

Abstract
Background
Familial hypercholesterolaemia (OMIM 143890) is most frequently caused by variations in the low-density lipoprotein receptor (LDLR) gene. Predicting whether novel variants are pathogenic may not be straightforward, especially for missense and synonymous variants. In 2013, the Association of Clinical Genetic Scientists published guidelines for the classification of variants, with categories 1 and 2 representing clearly not or unlikely pathogenic, respectively, 3 representing variants of unknown significance (VUS), and 4 and 5 representing likely to be or clearly pathogenic, respectively. Here, we update the University College London (UCL) LDLR variant database according to these guidelines.
Methods
PubMed searches and alerts were used to identify novel LDLR variants for inclusion in the database. Standard in silico tools were used to predict potential pathogenicity. Variants were designated as class 4/5 only when the predictions from the different programs were concordant and as class 3 when predictions were discordant.
Results
The updated database (http://www.lovd.nl/LDLR) now includes 2925 curated variants, representing 1707 independent events. All 129 nonsense variants, 337 small frame-shifting and 117/118 large rearrangements were classified as 4 or 5. Of the 795 missense variants, 115 were in classes 1 and 2, 605 in class 4 and 75 in class 3. 111/181 intronic variants, 4/34 synonymous variants and 14/37 promoter variants were assigned to classes 4 or 5. Overall, 112 (7%) of reported variants were class 3.
Conclusions
This study updates the LDLR variant database and identifies a number of reported VUS where additional family and in vitro studies will be required to confirm or refute their pathogenicity.
Keywords: Familial Hypercholesterolemia, LDLR, locus specific variant, database, in silico pathogenicity predictions
Introduction
Familial hypercholesterolaemia (FH) is an autosomal co-dominant disorder with a reported frequency in Europe of between 1 in 217 in Denmark1 to 1 in 600 in Finland.2 FH results from defective clearance of atherogenic low-density lipoprotein cholesterol (LDL-C) particles from the blood; consequently, patients with FH have a 5–8 times higher-than-average risk of premature coronary heart disease (CHD),3 which can be significantly reduced with statin treatment.4 5 FH is caused predominantly by variants in the low-density lipoprotein receptor (LDLR) gene.1 Pathogenic changes in the LDLR gene result in either fewer or functionally impaired LDL receptors and consequently impaired LDL-C particle uptake. All recent guidelines for the management of FH (reviewed in ref. 6) have emphasised the clinical utility of identifying the genetic variant in index patients to confirm the FH diagnosis, support the early commencement of intensive statin therapy and to use the information to test and unambiguously identify any relatives who may also carry the same variant,7 thereby allowing them to be offered lifestyle and therapeutic advice to reduce their CHD risk.
In the era of next-generation sequencing, there is an increasing demand for rapid genetic diagnosis; however, variant interpretation remains the most challenging part of the diagnostic process. The availability of an up-to-date open access locus-specific database for LDLR variants is a crucial tool for diagnostic and research laboratories. The UCL LDLR variant database embraces the principles of data sharing (variants and phenotypes), which ultimately will result in improved classification of novel LDLR variants (as pathogenic or not) as more data is collected. Clearly, accurate classification of such variants will be of benefit to patients and their relatives with regard to better diagnosis, prognosis, treatment and appropriate cascade testing. Since its inception in 1997, the UCL LDLR online variant database has undergone four major upgrades, with the addition of newly published variants, analysis and transition to improved web-based platforms.8–11 Predicting whether or not novel variants in LDLR are pathogenic may not always be straightforward, especially for synonymous and missense variants or for those occurring in intronic sequences or the promoter regions of the gene. Recently, guidelines for diagnostic laboratories reporting novel variants have been proposed by the Association for Clinical Genetic Science (ACGS),12 with classes 1 and 2 as clearly not or unlikely to be pathogenic, respectively, class 3 as variants of unknown significance (VUS), and 4 and 5 as likely to be or clearly pathogenic, respectively. The guidelines recommend a number of in silico programs for the prediction of the likely impact of missense variants on the resultant peptide, and intronic, missense and synonymous variants that may affect mRNA splicing. The guidelines state, “It is acceptable to predict the severity of an amino acid change using in-silico methods, but it is unacceptable to rely solely on these predictions to assign pathogenicity to a previously unclassified variant”. For variants affecting splicing, they state, “It is acceptable to assign nucleotide changes that disrupt the consensus dinucleotide splice sites (+/-1 and +/-2) as clearly pathogenic requiring no further investigation. It is acceptable to use in-silico splice site prediction; however, it is unacceptable to base an unequivocal clinical interpretation solely on this line of evidence”. Therefore, in vitro studies or ex vivo examination of RNA is required to look for the presence of abnormal splice products. Here, we update the UCL LDLR variant database with the addition of variants in the literature since 2012 and classification of all variants according to the ACGS guidelines. This should provide a standardised set of data regarding the pathogenicity of reported LDLR gene variants that have been identified in subjects with the clinical phenotype of FH. We believe that this will be of value to diagnostic laboratories and to physicians requesting DNA tests of index cases with FH.
