

Abstract
With the formidable growth in the volume of genetic information, it has become essential to identify and characterize mutations in macromolecules not only to predict contributions to disease processes but also to guide the design of therapeutic strategies. While mutations of certain residues have a predictable phenotype based on their chemical nature and known structural position, many types of mutations evade prediction based on current information. Described in this work are the crystal structures of two cancer variants located in the palm domain of DNA polymerase β (pol β), S229L and G231D, whose biological phenotype was not readily linked to a predictable structural implication. Structural results demonstrate that the mutations elicit their effect through subtle influences on secondary interactions with a residue neighboring the active site. Residues 229 and 231 are 7.5 and 12.5 Å, respectively, from the nearest active site residue, with a β-strand between them. A residue on this intervening strand, M236, appears to transmit fine structural perturbations to the catalytic metal-coordinating residue D256, affecting its conformational stability.
Graphical abstract
Maintenance of the human genome is an arduous task requiring the vigilance of key enzymes in the several repair pathways responsible for correcting the tens of thousands of damage sites that occur per cell per day caused by ultraviolet rays, oxygen used in normal cellular respiration, and other agents.1,2 Two fundamental phases of DNA maintenance are removal of damage followed by the faithful incorporation of the correct nucleotide(s) by the appropriate DNA polymerase. In the case of base excision repair and short patch repair processes, this role is served by pol β. The level of scrutiny of the impact of pol β on genome stability has increased as numerous mutations throughout the protein have been identified in many types of cancers.3 Understanding the structural impact of these mutations is of significant interest in both understanding their contribution to disease and ultimately predicting therapeutic outcomes.
Nucleic acid polymerases are machines that perform similar reactions using a related multidomain architecture, many adopting the long since described right-handed metaphor with fingers, palm, and thumb subdomains.4,5 Depending on the particular enzyme, differing degrees of conformational changes are observed during catalysis. Many replicative DNA polymerases undergo significant conformational events, particularly in the fingers domain, during each cycle of nucleotide incorporation. One such example is gp43 from bacteriophage RB69 in which mutations at the interface between the palm and fingers domains impact incorporation fidelity or inhibitor specificity strictly by affecting conformational dynamics independent of catalytic residues.6,7 At the other end of the spectrum are the RNA-dependent RNA polymerases like that from poliovirus, which rely on minor movements within the palm domain rather than large between domain motions, and where mutations within the palm subdomain distant from the active site can affect catalytic function and accuracy.8,9 While pol β is among the smallest of the DNA polymerases, it retains a multidomain structure analogous to that of the replicative DNA polymerases and experiences both domain-based conformational changes during the catalytic cycle and local residue rearrangements that follow domain repositioning.10 Because of this combination of events, the enzyme function can be perturbed at the global and local level of conformational dynamics.
Two separate mutations in the palm domain of pol β, S229L11 and G231D,12 have been phenotypically characterized subsequent to their identification in colon carcinomas as having impaired catalytic function yet contributing to chromosomal aberrations. The G231D variant exhibits a 140-fold decreased catalytic rate, compared to that of wild-type (WT) pol β, because of its 15-fold lower affinity for the dNTP substrate. The S229L variant binds to the DNA and the dNTP substrate with an affinity similar to that of WT pol β but exhibits an 8-fold decreased polymerization rate. Both mutations reside in β-strand 3,13 with strand 4 separating it from any direct interaction with active site residues (Figure 1). While S229 and G231 lie in the vicinity of the DNA duplex at the −3 and −4 template positions, the gel mobility shift shows that the decreased polymerase activity is independent of the affinity for the DNA duplex, suggesting that the source of the defect is either binding of the incoming nucleotide or the conformational events that cause the transition of pol β from the binary (pol + DNA) to ternary (pol + DNA + nucleotide) to prechemistry states of the enzyme.11,12
Figure 1.

Proximity of the G231 and S229 positions relative to the active site in DNA pol β. (A) Four β-strands (2–5) from the palm domain of the polymerase (pink cartoon) and the residues projecting into the minor groove of the DNA duplex. The catalytic (C) and nucleotide-binding (N) magnesium ions are colored green, and the incoming dNTP is shown as a ball-and-stick representation. The model shown is a representative WT structure of a ternary complex of pol β with DNA and nucleotide (Protein Data Bank entry 2FMS).17 (B) Topology diagram of the five β-strands that comprise the palm domain of pol β, along with key residues in this study. Residues in the topology diagram correspond to those shown in the structure in panel A. The nucleotide-binding metal (N) and the catalytic metal (C) are indicated in both panels. Numbers to the right of each panel indicate β-strand numbers within the palm domain. All molecular structure figures were made using PyMOL (Schrödinger, LLC).
