

Abstract
We have recently identified endothelial cell-secreted developmental endothelial locus-1 (Del-1) as an endogenous inhibitor of β2-integrin–dependent leukocyte infiltration. Del-1 was previously also implicated in angiogenesis. Here, we addressed the role of endogenously produced Del-1 in ischemia-related angiogenesis. Intriguingly, Del-1–deficient mice displayed increased neovascularization in two independent ischemic models (retinopathy of prematurity and hind-limb ischemia), as compared to Del-1–proficient mice. On the contrary, angiogenic sprouting in vitro or ex vivo (aortic ring assay) and physiological developmental retina angiogenesis were not affected by Del-1 deficiency. Mechanistically, the enhanced ischemic neovascularization in Del-1-deficiency was linked to higher infiltration of the ischemic tissue by CD45+ hematopoietic and immune cells. Moreover, Del-1-deficiency promoted β2-integrin–dependent adhesion of hematopoietic cells to endothelial cells in vitro, and the homing of hematopoietic progenitor cells and of immune cell populations to ischemic muscles in vivo. Consistently, the increased hind limb ischemia-related angiogenesis in Del-1 deficiency was completely reversed in mice lacking both Del-1 and the β2-integrin LFA-1. Additionally, enhanced retinopathy-associated neovascularization in Del-deficient mice was reversed by LFA-1 blockade. Our data reveal a hitherto unrecognized function of endogenous Del-1 as a local inhibitor of ischemia-induced angiogenesis by restraining LFA-1–dependent homing of pro-angiogenic hematopoietic cells to ischemic tissues. Our findings are relevant for the optimization of therapeutic approaches in the context of ischemic diseases.
Keywords: angiogenesis, integrins, leukocytes
Introduction
Postnatal neovascularization is triggered by tissue ischemia and hypoxia aiming at restoring vascularity and metabolic homeostasis of the insufficiently perfused tissue. Ischemia-triggered angiogenesis is therefore integral to peripheral artery disease and coronary heart disease; however, this compensatory angiogenesis is often not sufficient to meet the demands of the ischemic tissue. On the other hand, in cancer, psoriasis, arthritis, as well as in ischemic retinopathies (retinopathy of prematurity and diabetic retinopathy), the ischemia-induced angiogenic response is dysregulated leading to exuberant formation of pathologic vessels, thereby contributing to both development and exacerbation of the aforementioned pathologies (1, 2).
Postnatal neovascularization is regulated by a complex interplay between various angiogenic growth factors, vasoactive substances, hematopoietic cells (inflammatory leukocytes and bone marrow-derived progenitor cells) and cytokines thereof (3–6). In particular, hematopoietic cell infiltration of the ischemic area is a major regulator of ischemia-induced angiogenesis, as leukocytes produce angiogenic growth factors and angiogenesis-modulating cytokines such as VEGF, PlGF, PDGF, basic FGF, angiopoietin-2, MCP-1 and various interleukins and proteases (1, 5–7).
Recruitment of leukocytes and their progenitors to ischemic areas is mediated by integrins of the β2-family or α4β1-integrin, as we and others have previously shown (8–10). We have recently identified the endothelial cell-secreted developmental endothelial locus-1 (Del-1) as an endogenous inhibitor of leukocyte adhesion (11–14). Del-1 is a 52-kDa glycoprotein comprising three epidermal growth factor (EGF)-like repeats and two discoidin I-like domains (hence also known as EDIL3 for EGF-like repeats and discoidin I-like domains 3) (10, 15, 16). While the second EGF-like repeat contains an RGD-motif that allows Del-1 to interact with αvβ3-integrin (10, 16–19), we also found that Del-1 binds and antagonizes β2-integrins on leukocytes, including LFA-1 (CD11a/CD18) (11, 20), thereby inhibiting LFA-1–dependent leukocyte-endothelial adhesion and transendothelial migration in human and mouse experimental systems (11, 12, 15). Thus, Del-1 suppresses inflammatory cell recruitment, as unequivocally established in multiple in vitro and in vivo studies (11–13, 21, 22).
Although several studies have addressed the role of Del-1 in angiogenesis, the findings to date are not entirely conclusive. In the initial study that reported the discovery of Del-1, overexpression of Del-1 in yolk sac cells inhibited angiogenesis in vitro and in the chick chorioallantoic membrane (CAM) assay (17). However, in a latter study by the same group, treatment with recombinant Del-1 was reported to promote angiogenesis in the CAM assay (23). Further studies involving transient local exogenous application of Del-1 (either as free recombinant protein or via plasmid-mediated overexpression) suggested that Del-1 could promote angiogenesis (24–27). Yet, adenoviral overexpression of Del-1 in the cardiac muscle did not significantly affect blood flow and heart contractility in a porcine model of myocardial ischemia (28). Moreover, transgenic overexpression of Del-1 in the mouse skin did not alter neovascularization in the context of wound healing (29). In all these previous studies, the role of endogenously produced Del-1 in postnatal angiogenesis was not addressed, as Del-1-deficient mice have not been analysed in the angiogenesis models employed. This prompted us to engage Del-1–deficient mice and study the function of Del-1 (i) in physiologic sprouting angiogenesis (by assessing retina vascularization and the aortic ring assay), as well as (ii) in ischemia-induced angiogenesis in two ischemic models, retinopathy of prematurity (ROP) (30) and hind-limb ischemia (HLI) (31). Interestingly, we found that Del-1 deficiency increased the neovascularization response in both ROP and HLI models of ischemia-induced angiogenesis. Mechanistic experiments revealed that the enhanced ischemia-induced angiogenic response in Del-1 deficiency was associated with increased β2-integrin-mediated infiltration of hematopoietic and immune cells to the ischemic tissue. In contrast, physiologic angiogenesis was unaltered by Del-1 deficiency. In conclusion, endogenous Del-1 regulates angiogenesis in a context-dependent manner: While not affecting physiologic vascularization, Del-1 limits ischemia-induced angiogenesis, which is largely associated with inflammation, by blocking β2-integrin–dependent homing of hematopoietic cells to ischemic tissues.
Materials and Methods
Cells
Human umbilical vein endothelial cells (HUVEC) were purchased from Cambrex (Germany) and cultured in endothelial basal medium (EBM) supplemented with 1 μg/mL hydrocortisone, 12 μg/mL bovine brain extract, 50 μg/mL gentamicin, 50 ng/mL amphotericin-B, 10 ng/mL epidermal growth factor and 10% fetal calf serum until the third passage. For silencing human Del-1 we used ON-TARGETplus SMARTPool siRNAs and control siRNA from Dharmacon. For transfection we used Lipofectamine RNAiMAX (Invitrogen, Germany) according to the manufacturer’s protocol.
Peripheral blood-derived mononuclear cells (MNCs) were isolated by density-gradient centrifugation with Ficoll (Biochrom AG, Germany) from buffy coats from healthy human individuals as described previously. For homing experiments, bone marrow (BM)-derived Lin− progenitor cells were purified from WT mice by negative selection using a cocktail of biotinylated antibodies to lineage markers (Lineage cell depletion kit, mouse, Milteny Biotec, Bergisch-Gladbach, Germany) for 10 min at 4°C followed by anti-biotin microbeads for 15 min (Miltenyi Biotec) as described elsewhere (8, 32).
