

Abstract
Phenylketonuria's (PKU) treatment based on low natural protein diet may affect homocysteine (Hcys) metabolic pathway. Hcys alteration may be related to the methylation of arginine to asymmetric dimethylarginine (ADMA) and symmetric dimethylarginine (SDMA), which both modify nitric oxide production. The aim of this work is to evaluate the status of Hcys formation methylation cycle and ADMA and SDMA levels in patients with PKU in order to establish a potential relationship.
Forty-two early diagnosed PKU patients under dietary treatment and good adherence to their diets were enrolled in this cross-sectional study. Their nutritional and biochemical profile, as well as Hcys synthesis status, ADMA and SDMA levels were analyzed and compared with a control group of 40 healthy volunteers. ADMA and SDMA were determined by high-performance liquid chromatography system coupled to triple quadrupole mass spectrometer.
In this study, 23 classic PKU, 16 moderate PKU, and 3 mild HPA were enrolled. The median age was 10 years old. Median ADMA, SDMA, and Hcys concentration levels (5.1 μM [2.3–25.7], 0.35 μM [0.18–0.57], 0.43 μM [0.26–0.61], respectively) were lower in patients with PKU (P < .001 for ADMA and SDMA) whereas vitamin B12 and folate levels (616 pg/mL [218–1943] and 21 ng/mL [5–51], respectively) were higher comparing with controls. Statistically significant correlations were found between ADMA, and Phe (r = −0.504, P = .001) and Hcys (r = −0.458, P = .037) levels. Several nutrition biomarkers, such as prealbumin, 25-hydroxy vitamin D, selenium, and zinc, were below the normal range.
Our study suggests that patients with PKU suffer from poor methylation capacity. Restriction of natural proteins in addition to high intake of vitamin B12 and folic acid supplementation in the dietary products, produce an impairment of methylation cycle that leads to low Hcys and ADMA levels. As a result, methylated compounds compete for methyl groups, and there is an impairment of methylation cycle due to low Hcys levels, which is related to the lack of protein quality, despite of elevated concentrations of cofactors.
Keywords: asymmetric dimethylarginine, creatine, homocysteine, methylation cycle, phenylketonuria
1. Introduction
Phenylketonuria (PKU, MIM #261600) is the most common inborn error of amino acid metabolism. This disease results from a deficiency of the enzyme phenylalanine-4-hydroxylase (PAH), which catalyzes the conversion of the essential amino acid phenylalanine (Phe) to tyrosine.[1] Since PKU is included in most Newborn Screening programs, dietary treatment, which consists of a low-Phe diet, is usually initiated soon after birth to avoid neurological damage and developmental delay. As this treatment is mainly a vegan-like diet, it requires supplementation with Phe-free protein substitutes[2] and specially manufactured low-protein foods. (6R)-l-erythro-5,6,7,8-tetrahydrobiopterin (6R-BH4) is a cofactor of PAH used as therapy, which allows patients with PKU to partially or totally liberalize their diet maintaining blood Phe levels within safe limits. Specifically, this treatment may enable them to increase their intake of natural proteins and reduce the need for them to be supplemented with protein substitutes.[3] Therefore, 6R-BH4 therapy enables them to consume larger amounts of proteins and growth-related nutrients contained in protein-rich foods, such as essential fatty acids, minerals, and vitamins.
Low homocysteine (Hcys) levels in PKU population have been reported.[4] This compound is key in the methylation cycle, and since methionine (Met) mainly comes from diet this cycle may be affected by the PKU diet. The methylation cycle consists of the donation of a methyl group from S-adenosylmethionine to several methyl acceptors, such as DNA, myelin, hormones, phosphatidylethanolamine, cobalamin, or guanidinoacetate (GAA),[5] which receives the methyl group to generate creatine (Cr). Once S-adenosylmethionine losses its methyl group, it is converted to S-adenosylhomocysteine by transmethylation[6] (Fig. 1). Then, released Hcys can be degraded to glutathione and taurine by transsulfuration.[7] On the other hand, Hcys can be remethylated via folate and vitamin B12 as cofactors.[8,9]
Figure 1.
