

Abstract
Objective(s):
Chronic liver disease has become a major health problem that causes serious damage to human health. Since the existing treatment effect was not ideal, we need to seek new treatment methods.
Materials and Methods:
We utilized the gene recombination technology to obtain the human hair mesenchymal stem cells which overexpression of human hepatocyte growth factor (hHGF). Furthermore, we verified the property of transfected cells through detecting surface marker by flow cytometry.
Results:
We show here establishment of the hHGF-overexpressing lentivirus vector, and successfully transfection to human hair follicle mesenchymal stem cells. The verified experiments could demonstrate the human hair follicle mesenchymal stem cells which have been transfected still have the properties of stem cells.
Conclusion:
We successfully constructed human hair follicle mesenchymal stem cells which overexpression hHGF, and maintain the same properties compared with pro-transfected cells.
Keywords: Hair follicle, HGF, Lentivirus, Liver diseases, Stem cells
Introduction
Chronic hepatic injury includes liver diseases such as hepatic fibrosis, hepatic cirrhosis and liver cancer caused by viral infection, drugs, and alcohol. According to the World Health Organization, the three most prevalent etiologies of chronic liver disease are alcohol consumption and infection by hepatitis B and C viruses (1). More than 450 million people will develop cirrhosis as a consequence of these diseases (1), which demonstrates the great relevance of liver pathologies as causes of mortality and public health expenses. Therefore, newer strategies need to be developed to actively prevent and treat chronic liver disease in a timely and effective manner.
Orthotopic liver transplantation has been one of the most effective treatment for chronic liver disease, but its clinical application is limited due to a shortage of donated organs, the cost of treatment, and the need for life-long use of immunosuppressive drug therapy after the transplantation (2). Hepatocyte transplantation provides a new alternative for treating acute and
chronic hepatic failure as well as liver-related metabolic diseases (3-5). However, this application is limited by the lack of cellular origin and difficulty of large-scale production of primary cells because of their poor proliferative capacity. In addition, the use of bioartificial liver and liver tissue engineering, both of which are potential strategies for the treatment of liver diseases, are also limited by insufficient cellular sources and do not meet the needs of the large group of patient with chronic liver disease (6, 7).
Hepatocyte growth factor (HGF) was originally identified as a nutritional factor for hepatocytes in the plasma of partially hepatectomized rats in 1980. It is now believed that HGF promotes important biological processes such as mitosis, cell motility, and cell morphogenesis, and exhibits immunomodulatory activity in various cells such as epithelial and endo-thelial cells. In addition, it prevents the regeneration of tumor cells and mediates their apoptosis (8-12). Numerous studies have indicated that HGF exerts antagonistic activity against chronic hepatic injuries induced by various factors (13-15). There are three ways of obtaining HGF for therapeutic purposes. It can be extracted from the hepatocytes of mammal animals, which is widely used in clinical and commercial applications. However, their biological background and mechanism of action are still unknown and these extracts are not biologically stable. Another method of obtaining HGF is recombinant DNA technology. Recombinant HGF has been used in animal studies, and it has been confirmed that an intravenous injection of HGF results in evident antagonistic activity and exhibits therapeutic effects in liver fibrosis induced by various factors in rats (16). However, due to the low expression of recombinant HGF and blood-derived HGF, it is difficult to extract and purify a large amount of HGF using these methods for in vivo animal experiments. In addition, due to short in vivo half-life, HGF is frequently administered to-maintain a suitable serum concen-tration in vivo, and sometimes it fails to achieve an effective therapeutic concentration despite repeated administrations.
Since the large number of blood shunt from liver to extrahepatic organs, it is difficult to achieve the effective blood drug concentration. HGF continued direct access to the liver is the key to treatment. HGF gene therapy can overcome the limitations of protein therapy and has an excellent treatment efficacy.
Hair follicles are accessory organs of the skin (17) and are derived from mesenchymal-epithelial interac-tions. Mesenchymal stem cells have been isolated from hair follicles and these mesenchymal-like stem cells have the ability to not only auto-regenerate but also differentiate into bones, cartilage, adipose tissue, vascular smooth muscle cells, and nerve cells. Studies have shown that transplanted hair follicle stem cells participate in angiogenesis, skin reconstruction, and nerve repair. There are abundant sources for hair follicles, which are easily available. The collection of hair follicles is not effected by age or gender. Furthermore, it does not result in any functional or molecular changes in the human body. In addition, hair follicles have low immunogenicity, and studies have shown that allotransplanted hair follicles can remain in the human body for a long period without inducing tumor formation.
Gene therapy refers to the process of introducing a target gene into the patient’s body by using molecular biology techniques to enable the expression of the target gene product to treat the disease. The vector and receptor cell (target cell) are both key factors in determining the efficacy of gene therapy. Various vectors have been used in gene therapy, and viral vectors are the most widely used gene delivery tools due to the high transfection rate of the mediated gene and high target specificity (18).
Due to the high antagonistic activity of HGF against chronic hepatic injuries as well as the ease of access, high proliferative capacity, and low immunogenicity of human hair follicle mesenchymal stem cells (HFMSCs), the present study employed lentivirus as the vector for genetic transduction. The human HFMSCs were also used as the target cells to transduce the HGF-secreting gene to establish a human HFMSCs bank that continuously secretes the HGF transgene. This stem cell bank provides an effective strategy for treating chronic hepatic injuries and has biological and therapeutic functions in promoting hepatocyte regeneration and alleviating hepatic fibrosis.
Materials and Methods
Acquisition of the target gene hHGF
Extraction of total mRNA
Liver tissue was collected from patients with liver cancer (Bethune First Hospital of Jilin University) and then sliced and ground into a powder in liquid nitrogen. A spatula pre-cooled in liquid nitrogen was used to transfer 50-100 mg of tissue powder to an Eppendorf (EP) tube containing 1 ml of Trizol (Takara, Japan) (note: the total volume of the tissue powder should not exceed 10% of the volume of the Trizol used) and mixed evenly. This was followed by adding 200 μl of chloroform per ml of Trizol to extract the HGF-mRNA. The optical density (OD) values were measured at 260 nm and 280 nm using a UV spectrophotometer to obtain the OD260/OD280 ratio and RNA concentration.
RT-PCR amplification of hHGF gene fragment
Reverse transcription of the first-strand of cDNA
The reaction mixture consisted of 2 μg total mRNA, 10 μl 2× TS Reaction Mix, 1 μl anchored Oligo (dT) 18 (0.5 μg/μl), 1 μ TransScript RT/RI Enzyme Mi, and ddH2O was added to make the final volume up to 20 μl. The PCR conditions were: 1 μg RNA, denaturation at 72 °C for 5 min; incubation at 42°C for 5 min, heating at 70 °C for 15 min, and TransScript RT/RI Enzyme Mix deactivation. (Takara One Step RNA PCR Kit (AMV), Japan).
Primer design
Primers with specific enzyme cleavage sites for BamH1 and XbaI were designed based on the hHGF sequence (GenBank) by using premier 5.0. The sequence for the hHGF forward primer is 5’-CGCGGATCCATGTGGGTGACCAAACTCC-3’ and the sequence for the hHGF reverse primer is 5’-TGCTCTAGA AGACAGTTGTATTGGTGGGTGCTTC-3’. The primers were synthesized by Sangon Biotech (Shanghai) Co, Ltd.
Acquisition of the hHGF gene fragment using PCR
The reaction mixture consisted of 2 μl of forward primer (10 μM), 2 μl of reverse primer (10 μM), 10 μl 5× TransStart FastPfu buffer, 1.25 μl dNTPs, 1 μl TransStart FastPfu DNA polymerase, and 1 μl Template cDNA; ddH2O was also added to obtain a final volume of 32.75 μl. PCR conditions were as follows: initial denaturation at 95 °C for 5 min for 1 cycle, followed by 95 °C for 30 sec, annealing at 62°C for 30 sec, elongation for 35 cycles at 72 °C for 2.5 min, and final elongation for 1 cycle at 72 °C for 15 min, and finally holding at 4 °C.