Residues of neighboring β-strands 2 and 5 of the palm domain serve critical catalytic functions and typically do not undergo significant changes in position when undergoing the transition from the binary to ternary states. Conserved aspartates 190 and 192 of β-strand 2 function to bind the nucleotide-associated metal ion (metal N) for proper substrate positioning with only D192 undergoing a side-chain rotation in the binary to ternary transition. Once the substrate is bound, D190 and D192 also serve as two of the ligands for the catalytic metal (metal C), thus associating with both metals. The third critical residue is D256 on β-strand 5, which interacts with just the catalytic metal. Like that of D190, the conformational change is minor because it is anchored in position by R254. Perturbation of this interaction by mutation of either residue results in significant catalytic impairment of the enzyme.14,15
In this work, we utilize X-ray crystallography in combination with the more sensitive analytical tool of isomorphous difference Fourier map analysis16 to compare the binary states of these neighboring mutations. Solvent isotope effects and biochemical characterization of variants complemented the structural analyses. Our results demonstrate that the mutations in these palm domain residues elicit similar yet unique changes in local residue positions that distinguish their different cellular phenotypes. The observed positional rearrangements in the binary state impact the transition to the precatalytic conformation of the residues responsible for binding the metal ion needed for the chemical step.
MATERIALS AND METHODS
Crystallization and Data Collection
Binary complexes between pol β and a single-nucleotide gapped DNA duplex substrate were prepared as previously described17 at 10 mg/mL protein with a 3-fold excess of DNA with protein constructs expressed and purified as previously described.18 Initial crystallization was explored using established conditions with PEG 3350 as the precipitant along with a selection of buffers and additives. These conditions produce typical diffraction-quality crystals for the WT complex. In contrast, for the S229L and G231D variants, the resulting crystals while reasonably sized adopted a different morphology and diffracted poorly. Sparse-matrix screens were employed to explore alternative crystallization conditions, of which PEG 400 was identified as a suitable precipitant for both variants and the WT at 18 °C. The WT complex was optimized (typical dimensions of 420 μm × 260 μm × 50 μm) at 22–26% (v/v) PEG 400 with 30 mM potassium sodium tartrate and 1% (v/v) 1,4-dioxane or 1% (v/v) 2-propanol, whereas the S229L variant crystals were much smaller (140 μm × 60 μm × 20 μm) and were optimized using 22% (v/v) PEG 400 with 100 mM tartrate. The G231D crystals were modestly thicker than the S229L crystals and produced using similar conditions. For all crystals, cryoprotection was achieved by increasing the final PEG 400 concentration to 32% (v/v). Crystals were flash-cooled in liquid nitrogen and data collected on a Rigaku RUH-3R copper rotating anode system utilizing a Mar345 image plate detector (MarResearch) and data processed with HKL2000.19 For the S229L crystals, higher-resolution data were produced by merging a home source data set collected at 3 Å with a 2.45 Å data set collected at the Diamond Light Source (Didcot, U.K.). Data sets for WT (2.17 Å) and G231D (2.15 Å) crystals were collected on the home source described above.
Structure Solution and Refinement
Crystals grown in PEG 400 were of the space group P21 with cell parameters similar to those of the PEG 3350 condition. The crystallographic data sets, however, were poorly isomorphous between the two crystallization conditions, regardless of the choice of the unit cell a- and c-axes. The cross-R between the WT crystals was ~39% on intensity (26% on amplitude) for the two conditions (PEG 400 vs PEG 3350).16 A molecular replacement solution for the WT was obtained with Phaser20 within CCP421 using wild-type binary pol β [Protein Data Bank (PDB) entry 3ISB]22 as the search model, separated into the four structural subdomains (fingers, palm, thumb, and lyase).10 Crystals of the S229L variant produced data with a cross-R of 26% on intensity (18.3% on amplitude) compared to WT (also grown in PEG 400) and were thus solved by isomorphous replacement methods using the WTPEG 400 model. The G231D structure was determined similarly (cross-R values for intensities of 23 and 15% on amplitudes comparing G231D with WTPEG 400). Regions of pol β showing peaks in the isomorphous difference Fourier map between WT and S229L (residues 228–231, 234–238, and 253–257, corresponding to β-strands 3–5, respectively) were omitted from the model in the initial stages of refinement. Models were refined using a combination of Phenix23 and Refmac524 protocols, resulting in ~98% of the residues being in the preferred Ramachandran regions with final statistics determined by MolProbity.25 In the G231D model, peptide conformations 230 to 231 and 231 to 232 were refined to the less favored region of the Ramachandran plot to alleviate a clash between the side chain of D231 (replacing a Gly) and the DNA backbone. Data collection and refinement statistics for the WT (2.17 Å with Rfactor = 17.9% and Rfree = 22.5%), G231D (2.15 Å with Rfactor = 18.2% and Rfree = 23.4%), and S229L (2.45 Å with Rfactor = 18.4% and Rfree = 23.1%) crystals grown in PEG 400 are listed in Table 1. For the WT structure, residual electron density in the solvent accessible nucleotide-binding groove was observed, most likely pertaining to partial occupancy of the fragments of PEG 400 used in crystallization. However, as refinement results were ambiguous, the PEG 400 fragment was not built in the final model.