Cell adhesion
Ninety-six-well plates were coated overnight at 4 °C with 2.5 μg/mL soluble recombinant Del-1 (11) or BSA (2.5 μg/ml) in PBS and then blocked for 1 h at room temperature (RT) with 3 % (w/v) heat-inactivated (2 h, 56 °C) albumin (Sigma, Germany). MNC were stained with 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein acetoxymethyl ester (BCECF-AM) and after detachment with trypsin, cells were resuspended in RPMI1640 containing 0.05 % BSA. Cells were seeded at 100,000 cells/well in 100 μL and incubated for 20 min at 37 °C. After removal of non-adherent cells by washing, adherent cells were quantified in triplicates with a fluorescence plate reader (Synergy HT, Biotek, Germany).
Cell-cell adhesion
Cell-cell adhesion was performed as previously described (8, 32–35). Confluent HUVEC monolayers were used and MNC adhesion to them was assessed. HUVEC monolayers were stimulated, where indicated, with TNF (10 ng/ml) for 24 h. In the experiments with siRNA-mediated knockdown of Del-1 in HUVEC, endothelial cells were transfected with control siRNA or Del-1 siRNA 48 h prior to the experiments. MNC were stained with Cell Tracker Green-CMFDA (Molecular Probes, Eugene, Oregon) or with 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein acetoxymethyl ester (BCECF-AM) (Molecular Probes, Eugene, Oregon) and after detachment with trypsin, cells were resuspended in RPMI 1640 with 0.05 % bovine serum albumin. A total of 100,000 cells MNC/well (in 100 μL RPMI 1640 with 0.05 % albumin) was added to the HUVEC monolayers and after incubation for 20 minutes at 37°C, the plates were washed with warm RPMI 1640 to remove non-adherent cells. Adherent cells were quantified in triplicates with a fluorescence plate reader (Synergy HT, Biotek, Germany).
Spheroid-based angiogenic sprouting assay
Endothelial-cell spheroids were generated as described previously (36–39). Briefly, HUVEC were suspended in culture medium containing 0.2% (w/v) carboxymethylcellulose (Sigma-Aldrich) and seeded in round-bottom 96-well plates (Greiner, Germany) to form spheroids. When siRNA transfection was performed, spheroids were formed 24 h after the transfection of HUVEC with Del-1 siRNA or control siRNA. The following day, spheroids were embedded into rat collagen I (BD Biosciences, Germany) containing gels; after gel polymerization, cells were treated with bFGF-containing medium (bFGF, 30 ng/mL; PeproTech, Hamburg, Germany). After 24 h, images were acquired using an Axiocam MR digital camera with an Axiovert 100M inverted microscope using as objective a Plan-NEOFLUAR (at 10x/0.30) and were processed using AxioVision Rel 4.5 digital imaging software (all from Carl Zeiss, Jena, Germany). In vitro capillary sprouting was quantified by measuring the cumulative sprout length of each spheroid using a computer-assisted microscope (AxioVision 4.5 software, Carl Zeiss) and the mean cumulative sprout length of 10 spheroids/condition was calculated.
Murine model of retinopathy of prematurity (ROP, ischemia-induced retinal neovascularization)
The generation of Del-1−/− mice has been previously described (11). WT and Del-1−/− littermates from Del-1+/− heterozygous breedings were used in the model of retinopathy of prematurity (ischemia-induced retinal neovascularization) according to our previously described protocol (37, 40–43). Briefly, 7-day-old (P7) mice were exposed to 75 % oxygen for 5 days with their nursing mothers. The hyperoxia results in vessel regression in the developing retinas (37, 40, 41). On P12, the mice were returned to room air (21% O2). The resulting retinal hypoxia leads to a hypoxic response and pathologic neovascularization. Mice were sacrificed on P17 and the eyes were processed for quantification of epiretinal neovascular nuclei as previously described (37, 40, 41); moreover, flow cytometry analysis was performed for leukocyte populations in the blood. Specifically, after red blood cell lysis, leukocytes were stained with the following fluorophore-coupled antibodies: B220-FITC (eBioscience), CD4-APC (Miltenyi Biotec), CD8-PERCP (Miltenyi Biotec), and TCRβ-PECy7 (Biolegend) for B and T lymphocyte analysis; CD45-FITC (eBioscience), CD11b-PE (Invitrogen), and Ly6G-APC (BD Bioscience) for myeloid cell and neutrophil analysis. Hoechst 33258 (Life Technologies, Darmstadt, Germany) was used to distinguish live from dead cells. Thereafter, cell suspensions were diluted in FACS buffer and analyzed with a FACS Canto II flow cytometer (BD Bioscience). For the LFA-1 blocking experiments in vivo, anti-LFA-1 (clone: M17/4; Biolegend) was injected into the right eye, and isotype control antibody was injected into the left eye of Del-1−/− or WT mice on P14 of the ROP model. On P17 retinal neovascularization was quantified. Experiments were approved by the Landesdirektion Sachsen, Germany.
Retina histology
WT and Del-1–deficient (Del-1−/−) mice were sacrificed either on postnatal day 6 (P6) in order to assess physiological retinal vascularization or were subjected to the ROP model and sacrificed on day P15. For retina whole mounts, the eyes were enucleated and fixed in 4% PFA at 4°C overnight. The next day, the retinas were carefully removed and permeabilized for 1h at RT in PBS with 1% BSA and 0.5% TritonX-100 and then incubated in FITC conjugated isolectin B4 (10 μg/ml, Bandeiraea simplicifolia; Sigma-Aldrich, Germany) at 4°C overnight. For the analysis of CD45+ positive cells in the retina, whole mounts were additionally stained with PE-conjugated anti-CD45 antibody or isotype control (1:50, BD Pharmingen). After flat mounting the retinas were imaged with an Axiovert 200 Inverted Fluorescence Microscope and Axiovision image processing software (Zeiss, Germany). The assessment of the vascular area was performed with Axiovision software (Zeiss Germany).
The expression of Del-1 in the retina was analysed by staining for β-Galactosidase (β-Gal) in cross-sections of Del-1–LacZ knock-in mice (or WT mice as control) together with a rat anti-CD31 antibody (PharmingenTM, Germany) to identify vessels. Staining was performed as described (13). Images were acquired with an inverted Olympus IX 83 spinning disk microscope equipped with a Yokogawa CSU-X1 Spinning Disk Unit.
Aortic ring sprouting assay
The aortic ring assay was performed according to a previously published protocol (44). Briefly, after euthanasia of WT or Del-1−/− mice, the mouse aorta was dissected, flushed with serum-free Opti-MEM medium, cleared from surrounding adipose tissue, cut into small rings (each approximately 0.5 mm long) and incubated for 24 hours in serum-free Opti-MEM medium in a humidified incubator at 37°C and 5% CO2. The rings were subsequently incubated in collagen-containing gels with Opti-MEM medium supplemented with 2.5 % FBS in the absence or presence of VEGF (15 ng/ml) at 37°C and 5% CO2. After 5 days the plate was washed and fixed with 4% PFA. After permeabilization with 0.25% Triton X-100, BS1 lectin-FITC (0.1 mg/ml) was added to stain the endothelium for 24 hours at 4°C. After washing, Z-stack images were taken using a Leica LSI Macro confocal microscope and the individual images were analysed for the number of microvessels with the help of Image J software. Images of aortic rings displayed in the figure were acquired with an inverted Olympus IX 83 spinning disk microscope equipped with a Yokogawa CSU-X1 Spinning Disk Unit.