Materials and methods
Identification of LDLR variants
Newly reported variants were identified in the literature using the term ‘familial hypercholesterolemia mutation’ for PubMed searches and alerts. The nomenclature of variants was checked with Mutalzyer (https://mutalyzer.nl)13 to ensure that it adhered to Human Genome Variation Society recommendations (http://www.hgvs.org/mutnomen/).14 In a number of cases, the nomenclature of the variants has been changed due to updated recommendations (eg, single base pair insertions now being described as duplications where one of the adjacent bases is the same as the inserted base and large rearrangements where formerly unknown breakpoints were described, eg, as follows: c.67+1?_941-1?del are now described as c.(67+1_68-1)_(940+1_941-1)del). Nomenclature changes have also been made to correct inaccuracies in the literature in consultation with the originating authors wherever possible. All variants are described with respect to the reference sequences: LDLR LRG_274, based on NG_009060.1 with LRG_274t1 based on NM_000527.4. Variants were submitted to and made available through an LOVD-powered gene variant database15 that can be accessed via http://www.lovd.nl/LDLR .
In silico prediction of variant pathogenicity
Nonsense substitutions, frame-shifting small and large rearrangements were not subjected to in silico analyses as they are accepted to be pathogenic (classes 4 and 5). The predicted effects of missense variants on LDLR function were assessed using the following open access software packages: (a) PolyPhen2 (HumDiv and HumVar) (http://genetics.bwh.harvard.edu/pph2/index.shtml),16 (b) SIFT (Sorting Intolerant From Tolerant) (http://sift.jcvi.org/www/SIFT_seq_submit2.html),17 Refined SIFT (http://sift.jcvi.org/www/SIFT_aligned_seqs_submit.html)10 (for LDLR amino acid sequences used, see online supplementary table S1; for results of Refined SIFT analysis, see online supplementary table S2) and (c) Mutation Taster (http://www.mutationtaster.org).18
Accession numbers of peptide sequences used to generate LDLR refined SIFT analysis
jmedgenet-2016-104054supp001.pdf (346.1KB, pdf)
Refined SIFT Analasis of Human LDLR
jmedgenet-2016-104054supp002.pdf (1.8MB, pdf)
The effects of intronic and some synonymous variants on gene splicing were assessed using Berkeley Drosophila Genome Project, Splice Site Prediction by Neural Network, with the minimum scores for 5′ and 3′ splice sites set at 0.01 (http://www.fruitfly.org/seq_tools/splice.html)19 and SplicePort (http://spliceport.cbcb.umd.edu/).20 Both programs give predictive scores for splice acceptor and donor sequences for wild-type and variant sequences. The pathogenic impact of LDLR variants in the promoter and 5′ untranslated region of the gene have recently been published.21 Where appropriate, structural conservation scores have been given to variants (see online supplementary table S3).11
LDLR Structural Conservation Scores
jmedgenet-2016-104054supp003.pdf (558.6KB, pdf)
Pathogenicity class was assigned to each variant according to the ACGS guidelines as follows: all nonsense substitutions and frame-shifting variants were assigned to classes 4 or 5 (likely to be or clearly pathogenic) as were exon-deleting large rearrangements. Intronic variants affecting residues ±1 or 2 from intron/exon boundaries were assigned to class 4 (or 5 where in vitro evidence revealed frame-shifting splicing events). Pathogenicity scores were assigned to the remaining variants by taking into account information available in the literature including family segregation, in vitro studies and an overall assessment of the predictions from in silico analyses. Furthermore, the reported frequency of variants in large population studies such as the 1000 Genomes22 and Exome Aggregation Consortium (ExAC) database23 was also taken into account, such that a minor allele frequency (MAF) >0.0002 was considered to be suggestive of a non-pathogenic variant as this is the frequency of the most common single FH-causing variant, the p.R3527Q located in APOB.24 For missense variants with no supporting evidence from the literature, variants with three or more consistent in silico predictions were assigned as either class 4 (likely to be pathogenic) or 2 (unlikely to be pathogenic) and those with inconsistent in silico predictions were assigned as class 3 (VUS).