Table 1.
Crystallographic Data Collection and Refinement Statisticsa
| WT | G231D | S229L | |
|---|---|---|---|
| PDB entry | 5U8G | 5U8H | 5U8I |
| X-ray source | home source | home source | home source Diamond Light Source |
| space group | P21 | P21 | P21 |
| cell dimensions | |||
| a, b, c (Å) | 54.10, 78.66, 54.72 | 54.09, 78.77, 54.69 | 53.96, 78.76, 54.65 |
| β (deg) | 104.25 | 104.45 | 104.64 |
| resolution (Å) | 14–2.17 (2.25–2.17) | 14–2.15 (2.23–2.15) | 40–2.45 (2.54–2.45) |
| no. of unique reflections | 23445 | 23423 | 15069 |
| redundancy | 11.6 (4.3) | 5.9 (2.1) | 6.3 (4.0) |
| completeness (%) | 99.8 (98.1) | 97.6 (80.3) | 91.9 (76.6) |
| Rmeas (%) | 11.7 (70.7) | 10.3 (46.5) | 10.8 (21.2) |
| Rpim (%) | 3.3 (32.6) | 4.0 (28.1) | 4.0 (9.5) |
| CC1/2 | 0.994 (0.74) | 0.997 (0.81) | 0.99 (0.98) |
| RFriedel (%) intensity | 5.8 (66.3) | 7.7 (53.9) | 8.0 (19.6) |
| I/σ | 18.9 (2.0) | 15.9 (2.2) | 14.1 (4.9) |
| Wilson B factor (Å2) | 26.8 | 18.9 | 17.4 |
| Refinement | |||
| Rwork, Rfreeb (%) | 17.9, 22.5 | 18.2, 23.4 | 18.4, 23.1 |
| RMSD for bonds (Å) | 0.002 | 0.002 | 0.002 |
| RMSD for angles (deg) | 0.49 | 0.53 | 0.44 |
| Ramachandran favored (%) | 98.7 | 98.1 | 97.8 |
| Ramachandran outlier (%) | 0 | 0 | 0 |
| CC model map | 0.92 | 0.91 | 0.87 |
| B factor (Å2) | |||
| protein | 38.9 | 35.4 | 33.3 |
| DNA | 38.8 | 35.7 | 32.5 |
| solvent | 43.6 | 40.8 | 30.3 |
Values for the highest-resolution shell are shown in parentheses.
Rfree was calculated with 10% of the reflections not used in refinement.
Solvent Isotope Effect
DNA substrate, 32P-labeled, one-base gapped DNA (3C2M45) was prepared as previously described with the following exceptions for deuterium solutions.11 Oligos were annealed in deuterated annealing buffer (50 mM Tris-d11 and 250 mM NaCl in D2O) at a 1:1.2:2 ratio (primer:template:downstream). The kinetics of correct nucleotide incorporation under single-turnover conditions were measured as described above in buffer [50 mM Tris-HCl, 20 mM KCl, 20 mM NaCl, 10 mM MgCl2, 1 mM DTT, and 6% (v/v) glycerol (pH 8.6)]. In deuterium reactions, Tris and glycerol were replaced with Tris-d11 and glycerol-d3, respectively, and the buffer was prepared in D2O (pD 8.6, adjusted with a DCl solution). A 100 mM dCTP stock solution was prepared by dissolving Na-dCTP (Sigma-Aldrich) in D2O; this was used in place of the commercially available 100 mM dCTP in H2O (New England Biolabs, Ipswich, MA) used in the proton experiments. Rapid chemical quench kinetics experiments were performed using the RQF-3 Rapid Chemical Quench Flow (KinTek Corp.) apparatus with drive buffers replaced with H2O or D2O as appropriate. Quenched reactions were analyzed using 20% denaturing PAGE, visualized using a Storm860 phosphorimager, and quantified using ImageQuant software.
Biochemical Characterization of Mutants of Methionine 236
The M236 mutants were generated using polymerase chain reaction (PCR) and site-directed mutagenesis (QuikChange kit, Stratagene) using an untagged, WT human pol β plasmid. Proteins were expressed and purified as previously described.18 The 1 bp gapped DNA used for single-turnover experiments was made using a 32P radioactively labeled primer, phosphorylated downstream oligo, and template DNA via a protocol similar to that published previously.18 The DNA sequence used is described in Table 2.
Table 2.