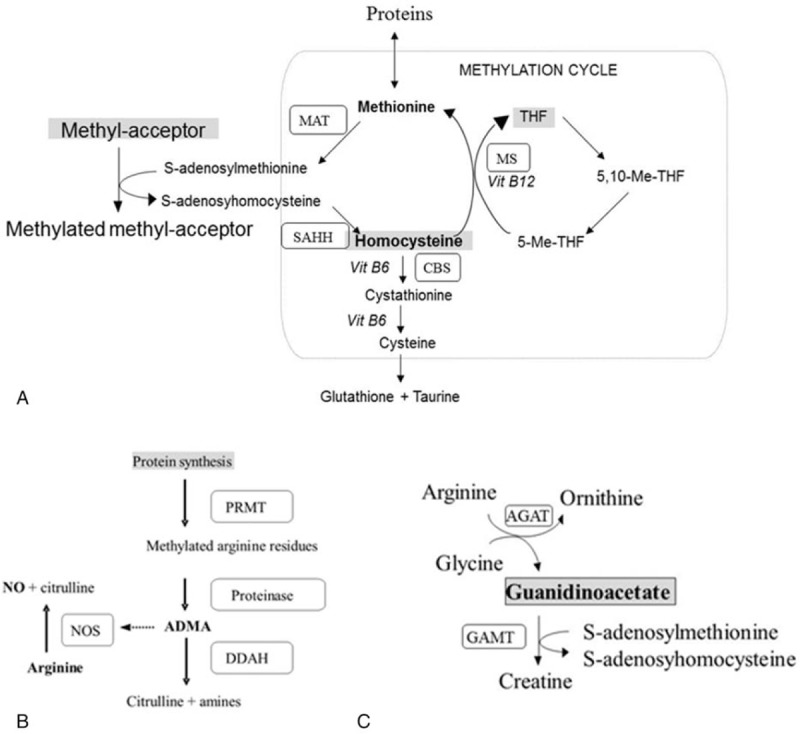
Methylation cycle pathway: (A) Methionine and homocysteine (Hcys) are generated by the following enzymes: CBS = cystathionine β-synthase, MAT = methionine adenosyltransferase, MS = methionine synthase, SAHH = S-adenosylhomocysteine hydrolasa. (B) Asymmetric dimethylarginine (ADMA) synthesis catalyzed by protein arginine methyltransferases (PRMT) and proteinases, and its involvement in the inhibition of nitric oxide by competition with nitric oxide synthase (NOS) for arginine. ADMA is degraded to citrulline and amines by dimethylarginine dimethylamino hydrolase (DDAH). (C) Relationship between the metabolic pathway from arginine to creatine, where guanidinoacetate acts as a methyl acceptor, and the methylation cycle. AGAT = l-arginine: glycine amidinotransferase, GAMT = guanidinoacetate N-methyltransferase.
Moreover, in protein methylation protein arginine (Arg) residues can be methylated in various ways, and therefore generates 3 types of compounds: asymmetric dimethylarginine (ADMA), symmetric dimethylarginine (SDMA), and N-monomethyl-l-arginine (NMMA). The methylation of protein Arg residues is catalyzed by type I and II protein arginine methyltransferase (PRMT), whose activity also depends on S-adenosylmethionine as a donor of methyl groups. Type I PRMT leads to the formation of ADMA, which is produced in all cells and competes with Arg for the nitric oxide synthase (NOS) isoforms, while type II PRMT produces NMMA and SDMA, which do not directly affect NOS activity, but may compete with Arg in the cationic amino acid transporter of endothelial cell membrane,[10] indirectly altering NO production.[11] Altered NO synthesis and ADMA levels is a result of reduced dimethylarginine dimethylaminohydrolase (DDAH) activity, which is distributed in the liver and kidneys,[12] and degrades ADMA, but not SDMA, to citrulline and dimethylamine (Fig. 1). In addition to lower levels of Hcys, lower ADMA levels in PKU population have been also described.[4] Moreover, there is a potential link between both compounds, because Hcys may increase ADMA levels by triggering protein arginine methylation, and the related increase in reactive oxygen species may impair ADMA release and degradation.[13]
We could hypothesize that levels of cardiovascular markers, such as dimethylated arginines, are related to methylation status and may be altered in patients with PKU due to low natural protein diet. The aim of this study was to evaluate the status of the methylation cycle and the levels of novel cardiovascular biomarkers (ADMA and SDMA) in patients with PKU in order to establish a potential relationship between both variables and the impact on the disease status.
2. Materials and methods
2.1. Patients
Forty-two patients with PKU from Cruces University Hospital and Santiago de Compostela Clinic University Hospital were enrolled in this cross-sectional study. The patients included in the study, 9 patients on 6R-BH4 (sapropterin dihydrochloride [KUVAN, Merck, Madrid, Spain]) and diet, and 33 treated exclusively with Phe-restricted diets, were diagnosed through Newborn Screening Programs and a definitive diagnosis was obtained by mutation analysis of the PAH gene. A defect in the synthesis or regeneration pathways of 6R-BH4 was ruled out by analyzing urinary pterin levels as well as by measuring the dihydropteridine reductase (DHPR) activity. Taking into account the classification proposed[14] depending on plasma Phe levels at time of diagnosis and the Phe tolerance and blood Phe levels, patients were classified into 1 of 3 phenotypic categories: mild hyperphenylalaninemia (mild HPA) (360–600 μmol/L), mild–moderate PKU (600–1200 μmol/L), and classic PKU (>1200 μmol/L). The inclusion criteria for the study were: early diagnosis of PKU; early and continuous treatment with Phe-restricted diet, supplemented with Phe-free substitutes and specially manufactured low-protein foods; absence of any other diseases known to affect physical development; and regular attendance to their scheduled clinical check-ups.
Control values for amino acids, ADMA, SDMA, GAA, and Cr in plasma were determined in 40 healthy volunteers matched by age and gender who had a minor surgery in our center. For the other variables, the reference ranges from our own population used in Biochemistry Laboratory at Cruces University Hospital were taken.
The study protocol was approved by Clinical Research Ethics Committees at both hospitals involved (code PI2012098); and performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Written informed consent was obtained from patients and parents or legal guardians of all the children included as patients or controls. Samples and follow-up data were collected over a period of 1 year (November 2014 to November 2015).