Murine model of hind limb ischemia
Experiments were approved by the Regional Board of Land Hessen, Darmstadt, Germany. The proximal femoral artery including the superficial and the deep branch as well as the distal saphenous artery were ligated in WT, Del-1−/−, Del-1−/−LFA-1−/− or LFA-1−/− mice. After 14 days, mice were sacrificed and the ischemic muscles were harvested. Immunofluoresence staining of cryosections was performed by a rabbit-anti-mouse-laminin antibody (Abcam, Germany) followed by an AlexaFluor 488-conjugated goat-anti-rabbit secondary antibody (Molecular Probes, Eugene, Oregon) to label the muscle fibers and a PE-conjugated anti-PECAM-1 antibody (BD Biosciences, Heidelberg, Germany) or Isolectin B4-Biotin and then with Streptavidin-Alexa488 (Life Technologies, Germany) to label vessels. The number of capillaries in relation to the number of muscle fibers was determined using 10 μm-cryosections. A total of 7 high power fields/mouse was evaluated by confocal microscopy (Zeiss LSM 510, Germany or Leica TCS SP8, Germany). In some experiments, we assessed the perfusion of ischemic limbs by calculating the ischemic (right) versus normal (left) limb blood flow ratio using a Laser Doppler blood flow imager (Moor Instruments, Wilmington, Delaware). To address the infiltration of the ischemic muscles with hematopoietic cells we performed immunofluorescence staining with an anti-CD45-Alexa Fluor 594 or anti-CD45-FITC antibody (clone: 30F11, BD Biosciences, Heidelberg, Germany) (pan-hematopoietic cell marker) and an anti-F4/80-Alexa Fluor 647 antibody (monocytes/macrophages, clone: Cl:A3-1, AbD Serotec, Germany) and anti-CD3 antibody (T-lymphocytes, Abcam, Germany, ab5690). Nuclei were stained by DAPI. A total of 5 high power fields/mouse was evaluated with confocal microscopy (Leica TCS SP8, Germany). In addition, we performed immunofluorescent staining of ischemic muscles with anti-Del-1 (Proteintech, Germany), PE-conjugated anti-PECAM-1 antibody (BD Biosciences, Heidelberg, Germany) to label endothelial cells, and anti-PDGFRB antibody (Neuromics, Germany) to label pericytes and smooth muscle cells.
For flow cytometry analysis of leukocyte infiltrates in muscles, we performed digestion of ischemic and non-ischemic muscles with the Skeletal Muscle Dissociation Kit and gentleMACS (Miltenyi, Germany) 4 days after the induction of hind limb ischemia in WT and Del-1−/− mice. After red blood cell lysis, leukocytes were stained with the following fluorophore-coupled antibodies: BV510-conjugated anti-CD45 (Biolegend, Germany), PE-conjugated anti-Ly6G (Biolegend, Germany), APC-conjugated anti-CD11b (Biolegend, Germany), FITC-conjugated anti-Ly6C (Biolegend, Germany), BV421-conjugated anti-F4/80, PE/Cy7-conjugated anti-I-Ab (MHCII), APC/Cy7 anti-CD3, and APC/Cy7-conjugated anti-CD45R/B220 (Biolegend, Germany). 7-AAD (Biolegend, Germany) was used to distinguish live from dead cells. For the quantification of absolute numbers of infiltrated cells, we employed the AccuCount Rainbow Fluorescent beads (Spherotech, Inc, Germany).
For the homing experiments, BM–derived Lin− progenitor cells were purified from WT mice and labelled with CellTracker Red (Molecular Probes, Eugene, Oregon). One million labelled cells were injected i.v. 24 h after induction of limb ischemia into WT and Del-1−/− mice. After further 24 h, the mice were sacrificed and the ischemic muscles were harvested. The number of Lin− BM progenitor cells per high power field was determined using 10 μm-cryosections and by employing confocal microscopy. Nuclei were stained with Topro III (Molecular Probes, Eugene, Oregon). Injected Lin− BM progenitor cells were identified by the CellTracker Red-labelling. A total of 10 high power fields/per mouse was evaluated with confocal microscopy (Zeiss LSM 510, Germany) for the presence of Lin− BM progenitor cells.
Quantitative real-time PCR (qPCR) analysis
RNA was isolated from tissues with Trizol (Life Technologies) according to the manufacturer’s instructions. Reverse transcription of RNA samples was performed using the iScript Reaction kit (Bio-Rad) and qPCR was performed with the SsoFastTM EvaGreen® Supermix (Bio-Rad). Sequence of primers used: Del-1 Fw: 5′ CCTGTGAGATAAGCGAAGC 3′ and Del-1 Rev: 5′ GAGCTCGGTGAGTAGATG 3′. For normalization, 18s RNA or PO was used. The 2-DDCT method was used to quantify the target gene expression.
Statistical analysis
Continuous variables are expressed as means ± SEM. Comparisons between two groups were analysed by t-test (2-sided) or Mann-Whitney test, whereas experiments with more than 2 groups were analysed by analysis of variance (ANOVA) (post-hoc test: Newman-Keuls) using GraphPad Prism version 5.0.
Results
Endogenous Del-1 is an inhibitor of ischemia-induced angiogenesis while not affecting physiological angiogenesis
Retinal neovascularization occurring during the first 2 postnatal weeks in mice represents an excellent model for the assessment of physiological developmental angiogenesis (45). We first verified that Del-1 is expressed in the retina, as evidenced by β-galactosidase staining in Del-1–LacZ knock-in mice (Supplementary Figure 1). In these mice, a LacZ transgene is controlled by the Del-1 promoter, thereby serving as a reporter of Del-1 expression (11). Del-1 expression was co-localized with endothelial cells of blood vessels in the retina; additionally, we observed β-galactosidase staining in non-endothelial cells in the retina, consistent with recent reports for additional cellular sources of Del-1 (13, 19). To explore the role of endogenous Del-1 in developmental angiogenesis, we analysed physiological angiogenesis of the retina in Del-1–deficient (Del-1−/−) mice and wild-type (WT) littermates, and found that endogenous Del-1 is not required for this function (Supplementary Figures 2A and 2B). In line with these results, Del-1−/− mice are viable, fertile and display no obvious embryonic vascular defects (29), suggesting that Del-1 is dispensable also for angiogenesis during embryonic development.
To address potential involvement of Del-1 in pathological angiogenesis, we employed the retinopathy of prematurity model (ROP), a murine model of ischemia-driven retinal angiogenesis (37, 41, 43). By comparing P17 retinas from ROP mice with P17 retinas from mice kept in room air, we observed a modest but not significant decrease in the Del-1 expression by qPCR (Supplementary Figure 3A). Interestingly, Del-1–deficient mice displayed enhanced formation of pathological neovessels, as compared to littermate Del-1–proficient mice (Figures 1A and 1B), suggesting that endogenous Del-1 regulates ischemia-related angiogenesis of the retina.
Figure 1. Del-1–deficiency increases pathological ischemia-induced angiogenesis.



(A) Quantification of neovascularization in the ROP model was performed by counting the epiretinal neovascular nuclei of vessels anterior to the inner limiting membrane (ILM) (on the vitreal side of ILM) in PAS stained retinal cross-sections of Del-1–deficient (Del-1−/−) and wild type (WT) mice on P17. Data are mean ± SEM (*P <0.05 vs. WT mice; n=11–22). (B) Representative images of epiretinal neovascular tufts (on the vitreal side of ILM) in PAS stained retinal cross-sections; scale bar=100 μm. The black arrows indicate pathological neovascular tufts; the black arrowheads indicate the ILM.
(C) Immunofluorescence staining in ischemic muscles for Del-1 (light blue fluorescence), PECAM1 (endothelial cell marker, red fluorescence), PDFGRB (pericyte and SMC marker, green fluorescence). White color indicates DAPI nuclear staining. The white arrows indicate colocalization of Del-1 with endothelial cells and pericytes/SMC and also Del-1 localization in perivascular space. (D and E) Hind limb ischemia was performed in WT and Del-1−/− mice. After 14 days, perfusion (E) of ischemic limbs was determined by laser Doppler imaging. The ischemic muscles were harvested and the capillary density (D) was assessed microscopically as described in Materials and Methods. (*P <0.05 vs. WT mice; n=8).