Identification of amplified products
Five microliters of the PCR product was collected and subjected to electrophoresis on a 1.1% agarose gel along with DL2000, and the bands of DNA were observed under an ultraviolet lamp.
Purification and recovery of PCR products of hHGF
The single target DNA band on the agarose gel was excised and the PCR products of hHGF were recovered by using the TransGen Biotech EasyPure Quick Gel Extraction Kit (Beijing, China) following manufacturer’s instructions.
Construction of the hHGF-overexpressing lentivirus vector
Acquisition and identification of hHGF-T1 by enzymatic digestion.
Ligation of hHGF with T1 vector
The PCR products of hHGF with specific restriction enzyme cleavage sites for BamH1 and XbaI were ligated to the T1 vector using a TransGen Biotech pEASY-T1 Simple Cloning Kit (Beijing China) following the manufacturer’s instructions.
Plasmid amplification
The ligated product was added to 50 μl of competent Escherichia coli and mixed evenly by gentle tapping, then placed on an ice bath for 30 min followed by incubation in a water bath at 42 °C for 30 sec for heat shock and immediately placed on ice for 2 min. The suspension was added to 250 μl of LB medium that has been equilibrated to room temperature (25 °C), and incubated at 37 °C on a rotary shaker at 300 rpm for 60 min. One hundred microliters of each of the bacterial liquid cultures was transferred onto a LB agar plate (containing 100 μg/ml ampicillin), and spread evenly over the surface of the agar with a sterile glass rod. The plates were inverted and incubated overnight at 37 °C. The transformant was streaked out, amplified, and identified the next day.
Plasmid extraction
The plasmid was extracted using a TransGen Biotech EasyPure HiPure Plasmid MiniPrep Kit (Beijing, China) following the manufacturer’s instructions.
PCR and identification of the hHGF-T1 plasmid
PCR identification of hHGF-T1 vector
PCR identification of the hHGF-T1 vector was performed using the TransGen Biotech 2× EasyTaq PCR SuperMix Kit (Beijing, China). The reaction mixture consisted of 1 μl forward primer (10 μM), 1 μl reverse primer (10 μM), 12.5 μl EasyTaq PCR SuperMix, and 1 μl hHGF-T1; ddH2O was also added to make the final volume to 9.5 μl. PCR conditions were as follows: initial denaturation at 95 °C for 5 min for 1 cycle, followed by 95 °C for 30 sec, annealing at 62 °C for 30 sec, elongation for 35 cycles at 72 °C for 2.5 min and final elongation for 1 cycle at 72 °C for 15 min and finally holding at 4 °C. cDNA and double distilled water were used as the positive and negative controls for duplicate PCR tests, respectively.
Identification of hHGF-T1 vector by single digestion and double digestion using the restriction enzymes BamHI and XbaI
A double digestion reaction mixture for HGF-T1 vector contained 1 μg of hHGF-T1, 0.5 μl BamHI, 0.5 μl XbaI, 0.2 μl of 10 μg/μl acetylated bovine serum albumin (BSA), and 2 μl 10× Buffer; ddH2O was also added to make the total volume of 20 μl. The reaction mixture was mixed gently and centrifuged for 5 to 10 sec. The single digestion reaction system for the HGF-T1 vector contained 1 μg hHGF-T1, 0.5 μl BamHI or 0.5 μl XbaI, 0.2 μl of 10 μg/μl acetylated BSA, and 2 μl 10× Buffer; ddH2O was also added to reach a total volume of 20 μl. The products of the double digestion and single digestion were subjected to agarose gel electrophoresis and the bands of DNA were visualized under UV light.
The Lv-BamHI-XbaI–GFP fragment was obtained by double digestion of the Lv-cMyc-GFP plasmid using BamHI and XbaI.
Thirty microliters of cryopreserved E. coli suspension containing the Lv-cMyc-GFP plasmid (9095-bp long, full-length Lv-cMyc-GFP plasmid with cleavage sites for the restriction enzymes BamHI and XbaI, purchased from Shanghai Genechem Co, Ltd) was added into LB medium (containing 100 μg/ml ampicillin), and incubated overnight at 37 °C in a 120 rpm shaking incubator.
Extraction of the Lv-cMyc-GFP plasmid
Refer to plasmid amplification. The Lv-cMyc-GFP plasmid was subjected to double digestion using BamHI and XbaI. The reaction mixture is the same as mentioned above.
Enzymatic digestion was performed overnight at 4 °C followed by agarose gel electrophoresis.
A long Lv-BamHI--XbaI-GFP fragment (7757 bp) with the cMyc fragment removed was obtained after double digestion. DNA bands were excised from the agarose gel and the target fragment was recovered.
HGF-T1 plasmid was subjected to double digestion using BamHI and XbaI to obtain the BamHI- hHGF- XbaI fragment
For the detailed procedure, refer to 2.2.
Construction of lentivirus vector plasmid Lv-hHGF-GFP
The Lv-hHGF-GFP4 fragment was obtained by using T4 ligase. T4 ligase was used to ligate the Lv-BamHI-XbaI–GFP and BamHI-hHGF-XbaI fragments. The reaction mixture consisted of 100 ng Lv-BamHI-XbaI–GFP, 60 ng BamHI-hHGF-XbaI, 1 U T4 DNA Ligase (Promega Madison, USA), 1 μl Ligase, and 10× Buffer; ddH2O was also added to make the total volume of 10 μl. The reaction mixture was incubated at 4 °C for 4 to 16 hr to obtain a recombinant Lv-hHGF-GFP fragment with the cMyc fragment removed.
The recombinant Lv-hHGF-GFP fragment obtained was transformed into competent E. coli and its plasmid was extracted as outlined before.
PCR and identification of the Lv-hHGF-GF plasmid was carried out as outlined before.
Gene sequencing
The constructed plasmid containing the target gene was sent to Sangon Biotech (Shanghai) Co, Ltd for DNA sequencing.
Packaging of viable virus particles in 293T cells after liposome-mediated cotransfection with four plasmids of the lentivirus vector
X-tremeGENE HP DNA Transfection Reagent (Roche, Switzerland) was used as the transducer for plasmid DNA to transfect three accessory plasmids of the lentivirus vector, pMD2.G, pMDL-g/p, and pSRV-rev, in addition to the expression plasmid containing hHGF (Lv- hHGF-GFP) into 293T cells and packaging into viable virus particles. The accessory plasmids and 293T cells were donated by Mr. Liu Jinyu, Key Laboratory of Pathobiology, Ministry of Education, Jilin University.
Liposome-mediated cotransfection with a four-plasmid system of the lentivirus vector.
Cell preparation
A day before transfection, the 293T cells were digested, counted, and transferred to 100 mm culture dishes at a concentration of 1 × 106 cells per dish and incubated overnight in 293T cell culture medium (293T cell culture medium: high glucose DMEM (Gibco, USA) containing 10% FBS (Gibco, USA), 100 U of penicillin, and 100 μg/ml streptomycin (Hyclone, USA). Transfection was performed after the cells reached 70–80% confluence.
Cotransfection of the 293T cells with the four-plasmid system
X-tremeGENE HP DNA Transfection Reagent and DNA diluents of four plasmids were equilibrated at -15 °C to -20 °C and mixed evenly by brief vortexing. The ratio of the X-tremeGENE HP DNA Transfection Regent to DNA (pMD2.G, 1.5 μg; pMDL-g/p, 5 μg; pSRV-rev, 3 μg; hHGF-GFP/GFP,15 μg) was 3:1. The transfection reagent/four-plasmid DNA mixture was incubated at 15 to 25 °C for 15 min. The culture dishes were then removed from the incubator and the transfection mixture was then added without discarding the growth medium. The expression of green fluorescent protein was observed after 24 hr.
Calculation of viral titer.
Viral titer was determined by 10-fold serial dilution of the virus stock. For cell preparation (Day 1), 293T cells in good growth state were digested, counted, and diluted to 1 × 105 cell/ml, and 100 μl of this preparation was added to each well of a 96-well plate; 10 wells were used for each virus. The 96-well plate was incubated at 37 °C in a 5% CO2 incubator. For virus inoculation (Day 2), 10-fold serial dilution was performed in EP tubes by preparing 10 successive dilutions. The dilution procedure was as follows: Ten 1.5 EP tubes were prepared for each virus; 90 μl of liquid culture was added into each tube. Then, 10 μl of the virus stock was added into the first tube and evenly mixed, followed by transferring 10 μl of the suspension from the first tube into the second tube. The same procedure was repeated to prepare ten successive dilutions (10–10-8).