2.2. Analytical methods
Biological data in blood samples correspond to single measurements carried out for a year in the PKU population. Blood samples were taken in the morning, after an overnight fasting, at the time of regular follow-up hospital visits. Blood samples were immediately centrifuged at 1000g for 5 minutes at 4°C. The platelet-poor plasmas were aliquoted and stored at −80°C until the assays were performed, usually within a few days. Cholesterol, triglycerides, glucose, insulin, vitamin B12, folate, calcium, vitamin D, selenium, zinc, prealbumin, total protein, Hcys, aspartate transaminase, alanine transaminase, gamma-glutamyltransferase, C-reactive protein, and creatinine were measured using standard laboratory techniques in Biochemistry Laboratories at both hospitals. Total protein intake and its natural protein fraction were calculated according to the 3-day food records completed by adult patients or parents/legal guardians for juvenile patients. Characteristics of growth were normalized by Z-score using web resources for Spanish population (“http://www.webpediatrica.com/endocrinoped/antropometria.php”). The plasma amino acids quantification was determined by a Biochrom 30 ionic chromatograph (Gomensoro, Madrid, Spain).
Arg levels and its derivatives (ADMA and SDMA) were determined by a 1100 high-performance liquid chromatography (HPLC) system coupled to a 6410 triple quadrupole mass spectrometer (both from Agilent Technologies, Madrid, Spain), as previously described.[15] Briefly, sample preparation plasma proteins were removed by ultrafiltration using centrifugal filters, avoiding derivatization, solid phase extraction, and organic solvents or acids. ADMA–SDMA chromatographic separation was performed by reverse phase chromatography in order to use the most sensitive mass transition (m/z 203.2–70.1). Positive electrospray ionization was performed and analytes were detected by multiple reaction monitoring.
Plasma GAA and Cr levels were quantified as previously described,[16] by liquid–liquid extraction with acetonitrile, using13C2-GAA and d3-Cr as internal standards. GAA and Cr butyl esters were separated and quantified on a HPLC system coupled to a triple quadrupole mass spectometer.
2.3. Statistical analysis
Statistical analysis was performed using SPSS 22.0 for Windows (IBM, Chicago, IL). Descriptive statistics are presented as median and 3rd–97th percentile range. Initially, all data were analyzed using the Kolmogorov–Smirnov test, to check the normal distribution of the data. Differences between groups were assessed using the Student t test. Pearson correlation coefficients (r) were calculated to assess bivariate correlations. Statistical significance was set at the P < .05 level. Linear regression models were characterized by R-squared and statistical significance (r2 and P, respectively).
3. Results
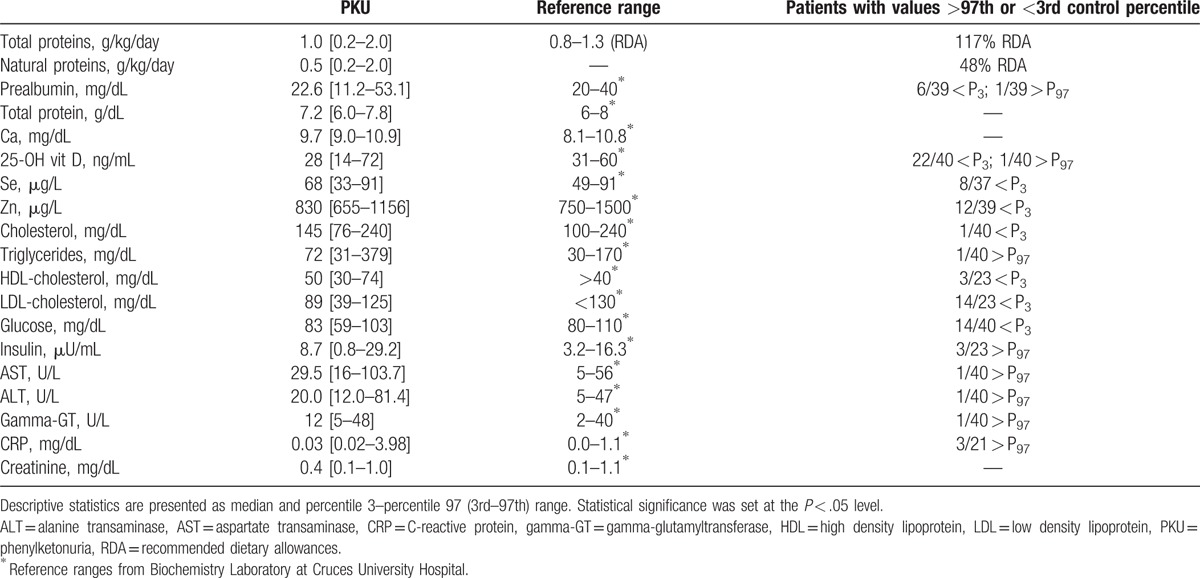
Forty-two patients with PKU (23 males, 19 females) were enrolled in this study: 23 classic PKU, 16 moderate PKU, and 3 mild HPA. The median age was 10 years old, P3 and P97 were 2 and 36 years old, respectively. Only 2 patients below 12 years of age had Phe values above 360 μM which is the maximum value to this age. Eight patients above 12 years of age displayed Phe values above 600 μM, which is the cut-off value for this age. Median and percentiles Z-score for weight, height, and body mass index (BMI) were −0.04 [−1.8 to 2.3], −0.8 [−2.3 to 1.1], and 0.4 [−1.4 to 2.4], respectively. Table 1 shows the biochemical plasma profile of our patients. These results indicate that the levels of several nutrition biomarkers, such as prealbumin (15%), 25-hydroxy vitamin D (55%), glucose (35%), Se (22%), and Zn (31%), were below the normal range. Other variables, such as transaminases or C-reactive protein, did not show a statistically significant tendency toward higher values comparing to the control range.
Table 1.
Nutritional and biochemical profile of patients with phenylketonuria (PKU).