To determine the general significance of this finding, we assessed the role of endogenous Del-1 for neovascularization in the murine model of hind limb ischemia (HLI). Immunofluorescence analysis in this model demonstrated that Del-1 co-localizes with endothelial cells and pericytes/smooth muscle cells (Figure 1C) and is moreover present in the perivascular space, consistent with its being an extracellularly secreted molecule. Del-1 mRNA expression was elevated in the ischemic limbs of WT mice, as compared to non-ischemic limbs (Supplementary figure 3B); however, this difference was not statistically significant. Similar to ischemia-driven pathological angiogenesis of the retina, Del-1−/− mice displayed an enhanced neovascularization response in comparison to WT mice, including both increased capillary density and perfusion of the ischemic limbs (Figures 1D and 1E). Together, while endogenous Del-1 is dispensable for physiological developmental angiogenesis, it functions as an inhibitor of ischemia-driven neovascularization.
Endogenous Del-1 affects angiogenesis in an endothelial cell-non-autonomous pathway
To delineate the role of endogenous Del-1 on endothelial cells during angiogenesis and specifically on angiogenic sprouting, we employed a three-dimensional angiogenic sprouting assay using endothelial cell spheroids embedded in collagen gels. Silencing of endogenous Del-1 with siRNA (Supplementary Figure 4) in HUVEC did not affect angiogenic sprouting under basal conditions or upon stimulation with bFGF as compared to control siRNA treatments (Figures 2A, and 2B). Since siRNAs may exert “off-target” effects, we further employed an independent approach, specifically, the angiogenic sprouting model of aortic rings. Analysis of aortic rings from WT and Del-1−/− mice showed that Del-1 deficiency did not affect angiogenic sprouting under basal or VEGF-stimulated conditions (Figures 2C and 2D). In conclusion, these data demonstrate that Del-1 deficiency does not affect angiogenic sprouting and that the inhibitory effect of endogenous Del-1 on ischemic angiogenesis (Figure 1) is likely not mediated by a direct effect of Del-1 on endothelial cells.
Figure 2. Del-1–deficiency affects angiogenesis in a non-endothelial cell autonomous way.

(A and B) Three-dimensional in vitro angiogenic sprouting in a spheroidal culture system with collagen–embedded spheroids is shown. (A) HUVEC were transfected with siRNAs targeting Del-1 or control siRNA. Three-dimensional in vitro angiogenic sprouting in a spheroidal culture system with collagen–embedded spheroids in the absence (control) or presence of bFGF (30 ng/mL). The mean cumulative length of sprouts per spheroid was assessed after 24 h. Silencing of endogenous Del-1 did not significantly affect angiogenic sprouting (n=3). (B) Representative images of spheroidal sprouting assay performed with endothelial cells transfected as indicated with control siRNA or Del-1 siRNA in the presence of bFGF (30 ng/mL). (C) Sprouting of aortic rings from WT and Del-1−/− mice in the absence (control) or presence of VEGF was performed. The number of angiogenic sprouts per aortic ring was assessed by confocal microscopy (*P<0.05 vs WT control and #P<0.05 vs Del-1−/− control; n.s.: not significant; n= 21–28 aortic rings per group). (D) Representative images of aortic rings from a WT and a Del-1−/− mouse.
Del-1 regulates hematopoietic cell infiltration of ischemic tissues
Hematopoietic cells (inflammatory cells and their progenitors) contribute to neovascularization of ischemic tissues by paracrine effects (5–7, 46). Since we have previously shown that Del-1 interferes with the recruitment of leukocytes to sites of acute or chronic inflammation (11, 12, 15), we explored the possibility that endogenous Del-1 inhibits neovascularization via regulating inflammatory cell infiltration of ischemic tissues in the ROP and HLI models. Indeed, Del-1–deficient mice displayed enhanced infiltration of CD45+ hematopoietic cells in ROP retinas, as compared to littermate Del-1–proficient mice (Figures 3A; representative images in Supplementary Figure 5A). In line with these results, Del-1–deficient mice showed increased infiltration of ischemic muscles with CD45+ hematopoietic cells, as compared to WT mice, 2 weeks after induction of the model of hind limb ischemia (Figure 3B).
Figure 3. Del-1 deficiency enhances leukocyte infiltration of ischemic tissues.


(A) Infiltration of ischemic retinas on postnatal day 15 with CD45+ leukocytes in WT and Del-1−/− mice in the ROP model is shown. Quantification of CD45+ leukocytes in ischemic retinas by fluorescence microscopy in WT and Del-1–deficient mice (*P<0.05 vs. WT, n=4). (B) Infiltration of ischemic muscles with CD45+ leukocytes (green fluorescence) in WT and Del-1−/− mice 2 weeks after the induction of hind limb ischemia, as assessed by confocal microscopy (40X objective). The red fluorescence indicates PECAM-1+ vessels. (C) Flow cytometry of digested muscles 4 days after induction of hind limb ischemia is shown. The infiltrated leukocytes were analysed by flow cytometry for leukocyte subset markets. Data are presented as infiltrated cells/mg tissue ± SEM. (*P<0.05 vs ischemic Del-1+/+, n=5–7/group).
In order to analyse in more detail the enhanced leukocyte recruitment to the ischemic limbs due to Del-1 deficiency, we performed multicolor flow cytometry analysis of ischemic muscles in WT and Del-1−/− mice and assessed the absolute numbers of infiltrating leukocytes/per mg muscle at an earlier time point, specifically 4 days after the induction of HLI. First, using this independent approach, we confirmed our earlier findings (Figure 3A and 3B) that Del-1 deficiency significantly increased the infiltration of ischemic muscles with leukocytes in comparison to the WT mice (Figure 3C). Interestingly, Del-1-deficiency was associated with an impressive and statistically significant increase of lymphocytes in ischemic muscles, while not significantly affecting the infiltration of ischemic muscles with granulocytes, monocytes and macrophages at this early time point (Figure 3C).
By performing flow cytometry of the blood in the course of the ROP model, we observed no difference in the number of myeloid cells, neutrophils, T cells or B cells in the peripheral blood (Supplementary Figure 5B) due to Del-1 deficiency. Similarly, Del-1 deficiency did not significantly affect the numbers of peripheral blood leukocytes, neutrophils, monocytes, T or B lymphocytes 4 days upon induction of HLI (Supplementary Figure 5C), further suggesting that Del-1 deficiency affects leukocyte infiltration of ischemic muscles through local regulatory effects.
Taken together, the enhanced angiogenesis observed in ischemic tissues of Del-1–deficient mice is associated with increased infiltration of the ischemic tissues with immune cells.
Endogenous Del-1 inhibits adhesion of hematopoietic and immune cells to endothelial cell monolayers and homing of progenitor cells to ischemic sites
To obtain further insight into the regulatory function of Del-1, which appeared to link leukocyte infiltration of the ischemic tissue with ischemia-driven angiogenesis, we addressed its role in the adhesion of leukocytes. In this regard, human mononuclear cells (MNC) were shown to bind to immobilized recombinant Del-1 in a β2-integrin–dependent manner (Figure 4A). Indeed, this binding interaction was significantly inhibited by neutralizing antibodies to Mac-1 (αMβ2-integrin) or LFA-1 (αLβ2-integrin) (Figure 4A), consistent with our previous findings (11, 20). Thus, inflammatory cells interact with Del-1 via β2-integrins, suggesting the possibility for inhibition of leukocyte recruitment by endothelial cell-derived Del-1.