Solvent Kinetic Isotope Effects
| WT | S229L | |
|---|---|---|
| kpol for protonic (s−1) | 27.9 s−1 | 5.45 s−1 |
| kpol for deuterated (s−1) | 3.2 s−1 | 1.15 s−1 |
| kpol for protonic/kpol for deuterated | 8.7 | 4.7 |
Pre-steady state burst kinetics experiments were performed with 100 nM pol β and 300 nM 1 bp gapped DNA at 37 °C in high-salt buffer [50 mM Tris-HCl (pH 8), 100 mM NaCl, and 10% glycerol]. The pol β/DNA mixture was combined with 10 mM MgCl2 and saturating dNTP (100 μM) from 0 to 3 s on the KinTek RQF-3 Rapid Chemical Quench Flow apparatus. Reactions were quenched with 0.25 mM EDTA. Products were separated on a denaturing polyacrylamide gel and visualized and quantified with Storm 860 phosphorimager and Image-Quant software, respectively. Data were fit with Prism 6 (Graphpad Software, Inc.) using the biphasic burst equation:
Correct incorporation of a nucleotide opposite template G under single-turnover conditions was performed using a KinTek RQF-3 instrument. Incorrect incorporation experiments were performed by hand in a 37 °C heat block. The concentration of pol β used in the experiments was determined empirically, and a 10:1 protein:DNA ratio was found to lead to saturation. Therefore, 500 nM pol β was preincubated with 50 nM radioactively labeled, 1 bp gapped DNA and mixed with various concentrations of either correct (0.5–100 μM) or incorrect (1–1500 μM) dNTP. Durations of reactions ranged from 0 to 10 s for correct incorporation or from 0 to 3600 s for incorrect incorporation, and reactions were visualized as described above. The results were fitted with Prism 6 (Graphpad Software, Inc.) to the following single-exponential equation:
where kobs is the observed rate constant for each concentration of dNTP. The calculated kobs is plotted against the varying dNTP concentration and fit to a hyperbolic equation:
where kpol is the maximal rate of polymerization and Kd(dNTP) is the equilibrium dissociation constant for the dNTP substrate.
RESULTS
The Nature of the PEG Precipitant Influences Crystal Packing
Subtle differences in protein models can be overlooked because an initial molecular replacement solution can often introduce model bias.26 Obtaining diffraction-quality crystals of the S229L variant necessitated changes in crystallization conditions from PEG 3350 (the polymer used for the vast majority of pol β crystals) to PEG 400. This change resulted in poor isomorphism comparing the variants crystals grown in PEG 400 to the WTPEG 3350 data set, thus requiring phasing by molecular replacement. This phasing method generated an initial model for the S229L variant with seemingly minor structural deviations that did not readily provide an explanation for the observed phenotype. We therefore collected a new reference data set for the WT grown in the same polymer, PEG 400, to apply isomorphous difference Fourier analyses. The difference in molecular orientation between models obtained with crystals grown in PEG 3350 and PEG 400 is illustrated in Figure 2.
Figure 2.

Change in the PEG precipitant that influences the protein conformation within the crystal. Shown in both panels are the asymmetric unit right, light tones) and nearest symmetry molecule (left, dark tones). (A) A comparison of the WTPEG 3350 (purple) and WTPEG 400 (gray) displays only minor differences in the asymmetric unit, but a shift in the adjacent symmetry-related molecule results in the two forms being poorly isomorphous. (B) WTPEG 400 (gray) is compared to S229LPEG 400 (cyan). The two models are more isomorphous, as exemplified by an RMSD of <0.4 Å. The RMSDs (angstroms) for each comparison are shown with mean and per residue values calculated using CNS version 1.2.30
The collection of data for WT and G231D on a home source rotating anode generator at the copper Kα wavelength allowed for verifcation of the position of sulfur atoms within the models using an anomalous difference Fourier map (Figure 3). Anomalous maps contoured at 3σ showed peaks for four of the six methionines and two of the three cysteines in pol β. The Fo − Fo isomorphous difference maps revealed that structural changes induced by the variants at positions 229 and 231 are propagated throughout the minor groove of the DNA duplex relative to the WT state prior to active site assembly and elicit effects on the catalytic metal-binding site through neighboring residues in the palm domain, as described below. Numerous attempts were made to obtain ternary complexes with the incoming nucleotides but were unsuccessful because of reagent incompatibility in the PEG 400 solution. Trials to exchange crystals in PEG 3350 resulted in dNTP compatibility, but with a significant loss of diffraction resolution complicated by high mosaicity.
Figure 3.

Position of several sulfur-containing residues in the palm domain, highlighted by anomalous difference maps calculated for WT (purple mesh) and G231D (orange mesh) pol β binary complexes crystallized in PEG 400 (maps contoured at 3σ). Residues of the WT are shown for the sake of simplicity.
The S229L Mutation Weakens the D256:R254 Salt Bridge and Induces a Shift in M236
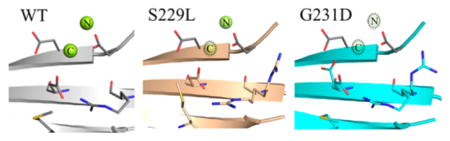
The S229L mutation results in minimal changes in the overall structure. The RMSD on C-α atoms compared with the WT is 0.37 Å. However, the Fo − Fo isomorphous difference map revealed a shift in the M236 position toward D256, likely due to the steric hindrance created by the introduction of two methyl groups at position 229. The final refinement also showed high B factors for the M236 side chain that were >2.5 times that of the backbone with a displacement at C-γ by 0.8 Å, S-δ by 1.3 Å, and C-ε by 3.0 Å relative to those of the WT. Peaks in the difference map indicated likely weakening of the D256:R254 salt bridge (Figure 4A), along with the loss of a water molecule between S229 OG and N2 of the nearby template strand dG of position −3. A second water molecule is also displaced by the methyl group of the leucine. Residual maps prior to side-chain inclusion showed that D256 is in the expected position, albeit at a lower occupancy, and R254 is more disordered (Figure S1A) and likely to adopt two conformations.