The original culture medium from the 96-well plate was transferred into the diluted virus suspension and each well was marked. For addition of the culture medium (Day 3), 100 μl of the complete culture medium was added to each well to facilitate cellular growth. Results, observations, and titer calculation (Day 4) was performed as follow: the results were observed under a fluorescent microscope and the numbers of fluorescent clones in the last two wells with fluorescence were counted. If the numbers of clones are X and Y, then the titer was calculated as titer (TU/ml)= (X+Y×1) ×1000/2/content of virus suspension of well X (μl).
Isolation, culture, and identification of human hair follicle mesenchymal-like stem cells
Isolation and culture of human hair follicle mesenchymal-like stem cells.
Around 20 hairs with intact hair follicles were pulled out from the occiput region of volunteers, and after approval of the protocol by the ethics committee of our university, they were placed in a watch glass, and washed several times with phosphate buffered saline (PBS) containing penicillin and streptomycin.
The full length of the hair follicle was cut using ophthalmic scissors. Each hair follicle was gently placed in one well of a 24-well plate. One-hundred microliters of the DMED/F12 culture medium was added to each well and the 24-well plate was incubated overnight in an incubator. (The culture medium for HFMSCs is the DMED/F12 culture medium containing 10% FBS, 100 μg/ml of each penicillin and streptomycin, and 2 ng/ml bFGF).
On day 2, 400 μl of culture medium was added into each well and the culture medium was changed every three days.
The 24-well plate was observed daily for cellular growth. Cells that looked like fibroblasts gradually grew out from the terminal portion of the hair follicles.
Cells that occupied the entire well volume were trypsinized and subcultured.
Cells were grown to a large quantity for subsequent identification.
HFMSCs were induced to differentiate into fat and bone cells. Surface markers for HFMSCs were detected using flowcytometry to identify HFMSCs.
Adipogenic experiment
The HFMSCs were transplanted in a 6-well plate at a density of 4 × 104/ml and cultured in adipogenic differentiation medium containing 1 μM dexamethasone, 10 μM recombinant human insulin, 0.5 mM IBMX, and 200 μM indomethacin (Sigma, USA). To confirm the results of the adipogenic experiment, the cells were stained with oil red O on day 15 of culture to detect the formation of fat droplets inside cells.
Osteogenic experiment
Cells were transplanted in a 6-well plate at a density of 4 × 104/ml and cultured in osteogenic differentiation medium containing 100 nM dexamethasone, 5 mM sodium β-glycerophosphate, and 50 mg/ml vitamin C (Sigma, USA).
Osteogenesis was confirmed by detecting the activity of alkaline phosphatase and calcium salt deposition by using cytochemical and histochemical methods on week 2 and week 4 of culture, respectively.
FITC-marked antibodies were used to identify the surface markers in the third passage of HFMSCs by flowcytometry. Cells were cultured following standard protocol. Cells were digested when they reached 80-90% confluence, then counted and diluted to 1 × 106 cells/sample. Cells were resuspended in PBS then centrifuged for 5 min at 1000 rpm and fixed with 4% paraformaldehyde for 10 min at room temperature. Fixed cells were washed three times with PBS and centrifuged for 5 min at 1000 rpm, followed by blocking for 30 min at room temperature in 2% BSA. Cells were then incubated with mouse anti-human primary antibodies CD44, CD73, CD90, CD105, CD31, CD34, and CD45 (eBioscience, USA) (with a dilution factor of 1:100) for 1 hr at room temperature. Only primary antibody was added to the control group. Cells were washed three times with PBS, centrifuged for 5 min at 1000 rpm and incubated with 0.5 μl of FITC-marked goat anti-mouse secondary antibody in the dark (CST, USA) (with a dilution factor of 1:400) and then incubated on ice for 30 min under dark conditions. After incubation, cells were centrifuged and the secondary antibodies were removed by suction. The cells were then washed with PBS and directly loaded into a flow cytometer to detect the expression of the surface markers (CDs) of human HFMSCs.
Cell proliferation experiment
Hair follicle mesenchymal-like stem cells were inoculated in a plastic 12-well plate at a constant cell density (1 × 104 cells/well) and cultured in DMEM medium containing 10% FBS and growth factors bFGF (10 ng/ml), IGF (10 ng/ml) and EGF(10 ng/ml) or DMEM medium containing no growth factors (control). The culture medium was changed every three days. On days 1, 3, 5, 7, and 10, three wells in each group were selected to determine the absorbance values at 490 nm with a microplate absorbance reader. A cell proliferation curve was plotted by using culture time as the horizontal axis and total number of harvested cells as the vertical axis. According to the exponential growth stage shown in the cell proliferation curve, the doubling times for cells under each culture condition were calculated and compared.
Preparation of transgenic hair follicle mesenchymal stem cells overexpressing hHGF (hHGF-HFMSCs)
Cell transfection
HF-MSCs were subcultured following standard protocol. When cells reached 80–90% confluence, the culture medium was discarded and virus-containing DMEM/F12 culture medium was added. After 24 hr of transfection, the expression of green fluorescent protein was observed.
Observation of hHGF expression in HFMSCs under different viral titers
Cells from the third passage of HFMSCs were inoculated with the virus suspension. On day 7 of transduction, the expression of GFP was observed under a fluorescence microscope. Cells from the third passage of HFMSCs were inoculated with virus suspension containing the hHGF-overexpressing lentivirus, and the expression of GFP in HFMSCs under different viral titers was observed after 48 hr of viral transfection.
Western blot detection of hHGF expression in hair follicle mesenchymal tissue stem cells
hHGF-HFMSCs were collected 48 hr after transfection, mixed with a homogenization buffer and mechanically or ultrasonically homogenized at room temperature for 0.5 to 1 min. This was followed by centrifugation for 15 min at 4 °C at 13000 rpm. The supernatant was collected as the sample. (Homogenization buffer: 1.0 ml 1.0 M Tris-HCl (pH 6.8); 6.0 ml 10% SDS; 0.2 ml β- mercaptoethanol; 2.8 ml ddH2O). Western blot was used to detect hHGF using anti-HGF antibody (ab83760).
Detection of HGF expression in cell culture supernatant at different time points by ELISA
The secretion of hHGF into the supernatant of the hHGF-HFMSC culture medium was detected on days 0, 1, 3, 5, and 7 using a commercially available ELISA Kit (Invitrogen, Catalog#KAC2211). Result interpretation involved visual observation of results against a white background; the darker color in reaction well indicated the stronger positive response. No color change or very light color indicated a negative response. The degree of positivity or negativity was indicated by “+” or “-” according to the degree of lightness or darkness of the color. For OD value measurement, the zero point of the ELISA plate reader was adjusted using the solution of the blank well. The OD values of various wells were measured at 450 nm (or 410 nm if ABTS showed color change). An OD value greater than 2.1 times the OD value of the negative control indicated a positive response.
Verification of the biological activity of transgenic hHGF-HFMSCs
The transgenic hHGF-HFMSCs were re-induced to differentiate into fat and bone cells. The surface markers in the transgenic hHGF-HFMSCs were analyzed by flowcytometry to verify the biological activity of the transgenic hHGF-HFMSCs.
Results
Acquisition of the target of hHGF gene
The RNA concentration and absorbance values were determined as 520 ng/μl and 1.92, respectively, using a nucleic acid analyzer. The ratio of absorbance indicated that the RNA was not decomposed. In addition, the extracted RNA was subjected to agarose gel electrophoresis (as shown in Figure 1). Two bright bands were observed under UV light and the second band was brighter without a tail. These findings confirmed that the RNA extracted from the tissue did not decompose and could be used for subsequent experiments.
Figure 1.