Figure 4. Del-1 inhibits adhesion of leukocytes on endothelial cell monolayers.

(A) Binding of PMA-stimulated human MNC to immobilized Del-1 in the presence of isotype control antibody or neutralizing monoclonal antibodies against β2-integrins: against αMβ2-integrin (Mac1) (clone: CBRM1/5) or against αLβ2-integrin (LFA-1) (clone 38). Data are presented as adhesion % of cell input ± SEM (*P<0.05 vs. PMA+ control IgG, n=6). (B) Adhesion of human MNC cells onto TNF-stimulated or non-stimulated HUVEC monolayers 48 h after transfection of HUVEC with control siRNA or Del-1 siRNA. Data are presented as adhesion % of cell input ± SEM (*P<0.05 vs. control siRNA+TNF; n=4). (C) Cell tracker Red-labelled bone marrow–derived Lin- hematopoietic progenitor cells were intravenously injected in WT or Del-1−/− mice one day after the induction of hind limb ischemia. The ischemic muscles were harvested 24h thereafter. Quantification of the infiltration of ischemic muscles with Cell tracker Red-labelled bone marrow–derived Lin- hematopoietic progenitor cells. Data are presented as infiltrated Lin- in % of WT ± SEM (*P<0.05 vs. WT; n=3/group).
To further delineate the role of endogenous Del-1 on the adhesion of MNC onto HUVEC monolayers, we transfected HUVEC with Del-1 siRNA or control siRNA and then performed cell-cell adhesion assays with MNC. Interestingly, silencing of endogenous Del-1 (Supplementary figure 4) led to increased adhesion of MNC onto TNF-pre-stimulated HUVEC monolayers (Figure 4B). In summary, endogenous Del-1 inhibits leukocyte adhesion to endothelial cells.
We next questioned whether endogenous Del-1 could affect hematopoietic progenitor cell homing to sites of ischemia in vivo. To this end, BM-derived Lin− hematopoietic progenitor cells from WT mice that express the β2-integrin LFA-1 (8, 32) were i.v. injected into WT or Del-1−/− mice 24 h after the induction of HLI. After further 24 h, the ischemic muscles were harvested. Strikingly, homing of Lin− hematopoietic progenitor cells to ischemic muscles of Del-1–deficient mice was significantly higher, as compared to homing to ischemic muscles of WT mice (Figure 4C).
Endogenous Del-1 limits ischemia-induced neovascularization via inhibiting leukocyte integrin LFA-1–dependent hematopoietic cell recruitment
Our data so far demonstrated that Del-1–deficiency enhances ischemia-induced angiogenesis, which is associated with enhanced recruitment of hematopoietic and immune cells into the ischemic muscles and that endogenous Del-1 inhibits leukocyte adhesion and homing, which is mediated by the LFA-1-integrin (11).
We therefore assessed the role of LFA-1 integrin on the enhanced ischemia-induced neovascularization due to Del-1 deficiency. First, we addressed if LFA-1 blockade could reverse the increased angiogenesis of Del-1 deficient mice in the ROP model. We injected anti-LFA-1 antibody into the right eye and a control antibody into the left eye of WT or Del-1-deficient mice at P14 of the ROP model. Antibody blockade of LFA-1 reversed the enhanced neovasculaization seen in Del-1−/− mice (as compared to littermate Del-1-proficient mice) (Figure 5A), thus firmly establishing that Del-1 acts through an LFA-1-dependent mechanism.
Figure 5. Genetic or pharmacological ablation of LFA-1 reverses the enhanced ischemia-related angiogenesis observed in Del-1−/− mice.

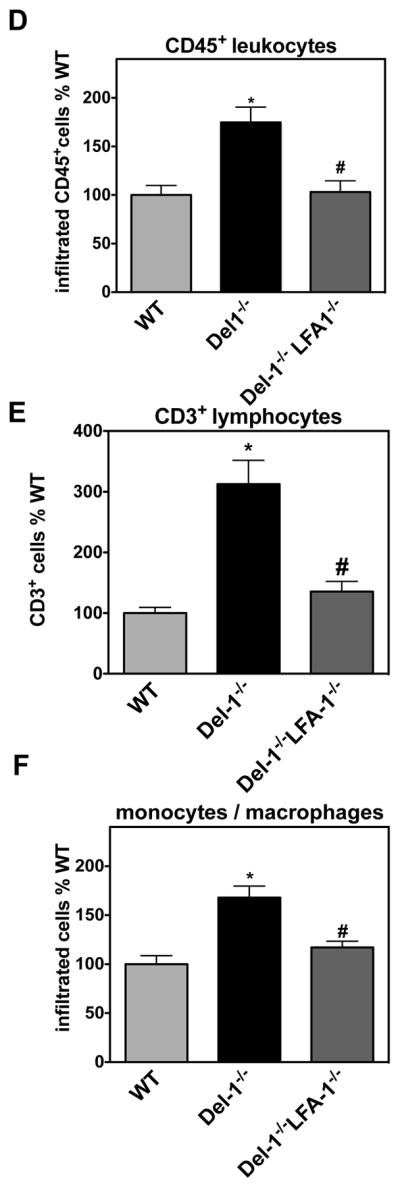
(A) Quantification of neovascularization in the ROP model was performed by counting epiretinal nuclei in retinal cross sections of Del-1–deficient (Del-1−/−) and wild type (WT) mice on P17. On day P14 an intraocular injection of an anti-LFA-1 (right eye) or isotype control (left eye) antibody was performed. Data are mean ± SEM (*P <0.05, n=12–14). (B–E) Hind limb ischemia was performed in WT, Del-1−/− and Del-1−/−LFA-1−/− mice. After 14 days, the ischemic muscles were harvested and the capillary density (B) was assessed by microscopy as described in Materials and Methods. (#P <0.05 vs. WT mice; *P <0.05 vs. Del-1−/− mice, n=6/group). (C) Representative images of ischemic muscles of WT, Del-1−/− and Del-1−/−LFA-1−/− mice 14 days after the induction of hind limb ischemia as assessed by confocal microscopy (40X objective) are shown. Green fluorescence indicates Lectin IB4 staining of vessels, grey colour indicates anti-laminin staining (displaying muscle fibers) and blue colour indicates nuclear staining (DAPI). (D–F) Quantification of the infiltration of ischemic muscles with CD45+ leukocytes (D), CD3+ T cells (E) and F4/80+ monocytes/macrophages (F) in WT, Del-1−/− and Del-1−/−LFA-1−/− mice. Data are presented as infiltrated cells in % of WT ± SEM (*P <0.05 vs. WT mice; #P <0.05 vs. Del-1−/− mice, n=5–6/group).