Figure 4.

Disruption of the precatalytic active site stability in DNA pol β S229L and G231D variants demonstrated using isomorphous difference Fourier maps. (A) Shown are the palm domain of the WT binary structure (gray) and corresponding residues from the S229L structure (tan). The isomorphous difference Fourier map shown was calculated between the WT and S229L. The displaced water molecules from the WT structure are shown as blue spheres. (B) Shown are the WT binary (gray) and G231D (cyan) structures. The isomorphous difference Fourier map shown was calculated between the WT and the G231D variant. All maps shown are contoured at 3σ In this figure and all subsequent figures, a green mesh signifies positive density whereas a red mesh signifies negative density. (C) Shown are the S229L (orange) and G231D (cyan) variants of DNA pol β. The isomorphous difference Fourier FoG231D − FoS229L map (±3σ) was calculated between the two variants such that residue positions of G231Dare indicated by a positive contour (green) and positions for S229L are shown as the negative contour (red). The map has been drawn to cover only M236 for the sake of clarity.
The G231D Mutation Also Induces a Shift in the Position of M236, Albeit in the Opposite Direction
Like those of the S229L variant, peaks in the isomorphous difference map indicated movement of residues near the active site along with a shift in the D231-containing β-strand. This is accompanied by a rotation of M236 away from D256. The value of calculating isomorphous difference Fourier maps is especially evident in the case of the G231D variant (Figure 4B). Whereas the overall displacement of M236 relative to the WT when relying on final models and Fo − Fc maps is minor, changes are striking in the Fo − Fo electron density map. Displacement of both C-γ and S-δ (see anomalous diffraction in Figure 3) for the final model is 0.5 Å but 1.43 Å for C-ε because of the side-chain rotation away from D256. The M236 positions are even more readily distinguishable between S229L and G231D upon calculation of the associated difference map between the two variants (Figure 4C). The WT difference map with G231D also revealed a clear disruption of the D256:R254 salt bridge, and a second rotamer of D256 is evident. The secondary D256 conformation positions the residue between D192 and D190 in the binary state void that exists in the absence of the nucleotide-binding metal prior to rotation of D192 into position. Residual maps prior to side-chain inclusion indicated that like D256, two conformations were also likely for R254 (Figure S1B).
More discernible structural changes were observed for the G231D-containing β-strand (residues L228–E232) than for the S229L variant (Figure 5). Initial Fo − Fo maps suggested a change in the conformation of the strand. Upon initial refinement, residual densities and high B factors suggested that the segment of the β-strand was in a conformation significantly different from that of the wild type and likely has increased the flexibility in the region. Simulated annealing and composite omit maps displayed weak residual density about the WT-like conformation, while the Feature Enhanced Map (Phenix27) provided improvements to aid in placement of the new conformation. The disruption of the strand is likely caused by the introduction of the Asp at position 231 adjacent to E232 at the interface at the phosphate backbone of the template strand of the DNA duplex. While the new conformation projects D231 away from the DNA duplex, the backbone contacts from the amide groups of K230 and E232 to the nonbridging oxygens of the DNA backbone appear to be relatively retained (3.0 and 2.9 Å for the WT and 3.2 and 3.2 Å for G231D, respectively). This likely explains the minimal impact observed for the KD of the DNA duplex, 4.1 nM for the WT versus 1.7 nM for G231D.12
Figure 5.

G231D mutation induces a β-strand shift. Close-up on β-strand 4 with an overlaid isomorphous difference Fourier map (contoured at 3σ) calculated between the WT (gray) and G231D (cyan) binary complexes. Shown are two conformations of the end of the β-strand, from residue 228 to 232.
Solvent Kinetic Isotope Effect
Single-turnover kinetics were conducted in protonic, H2O-based, and deuteronic, D2O-based, buffers (shown in Table 2). For both the WT and S229L in protonic buffer, the maximal rates of polymerization (kpol) were similar to the original single-turnover data (27.9 and 5.5 s−1, respectively). In the presence of deuteronic buffer, the kpol for WT dropped dramatically (8.7-fold). The kpol for S229L was reduced 4.7-fold in deuteronic buffer (from 5.45 to 1.15 s−1), which is 2-fold lower than the large decrease seen with the WT. Interestingly, the maximal rates of polymerization for the WT and S229L are more similar in the deuteronic versus protonic buffer (3.2 s−1 vs 1.0 s−1, respectively). In light of disruption of the metal-binding residues and displacement of two water molecules in the minor groove of the DNA for the S229L structure, these data suggest that the protons involved in the transition state may be handled differently in S229L and in WT pol β.
Kinetic Characterization of M236 Substitutions
On the basis of the observed impact of M236 on both variants, the effect of substitution at the methionine position was explored. Under multiple-turnover conditions, the Ala substitution produced biphasic behavior similar to that of WT with an only 50% reduction in kobs in the burst phase (6.2 s−1 original value of 13 s−1) but had no impact on the steady state rate (1.1 s−1 vs an original value of 0.9 s−1) as shown in Figure 6. However, substitution with a Leu did not display biphasic behavior with only a linear single phase of 3.3 s−1 determined indicating a more significant decrease in the burst phase and likely coincident with an increase in the steady state rate. The absence of a burst phase has also been previously observed for both S229L11 and G231D.12 The trend was recapitulated under single-turnover conditions where the greater detriment on both kpol and catalytic efficiency was imposed by the Leu rather than Ala substitution (Table 3). With enzyme in vast excess over substrate, M236A showed an efficiency nearly identical to that of the WT while M236L showed a 2.4-fold reduction relative to that of the WT due to the reduction in kpol from 18.5 to 7.1 s−1.
Figure 6.

Burst kinetics of M236L and M236A variants of DNA pol β. The level of product formation as a function of time was measured using a 3-fold excess of DNA to enzyme. Data were fit to a biphasic burst equation (see Materials and Methods). The observed rates for WT, M236L, and M236A are 13 s−1, undefined, and 6.2 s−1, respectively.
Table 3.
Single-Turnover Kinetics for M236 Mutations of DNA pol β
| Protein | DNAa | kpol (s−1) | Kd (μM) | efficiencyb |
|---|---|---|---|---|
| M236A | G:dCTP | 12 ± 0.3 | 2.2 ± 0.2 | 5454 × 103 |
| M236A | G:dATP | 0.23 ± 0.02 | 577 ± 82 | 0.40 × 103 |
| M236L | G:dCTP | 7.1 ± 0.3 | 3.2 ± 0.7 | 2219 × 103 |
| M236L | G:dATP | 0.086 ± 0.003 | 94 ± 10 | 0.91 × 103 |
| WT | G:dCTP | 18.5 ± 0.7 | 3.5 ± 0.5 | 5286 × 103 |
| WT | G:dATP | 0.086 ± 0.003 | 114 ± 11 | 0.75 × 103 |
DNA sequence 3C2M-45G
5′ GCCTCGCAGCCGGCAGATGCGC GTCGGTCGATCCAATGCCGTCC CGGAGCGTCGGCCGTCTACGCGCGGCAGCCAGCTAGGTTACGGCAGG 5′
kpol/Kd in units of M−1 s−1.
DISCUSSION
Mutations to catalytically competent or substrate-binding residues often have a predictable impact on a particular enzyme function. The challenge in understanding a particular structure–function relationship is interpreting functional defects induced by residues not directly involved in a critical function. For the cancer variants G231D and S229L of DNA pol β, the structural impacts observed are similar but occur through different mechanisms. Isomorphous difference maps between the two variants showed clear differential repositioning of M236 but in opposite directions (Figure 4C) dictated by the specific impact of each mutation. Little overall structural impact was observed for S229L, with the bulkier methyl groups of the Leu putting van der Waals pressure on M236 and pushing it toward the catalytic metal-binding residue D256. Conversely, the G231D mutation has a greater impact on local structure because of the reorientation of the β-strand, thereby pulling M236 away from D256. This result suggests that the directionality, not necessarily magnitude, of the M236 conformational shift plays a significant role in the phenotypic result.
The net result for both variants is a perturbation of D256, which is exhibited phenotypically by the decrease in kpol of near 8-fold for S229L11 and 140- 3.5-fold depending on multiple-turnover or single-turnover conditions for G231D.12 The M236 position oriented toward the D256:R254 salt bridge likely explains the impact on kpol observed for S229L. A distinguishing characteristic of G231D is the stronger second conformer of D256 in the vicinity of the nucleotide-binding metal residue D192. This is likely to contribute to the 16-fold increase in the Kd for dNTP unique to G231D, compared to no impact on Kd for S229L. Despite these effects on catalysis, previous studies have shown that neither S229L nor G231D negatively influences the fidelity of the enzyme.11,12 This suggests that the cellular phenotype for these two cancer variants is unlikely to be the result of pol β-induced mutagenesis via dNTP misincorporation.
The structures here suggest that the distinguishing effect between the G231D and S229L variants is due to the alternate conformation of D256 in G231D, which provides an energetic barrier for the rotation of D192 required to bind the nucleotide metal upon undergoing the transition from the binary to ternary state. This results in the elevated Kd for dNTP unique to G231D. The disruption of the salt bridge between R254 and D256 is likely to affect water shell distribution during conformational changes leading from the binary to ternary states and affect binding of the catalytic metal for both variants. The diffraction limits of the crystals preclude assignment of some water molecules compared to higher-resolution structures, but it is reasonable to expect that positional disorder of the catalytic metal-binding residue D256 could impact the assembly of critical water molecules around the nucleotide substrate. While we do not anticipate there to be a loss of a critical water molecule required for the transition state during chemistry, it is possible that the rate of positioning is what is being observed in the kinetic isotope effect data for S229L.
While the β-strand harboring both S229 and G231 sits in the vicinity of the minor groove of the DNA closest to the template (−3) position, M236 lies within van der Waals distance of the backbone of the primer terminus (Figure 1). NMR studies indicate that the transition from binary to ternary states of pol β shifts M236 from its position near the primer terminus closer to the penultimate backbone position of the primer strand.28 This trajectory would essentially move M236 farther from the R254:D256 salt bridge. Interestingly, the M236L mutation was among the mutations, including E232K, identified in sampling of somatic mutations in prostate cancer.29 While modest effects on kinetic parameters were noted with the M236 variants, the values were generated via steady state conditions, which are heavily influenced by the koff of the DNA duplex making phenotypic interpretation difficult. Under pre-steady state kinetics, a decrease in kpol is observed for the M236L variant. Like for S229L, the change in kpol is modest but significant for M236L (7.7-, 3.5-, and 2.6-fold). The absence of an impact on the Kd of the incoming nucleotide most likely indicates an impact on the chemical step or prechemistry state independent of substrate binding.
Mechanistic studies involving both molecular dynamics simulations and X-ray crystal structures for D256 variants were recently published and showed a similar impact on the observed position of M236.14 The isomorphous difference Fourier maps between these variants and the WT (PDB entry 2FMS)17 produce similar M236 peaks consistent with the observations for the cancer variants reported here. The catalytically deficient D256A mutant (PDB entry 4JWN)14 model showed a significant displacement of M236 into the void created by the loss of the salt bridge involving R254 and D256, whereas the D256E mutation (PDB entry 4JWM)14 resulted in a less dramatic shift in the methionine residue (Figure 7). For D256A, the shift in M236 is most analogous to that observed in the S229L variant. As the D256A and D256E models represent ternary structures, this fact demonstrates that the mobility of M236 in the binary state will likely propagate the impact between substrate-bound and unbound states of the enzyme.
Figure 7.

Another palm variant at position 256 also alters the position of M236. (A) Superposition of the WT (PDB entry 2FMS)17 in light gray and the D256E variant (PDB entry 4JWM)14 in pink with an isomorphous difference Fourier map overlaid. (B) Similar superposition, but with the WT and D256A mutant (PDB entry 4JWN) colored blue.14 Structure factors for each were retrieved from the PDB and maps calculated using Phenix.23 Maps shown are contoured at 3σ The maps illustrate differences caused by the mutations at position 256 and indicate a structural impact on the position of M236.
While the S229L and G231D variants described here were identified in colon cancer, earlier studies of solid tumor samples identified residues throughout the protein, including several in the palm domain distant from catalytic residues3 in other cancers. Combined with the study identifying M236L, a collection of four different variants map onto β-strands 3 and 4 of the palm domain (S229L, G231D, and E232K in prostate cancer and C239R in gastric cancer). Additionally, K248Q and P242R mutations reside in a loop connecting β-strand 4 to β-strand 5, whereas the V215P mutation lies in the helix at the packing interface of β-strand 3. The high concentration of mutations in a small area of the palm subdomain, where large conformational changes are not observed, indicates that relatively small movements induced by these mutations are enough to trigger local changes that have a significant impact on the polymerase activity and thus explain the resulting phenotype.
Supplementary Material
Acknowledgments
We thank April M. Averill and Brittany L. Carroll for protein expression and purification and Dr. Pierre Aller of Diamond Light Source beamline I04 for collection of a higher-resolution data set of the S229L variant.
Funding
This work was supported by National Institutes of Health Grant R01 CA080830 awarded to J.B.S.
ABBREVIATIONS
- pol β
DNA polymerase β
- PEG
polyethylene glycol
- dNTP
deoxyribonucleoside triphosphate
- CNS
crystallography and NMR system
- RMSD
root-mean-square deviation
- WT
wild-type
- kpol
rate of nucleotide incorporation determined by burst kinetics
- Kd
dissociation constant for dNTP
- KD
dissociation constant for the DNA duplex
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.6b01287.
Additional supporting refinement residual maps (PDF)
References
- 1.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 2.Wallace SS, Murphy DL, Sweasy JB. Base excision repair and cancer. Cancer Lett. 2012;327:73–89. doi: 10.1016/j.canlet.2011.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase beta and cancer? Cell Cycle. 2004;3:996–999. [PubMed] [Google Scholar]
- 4.Ollis DL, Brick P, Hamlin R, Xuong NG, Steitz TA. Structure of large fragment of Escherichia coli DNA polymerase I complexed with dTMP. Nature. 1985;313:762–766. doi: 10.1038/313762a0. [DOI] [PubMed] [Google Scholar]
- 5.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 6.Jacewicz A, Trzemecka A, Guja KE, Plochocka D, Yakubovskaya E, Bebenek A, Garcia-Diaz M. A remote palm domain residue of RB69 DNA polymerase is critical for enzyme activity and influences the conformation of the active site. PLoS One. 2013;8:e76700. doi: 10.1371/journal.pone.0076700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zahn KE, Tchesnokov EP, Götte M, Doublié S. Phosphonoformic acid inhibits viral replication by trapping the closed form of the DNA polymerase. J Biol Chem. 2011;286:25246–5255. doi: 10.1074/jbc.M111.248864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong P, Peersen OB. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A. 2010;107:22505–22510. doi: 10.1073/pnas.1007626107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moustafa IM, Korboukh VK, Arnold JJ, Smidansky ED, Marcotte LL, Gohara DW, Yang X, Sanchez-Farran MA, Filman D, Maranas JK, Boehr DD, Hogle JM, Colina CM, Cameron CE. Structural dynamics as a contributor to error-prone replication by an RNA-dependent RNA polymerase. J Biol Chem. 2014;289:36229–36248. doi: 10.1074/jbc.M114.616193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beard WA, Wilson SH. Structure and mechanism of DNA polymerase Beta. Chem Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 11.Nemec AA, Murphy DL, Donigan KA, Sweasy JB. The S229L colon tumor-associated variant of DNA polymerase beta induces cellular transformation as a result of decreased polymerization efficiency. J Biol Chem. 2014;289:13708–13716. doi: 10.1074/jbc.M114.550400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nemec AA, Donigan KA, Murphy DL, Jaeger J, Sweasy JB. Colon cancer-associated DNA polymerase beta variant induces genomic instability and cellular transformation. J Biol Chem. 2012;287:23840–23849. doi: 10.1074/jbc.M112.362111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sawaya MR, Pelletier H, Kumar A, Wilson SH, Kraut J. Crystal structure of rat DNA polymerase beta: evidence for a common polymerase mechanism. Science. 1994;264:1930–1935. doi: 10.1126/science.7516581. [DOI] [PubMed] [Google Scholar]
- 14.Batra VK, Perera L, Lin P, Shock DD, Beard WA, Pedersen LC, Pedersen LG, Wilson SH. Amino acid substitution in the active site of DNA polymerase beta explains the energy barrier of the nucleotidyl transfer reaction. J Am Chem Soc. 2013;135:8078–8088. doi: 10.1021/ja403842j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menge KL, Hostomsky Z, Nodes BR, Hudson GO, Rahmati S, Moomaw EW, Almassy RJ, Hostomska Z. Structure-function analysis of the mammalian DNA polymerase beta active site: role of aspartic acid 256, arginine 254, and arginine 258 in nucleotidyl transfer. Biochemistry. 1995;34:15934–15942. doi: 10.1021/bi00049a008. [DOI] [PubMed] [Google Scholar]
- 16.Rould MA, Carter CW., Jr Isomorphous difference methods. Methods Enzymol. 2003;374:145–163. doi: 10.1016/S0076-6879(03)74007-5. [DOI] [PubMed] [Google Scholar]
- 17.Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, Wilson SH. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eckenroth BE, Towle-Weicksel JB, Sweasy JB, Doublié S. The E295K cancer variant of human polymerase beta favors the mismatch conformational pathway during nucleotide selection. J Biol Chem. 2013;288:34850–34860. doi: 10.1074/jbc.M113.510891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 20.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beard WA, Shock DD, Batra VK, Pedersen LC, Wilson SH. DNA polymerase beta substrate specificity: side chain modulation of the “A-rule”. J Biol Chem. 2009;284:31680–31689. doi: 10.1074/jbc.M109.029843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Terwilliger TC. Using prime-and-switch phasing to reduce model bias in molecular replacement. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2144–2149. doi: 10.1107/S0907444904019535. [DOI] [PubMed] [Google Scholar]
- 27.Afonine PV, Moriarty NW, Mustyakimov M, Sobolev OV, Terwilliger TC, Turk D, Urzhumtsev A, Adams PD. FEM: feature-enhanced map. Acta Crystallogr, Sect D: Biol Crystallogr. 2015;71:646–666. doi: 10.1107/S1399004714028132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bose-Basu B, DeRose EF, Kirby TW, Mueller GA, Beard WA, Wilson SH, London RE. Dynamic characterization of a DNA repair enzyme: NMR studies of [methyl-13C]methionine-labeled DNA polymerase beta. Biochemistry. 2004;43:8911–8922. doi: 10.1021/bi049641n. [DOI] [PubMed] [Google Scholar]
- 29.An CL, Chen D, Makridakis NM. Systematic biochemical analysis of somatic missense mutations in DNA polymerase beta found in prostate cancer reveal alteration of enzymatic function. Hum Mutat. 2011;32:415–423. doi: 10.1002/humu.21465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.