With regard to variables which are directly related to Hcys formation (methylation cycle), median plasma Hcys (5.1 μM) was clearly decreased, and therefore 11 patients displayed values below the 3rd percentile of controls. However, median levels of the cofactors of remethylation vitamin B12 and folate (616 pg/mL and 21 ng/mL, respectively) were increased comparing with controls (Table 2).
Table 2.
Methylation cycle parameters of patients with phenylketonuria, including cofactors and related pathways, such as asymmetric dimethylarginine and creatine synthesis.
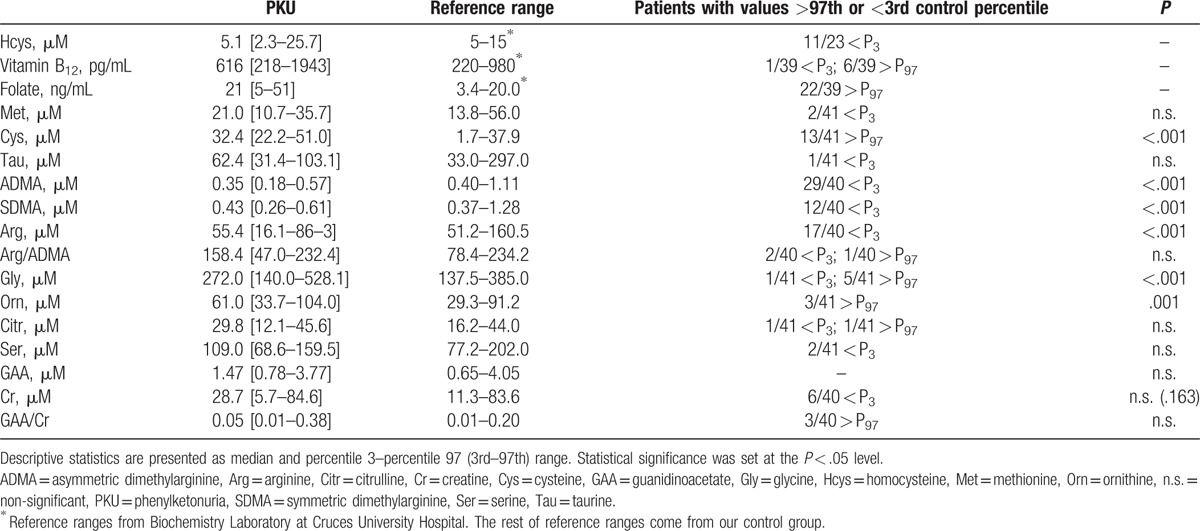
Patients with PKU had statistically significant lower plasma ADMA (P < .001) and SDMA (P < .001) levels (Fig. 2), as well as Arg levels (P < .001), than the control group. It should be noted that although plasma Cr levels were not significantly lower in patients comparing with the healthy population (P = .163), there was a tendency toward lower Cr values and higher GAA/Cr ratio (Table 2), while maintaining GAA values in the normal range.
Figure 2.
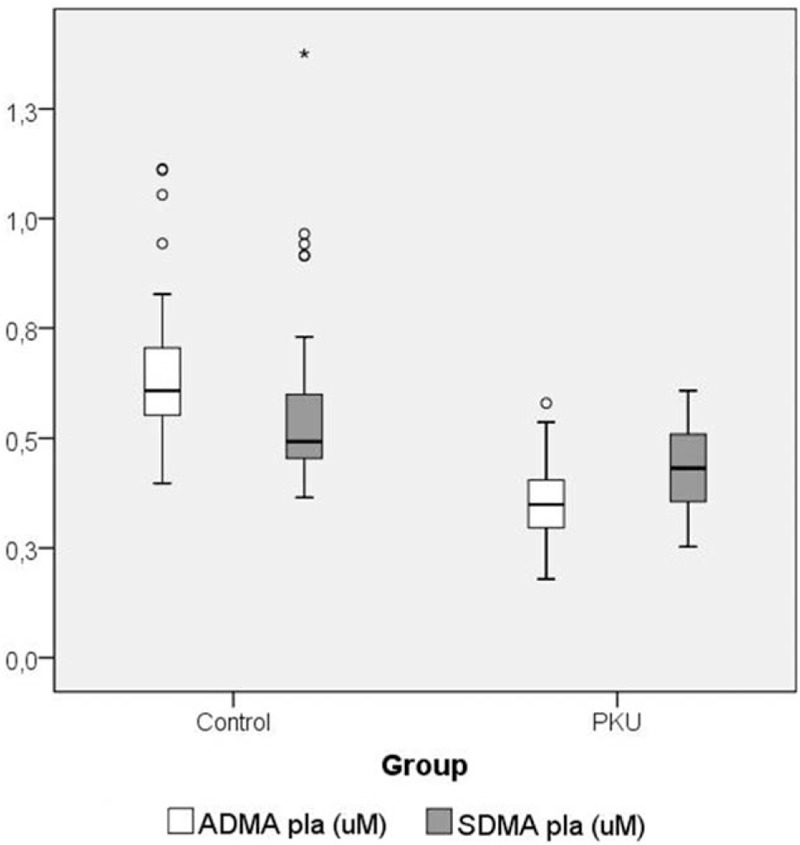
Plasma levels of asymmetric dimethylarginine (ADMA) and symmetric dimethylarginine (SDMA) in patients with PKU comparing with the control group.