Moreover, we addressed the role of the Del-1–LFA-1-integrin interaction in ischemia-driven angiogenesis by engaging Del-1/LFA-1-double–deficient mice in the HLI model. To this end, we induced HLI in WT, Del-1–deficient and Del-1/LFA-1-double–deficient mice. After 14 days, we assessed capillary density in the ischemic muscles. Strikingly, the significantly increased capillary density in ischemic muscles due to Del-1 deficiency, as compared to wild-type mice, was completely reversed in Del-1/LFA-1 double–deficient mice, reaching a similar level to that of WT mice (Figures 5B and 5C). In contrast, LFA-1–deficiency alone did not significantly alter capillary density in comparison to the WT mice (data not shown). Furthermore, we assessed the infiltration of ischemic muscles with CD45+ leukocytes, T cells and monocytes/macrophages. In contrast to an earlier time point (4 days after the induction of HLI) when Del-1-deficiency caused a significant increase of lymphocytes in ischemic muscles without significantly affecting the infiltration of monocytes/macrophages (Figure 3C), at 14 days after induction of HLI, Del-1-deficiency caused enhanced infiltration of both T cells and macrophages in the ischemic muscles (Figure 5E,F). The observed increase in the infiltration of ischemic muscles on day 14 post-HLI with CD45+ leukocytes, T lymphocytes and F4/80+ macrophages in Del-1–deficiency was reversed in the simultaneous absence of LFA-1, that is, in Del-1/LFA-1 double–deficient mice (Figures 5D–F). Therefore, the inhibitory action of Del-1 in ischemia-driven inflammation-associated angiogenesis is mediated by the blocking effect of endogenous Del-1 on LFA-1-integrin–dependent leukocyte cell recruitment.
Discussion
The present study underscores the relevance of endogenous Del-1 as a regulator of angiogenesis in a context-dependent manner: While not affecting physiological angiogenesis (as assessed in developmental retina angiogenesis and the aortic ring assay), Del-1 inhibits ischemia-induced angiogenesis. Specifically, our findings revealed that Del-1 deficiency enhanced ischemia-induced inflammation-associated angiogenesis in ischemic retinopathy and in hind-limb ischemia, associated with increased LFA-1–mediated leukocyte infiltration of ischemic tissues. Our data therefore reveal a hitherto unrecognized function of endogenous Del-1 as a negative regulator of ischemia-driven angiogenesis.
Del-1 knockdown or deficiency did not alter angiogenic sprouting of endothelial cells in vitro and ex vivo in the aortic ring assay. Consistently, developmental angiogenesis of the retina was also not affected by Del-1-deficiency. Our data that endogenous Del-1 does not regulate physiological angiogenesis are in line with a previous study that showed that Del-1–deficient mice display no obvious developmental vascular defects (29). Moreover, transgenic Del-1 overexpression in the same study did not promote neovascularization (29).
Our present work, however, demonstrates that in the context of ischemia-driven inflammation, deficiency of endogenous Del-1 enhanced angiogenesis in two independent ischemic models (ROP and HLI). Our work is the first to assess the function of endogenous Del-1 in this context by engaging Del-1-deficient mice. Previous reports addressing the role of exogenous Del-1 application on ischemia-related angiogenesis yielded controversial results. Specifically, some previous studies indicated that local transient overexpression of exogenous Del-1 via plasmid- or viral-mediated delivery or local injections of recombinant Del-1 could increase neovascularization (24–27), whereas other studies demonstrated that overexpression of Del-1 does not promote (17, 28, 29) or even inhibited angiogenesis (17). A Phase II multicentre, double-blind, placebo-controlled trial (DELTA-1 trial) in subjects with intermittent claudication secondary to moderate to severe peripheral arterial disease demonstrated that intramuscular delivery of a Del-1–expressing plasmid did not display any significant clinical benefit over control treatment (47). Although studies with exogenous Del-1 (administration or overexpression) have yielded different results on the role of Del-1 in angiogenesis, ranging from stimulation to no effect or even to inhibition of angiogenesis, the majority of these studies pointed to a rather pro-angiogenic effect of the molecule. However, none of these studies addressed the role of endogenous Del-1, by using Del-1-deficient mice, as we did here. This is a biologically more relevant system since it depends on physiological levels of Del-1, the overexpression of which may have dose-dependent differential results. Our results unequivocally demonstrated that endogenous Del-1 inhibits ischemia-driven neovascularization, which is linked with inflammation. In particular, Del-1 regulates recruitment of hematopoietic and inflammatory cells to the ischemic tissue, while it does not affect physiological developmental retina angiogenesis (driven by low-grade ischemia and not accompanied by leukocyte recruitment) or sprouting angiogenesis in the aortic ring assay (not ischemia-driven and not accompanied by acute inflammatory cell recruitment). Therefore, the regulation of angiogenesis by Del-1 is context-dependent. Indeed, since endogenous Del-1 has a well-established role in inhibition of extravasation of inflammatory cells from the circulation to the tissue (11, 12, 19), it follows that recruitment of hematopoietic and inflammatory cells, exerting proangiogenic actions (5–7), will be disinhibited in Del-1-deficient mice. Therefore, in the context of inflammation-associated ischemic angiogenesis, the above-stated facts explain the enhanced ischemic angiogenesis in Del-1-deficiency. In contrast, the studies demonstrating a proangiogenic effect of exogenous Del-1 administered or overexpressed this factor within the ischemic tissue, which bears obvious differences, as compared to our present study. In addition to possible dose-dependent differences (see above), the localization of overexpressed/administered Del-1 in the tissue may not necessarily be the same as that of endogenous Del-1. Thus, the overexpressed/administered Del-1 may have been unavailable to block leukocyte extravasation from the circulation to the ischemic site, as such a function would strictly require intravascular Del-1. Additionally, overexpressed/administered Del-1 likely achieved supraphysiological levels, much higher than any endogenous levels, which may exert other, yet unidentified, effects on tissue-resident cells, thereby indirectly promoting angiogenesis.
An essential mechanistic question addressed here was how endogenous Del-1 restricts ischemia-driven angiogenesis. Del-1 deficiency was accompanied by increased leukocyte infiltration of ischemic tissues, indicating that the well-established anti-inflammatory role of Del-1 (11, 21) is also relevant for inflammation of ischemic tissues. In support of our present findings for an in vivo LFA-1–dependent anti-angiogenic role of Del-1, CD18−/− mice (deficient in all β2-integrins, including the LFA-1-integrin) exhibit reduced angiogenesis in the HLI model (8). In addition, leukocyte β2-integrins contribute to tumour angiogenesis by promoting myeloid cell infiltration into tumours (48). Consistent with our proposed mechanism, combined LFA-1-integrin and Del-1 deficiency (Del-1/LFA-1-double–deficient mice) completely reversed the pro-angiogenic phenotype and the increased leukocyte infiltration of ischemic muscles observed in Del-1 deficiency. Similarly, pharmacologic LFA-1 inhibition reversed the pro-angiogenic phenotype of Del-1 deficiency in proliferative retinopathy. Our data therefore indicate that endogenous Del-1 restrains ischemia-driven neovascularization associated with inflammation by inhibiting the LFA-1-integrin–mediated leukocyte infiltration of ischemic tissues rather than by directly regulating endothelial cells. Consistent with this notion, leukocytes are well-established players in angiogenesis. Specifically, myeloid cells contribute via paracrine effects and cell-cell interactions to the pro-angiogenic phenotype in endothelial cells (5–7). Besides inhibiting the infiltration of mature leukocytes, Del-1 also blocked the homing of intravenously administered bone marrow–derived hematopoietic progenitors known to therapeutically promote angiogenesis of ischemic tissues (6, 8, 46, 49). This finding is in keeping with the established function of β2-integrins to promote the homing of hematopoietic progenitor cells to ischemic tissues (5, 8, 46, 49).
The downstream signaling pathways enhanced by Del-1–deficiency in ischemic tissues leading to increased angiogenesis are not entirely clear and may involve additional mechanisms to those investigated here. Interestingly, our findings suggest that enhanced LFA-1-dependent lymphocyte infiltration due to Del-1 deficiency at early points in the HLI model may trigger further infiltration of monocytes/macrophages at later time points of the model. The exact mechanistic underpinnings of this early lymphocyte infiltration to the ischemic tissue due to Del-1 deficiency merit further investigation. Del-1 was previously shown to downregulate the expression of interleukin-17 (IL-17) in models of chronic inflammation such as periodontitis and neuroinflammation (12, 13). In this regard, IL-17 may potentially contribute to angiogenesis (50). Therefore, the contribution of the IL-17/IL-17R pathway as a potential intermediate of the Del-1–dependent inhibition of ischemia-driven angiogenesis is a possible scenario that merits assessment in future investigations.