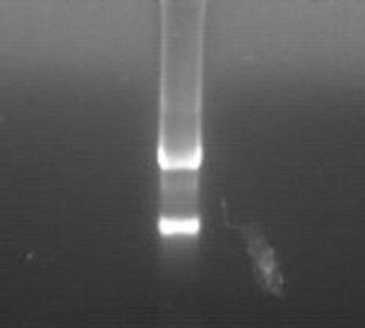
Gel electrophoresis pattern of total RNA
Reverse transcription was performed using this RNA as a template to obtain the cDNA. PCR was then performed on the cDNA using the pre-designed forward and reverse primers for hHGF with enzyme cleavage sites for BamHI and XbaI to obtain the target hHGF gene fragment (2218 bp). The PCR products were then subjected to agarose gel electrophoresis as shown in Figure 2.
Figure 2.
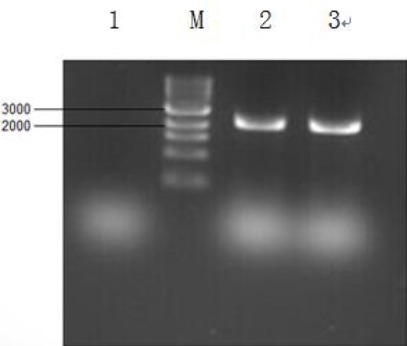
Gel electrophoresis pattern for hepatocyte growth factor (HGF) gene obtained by PCR. Reverse transcription was performed to obtain the cDNA of HGF by using total RNA extracted from liver tissue as template, followed by PCR to obtain the HGF gene. M is marker 10000; 1 is negative control, 2 and 3 are positive controls with cDNA as the template Translation for embedded text:
This experiment confirmed that the hHGF gene could be obtained from human liver tissues. Thus, a large amount of HGF gene could be obtained by repeating this experiment and scaling up the reaction system. Target hHGF fragment was recovered for subsequent experiments using a commercially available gel extraction kit by following the manufacturer’s instructions.
Construction of the hHGF-overexpressing lentivirus vector
For subsequent construction of the target plasmid, hHGF obtained from PCR was inserted into the cloning vector T1 for amplification to obtain more target gene for future experiments. The plasmid was extracted from the bacterial suspension and identified using a commercially available kit following the manufacturer’s instructions.
Briefly, the existing hHGF primer was used for PCR identification of the extracted plasmid from the E. coli mentioned above to verify the presence of HGF gene, as shown in Figure 3. Subsequently, endonucleases BamHI and XbaI were used to perform single digestion and double digestion on the plasmid to prove that the hHGF fragment has been inserted into the T1 vector. The digestion products were subjected to agarose gel electrophoresis and the results are shown in Figure 4.
Figure 3.
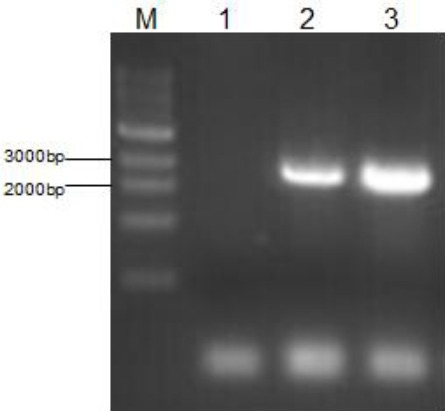
Gel electrophoresis pattern for PCR identification after binding with hepatocyte growth factor (HGF)-T1 vector
Escherichia coli plasmid infected with HGF-T1 was used as the template to perform PCR identification, verifying the presence of HGF gene in the E. coli. M is marker 10000; 1 is negative control;2 is the cDNA with HGF as the template; 3 is the plasmid extracted from infected E. coli
Figure 4.
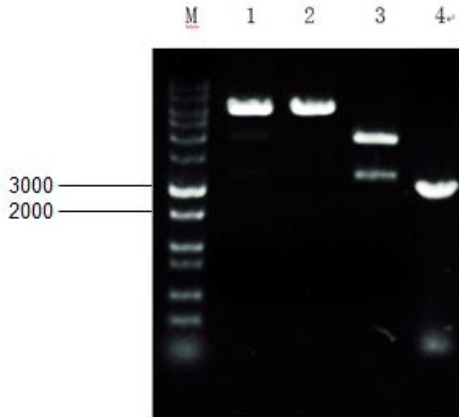
Gel electrophoresis pattern for digestion products after transduction and T1 vector binding. 1 and 2 are products of single digestion by BamHI and XbaI after T1 vector binding; 3 is the products of double digestion. M is marker 10000 Restriction Enzymes That Cleave Lv- cMyc -GFP Once
According to the enzyme cleavage sites shown in the existing Lv-cMyc-GFP plasmid structure diagram (as shown in Figure 5), two cleavage sites for restriction endonucleases BamHI and XbaI were selected and the two corresponding restriction endonucleases were used to perform double digestion on the plasmid. The digestion products were subjected to agarose gel electrophoresis and then gel extraction was performed to obtain Lv-BamHI--XbaI–GFP fragment for subsequent experiments.
Figure 5.
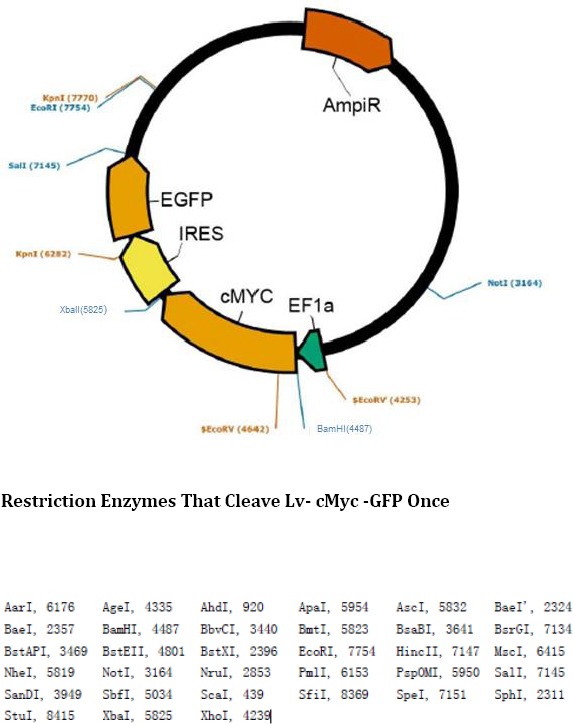
Existing Lenti- EF1a-cMyc-IRES-eGFP plasmid diagram and enzyme cleavage sites. According to the enzyme cleavage sites on the plasmid diagram, two common enzyme cleavage sites for BamH1 (4487) and XbaI (5825) were selected for double digestion
Further, the plasmid was extracted from E. coli infected with the HGF-T1 vector and subjected to double digestion using the two endonucleases BamHI and XbaI, and the digestion products were subjected to agarose gel electrophoresis followed by gel extraction to obtain the BamHI-hHGF-XbaI fragment.
Subsequently, the recovered BamHI-XbaI–GFP and the BamHI-hHGF-XbaI fragments were ligated by T4 ligase to construct the final Lv-hHGF-GFP fragment. To ensure successful construction of plasmid, the plasmid was subjected to PCR followed by single and double digestions and finally agarose gel electrophoresis, as shown in Figure 6. The results show that the length of PCR product is consistent with that of the hHGF fragment mentioned above, and that single digestion with BamHI yielded identical bands to those obtained by XbaI and indicate accurate construction of the Lv-hHGF-GFP plasmid. E. coli containing Lv-hHGF-GFP plasmid was sent to Sangon Biotech (Shanghai) Co, Ltd for gene sequencing. The sequence map is shown in Figure 7.
Figure 6.
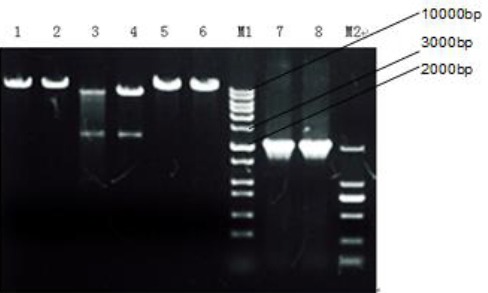
Gel electrophoresis pattern for identification of plasmid after binding with T4 ligase. 1 and 2 show bands after single digestion with XbaI while, 3 and 4 show bands after double digestion with XbaI and BamHI, 5 and 6 show bands after single digestion with BamHI, and 7 and 8 show PCR products. M1 and M2 represent marker 10000 and marker 2000, respectively
Figure 7.