Results
A total of 2925 variants, representing 1707 unique events, have now been added to the UCL LDLR variant database on the LOVD3 platform (http://www.lovd.nl/LDLR). The number of unique events reported on the database homepage differs from 1707 as the compiling software assumes that large rearrangements involving the same exons are identical, whereas they may well have different breakpoints as defined by the differing sizes of the fragments deleted or duplicated. For the purpose of analysis, the variants were subdivided into the following categories: promoter (n=37), intronic (n=181), synonymous substitutions (n=34), nonsense substitutions (n=129), missense substitutions (n=795), large rearrangements (>100 bp) (n=118), small frame-shifting rearrangements (<100 bp) (n=337) and small non-frame-shifting rearrangements (<100 bp) (n=76). A summary of pathogenicity classifications for each variant category is shown in figure 1. Overall, 81% of variants were classified as pathogenic (n=800 class 4 and n=585 class 5) and 12% as non-pathogenic (n=26 class 1 and n=184 class 2), with 7% (n=112) being classed as VUSs.
Figure 1.
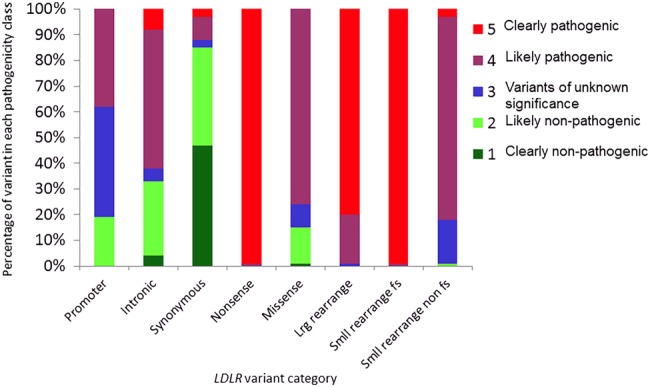
A summary of pathogenicity classifications for each variant category. The percentage of variants in each of the outlined categories according to their pathogenicity classification is shown. fs, frame-shifting; LDLR, low-density lipoprotein receptor; lrg, large; smll, small.
All nonsense substitutions (1 class 4, 128 class 5), small frame-shifting rearrangements (3 class 4, 334 class 5) and 117/118 large rearrangements (22 class 4, 95 class 5) were considered to be pathogenic according to the criteria set out in the ACGS guidelines.12 One large rearrangement was classified as a VUS as it was an in-frame duplication of exons 16–18, which may or may not impact on the correct transcription of the LDLR gene.
Missense substitutions represent the largest category of variants in the database (n=795), of which 605 (76%) were classified as likely pathogenic (class 4), 115 (15%) as clearly not or probably not pathogenic (classes 1 and 2) and 75 (9%) as VUSs. In vitro functional analysis was only available for 73 (9%) missense variants, and so the majority of the pathogenicity classifications were made using in silico predictions. In an attempt to gauge the reliability of the in silico tools used in this study, pathogenicity classifications based on in vitro functional studies and in silico predictions were compared where possible. The results were in agreement for 63/73 (86%) of the variants, and those with discordant results are shown in table 1. Since the functional studies provide the more reliable evidence, we have assigned these variants to the categories suggested by the in vitro and not the in silico data.
Table 1.
Low-density lipoprotein receptor (LDLR) missense variants with discordant in silico and in vitro functional studies
| DNA | Peptide | Refined SIFT | SIFT | PolyPhen 2 HumDiv | PolyPhen 2 HumVar | Mutation taster | Structural conservation | In silico prediction (ACGS classification) | In vitro classification (ACGS classification) | In vitro evidence (LDLR defect classification)25 |
|---|---|---|---|---|---|---|---|---|---|---|
| c.226G>T | p.(Gly76Trp) | Not tolerated | Not tolerated | Probably damaging, 1 | Probably damaging, 1 | Disease causing | 0.922 | Likely to be pathogenic4 | Probably not pathogenic2 | Normal LDLR expression, LDL-binding and internalisation26 |
| c.344G>A | p.(Arg115His) | Not tolerated | Tolerated | Benign, 0.157 | Benign, 0.128 | Disease causing | 0.54 | VUS3 | Likely to be pathogenic4 | Retained in ER, 36% reduced LDLR on cell surface, 36% reduced LDL binding and internalisation (class 2)27 |
| c.768C>G | p.(Asp256Glu) | Not tolerated | Not tolerated | Benign, 0.388 | Benign, 0.329 | Disease causing | 0.721 | VUS3 | Likely to be pathogenic4 | 50–70% reduced LDL binding and internalisation, co-segregated with FH in large family (class 3)28 |