In conclusion, our present data reveal a hitherto unrecognized mechanism, by which Del-1 regulates angiogenesis in the context of ischemia-driven inflammation. Del-1 restricts the pro-angiogenic action of mature leukocytes and progenitors by limiting their recruitment to ischemic tissues. In addition, our findings extend and improve the current models for understanding inflammation-related angiogenesis. Importantly, since hematopoietic progenitors are used in clinical studies for the treatment of patients with ischemic diseases (5, 6, 46), our data have tremendous clinical relevance for appreciating the benefits and limitations of such therapeutic approaches. In particular, our data imply the necessity for optimized therapeutic strategies that bypass endogenous inhibitors of homing, such as Del-1, so that hematopoietic progenitor-based therapies succeed in promoting therapeutic angiogenesis.
Supplementary Material
Table.
| What is known about the topic ? |
|
| What does this paper add ? |
|
Acknowledgments
This work was supported by the Else Kröner-Fresenius-Stiftung (2013_A2 to E.C.). E.C. and S.D. are members of the Excellence Cluster Cardiopulmonary System (DFG; Exc147-1), the German Centre for Cardiovascular Research (BMBF) and the LOEWE Center for Gene and Cell Therapy (Hessen, Germany). S.C. is also supported by a grant from the LOEWE Center for Gene and Cell Therapy. T.C. was supported by the ERC (ENDHOMRET), the DFG (INST 515/11-1) and DE026152 from the NIH. M.E. was supported by the Else Kröner-Fresenius-Stiftung. G.H. was supported by DE026152, DE024716 and DE015254 from the NIH. S.K. was supported by a Grant of DAAD (Deutscher Akademischer Austauschdienst).
We thank Bettina Gercken and Sylvia Grossklaus for technical assistance and the MTZ imaging facility of the TU Dresden for their support. Moreover, we thank Guillaume Carmona for critical reading of the manuscript.
Footnotes
Conflict of interest
The authors have not any direct or indirect conflict of interests to declare.
References
- 1.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011 Sep 16;146(6):873–87. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 2.Sapieha P, Joyal JS, Rivera JC, et al. Retinopathy of prematurity: understanding ischemic retinal vasculopathies at an extreme of life. J Clin Invest. 2010 Sep;120(9):3022–32. doi: 10.1172/JCI42142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kopp HG, Ramos CA, Rafii S. Contribution of endothelial progenitors and proangiogenic hematopoietic cells to vascularization of tumor and ischemic tissue. Curr Opin Hematol. 2006 May;13(3):175–81. doi: 10.1097/01.moh.0000219664.26528.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chambers SE, O’Neill CL, O’Doherty TM, et al. The role of immune-related myeloid cells in angiogenesis. Immunobiology. 2013 Nov;218(11):1370–5. doi: 10.1016/j.imbio.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 5.Chavakis E, Dimmeler S. Homing of progenitor cells to ischemic tissues. Antioxidants & redox signaling. 2011 Aug 15;15(4):967–80. doi: 10.1089/ars.2010.3582. [DOI] [PubMed] [Google Scholar]
- 6.Chavakis E, Koyanagi M, Dimmeler S. Enhancing the outcome of cell therapy for cardiac repair: progress from bench to bedside and back. Circulation. 2010 Jan 19;121(2):325–35. doi: 10.1161/CIRCULATIONAHA.109.901405. [DOI] [PubMed] [Google Scholar]
- 7.Murdoch C, Muthana M, Coffelt SB, et al. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008 Aug;8(8):618–31. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 8.Chavakis E, Aicher A, Heeschen C, et al. Role of beta2-integrins for homing and neovascularization capacity of endothelial progenitor cells. J Exp Med. 2005 Jan 3;201(1):63–72. doi: 10.1084/jem.20041402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin H, Su J, Garmy-Susini B, et al. Integrin alpha4beta1 promotes monocyte trafficking and angiogenesis in tumors. Cancer Res. 2006 Feb 15;66(4):2146–52. doi: 10.1158/0008-5472.CAN-05-2704. [DOI] [PubMed] [Google Scholar]
- 10.Mitroulis I, Alexaki VI, Kourtzelis I, et al. Leukocyte integrins: role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol Ther. 2015 Mar;147:123–35. doi: 10.1016/j.pharmthera.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi EY, Chavakis E, Czabanka MA, et al. Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science. 2008 Nov 14;322(5904):1101–4. doi: 10.1126/science.1165218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eskan MA, Jotwani R, Abe T, et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nature immunology. 2012 May;13(5):465–73. doi: 10.1038/ni.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi EY, Lim JH, Neuwirth A, et al. Developmental endothelial locus-1 is a homeostatic factor in the central nervous system limiting neuroinflammation and demyelination. Mol Psychiatry. 2015 Jul;20(7):880–8. doi: 10.1038/mp.2014.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maekawa T, Hosur K, Abe T, et al. Antagonistic effects of IL-17 and D-resolvins on endothelial Del-1 expression through a GSK-3beta-C/EBPbeta pathway. Nature communications. 2015;6:8272. doi: 10.1038/ncomms9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chavakis E, Choi EY, Chavakis T. Novel aspects in the regulation of the leukocyte adhesion cascade. Thromb Haemost. 2009 Aug;102(2):191–7. doi: 10.1160/TH08-12-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hajishengallis G, Chavakis T. Endogenous modulators of inflammatory cell recruitment. Trends Immunol. 2013 Jan;34(1):1–6. doi: 10.1016/j.it.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hidai C, Zupancic T, Penta K, et al. Cloning and characterization of developmental endothelial locus-1: an embryonic endothelial cell protein that binds the alphavbeta3 integrin receptor. Genes Dev. 1998 Jan 1;12(1):21–33. doi: 10.1101/gad.12.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dasgupta SK, Le A, Chavakis T, et al. Developmental endothelial locus-1 (Del-1) mediates clearance of platelet microparticles by the endothelium. Circulation. 2012 Apr 3;125(13):1664–72. doi: 10.1161/CIRCULATIONAHA.111.068833. [DOI] [PubMed] [Google Scholar]
- 19.Shin J, Maekawa T, Abe T, et al. DEL-1 restrains osteoclastogenesis and inhibits inflammatory bone loss in nonhuman primates. Sci Transl Med. 2015 Sep 30;7(307):307ra155. doi: 10.1126/scitranslmed.aac5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitroulis I, Kang YY, Gahmberg CG, et al. Developmental endothelial locus-1 attenuates complement-dependent phagocytosis through inhibition of Mac-1-integrin. Thromb Haemost. 2014 May 5;111(5):1004–6. doi: 10.1160/TH13-09-0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanczkowski W, Chatzigeorgiou A, Grossklaus S, et al. Role of the endothelial-derived endogenous anti-inflammatory factor Del-1 in inflammation-mediated adrenal gland dysfunction. Endocrinology. 2013 Mar;154(3):1181–9. doi: 10.1210/en.2012-1617. [DOI] [PubMed] [Google Scholar]