Gene sequencing data after hHGF insertion and positive expression. This indicates successful construction of lentivirus plasmid expressing hHGF
To verify whether the target gene was expressed, the sequenced target plasmid containing hHGF was transfected into 293T cells using a transfection reagent, identified by western blot. Fluorescence microscopic observation showed expression of green fluorescent protein in cells at a level up to approximately 70%. Western blot showed that the protein expression gradually increased in a time dependent manner. Quantitative real time PCR indicated increased mRNA expression (P<0.001); the results are shown in Figures 8, 9 and 10. These results further validate our data on the successful construction of the hHGF-overexpressing lentivirus plasmid.
Figures 8.
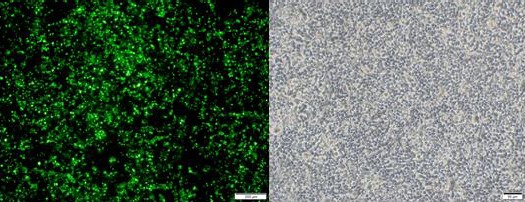
A and B are the images of 293T cells transfected with hHGF seen under a fluorescence microscope (A) and regular microscope (B) (40×)
Figure 9.
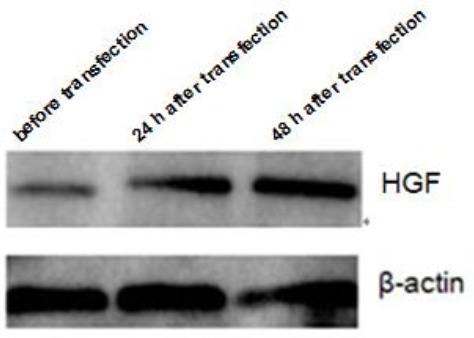
Expression level of hepatocyte growth factor (HGF) protein after transfection of target plasmid into 293T cells
Figure 10.
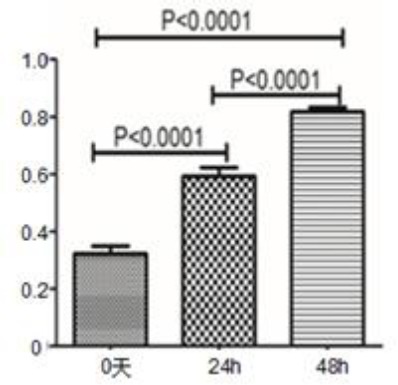
Expression of hepatocyte growth factor (HGF) mRNA after 24 hr and 48 hr following transfection of target plasmid into 293T cells
Packaging of 293T cells into viable virus particles after liposome-mediated cotransfection with four plasmids of the lentivirus vector
Since the four-plasmid system of lentivirus is safer than the three-plasmid system, we used the four-plasmid system for viral packaging. The 293T cells were infected with a certain ratio of the accessory plasmids and the target plasmid. The viral supernatant was collected after 48 and 72 hr. Figure 11 shows the images of 293T cells under bright field and fluorescence microscopes, 48 hr after transfection. This data indicates a high transfection efficiency of cells. Subsequently, based on the existing viral titer determination method, the viral titer of the packaged virus was determined to be 108 TU/ml.
Figure 11.
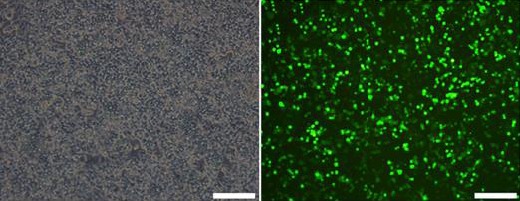
Lentivirus transfected 293 T cells that secreted the overexpressed hepatocyte growth factor (HGF) (48 hr after transfection, 40×)
Isolation, culture, and identification of human hair follicle mesenchymal-like stem cells
Human HFMSCs were cultured to the third passage using tissue digestion method. The third passage was characterized by cell adhesion, long spindle-shaped appearance, and fish school-like distribution that matched the characteristics of HFMSCs. The third passage with good activity was selected for adipogenic and osteogenic experiments to further verify successful isolation and culture of cells. After 3 weeks of induction with osteogenic differentiation medium, alizarin red staining revealed mineralized cells in reddish-brown color as shown in Figures 12 A and B. After 2 weeks of induction with adipogenic differentiation medium, oil red O stained intracellular fat droplets in bright red color, as shown in Figures 12 C and D. The cellular phenotype was verified using flowcytometry, and the results indicated that the isolated and cultured cells could express surface markers CD44, CD105, and CD90, while they hardly expressed CD34, CD45, and CD32. These characteristics matched the phenotypic characteristics of HFMSCs as shown in Figure 13. These findings prove successful isolation and culture of HFMSCs.
Figure 12.
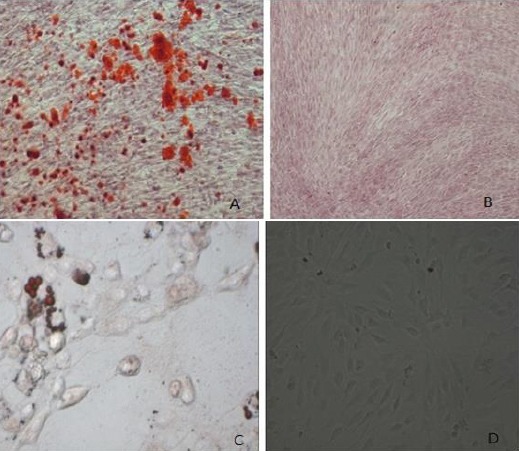
Identification of adipogenesis and osteogenesis of hair follicle mesenchymal stem cells (HFMSCs).
A: After 3 weeks of induction of HFMSCsin osteogenic differentiation medium, staining with alizarin red revealed mineralized cells in a reddish-brown color; C: After 2 weeks of induction in adipogenic differentiation medium, oil red O staining showed bright fat droplets in bright red color; B and D: Cells cultured for 2 weeks in basic medium instead of osteogenic and adipogenic differentiation media were used as the negative control. Alizarin red and oil red O staining revealed that cells did not change color. (200×)
Figure 13.
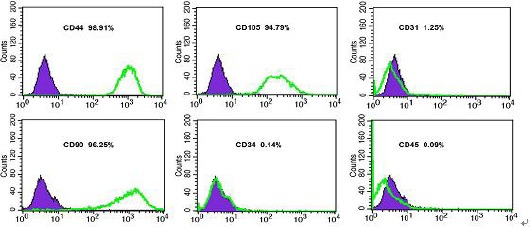
Determination of phenotype characteristics of hair follicle mesenchymal stem cells by flowcytometry
Cell proliferation experiment
To compare the effects of different growth factors on the proliferative capacity of the cells, the cells were grown in a 12-well plate and divided into three groups, which were then cultured in regular culture medium supplemented with growth factors bFGF (10 ng/ml), IGF (10 ng/ml), EGF (10 ng/ml) or culture medium containing no growth factors. On days 1, 3, 5, 7 and 10 of culture, three wells in each group were selected to perform the MTT assay, and the absorbance values (A) were determined at 490 nm with a microplate absorbance reader. The results are shown in Figure 14, indicating that the group with bFGF displayed a faster proliferation rate than the other two groups.
Figure 14.
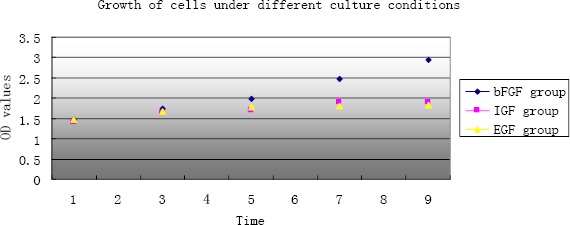
The Growth status of cells under different culture conditions
Preparation of transgenic hair follicle mesenchymal stem cells overexpressing hHGF (hHGF-HFMSCs)
HFMSCs were created for verification of the hHGF overexpression in transgenic HFMSCs. Figure 15 shows the expression of GFP 48 hr after the transfection of human HFMSCs with 100-fold dilution of packaged hHGF lentivirus. Cells from the third passage of HFMSCs were transferred into the virus suspension and subcultured. On day 7 of transfection, no change to the long spindle-shaped appearance of cells was observed under bright field microscope, and the GFP expression was found under a fluorescence microscope.