As far as the correlations among variables is concerned, there was a statistically significant inverse correlation between ADMA levels and Hcys levels (r = −0.458, P = .037). Hcys levels inversely correlated with folate levels (r = −0.483, P = .020). However, no correlation was observed between Phe plasma levels and cholesterol fractions. Serum total protein significantly correlated with cholesterol (r = 0.485, P = .002) and low density lipoprotein (LDL)-cholesterol (r = 0.523, P = .011). Phe plasma concentrations significantly correlated with prealbumin (r = 0.479, P = .002) and Hcys (r = 0.664, P = .001) (Fig. 3), but inversely with ADMA (r = −0.504, P = .001) (Fig. 4), Arg (r = −0.391, P = .011), and Cr (r = −0.377, P = .018).
Figure 3.
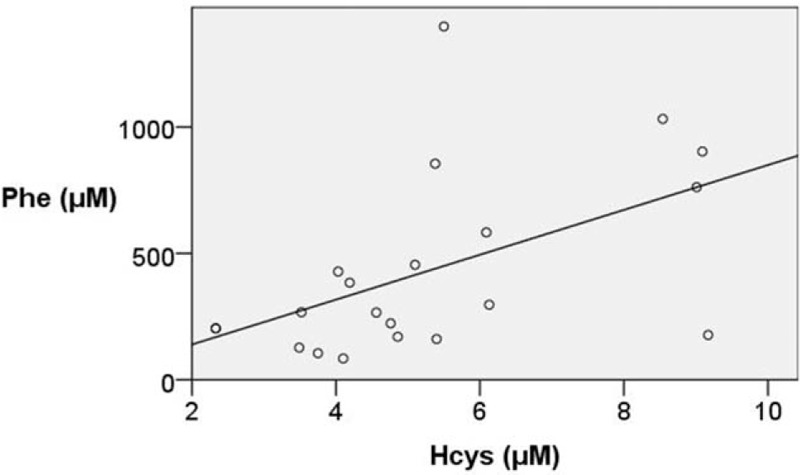
Correlation between asymmetric Phe (μM) and Hcys (μM) levels by linear regression (corrected r2 = 0.265, P = .017).
Figure 4.
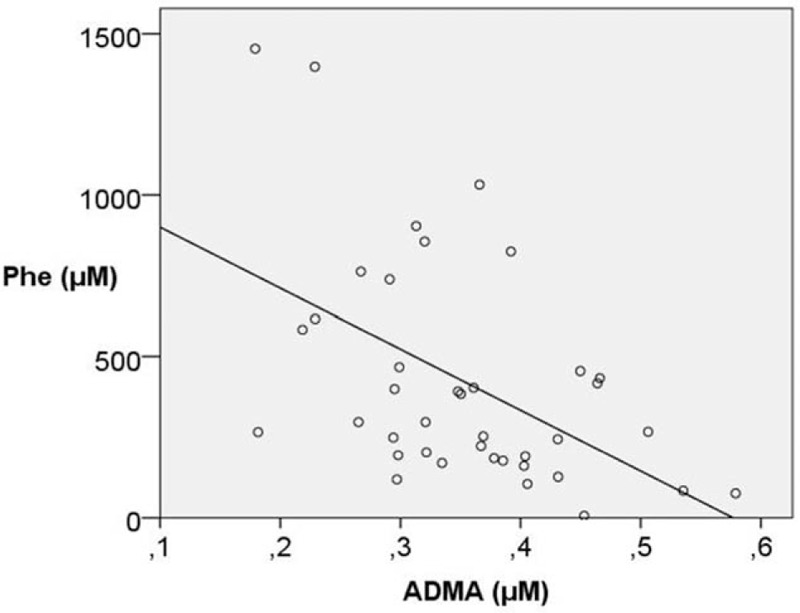
Correlation between Phe (μM) and ADMA (μM) levels by linear regression (corrected r2 = 0.302, P < .001).
Patients with PKU under 6R-BH4 showed a tendency toward higher levels of ADMA (P = .170), glucose (P = .083), and insulin (P = .057) comparing with the group of patients with protein-restricted diet. On the other hand, Cr levels (P = .008), vitamin B12 (P < .001), cysteine (P = .037), and folate (P = .093) were significantly lower in the 6R-BH4 therapy group than in the protein-restricted diet group.
4. Discussion
Nowadays, novel aspects of Hcys metabolism and cardiovascular risk are investigated in PKU patients with low-Phe diets. This work aimed to check the status of Hcys levels (methylation cycle) and cardiovascular biomarkers, such as ADMA and SDMA, in patients with PKU in order to establish a relationship between both variables and nutritional status.
Calcium and micronutrients are key elements during growth; and therefore they should meet the recommended dietary allowances (RDA) in order to prevent nutritional deficiency.[17] However, in our PKU population, while calcium concentrations were found in the normal range, vitamin D was 55% below the control range; a common fact that has been reported in the general population living in less sunlight-hours areas.[18]
With regard to zinc and selenium levels, both were remarkably decreased in our population comparing with the control group, as previously reported.[19,20] These deficits are due to the specific protein-restricted diet which could affect growth development. Indeed, the low micronutrients levels found in PKU population may also alter the antioxidant capacity, and therefore it should be strongly advisable to adjust their intake to the recommended doses by means of nutritional supplements.[21]
It should be pointed out that hypocholesterolemia has been related to patients with PKU who had good adherence to the diet.[22] However, in our study there was no significant correlation between Phe levels and LDL or high density lipoprotein-cholesterol. This result suggests that low cholesterol intake due to low-protein diet may be the cause of hypocholesterolemia (olive oil and cream cheese were the only sources of lipid intake), rather than a role of Phe in the inhibition of cholesterol biosynthesis.[23] Consistent with this hypothesis, in our study there was a clear correlation between total serum protein levels and cholesterol and LDL-cholesterol.