- 22.Kourtzelis I, Kotlabova K, Lim JH, et al. Developmental endothelial locus-1 modulates platelet-monocyte interactions and instant blood-mediated inflammatory reaction in islet transplantation. Thromb Haemost. 2015 Dec 17;115(4) doi: 10.1160/TH15-05-0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Penta K, Varner JA, Liaw L, et al. Del1 induces integrin signaling and angiogenesis by ligation of alphaVbeta3. J Biol Chem. 1999 Apr 16;274(16):11101–9. doi: 10.1074/jbc.274.16.11101. [DOI] [PubMed] [Google Scholar]
- 24.Zhong J, Eliceiri B, Stupack D, et al. Neovascularization of ischemic tissues by gene delivery of the extracellular matrix protein Del-1. J Clin Invest. 2003 Jul;112(1):30–41. doi: 10.1172/JCI17034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho HK, Jang JJ, Kaji S, et al. Developmental endothelial locus-1 (Del-1), a novel angiogenic protein: its role in ischemia. Circulation. 2004 Mar 16;109(10):1314–9. doi: 10.1161/01.CIR.0000118465.36018.2D. [DOI] [PubMed] [Google Scholar]
- 26.Fan Y, Zhu W, Yang M, et al. Del-1 gene transfer induces cerebral angiogenesis in mice. Brain Res. 2008 Jul 11;1219:1–7. doi: 10.1016/j.brainres.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciucurel EC, Sefton MV. Del-1 overexpression in endothelial cells increases vascular density in tissue-engineered implants containing endothelial cells and adipose-derived mesenchymal stromal cells. Tissue Eng Part A. 2014 Apr;20(7–8):1235–52. doi: 10.1089/ten.tea.2013.0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kown MH, Suzuki T, Koransky ML, et al. Comparison of developmental endothelial locus-1 angiogenic factor with vascular endothelial growth factor in a porcine model of cardiac ischemia. Ann Thorac Surg. 2003 Oct;76(4):1246–51. doi: 10.1016/s0003-4975(03)00721-5. [DOI] [PubMed] [Google Scholar]
- 29.Hsu GP, Mathy JA, Wang Z, et al. Increased rate of hair regrowth in mice with constitutive overexpression of Del1. J Surg Res. 2008 May 1;146(1):73–80. doi: 10.1016/j.jss.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 30.Connor KM, Krah NM, Dennison RJ, et al. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nature protocols. 2009;4(11):1565–73. doi: 10.1038/nprot.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Limbourg A, Korff T, Napp LC, et al. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat Protoc. 2009;4(12):1737–46. doi: 10.1038/nprot.2009.185. [DOI] [PubMed] [Google Scholar]
- 32.Chavakis E, Carmona G, Urbich C, et al. Phosphatidylinositol-3-kinase-gamma is integral to homing functions of progenitor cells. Circ Res. 2008 Apr 25;102(8):942–9. doi: 10.1161/CIRCRESAHA.107.164376. [DOI] [PubMed] [Google Scholar]
- 33.Choi EY, Orlova VV, Fagerholm SC, et al. Regulation of LFA-1-dependent inflammatory cell recruitment by Cbl-b and 14–3–3 proteins. Blood. 2008 Apr 1;111(7):3607–14. doi: 10.1182/blood-2007-07-103077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carmona G, Chavakis E, Koehl U, et al. Activation of Epac stimulates integrin-dependent homing of progenitor cells. Blood. 2008 Mar 1;111(5):2640–6. doi: 10.1182/blood-2007-04-086231. [DOI] [PubMed] [Google Scholar]
- 35.Chavakis E, Hain A, Vinci M, et al. High-mobility group box 1 activates integrin-dependent homing of endothelial progenitor cells. Circ Res. 2007 Feb 2;100(2):204–12. doi: 10.1161/01.RES.0000257774.55970.f4. [DOI] [PubMed] [Google Scholar]
- 36.Carmona G, Gottig S, Orlandi A, et al. Role of the small GTPase Rap1 for integrin activity regulation in endothelial cells and angiogenesis. Blood. 2009 Jan 8;113(2):488–97. doi: 10.1182/blood-2008-02-138438. [DOI] [PubMed] [Google Scholar]
- 37.Manavski Y, Carmona G, Bennewitz K, et al. Brag2 differentially regulates beta1- and beta3-integrin-dependent adhesion in endothelial cells and is involved in developmental and pathological angiogenesis. Basic Res Cardiol. 2014 Mar;109(2):404. doi: 10.1007/s00395-014-0404-2. [DOI] [PubMed] [Google Scholar]
- 38.Athanasopoulos AN, Schneider D, Keiper T, et al. Vascular endothelial growth factor (VEGF)-induced up-regulation of CCN1 in osteoblasts mediates proangiogenic activities in endothelial cells and promotes fracture healing. J Biol Chem. 2007 Sep 14;282(37):26746–53. doi: 10.1074/jbc.M705200200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Athanasopoulos AN, Economopoulou M, Orlova VV, et al. The extracellular adherence protein (Eap) of Staphylococcus aureus inhibits wound healing by interfering with host defense and repair mechanisms. Blood. 2006 Apr 1;107(7):2720–7. doi: 10.1182/blood-2005-08-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Economopoulou M, Bdeir K, Cines DB, et al. Inhibition of pathologic retinal neovascularization by alpha-defensins. Blood. 2005 Dec 1;106(12):3831–8. doi: 10.1182/blood-2005-03-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Economopoulou M, Langer HF, Celeste A, et al. Histone H2AX is integral to hypoxia-driven neovascularization. Nat Med. 2009 May;15(5):553–8. doi: 10.1038/nm.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langer HF, Chung KJ, Orlova VV, et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood. 2010 Nov 25;116(22):4395–403. doi: 10.1182/blood-2010-01-261503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Economopoulou M, Avramovic N, Klotzsche-von Ameln A, et al. Endothelial-specific deficiency of Junctional Adhesion Molecule-C promotes vessel normalisation in proliferative retinopathy. Thromb Haemost. 2015 Nov 25;114(6):1241–9. doi: 10.1160/TH15-01-0051. [DOI] [PubMed] [Google Scholar]
- 44.Baker M, Robinson SD, Lechertier T, et al. Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc. 2012 Jan;7(1):89–104. doi: 10.1038/nprot.2011.435. [DOI] [PubMed] [Google Scholar]
- 45.Pitulescu ME, Schmidt I, Benedito R, et al. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nature protocols. 2010 Sep;5(9):1518–34. doi: 10.1038/nprot.2010.113. [DOI] [PubMed] [Google Scholar]
- 46.Chavakis E, Urbich C, Dimmeler S. Homing and engraftment of progenitor cells: a prerequisite for cell therapy. J Mol Cell Cardiol. 2008 Oct;45(4):514–22. doi: 10.1016/j.yjmcc.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 47.Grossman PM, Mendelsohn F, Henry TD, et al. Results from a phase II multicenter, double-blind placebo-controlled study of Del-1 (VLTS-589) for intermittent claudication in subjects with peripheral arterial disease. Am Heart J. 2007 May;153(5):874–80. doi: 10.1016/j.ahj.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 48.Soloviev DA, Hazen SL, Szpak D, et al. Dual Role of the Leukocyte Integrin alphaMbeta2 in Angiogenesis. J Immunol. 2014 Nov 1;193(9):4712–21. doi: 10.4049/jimmunol.1400202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu Y, Ip JE, Huang J, et al. Essential role of ICAM-1/CD18 in mediating EPC recruitment, angiogenesis, and repair to the infarcted myocardium. Circ Res. 2006 Aug 4;99(3):315–22. doi: 10.1161/01.RES.0000235986.35957.a3. [DOI] [PubMed] [Google Scholar]
- 50.Numasaki M, Fukushi J, Ono M, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003 Apr 1;101(7):2620–7. doi: 10.1182/blood-2002-05-1461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.