| c.769C>T | p.(Arg257Trp) | Not tolerated | Not tolerated | Probably damaging, 0.993 | Probably damaging, 0.916 | Polymorphism | 0.568 | Likely to be pathogenic4 | Probably not pathogenic2 | Normal LDLR expression, LDL-binding and internalisation29 |
| c.895G>A | p.(Ala299Thr) | Tolerated | Tolerated | Benign, 0.192 | Benign, 0.068 | Polymorphism | 0.28 | Probably not pathogenic2 | Likely to be pathogenic4 | 50% reduced LDL binding and internalisation (class 3)30 |
| c.1285G>C | p.(Val429Leu) | Tolerated | Tolerated | Benign, 0.307 | Benign, 0.225 | Disease causing | 0.672 | Probably not pathogenic2 | Likely to be pathogenic4 | Retained in ER, 75% reduced LDLR on cell surface, >80% reduced LDL binding and internalisation (class 2)31 |
| c.1361C>A | p.(Thr454Asn) | Tolerated | Not tolerated | Possibly damaging, 0.475 | Possibly damaging, 0.738 | Polymorphism | 0.711 | VUS3 | Likely to be pathogenic4 | Normal LDLR expression, 40% reduced LDLR on cell surface, 36%, reduced LDL binding and internalisation. Recycling defect (class 5)32 |
| c.1942T>C | p.(Ser648Pro) | Tolerated | Tolerated | Benign, 0.12 | Benign, 0.083 | Polymorphism | 0.537 | Probably not pathogenic2 | Likely to be pathogenic4 | Retained in ER, 80% reduced LDLR on cell surface, 75% reduced LDL binding and internalisation (class 2)31 |
| c.2389G>A | p.(Val797Met) | Not tolerated | Tolerated | Benign, 0.448 | Benign, 0.085 | Disease causing | 0.545 | Probably not pathogenic2 | Likely to be pathogenic4 | RT-PCR disruption of normal gene splicing33 |
| c.2389G>T | p.(Val797Met) | Tolerated | Tolerated | Benign, 0.002 | Benign, 0.007 | Disease causing | 0.545 | Probably not pathogenic2 | Likely to be pathogenic4 | RT-PCR disruption of normal gene splicing, exon skipping34 |
ACGS, Association for Clinical Genetic Science; FH, familial hypercholesterolaemia; LDL, low-density lipoprotein; SIFT, Sorting Intolerant From Tolerant; VUS, variants of unknown significance.
From the total of 181 intronic variants, the 98 involving bases at positions ±1 or 2 from intron/exon boundaries were considered to be pathogenic. Seventeen of these were classed as clearly pathogenic (class 5) as in vitro evidence was available from the literature that demonstrated the disruption of normal mRNA splicing. In vitro functional analysis was available for 15 of the variants that involved bases beyond ±2 in the intron; as a result, 13 were classified as pathogenic (8 class 4, 5 class 5) (table 2). Of the remaining 70 intronic variants 11 were classified as class 3 as in silico predictions suggested that they would disrupt normal splicing, but no in vitro evidence was available.
Table 2.
Low-density lipoprotein receptor intronic variants affecting residues >±1 or 2
| Intron | Variant | Pathogenicity classification | In vitro evidence | Mechanism |
|---|---|---|---|---|
| 2 | c.190+2_190+3dup | Clearly pathogenic5 | cDNA sequencing revealed use of novel splice donor site (GT) at c.190+3_190+4, resulting in inclusion 2 bp from 5′ end of intron 2, a frame shift and premature truncation of the peptide—p.(Leu64Cysfs*143).35 | Use of novel splice site |
| 3 | c.313+2dup | Likely to be pathogenic4 | RT-PCR sequencing revealed skipping of exon 3, resulting in an in-frame deletion—p.(Leu64_Pro105delinsSer).30 | Inactivation of wild-type splice site |
| 3 | c.313+5G>A | Likely to be pathogenic4 | cDNA sequencing revealed skipping of exon 3, resulting in an in-frame deletion—p.(Leu64_Pro105delinsSer).36 | Inactivation of wild-type splice site |
| 3 | c.313+6T>C | Likely to be pathogenic4 | cDNA sequencing revealed skipping of exon 3, resulting in an in-frame deletion—p.(Leu64_Pro105delinsSer).34 | Inactivation of wild-type splice site |
| 6 | c.941-12G>A | Likely to be pathogenic4 | RT-PCR sequencing revealed abnormal splicing of intron 6 (no further information provided by authors).37 | Unknown |
| 7 | c.1061-8T>C | Clearly not pathogenic1 | RT-PCR sequencing reveals normal splicing of intron 7.34 | Normal splicing |
| 8 | c.1186+5G>A | Clearly pathogenic5 | RT-PCR sequencing revealed inclusion of intron 8, resulting in a frame shift and premature truncation of the peptide—p.(Gly396fs*26).30 | Use of novel splice site |
| 8 | c.1187-10G>A | