parameters.
Survival of Connections Is Associated with Baseline Activity at the Responding Electrode
In normoxic control experiments, there was no significant difference between the baseline strength of persisting/recovered connections and lost connections (two-way ANOVA: P > 0.21), and the baseline strengths of both sets remained unchanged during the 60 h of recording (P > 0.97; Fig. 5B). In experiments that included 48 h at 70% of normoxia, baseline strengths of surviving connections were higher than those of lost connections (two-way ANOVA: P < 0.01; Fig. 5D) during baseline, as well as during all subsequent periods. The average baseline strength of the sets of surviving and lost connections did not change during either period of recovery (6–12 h after the onset of hypoxia or after return to normoxia) compared with baseline (P > 0.98).
In control experiments, the ratio between baseline activity of electrodes associated by surviving and lost connections did not change significantly in the later phases (hypox6–12, hypox24–30, and post hypox) compared with baseline (Fig. 6; paired t-test: P > 0.24 in all cases).
Fig. 6.
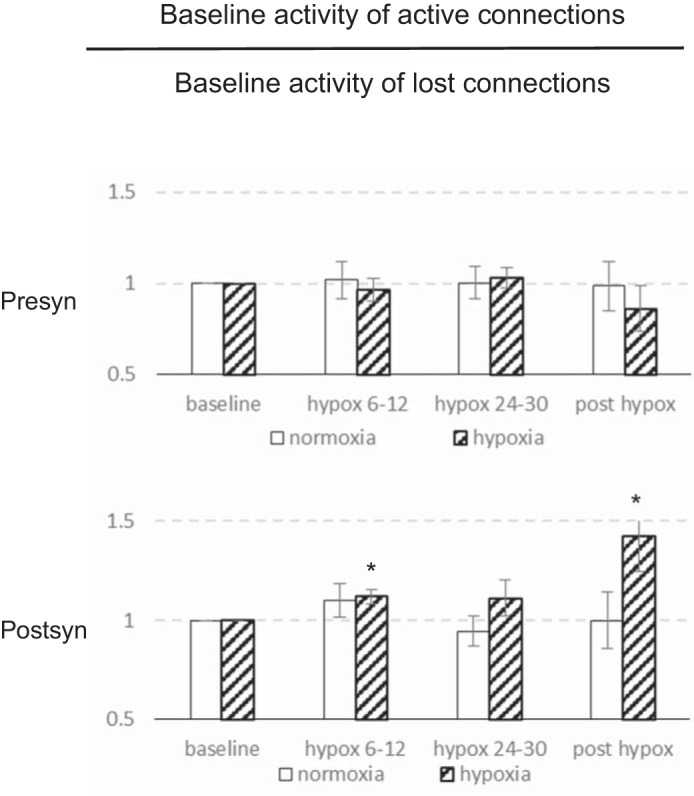
Baseline activity of electrodes in active vs. lost connections. Under control conditions, we determined the set of baseline connections, as well as the baseline activity of all associated initiating and responding electrodes. Bars indicate the baseline activity of electrodes of persisting or recovered connections (active connections), divided by the baseline activity of electrodes in lost connections. Persisting connections had more active responding electrodes and less active initiating electrodes than connections that were lost. Under control conditions (open bars) there were no significant changes with time (paired t-test: P > 0.24 in all cases). During and after hypoxia (at 60–70% of normoxia; hatched bars), persisting or recovered connections had initiating electrodes with baseline activity at the same level as during baseline, but responding electrodes had significantly higher baseline activity in persisting/recovered connections than in lost ones. Error bars reflect SE; *P < 0.05 (2-tailed paired t-test).
In two phases during (hypox6–12) and after (post hypox) hypoxia, connections with relatively active postsynaptic electrodes were preserved/recovered significantly better than those with less-active responding electrodes (paired t-test, P < 0.05). The phase in between (hypox24–30) showed a similar trend, but here, differences were not significant (P = 0.10). This analysis yielded the same results when initiating and responding electrodes were weighed by the number of functional connections that they constituted (data not shown).
Similar findings were obtained at 50% of normoxia, but only the difference in hypox6–12 was significant. At 10% of normoxia, responding electrodes of persisting connections were more active than those in lost connections, but there were no significant changes during and after hypoxia (P > 0.2).
Ratio Astrocytes:Neurons
Ten cultures were cultured on coverslips under normoxic conditions, fixed after 19–26 days in vitro, and stained for GFAP and MAP2. The ratio between GFAP-positive cells (astrocytes) and MAP2-positive cells (neurons) showed no significant trend with aging during this period. Values fluctuated between and within cultures, but on average, the ratio remained fairly constant, at (4.1 ± 2.9):1 (astrocytes:neurons).
DISCUSSION
Measurements in an in vitro model of the ischemic penumbra showed that synaptic functioning was severely hampered by the lack of oxygen, which led to rapidly decreasing activity. This activity drop was, in principle, reversible if depth and duration of the hypoxic incident were sufficiently mild. However, recovery of the mean firing rate in the penumbra is not sufficient to restore brain functioning, as this also requires restoration of functional connectivity (Park and Friston 2013; Rychwalska 2013). We investigated how hypoxia-induced reversible synaptic failure affected functional connectivity in the in vitro model.
Recovery
We observed two periods of recovery: the first between 6 and 18 h after induction of hypoxia and the second, after return to normoxia. The latter only occurred if the hypoxic period was sufficiently mild in depth and duration. Recovery of synaptic function after return to normoxia confirms the reversibility of synaptic failure upon restoration of the availability of ATP, in agreement with previous observations in hippocampal neurons (Jalini et al. 2016; Khazipov et al. 1995), in vivo hippocampus (Gao et al. 1999), and cortex (Bolay et al. 2002).
Recovery during hypoxia probably reflects some compensatory mechanism to maintain activity homeostasis. Such a mechanism is crucial for network viability, because electrical activity promotes the calcium influx-dependent survival of neurons (Ghosh et al. 1994; Mao et al. 1999). Activity homeostasis might be achieved by decreasing inhibitory activity or by rescaling of synaptic weights. Inhibitory activity might decrease due to dysfunction of inhibitory neurons and/or synapses. Although recent work (le Feber et al. 2016) suggested that inhibitory neurons are more vulnerable to hypoxia than excitatory ones, inhibitory as well as excitatory neurons remained functional and active during at least 16 h after the onset of severe hypoxia in that study. It thus seems unlikely that the inhibitory neurons lose their functionality in the period of recovery during hypoxia. The possibility that inhibition decreases, due to selective failure of inhibitory synapses, is unlikely, as experimental evidence suggests that excitatory synapses are more vulnerable to hypoxia than inhibitory ones (Khazipov et al. 1995). Still, inhibition might decrease due to dropping activation of inhibitory neurons, as suggested in a recent modeling study (Tjepkema-Cloostermans et al. 2014).
Another possible compensatory mechanism is synaptic scaling, which in agreement with current findings, occurs within hours and has an immediate effect on postsynaptic firing rates (Turrigiano 2008). The synaptically mediated phase of responses to electrical stimulation increased during perihypoxia recovery, supporting the hypothesis of upscaling of excitatory synapses. Thus synaptic scaling in response to hypoxia-induced low activity seems a plausible mechanism to explain the first period of partial recovery. Synaptic scaling involves protein synthesis and relocation for the formation of AMPA and possibly N-methyl-d-aspartate receptors (Turrigiano et al. 1998) and requires ATP. Apparently, these processes take place, even despite the energy scarcity during hypoxia. Because the origin of synaptic failure during hypoxia lies, at least partially, at the presynaptic side of the synapse (Bolay et al. 2002; Khazipov et al. 1995), synaptic scaling seems to provide postsynaptic compensation for disturbed presynaptic functioning.
Determinants of Recovery
In principle, random upregulation of excitatory synapses or downregulation of inhibitory ones might suffice to achieve activity homeostasis. The generally accepted view that low activity triggers upscaling of excitatory receptors implies that upscaling occurs in most or all excitatory synapses. Recent work, however, showed that infrequent receptor activation, rather than low network-wide activity, triggers upscaling (Fong et al. 2015). This suggests a more local phenomenon and leaves open the possibility that certain synapses are upscaled more than others. Certain preferred synapses for upscaling would lead to distinctive sets of functional connections that are lost or may recover. Formation of new, functional connections during the first recovery period occurred at an apparent constant rate, seemingly independent of the hypoxic conditions. Thus significant recovery occurred during hypoxia in connections that already existed during baseline. Still, baseline connectivity was not recovered completely, leaving the question: what determined which connections were restored and which ones were not?
Baseline strength of functional connections did not differ between persisting/recovered connections and lost connections nor did baseline activity of initiating electrodes. In contrast, baseline activity of responding electrodes of persisting/recovered connections was significantly higher than that of lost connections. Restoration of connections with the most active responding electrodes seems an effective mechanism to restore network activity and thus agrees with synaptic scaling as a presumed compensatory mechanism. The physiological mechanism behind this selection, however, remains unclear.
Implications
Neuronal networks may respond to hypoxia-induced low activity by rescaling their synapses, which may have an impact on functional connectivity. Furthermore, synaptic upscaling during hypoxia implies that a compensatory mechanism is activated, which is energy consuming. This may be countereffective under hypoxic conditions and the associated energy scarcity. Additional energy consumption further depletes the amount of ATP available for synaptic transmission. In our experiments, the beneficial effects of synaptic rescaling wore off within 18 h.
Partial recovery after 24 h of severe hypoxia was often lost during the following 24 h of normoxia. Possibly, apoptosis had been initiated during hypoxia, leading to the eventual cell death. This is in agreement with the notion that many neurons in the ischemic penumbra may undergo apoptosis after several hours or days (Broughton et al. 2009). Alternatively, the ongoing process of synaptic scaling may consume a major part of the ATP that becomes available after return to normoxia. This might severely limit the amount of ATP available for neurotransmitter release and might thus sustain the vicious circle of presynaptic failure, associated low activity, and synaptic scaling. The interaction among synaptic failure, inactivity, activity homeostasis, and apoptosis may reveal additional opportunities for therapy and needs further investigation.
Limitations
The experimental approach followed here has several limitations, including the difficult translation of partial oxygen pressures (Po2) to the in vivo situation. Po2 during normoxia in the in vivo rat brain lies at Po2 ≈ 30–35 mmHg (Grote et al. 1996; Nair et al. 1987), five times lower than normoxia in the current study and even lower than the most hypoxic oxygen pressures. Networks of dissociated cortical neurons are usually cultured under high “normoxic” conditions, and it is unclear how this relatively high Po2 under in vitro conditions relates to physiological oxygen pressure in vivo. Still, all cultures responded immediately to decreasing Po2, far before it dropped below 30–35 mmHg, indicating a clear effect of Po2 variations in the applied range, as discussed before (le Feber et al. 2016). Primary neurons obtained from the striatum have been cultured under conditions of low oxygen (down to 2%) and were shown to survive. Neurons, cultured at physiological oxygen levels found in the brain, showed larger mitochondrial networks, greater cytoplasmic fractions of mitochondria, and larger mitochondrial perimeters than those cultured at higher oxygen levels (Tiede et al. 2011), illustrating that cells adapted to low oxygen. Growing cultures under hypoxic conditions from the time of plating may lead to closer resemblance between normoxic Po2 in vivo and in vitro.
In the penumbra of a brain infarct, the availability of both glucose and oxygen is compromised. In vivo, additional processes, possibly related to the limited availability of glucose, may occur in parallel to the processes observed in our model system.
Astrocytes
Astrocytes occupy a substantial amount of space in the brain, covering for up to 50% of cerebral volume (Magistretti and Pellerin 1999). This reflects a roughly estimated average ratio of astrocytes:neurons close to one in the in vivo brain (Azevedo et al. 2009). However, estimated ratios vary widely between brain areas and even within specific areas. In the cortex, a ratio of ~4:1 has been reported (Azevedo et al. 2009). Neurons are far more susceptible to ischemic damage than astrocytes, but astrocytes provide essential metabolic support to neurons during transient ischemia (Rossi et al. 2007; Takano et al. 2009). Experiments in the hippocampus showed that during hypoxia, astrocytes contribute to regulate the excitatory synaptic transmission through the release of adenosine, which by acting on A1 adenosine receptors, reduces presynaptic transmitter release (Martín et al. 2007). Conversely, astrocytes were able to restore neuronal activity under conditions of glucose deprivation, due to lactate provided by the astrocytes (Rouach et al. 2008).
Cultures in the current study had an astrocyte:neuron ratio of ~4:1, similar to the estimated value in the in vivo cortex. Glial growth was limited by the use of a chemically defined culture medium without serum. In comparison, a ratio of (174 ± 24):1 was found in a recent study that added 10% fetal calf serum to the medium (Hacimuftuoglu et al. 2016). Still, we did not fully control the astrocyte:neuron ratio, and consequently, it varied among cultures. Given the importance of astrocytes for synaptic and neuronal functioning, this might have affected results. Astrocyte-released adenosine may have played a role in the observed excitatory synaptic depression during hypoxia. However, it seems unlikely that possible additional energy in the form of lactate produced by astrocytes could restore normoxic energy levels in the current experiments, because hypoxia hampered the oxidation of lactate in neurons as well.
In summary, the magnitude and reversibility of hypoxia-induced connectivity changes depend on the combination of hypoxic depth and duration. Posthypoxia connectivity was dominated by a subset of baseline connectivity, with set size depending on the severity of hypoxia. Functional connectivity partly recovered during hypoxia. Functional connections with relatively active postsynaptic electrodes were, most likely, to be restored, suggesting that recovery occurs locally and that it reflects an effective mechanism to restore network activity.
This mechanism appears effective during a limited period of time, where after, it may interfere with synaptic recovery. As it may also diffuse functional connectivity, this mechanism seems an ultimate effort to survive at substantial collateral costs. As such, possible therapy might aim to avoid initiation of this recovery mechanism, e.g., by electrical or pharmacological neuronal activation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.l.F., M.v.P., and J.H. conceived and designed research; N.E. performed experiments; J.l.F. and N.E. analyzed data; J.l.F. and N.E. interpreted results of experiments; J.l.F. prepared figures; J.l.F. drafted manuscript; J.l.F., M.v.P., and J.H. edited and revised manuscript; J.l.F., N.E., M.v.P., and J.H. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Gerco Hassink for the preparation of cultures and Anneloes Dummer for cell staining.
REFERENCES
- Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, Jacob Filho W, Lent R, Herculano-Houzel S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol 513: 532–541, 2009. doi: 10.1002/cne.21974. [DOI] [PubMed] [Google Scholar]
- Bolay H, Gürsoy-Ozdemir Y, Sara Y, Onur R, Can A, Dalkara T. Persistent defect in transmitter release and synapsin phosphorylation in cerebral cortex after transient moderate ischemic injury. Stroke 33: 1369–1375, 2002. doi: 10.1161/01.STR.0000013708.54623.DE. [DOI] [PubMed] [Google Scholar]
- Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke 40: e331–e339, 2009. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- Fedorovich S, Hofmeijer J, van Putten MJ, le Feber J. Reduced synaptic vesicle recycling during hypoxia in cultured cortical neurons. Front Cell Neurosci 11: 32, 2017. doi: 10.3389/fncel.2017.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florence SL, Taub HB, Kaas JH. Large-scale sprouting of cortical connections after peripheral injury in adult macaque monkeys. Science 282: 1117–1121, 1998. doi: 10.1126/science.282.5391.1117. [DOI] [PubMed] [Google Scholar]
- Fong MF, Newman JP, Potter SM, Wenner P. Upward synaptic scaling is dependent on neurotransmission rather than spiking. Nat Commun 6: 6339, 2015. doi: 10.1038/ncomms7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao TM, Pulsinelli WA, Xu ZC. Changes in membrane properties of CA1 pyramidal neurons after transient forebrain ischemia in vivo. Neuroscience 90: 771–780, 1999. doi: 10.1016/S0306-4522(98)00493-X. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science 263: 1618–1623, 1994. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Goyal M, Menon BK, van Zwam WH, Dippel DW, Mitchell PJ, Demchuk AM, Dávalos A, Majoie CB, van der Lugt A, de Miquel MA, Donnan GA, Roos YB, Bonafe A, Jahan R, Diener H-C, van den Berg LA, Levy EI, Berkhemer OA, Pereira VM, Rempel J, Millán M, Davis SM, Roy D, Thornton J, Román LS, Ribó M, Beumer D, Stouch B, Brown S, Campbell BC, van Oostenbrugge RJ, Saver JL, Hill MD, Jovin TG; HERMES Collaborators . Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet 387: 1723–1731, 2016. doi: 10.1016/S0140-6736(16)00163-X. [DOI] [PubMed] [Google Scholar]
- Grond M, Stenzel C, Schmülling S, Rudolf J, Neveling M, Lechleuthner A, Schneweis S, Heiss W-D. Early intravenous thrombolysis for acute ischemic stroke in a community-based approach. Stroke 29: 1544–1549, 1998. doi: 10.1161/01.STR.29.8.1544. [DOI] [PubMed] [Google Scholar]
- Grote J, Laue O, Eiring P, Wehler M. Evaluation of brain tissue O2 supply based on results of PO2 measurements with needle and surface microelectrodes. J Auton Nerv Syst 57: 168–172, 1996. doi: 10.1016/0165-1838(95)00096-8. [DOI] [PubMed] [Google Scholar]
- Hacimuftuoglu A, Tatar A, Cetin D, Taspinar N, Saruhan F, Okkay U, Turkez H, Unal D, Stephens RL Jr, Suleyman H. Astrocyte/neuron ratio and its importance on glutamate toxicity: an in vitro voltammetric study. Cytotechnology 68: 1425–1433, 2016. doi: 10.1007/s10616-015-9902-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmeijer J, van Putten MJ. Ischemic cerebral damage: an appraisal of synaptic failure. Stroke 43: 607–615, 2012. doi: 10.1161/STROKEAHA.111.632943. [DOI] [PubMed] [Google Scholar]
- Jalini S, Ye H, Tonkikh AA, Charlton MP, Carlen PL. Raised intracellular calcium contributes to ischemia-induced depression of evoked synaptic transmission. PLoS One 11: e0148110, 2016. doi: 10.1371/journal.pone.0148110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazipov R, Congar P, Ben-Ari Y. Hippocampal CA1 lacunosum-moleculare interneurons: comparison of effects of anoxia on excitatory and inhibitory postsynaptic currents. J Neurophysiol 74: 2138–2149, 1995. [DOI] [PubMed] [Google Scholar]
- Kilman V, van Rossum MC, Turrigiano GG. Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABA(A) receptors clustered at neocortical synapses. J Neurosci 22: 1328–1337, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Feber J, Rutten WL, Stegenga J, Wolters PS, Ramakers GJ, van Pelt J. Conditional firing probabilities in cultured neuronal networks: a stable underlying structure in widely varying spontaneous activity patterns. J Neural Eng 4: 54–67, 2007. doi: 10.1088/1741-2560/4/2/006. [DOI] [PubMed] [Google Scholar]
- le Feber J, Tzafi Pavlidou S, Erkamp N, van Putten MJ, Hofmeijer J. Progression of neuronal damage in an in vitro model of the ischemic penumbra. PLoS One 11: e0147231, 2016. doi: 10.1371/journal.pone.0147231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Feber J, van Pelt J, Rutten WL. Latency-related development of functional connections in cultured cortical networks. Biophys J 96: 3443–3450, 2009. doi: 10.1016/j.bpj.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay J, Mensah G.. The Atlas of Heart Disease and Stroke. Geneva, Switzerland: World Health Organization, 2004, p. 112. [Google Scholar]
- Magistretti PJ, Pellerin L. Astrocytes couple synaptic activity to glucose utilization in the brain. News Physiol Sci 14: 177–182, 1999. [DOI] [PubMed] [Google Scholar]
- Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 286: 785–790, 1999. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- Marom S, Shahaf G. Development, learning and memory in large random networks of cortical neurons: lessons beyond anatomy. Q Rev Biophys 35: 63–87, 2002. doi: 10.1017/S0033583501003742. [DOI] [PubMed] [Google Scholar]
- Martín ED, Fernández M, Perea G, Pascual O, Haydon PG, Araque A, Ceña V. Adenosine released by astrocytes contributes to hypoxia-induced modulation of synaptic transmission. Glia 55: 36–45, 2007. doi: 10.1002/glia.20431. [DOI] [PubMed] [Google Scholar]
- Nair PK, Buerk DG, Halsey JH. Comparisons of oxygen metabolism and tissue PO2 in cortex and hippocampus of gerbil brain. Stroke 18: 616–622, 1987. doi: 10.1161/01.STR.18.3.616. [DOI] [PubMed] [Google Scholar]
- Park H-J, Friston K. Structural and functional brain networks: from connections to cognition. Science 342: 1238411, 2013. doi: 10.1126/science.1238411. [DOI] [PubMed] [Google Scholar]
- Rodrigues FB, Neves JB, Caldeira D, Ferro JM, Ferreira JJ, Costa J. Endovascular treatment versus medical care alone for ischaemic stroke: systematic review and meta-analysis. BMJ 353: i1754, 2016. doi: 10.1136/bmj.i1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J, Roger VL, Turner MB; American Heart Association Statistics Committee and Stroke Statistics Subcommittee . Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation 123: e18–e209, 2011. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romijn HJ, van Huizen F, Wolters PS. Towards an improved serum-free, chemically defined medium for long-term culturing of cerebral cortex tissue. Neurosci Biobehav Rev 8: 301–334, 1984. doi: 10.1016/0149-7634(84)90055-1. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Brady JD, Mohr C. Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci 10: 1377–1386, 2007. doi: 10.1038/nn2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322: 1551–1555, 2008. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- Rychwalska A. Understanding Cognition Through Functional Connectivity. Berlin: Springer-Verlag, 2013. doi: 10.1007/978-3-642-31436-0_2. [DOI] [Google Scholar]
- Schmitz Y, Luccarelli J, Kim M, Wang M, Sulzer D. Glutamate controls growth rate and branching of dopaminergic axons. J Neurosci 29: 11973–11981, 2009. doi: 10.1523/JNEUROSCI.2927-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh J, Radojicic M, Pesce LL, Bhansali A, Wang J, Tryba AK, Marks JD, van Drongelen W. Network burst activity in hippocampal neuronal cultures: the role of synaptic and intrinsic currents. J Neurophysiol 115: 3073–3089, 2016. doi: 10.1152/jn.00995.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Oberheim N, Cotrina ML, Nedergaard M. Astrocytes and ischemic injury. Stroke 40, Suppl: S8–S12, 2009. doi: 10.1161/STROKEAHA.108.533166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiede LM, Cook EA, Morsey B, Fox HS. Oxygen matters: tissue culture oxygen levels affect mitochondrial function and structure as well as responses to HIV viroproteins. Cell Death Dis 2: e246, 2011. doi: 10.1038/cddis.2011.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjepkema-Cloostermans MC, Hindriks R, Hofmeijer J, van Putten MJ. Generalized periodic discharges after acute cerebral ischemia: reflection of selective synaptic failure? Clin Neurophysiol 125: 255–262, 2014. doi: 10.1016/j.clinph.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol 4: a005736, 2012. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135: 422–435, 2008. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391: 892–896, 1998. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Wagenaar DA, DeMarse TB, Potter SM. MeaBench: a toolset for multi-electrode data acquisition and on-line analysis. In: 2nd International IEEE EMBS Conference on Neural Engineering, Arlington, VA, 2005. [Google Scholar]
- Wagenaar DA, Pine J, Potter SM. Effective parameters for stimulation of dissociated cultures using multi-electrode arrays. J Neurosci Methods 138: 27–37, 2004. doi: 10.1016/j.jneumeth.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Warlow CP, Counsell C. Systematic review of evidence on thrombolytic therapy for acute ischaemic stroke. Lancet 350: 607–614, 1997. doi: 10.1016/S0140-6736(97)03022-5. [DOI] [PubMed] [Google Scholar]
- Wong RO, Ghosh A. Activity-dependent regulation of dendritic growth and patterning. Nat Rev Neurosci 3: 803–812, 2002. doi: 10.1038/nrn941. [DOI] [PubMed] [Google Scholar]