Figure 15.
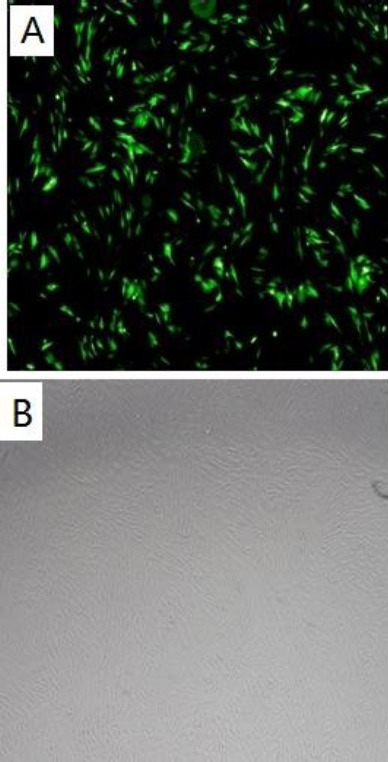
A: Image of hair follicle mesenchymal stem cells after viral transfection seen under a fluorescence microscope, B: Image of cells after transfection as seen under an inverted microscope (40×)
Western blots to detect hHGF expression in hair follicle mesenchymal tissue stem cells
Proteins from regular human HFMSCs, control group of human HFMSCs, and overexpressing human HFMSCs were collected for western blot to determine the protein expression of hHGF, as shown in Figure 16. The results show that the expression level of hHGF in overexpressing human HFMSCs was evidently higher than that in the other two groups, indicating that the hHGF-overexpressing HFMSCs constructed in this study had the ability to express hHGF.
Figure 16.
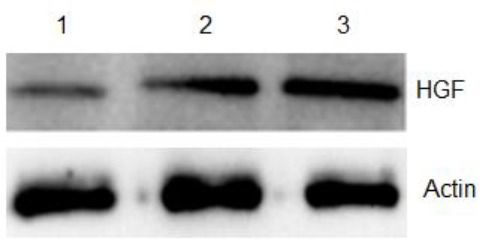
Expression level of hHGF in human hair follicle mesenchymal stem cells. 1, 2, and 3 show the protein expression of the control group, only GFP transfection G-HFMSCs group, and human hepatocyte growth factor (hHGF) lentivirus transfection hHGF-HFMSCs group, respectively
Detection of HGF in cell culture supernatant at different time points by ELISA
An ELISA assay was performed to detect the secretion and expression levels of hHGF in the supernatant of overexpressing transfected human HFMSCs. The supernatants of cells of various groups were collected for 28 days, and the hHGF levels were determined on days 0, 1, 3, 5, and 7. The secreted volume is shown in Figure 17. The results show that the expression level of hHGF in cell supernatant increased over time indicating that the human HFMSCs overexpressing hHGF created in this study could express HGF.
Figure 17.
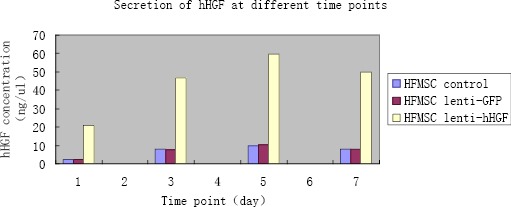
Data from ELISA assay showing the amount of human hepatocyte growth factor (hHGF) secreted into the cell culture supernatant at different time points
MTT assay was used to detect cell proliferation after transfection
The MTT assay was performed to detect the proliferation of transgenic human HFMSCs transfected by the constructed lentivirus following procedure mentioned above. The data showed no evident difference in cell proliferation between normal human HFMSCs and transgenic cells at any time point, as shown in Figure 18. This indicates that the transgenic HFMSCs retained proliferative ability following transfection.
Figure 19.
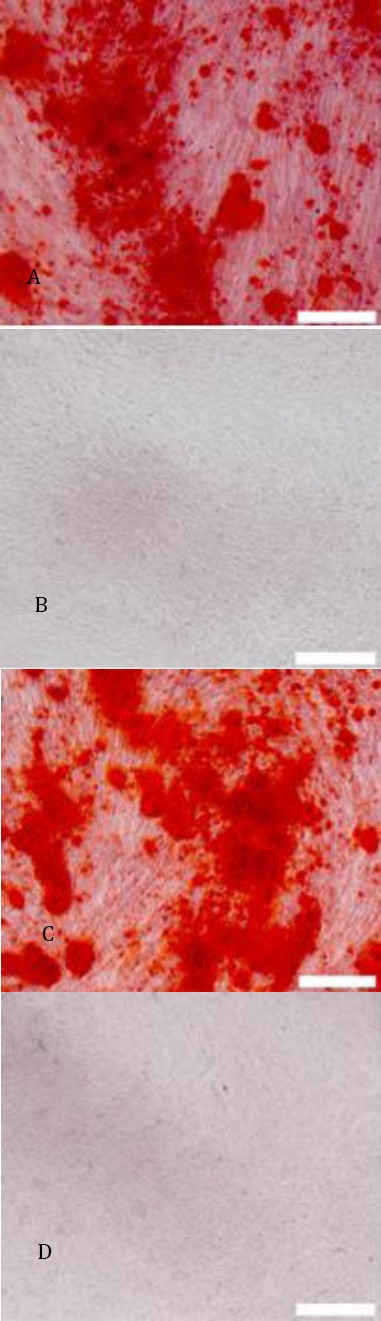
A C:hHGF-HFMSCs group and GFP-HFMSCs guoup; alizarin red staining revealed red cells at the deposition sites of calcium salt after 4 weeks of induction(40×); B D: Cells cultured for 4 weeks in basic medium instead of osteogenic differentiation media were used as the negative control. Alizarin red staining revealed that cells did not change color (40×)
Verification experiment
These experiments confirmed that human HFMSCs overexpressing HGF constructed herein possess the ability to secrete hHGF. However, it is still unknown if the lentiviral transfection affected the cells or changed the properties of the cells. Therefore, the transfected HFMSCs were induced to differentiate into bone and fat cells, and the surface markers of hHGF-HFMSCs were detected by flowcytometry to verify the bioactivity of transgenic hHGF-HFMSCs. Alizarin red staining after 4 weeks of induction of transgenic human HFMSCs with osteogenic differentiation medium revealed red cells at the deposition sites of calcium salt. After 2 weeks of induction with adipogenic differentiation medium, oil red O staining indicated the presence of red fat droplets. The phenotype of the cells was identified by flowcytometry, indicating that the transgenic cells could express surface markers CD44, CD105, and CD90, but did not express CD34, CD45, and CD31, as shown in Figures 19, 20, and 21. These results are the same as those obtained from untreated cells, indicating that the transfected cells did not change their properties and retained the characteristics of stem cells.
Figure 20.
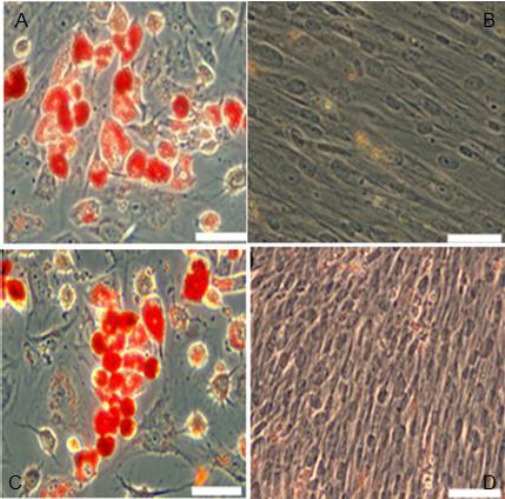
AC: hHGF-HFMSCs group and GFP-HFMSCs guoup change in cellular morphology was observed and oil red O staining revealed the presence of orange fat droplets inside cells after 2 weeks of induction; BD: Cells cultured for 2 weeks in basic medium instead of adipogenic differentiation media were used as the negative control. Oil red O staining revealed that cells did not change color (100×)
Figure 21.
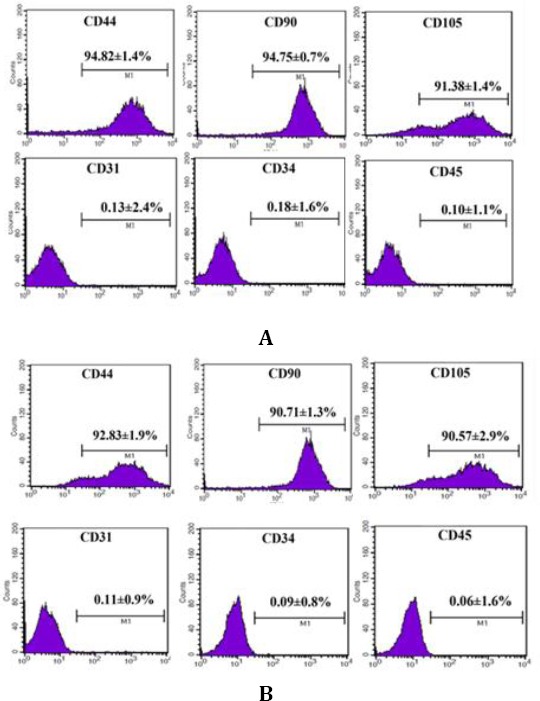
Detection of surface markers for hHGF-HFMSCs group and GFP-HFMSCs group by flow cytometry:
A: Detection of surface markers for HGF-HFMSCs group; B: Detection of surface markers for GFP-HFMSCs group
Discussion
Degeneration, necrosis, and fibrosis of liver cells are the main morphological types of chronic liver diseases. When the lesion becomes irreversible, it severely affects the liver functions and the life quality of the patients.
In the early 1980s, Nakamura et al (8) discovered that hepatocytes from adult rats had the ability to proliferate in conditioned culture medium containing low concentrations of insulin and endothelial growth factor. Subsequently, a hepatocyte nutrition factor capable of stimulating proliferation and DNA synthesis of the primary adult rat hepatocytes in plasma was identifed from 70% hepatectomized rats. This nutrition factor was named HGF (19). In 1989, the complete rat and human HGF cDNA fragments were successfully cloned (20-23). The nucleotide sequence of HGF comprises an open reading frame containing 2184 nucleotides and a 3’ non-coding region containing 3580 nucleotides. The precursor of HGF comprises 728 amino acids, which is subjected to proteolytic cleavage, resulting in mature HGF.
HGF is generated by stromal cells and has the ability to stimulate the proliferation of epithelial cells through the tyrosine phosphorylation of c-Met on the HGF receptor; it also participates in the morphogenesis of various tissues and organs, as well as in angiopoiesis (24-26). These biological actions of HGF promote the repair of injured tissues and organs.
HGF inhibits apoptosis, inflammation, and fibrosis enabling it to play a key role in the repair of hepatic injuries (27). The results of a study (28) indicated that in a model of partial hepatectomy for hepatic fibrosis introduction of HGF gene in vitro via electroporation, hepatocyte regeneration and collagen dissolution is induced accompanied by evident recovery of liver functions.
Initiation of live regeneration by blood-derived HGF was first observed in 1992. When recombinant human HGF was administrated intravenously into a mouse model of partial hepatectomy, the proliferative abilities of hepatocytes were significantly increased (29).
We extracted mRNA from human liver carcinoma tissue, obtained the HGF cDNA through RT-PCR, and performed PCR using cDNA as the template to obtain the hHGF target gene. The hHGF obtained was ligated into a cloning vector using restriction enzyme cleavage sites for amplification to obtain a large amount of target gene for subsequent experiments. In addition, we modified existing plasmids through enzymatic cleavage and selected two commonly used cleavage sites for the restriction endonucleases BamHI and XbaI to perform double digestion to cleave the cMYC fragment. The hHGF fragment obtained from PCR was ligated into the vector that had been double digested to construct the target plasmid containing the hHGF fragment. After ligation of hHGF into the vector and its positive expression, gene sequencing was performed to confirm the successful construction of the lentivirus plasmid expressing hHGF. At 24 hr and 48 hr after transfection, the analysis of protein and mRNA levels indicated that the expression level of hHGF in the constructed target plasmid continuously increased and the target plasmid had the ability to express hHGF, confirming the successful construction of the plasmid.
Common viral vectors include adenoviruses, retroviruses, and lentiviruses. Lentiviruses have significant advantages over other viral vectors. They have the ability to infect non-dividing cells, accommodate large exogenous gene fragments, and allow for long-term expression. In addition, lentiviruses do not cause any effective cellular immune response, so they can be used as an in vitro gene delivery tool. Lentivirus vector-mediated transgenic expression can last up to several months without any observable pathological signs. Furthermore, most of the previous lentivirus vectors were three-plasmid systems. The packaging plasmids of a three-plasmid system contain all accessory genes (vif, vpr, vpu and nef genes) of HIV; under certain circumstances, all the viral proteins might be transcribed into RNA, putting the operator at a risk of infection. The four-plasmid system does not have such risk. Therefore, a lentivirus four-plasmid system was selected as the gene vector for experiments.
Target cell selection is an important part of gene therapy. The frequently studied target cells include mesenchymal stem cells derived from the bone marrow, blood, and fat. Hu JJ et al. (30) constructed lentiviral plasmids containing the HGF gene and packaged the plasmid into viable viral particles. The plasmid was then transduced into rat bone marrow mesenchymal stem cells. Lentiviral particles can continuously express HGF at high levels in bone marrow mesenchymal stem cells. Transplantation of these transgenic mesenchymal stem cells into a mouse model of liver fibrosis promoted the recovery and alleviation of hepatic fibrosis, suggesting the feasibility of the gene therapy platform systems and the overall effectiveness of gene therapy. However, due to limited sources, low availability, low proliferative capacity, and immune response initiated by the bone marrow, and hematopoietic and fat mesenchymal stem cells, the use of these mesenchymal stem cells as target cells to treat chronic hepatic injuries has resulted in unsatisfactory results in practical applications. Therefore, a critical issue that needs to be addressed for successful gene therapy is to identify target cells that have a high proliferative capacity, low immunogenicity, and are suitable for transplantation treatment of different individuals.
Some essential characteristics of such cells would be an abundant source, high availability, high proliferative capacity, and low immunogenicity. Using hair follicle stem cells as seed cells for regenerative medicine studies can solve the practical problems associated with the studies based on embryonic or other adult stem cells such as limited source, low availability, ethical issues, and induction of neoplasia. Therefore, as a kind of novel seed cell, hair follicle stem cells have irreplaceable features and advantages over other stem cells and have great potential for use in the study and application of regenerative medicine.
We have successfully isolated and cultured human HFMSCs in this study. The expression of CD44, CD90, CD105, CD31, CD34, and CD45 in human HFMSCs was detected by using immunofluorescence in combination with flowcytometry, proving that the cultured human HFMSCs had phenotypic characte-ristics similar to human HFMSCs. Using histochemi-cal and immunochemical methods, we determined the osteogenic and adipogenic capacity of human hair follicle stem cells. This confirmed that human HFMSCs possess the capacity to proliferate as well as to differentiate. The hHGF-containing virus was transfected into HFMSCs and its protein expression was determined to be higher than that before transfection. On days 0, 1, 3, 5, and 7, the cell culture supernatant was assayed and the expression of HGF in the supernatant also increased. This indicated successful transfection and the ability of the infected cells to overexpress hHGF.