Moreover, in this study we found low prealbumin levels, which positively correlated with Phe concentrations. This finding suggests that despite the fact that total protein intake reached 100% of RDA, there is still a protein necessity or an increase in its quality. In this regard, these low natural proteins levels as well as total protein quality may affect the methylation capacity as well as pathways related to the methylation cycle. It should be pointed out that amino acids supplements may suffer from oxidation, and consequently these amino acids would not be efficient enough.
As previously reported, low-Phe PKU diet supplemented with protein substitutes implies that vitamin B12 and folate concentrations are above control ranges.[24,25] Therefore, protein substitutes should be adjusted in adult PKU patients in order to follow vitamins B recommendations.[26] In this regard, it has been suggested that high intake (above the recommended levels) of folic acid and vitamin B12 in PKU patients could have side effects such as increased tumor risk.[27]
As far as the metabolic pathways is concerned, it should be noted that Hcys synthesis is partially ensured due to polyvitamin status provided in the amino acids supplements, which are enriched with vitamin B12 and folic acid, cofactors of remethylation and transsulfuration. To the best of our knowledge, there is no evidence of a reduced methyl-group supply. However, as it has been previously described[4,28,29] we have observed decreased ADMA and Hcys levels in our PKU population with amino acid supplementation. Accordingly, Hcys range in our PKU population was altered due to low-quality protein intake. Indeed, we found a statistically significant inverse correlation between Hcys and folate levels, suggesting that adjusting folic acid intake, may normalize Hcys levels.
It should be highlighted that hyperhomocysteinemia may also cause accumulation of ADMA, because Hcys has an effect on the enzymatic activity of DDAH, which degrades ADMA to citrulline and amines. In particular, the Hcys links to DDAH modifying the enzymatic configuration and inhibiting its activity.[30] Moreover, the high biochemical reactivity of Hcys could release methylated Arg from proteins and generates ADMA. However, in this study we found that Hcys levels were low and inversely correlated with ADMA levels. This result indicates that under this methylation deficient situation there is a strong competition for methyl group between Arg methylation to produce ADMA and Cr synthesis from GAA; and therefore, if a certain amount of Hcys is generated by transmethylation, there would be few methyl groups left for the methylation. In this regard, by linear regression models we found that higher Phe values in severe episodes of the disease correlated with lower ADMA levels. Moreover, under low ADMA levels, Hcys competes for methyl groups and inversely correlates with ADMA.
It has been reported that since urinary and plasma nitrate and nitrite levels were normal, low ADMA levels seemed not to be altered due to a poor NO synthesis in patients with PKU or homocystinuria.[29] Consistent with our findings, patients with PKU also displayed lower ADMA and Arg levels than the control group.[29] Therefore, taking into account that total antioxidant status is higher in children with PKU due to dietary antioxidants intake, cardiovascular risk could be caused by low NO bioavailability.[31] On the other hand, one could hypothesized that low ADMA levels are linked to low glucose levels, since it has been reported that low blood glucose levels in PKU are related to an increased DDAH enzymatic activity.[32] In this regard, a relationship between ADMA and insulin resistance has been suggested, since low glycemic index seemed to upregulate DDAH activity.[4] However, in our modest opinion we do not consider that glucose levels in our PKU group were too low (only 1 patient had them below 60 mg/dL) to have an effect on DDAH activity. Moreover, in this study no correlation between ADMA and glucose was found.
With regard to the process to obtain energy, low Cr levels in patients with PKU may have an effect on phosphocreatine (PCr) synthesis, and therefore ATP production, which is essential in high energy consumption tissues, such as muscle and brain.[33] In fact, these alterations in Cr metabolism reduce Cr levels in the central nervous system, and eventually lead to poor neuronal growth, with hypotonia and mental delay as clinical symptoms. This could also be caused by high Phe levels at first stages of the disease. Furthermore, patients with PKU have an impaired conversion of Phe to tyrosine, an essential precursor of a wide variety of neurotransmitters, including dopamine, which is critically involved in higher-order cognitive operations subserved by the prefrontal cortex of the brain.[34] It should be noted that PCr produces energy faster than glycolysis and oxidative phosphorylation. Its cellular expression is highly regulated by Cr and metabolic enzymes; so, Cr/PCr ratio plays a crucial role in the brain energetic homeostasis through novel neuron-glial relationship.[35]
Several therapeutic possibilities could be suggested in order to improve PKU diet and treatment. Firstly, we consider that since Met is an essential amino acid and methyl-donor, Met may be able to restore methylation cycle dysfunction as well as the poor capacity to give methyl groups to acceptors. Secondly, Cr could increase intracellular levels of PCr and ATP, preventing neurological damage in infants and children with PKU. A recent study in a PKU animal rat model concluded that Cr may help prevent brain damage in the brain of patients with restrictive diets.[36] Thirdly, taking into account that NO synthesis and low ADMA and SDMA levels, supplementation of the diet with Arg may improve NO formation, and therefore, endothelial dysfunction. However, further investigations about methylation in PKU would be required in order to design the characteristics a supplement which would increase methylation capacity in patients with PKU.
In the present study, we observed that patients under 6R-BH4 therapy and with a less stricted vegan-like diet displayed a clear tendency toward higher ADMA and glucose values, so these parameters tend to normalize reaching normal values. Similarly, vitamin B12, cysteine, and folate were significantly lower in patients under 6R-BH4 treatment and therefore close to the normal range. These findings suggest that 6R-BH4 has a positive effect on NO synthesis and methylation capacity. However, we are aware of the limitations of this study, since high number of patients and more parameters should be analyzed in order to evaluate this treatment and NO-Hcys metabolic pathways, such as choline, betaine, or S-adenosylmethionine.
5. Conclusions
Our study suggests that patients with PKU suffer from poor methylation capacity and hypohomocysteinemia. As a result, methylated compounds levels, such as ADMA and Cr, were below the control range, and therefore they compete for methyl groups. ADMA analysis would be useful for a follow-up test among PKU to assess long-term methylation capacity and its relationship with nutritional status. In addition, there is an impairment of methylation cycle due to low Hcys levels despite of elevated concentrations of cofactors, which is related to the lack of protein quality. Vitamin B12 and folate in patients with PKU were higher than in the controls due to enrichment in the protein substitutes, and accordingly their amounts should be readjusted. Moreover, analysis of minerals, trace elements, and micronutrients levels should be incorporated in routine laboratory tests for the follow-up of patients with PKU.
Acknowledgment
We thank all patients for kindly participating in the study.
Footnotes
Abbreviations: ADMA = asymmetric dimethylarginine, BH4 = (6R)-l-erythro-5,6,7,8-tetrahydrobiopterin, BMI = body mass index, Cr = creatine, DDAH = dimethylarginine dimethylaminohydrolase, GAA = guanidinoacetate, Hcys = homocysteine, HPA = hyperphenylalaninemia, NO = nitric oxide, NOS = nitric oxide synthase, PCr = phosphocreatine, Phe = phenylalanine, PKU = phenylketonuria, RDA = recommended dietary allowances, SD = standard deviation, SDMA = symmetric dimethylarginine.
Funding: This work was supported in part by NUTRICIA S.L.R. (Madrid, Spain). Research in this paper was partially financed by BioCruces Health Research Institute and Carlos III Health Research Institute. Article-processing charge was covered by the Fundación Ramón Dominguez-C012.
The authors have no conflicts of interest to disclose.
References
- [1].Donlon J, Sarkissian C, Levy H. Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, et al. Hyperphenylalaninemia: Phenylalanine Hydroxylase Deficiency. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2014;Available at: http://ommbid.mhmedical.com/content.aspx?bookid=971&Sectionid=62673211 [accessed December 29, 2015]. [Google Scholar]
- [2].Feillet F, Agostoni C. Nutritional issues in treating phenylketonuria. J Inherit Metab Dis 2010;33:659–64. [DOI] [PubMed] [Google Scholar]
- [3].Ziesch B, Weigel J, Thiele A, et al. Tetrahydrobiopterin (BH4) in PKU: effect on dietary treatment, metabolic control, and quality of life. J Inherit Metab Dis 2012;35:983–92. [DOI] [PubMed] [Google Scholar]
- [4].Huemer M, Simma B, Mayr D, et al. Free asymmetric dimethylarginine (ADMA) is low in children and adolescents with classical phenylketonuria (PKU). J Inherit Metab Dis 2012;35:817–21. [DOI] [PubMed] [Google Scholar]
- [5].Finkelstein JD. The metabolism of homocysteine: pathways and regulation. Eur J Pediatr 1998;157:S40–4. [DOI] [PubMed] [Google Scholar]
- [6].Ueland PM. Pharmacological and biochemical aspects of S-adenosylhomocysteine and S-adenosylhomocysteine hydrolase. Pharmacol Rev 1983;34:223–53. [PubMed] [Google Scholar]
- [7].Finkelstein JD. Metabolic regulatory properties of S-adenosylmethionine and S-adenosylhomocysteine. Clin Chem Lab Med 2007;45:1694–9. [DOI] [PubMed] [Google Scholar]
- [8].Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem 1990;1:228–37. [DOI] [PubMed] [Google Scholar]
- [9].McKeever MP, Weir DG, Molloy A, et al. Betaine-homocysteinemethyltransferase: organ distribution in man, pig and rat and subcellular distribution in the rat. Clin Sci 1991;81:551–6. [DOI] [PubMed] [Google Scholar]
- [10].Vallance P, Leiper J. Cardiovascular biology of the asymmetric dimethylarginine: dimethylargininedimethylaminohydrolase pathway. Arterioescler Thromb Vasc Biol 2004;24:1023–30. [DOI] [PubMed] [Google Scholar]
- [11].Böger RH, Bode-Böger SM, Szuba A, et al. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction. Circulation 1998;98:1842–7. [DOI] [PubMed] [Google Scholar]
- [12].Nijveldt RJ, Teerlink T, van Guldener C, et al. Handling of asymmetrical dimethylarginine and symmetrical dimethylarginine by the rat kidney under basal conditions and during endotoxaemia. Nephrol Dial Transpl 2003;18:2542–50. [DOI] [PubMed] [Google Scholar]
- [13].Antoniades C, Tousoulis D, Marinou K, et al. Asymmetrical dimethylarginine regulates endothelial function in methionine-induced but not in chronic homocystinemia in humans: effect of oxidative stress and proinflammatory cytokines. Am J Clin Nutr 2006;84:781–8. [DOI] [PubMed] [Google Scholar]
- [14].Camp KM, Parisi MA, Acosta PB, et al. Phenylketonuria Scientific Review Conference: state of the science and future research needs. Mol Genet Metab 2014;112:87–122. [DOI] [PubMed] [Google Scholar]
- [15].Andrade F, Llarena M, Lage S, et al. Quantification of arginine and its methylated derivatives in healthy children by liquid chromatography-tandem mass spectrometry. J Chromatogr Sci 2015;53:787–92. [DOI] [PubMed] [Google Scholar]
- [16].Bodamer OA, Bloesch SM, Gregg AR, et al. Analysis of guanidinoacetate and creatine by isotope dilution electrospray tandem mass spectrometry. Clin Chim Acta 2001;308:173–8. [DOI] [PubMed] [Google Scholar]
- [17].Mirás A, Bóveda MD, Leis MR, et al. Risk factors for developing mineral bone disease in phenylketonuric patients. Mol Genet Metab 2013;108:149–54. [DOI] [PubMed] [Google Scholar]
- [18].Pela I. How much vitamin D for children? Clin Cases Miner Bone Metab 2012;9:112–7. [PMC free article] [PubMed] [Google Scholar]
- [19].Robert M, Rocha JC, van Rijn M, et al. Micronutrient status in phenylketonuria. Mol Genet Metab 2013;110:S6–17. [DOI] [PubMed] [Google Scholar]
- [20].Crujeiras V, Aldámiz-Echevarría L, Dalmau J, et al. Micronutrient in hyperphenylalaninemia. Data Brief 2015;4:614–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lammardo AM, Robert M, Rocha JC, et al. Main issues in micronutrient supplementation in phenylketonuria. Mol Genet Metab 2013;110:S1–5. [DOI] [PubMed] [Google Scholar]
- [22].Schulpis KH, Karakonstantakis T, Bartzeliotou A, et al. The association of serum lipids, lipoproteins and apolipoproteins with selected trace elements and minerals in phenylketonuric patients on diet. Clin Nutr 2004;23:401–7. [DOI] [PubMed] [Google Scholar]
- [23].Colomé C, Artuch R, Lambruschini N, et al. Is there a relationship between plasma phenylalanine and cholesterol in phenylketonuric patients under dietary treatment? Clin Biochem 2001;34:373–6. [DOI] [PubMed] [Google Scholar]
- [24].Evans S, Daly A, MacDonald J, et al. The micronutrient status of patients with phenylketonuria on dietary treatment: an ongoing challenge. Ann Nutr Metab 2014;65:42–8. [DOI] [PubMed] [Google Scholar]
- [25].Crujeiras V, Aldámiz-Echevarría L, Dalmau J, et al. Vitamin and mineral status in patients with hyperphenylalaninemia. Mol Genet Metab 2015;115:145–50. [DOI] [PubMed] [Google Scholar]
- [26].Wiig I, Motzfeldt K, Løken EB, et al. Nutritional consequences of adhering to a low phenylalanine diet for late-treated adults with PKU: low Phe diet for adults with PKU. JIMD Rep 2013;7:109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chiang FF, Huang SC, Wang HM, et al. High serum folate might have a potential dual effect on risk of colorectal cancer. Clin Nutr 2015;34:986–90. [DOI] [PubMed] [Google Scholar]
- [28].Özcan Ö, Ipcioglu OM, Gultepe M. Unexpectedly low asymmetric dimethylarginine (ADMA) and homocysteine levels in patients with phenylketonuria (PKU). J Inherit Metab Dis 2012;35:1153. [DOI] [PubMed] [Google Scholar]
- [29].Kanzelmeyer N, Tsikas D, Chobanyan-Jürgens K, et al. Asymmetric dimethylarginine in children with homocystinuria or phenylketonuria. Amino Acids 2012;42:1765–72. [DOI] [PubMed] [Google Scholar]
- [30].Stühlinger MC, Tsao PS, Her JH, et al. Homocysteine impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine. Circulation 2001;104:2569–75. [DOI] [PubMed] [Google Scholar]
- [31].Verduci E, Banderali G, Moretti F, et al. Diet in children with phenylketonuria and risk of cardiovascular disease: a narrative overview. Nutr Metab Cardiovasc Dis 2016;26:171–7. [DOI] [PubMed] [Google Scholar]
- [32].Ellger B, Richir MC, van Leeuwen PA, et al. Glycemic control modulates arginine and asymmetrical-dimethylarginine levels during critical illness by preserving dimethylarginine-dimethylaminohydrolase activity. Endocrinology 2008;149:3148–57. [DOI] [PubMed] [Google Scholar]
- [33].Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev 2000;80:1107–213. [DOI] [PubMed] [Google Scholar]
- [34].Gropman AL. Patterns of brain injury in inborn errors of metabolism. Semin Pediatr Neurol 2012;19:203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tachikawa M, Fukaya M, Terasaki T, et al. Distinct cellular expressions of creatine synthetic enzyme GAMT and creatine kinases uCK-Mi and CK-B suggest a novel neuron-glial relationship for brain energy homeostasis. Eur J Neurosci 2004;20:144–60. [DOI] [PubMed] [Google Scholar]
- [36].Dos Reis EA, Rieger E, de Souza SS, et al. Effects of a co-treatment with pyruvate and creatine on dendritic spines in rat hippocampus and posterodorsal medial amygdala in a phenylketonuria animal model. Metab Brain Dis 2013;28:509–17. [DOI] [PubMed] [Google Scholar]