We successfully used a lentivirus vector that has high transfection efficiency, high target specificity, mild immune response, and the ability to express HGF for a long period as the vector for gene transduction. Further, we employed human HFMSCs that have abundant sources, high availability, and low immunogenicity, and that can be used as a universal source of seed cells as target cells for gene therapy to construct a human HFMSC bank that continuously secretes transgenic hHGF. This can be used for future stem cell-based gene therapy studies. This study provided a solid foundation for creating transgenic human HFMSCs with biological and therapeutic functions such as secreting HGF to promote liver cell regeneration and alleviating hepatic fibrosis.
Conclusion
We successfully constructed human hair follicle mesenchymal stem cells which overexpression hHGF, and maintain the same properties compared with pro-transfected cells.
Acknowledgment
This project was supported by grants from the Provincial Science & Technology Department, China (No.201201042).
Conflict of interests
The authors declare that there are no conflicts of interests related to the trademarks mentioned in this paper.
References
- 1.Neuberger J. An update on liver transplantation: A critical review. J Autoimmun. 2016;66:51–59. doi: 10.1016/j.jaut.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 2.Desai R, Neuberger J. Donor transmitted and de novo cancer after liver transplantation. World J Gastroentrol. 2014;20:6170–6179. doi: 10.3748/wjg.v20.i20.6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan L, Cai C, Li J, Xu S, Chang Q, Li Y, et al. Present status and perspectives of stem cell- based therapies for gastrointestinal diseases. Stem Cell Rev. 2009;5:278–282. doi: 10.1007/s12015-009-9070-4. [DOI] [PubMed] [Google Scholar]
- 4.Ferrer JR, Chokechanachaisakul A, Wertheim JA. New tools in experimental cellular therapy for the treatment of liver diseases. Curr Transplant Rep. 2015;2:202–210. doi: 10.1007/s40472-015-0059-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou H, Liu H, Ezzelarab M, Schmelzer E, Wang Y, Gerlach J, et al. Experimental hepatocyte xenotransplantation-a comprehen-sive review of the literature. Xenotransplantation. 2015;22:239–48. doi: 10.1111/xen.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatia SN, Underhill GH, Zaret KS, Fox IJ. Cell and tissue engineering for liver disease. Sci Transl Med. 2014;6:245sr2. doi: 10.1126/scitranslmed.3005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ebrahimkhani MR, Neiman JA, Raredon MS, Hughes DJ, Griffith LG. Bioreactor technologies to support liver function in vitro. Adv Drug Deliv Rev. 2014;69-70:132–157. doi: 10.1016/j.addr.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamura T, Mizuno S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:588–610. doi: 10.2183/pjab.86.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gómez-Aristizábal A, Keating A, Davies JE. Mesenchymal stromal cells as supportive cells for hepatocytes. Mol Ther. 2009;17:1504–1508. doi: 10.1038/mt.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forte TM. Primary hepatocytes in mono-layer culture: a model for studies on lipoprotein metabolism. Annu Rev Physiol. 1984;46:403–415. doi: 10.1146/annurev.ph.46.030184.002155. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura T1, Sakai K, Nakamura T, Matsumoto K. Hepatocyte growth factor twenty years on: Much more than a growth factor. J Gastroenterol Hepatol. 2011;1:188–202. doi: 10.1111/j.1440-1746.2010.06549.x. [DOI] [PubMed] [Google Scholar]
- 12.Kaido T, Oe H, Yoshikawa A, Okajima A, Imamura M. Expressions of molecules associated with hepatocyte growth factor activation after hepatectomy in liver cirrhosis. Hepatogastroenterology. 2004;51:547–551. [PubMed] [Google Scholar]
- 13.Atta H, El-Rehany M, Hammam O, Abdel-Ghany H, Ramzy M, Roderfeld M, et al. Mutant MMP-9 and HGF gene transfer enhance resolution of CCl4-induced liver fibrosis in rats: role of ASH1 and EZH2 methyltrans-ferases repression. PLoS One. 2014;9:e112384. doi: 10.1371/journal.pone.0112384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim MD, Kim SS, Cha HY, Jang SH, Chang DY. Therapeutic effect of hepatocyte growth factor-secre-ting mesenchymal stem cells in a rat model of liver fibrosis. Exp Mol Med. 2014;22:46–e110. doi: 10.1038/emm.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seo KW, Sohn SY, Bhang DH, Nam MJ, Lee HW, Youn HY. Therapeutic effects of hepatocyte growth factor-overexpre ssing human umbilical cord blood-derived mesenchymal stem cells on liver fibrosis in rats. Cell Biol Int. 2014;38:106–116. doi: 10.1002/cbin.10186. [DOI] [PubMed] [Google Scholar]
- 16.Xia JL, Dai C, Michalopoulos GK, Liu Y. Hepatocyte growth factor attenuates liver fibrosis induced by bile duct ligation. Am J Pathol. 2006;168:1500–1512. doi: 10.2353/ajpath.2006.050747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waters James M, Richardson Gavin D, Jahoda Colin AB. Hair follicle stem cells. Semin Cell Dev Biol. 2007;18:245–254. doi: 10.1016/j.semcdb.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Anas El-Aneed. An overview of current delivery systems in cancer gene therapy. J Control Release. 2004;94:1–14. doi: 10.1016/j.jconrel.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura T, Nawa K, and Ichihara A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem Biophys Res Commun. 1984;122:1450–1459. doi: 10.1016/0006-291x(84)91253-1. [DOI] [PubMed] [Google Scholar]
- 20.Gohda E, Tsubouchi H. Nakayama H, Hirono S, Sakiyama O, Takahashi K, et al. Purification and partial characterization of hepatocyte growth factor from plasma of a patient with fulminant hepatic failure. J Clin Invest. 1988;81:414–419. doi: 10.1172/JCI113334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zarnegar R, Michalopoulos G. Purification and biological characterization of human hepatopoietin A, a polypeptide growth factor forhepatocytes. Cancer Res. 1989;49:3314–3320. [PubMed] [Google Scholar]
- 22.Nakamura T, Nishizawa T, Hagiya M, Seki T, Shimonishi M, Sugimura A, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342:440–443. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 23.Tashiro K, Hagiya M, Nishizawa T, Seki T, Shimonishi M, Shimizu S, et al. Deduced primary structure of rat hepatocyte growth factor and expression of the mRNA in rat tissues. Proc Natl Acad Sci U S A. 1990;87:3200–3204. doi: 10.1073/pnas.87.8.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakamura T. Structure and function of hepatocyte growth factor. Prog Growth Factor Res. 1991;3:67–85. doi: 10.1016/0955-2235(91)90014-u. [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto K, Nakamura T. Hepatocyte growth factor: molecular structure, roles in liver regeneration, and other biological functions. Crit Rev Oncog. 1992;3:27–54. [PubMed] [Google Scholar]
- 26.Birchmeier C, Gherardi E. Develop-mental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol. 1998;8:404–410. doi: 10.1016/s0962-8924(98)01359-2. [DOI] [PubMed] [Google Scholar]
- 27.Kellermann G, Boudechiche L, Weber A, Hadchouel M. Increased engraftment of hepatic progenitors after activation of the hepatocyte growth factor signaling pathway by protein transduction. Exp Biol Med. 2009;234:1102–1108. doi: 10.3181/0901-RM-32. [DOI] [PubMed] [Google Scholar]
- 28.Xue F, Takahara T, Yata Y, Kuwabara Y, Shinno E, Nonome K, et al. Hepatocyte growth factor gene therapy accelerates regeneration in cirrhotic mouse livers after hepatectomy. Gut. 2003;52:694–700. doi: 10.1136/gut.52.5.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yanagita K, Nagaike M, Ishibashi H, Niho Y, Matsumoto K, Nakamura T. Lung may have an endocrine function producing hepatocyte growth factor in response to injury of distal organs. Biochem Biophys Res Commun. 1992;182:802–809. doi: 10.1016/0006-291x(92)91803-x. [DOI] [PubMed] [Google Scholar]
- 30.Hu JJ, Sun C, Lan L, Chen YW, Li DG. Therapeutic effect of transplanting β2m-/Thy1+ bone marrow-derived hepatocyte stem cells transduced with lentiviral-mediated HGF gene into CCl4-injured rats. J Gene Med. 2010;12:244–254. doi: 10.1002/jgm.1439. [DOI] [PubMed] [Google Scholar]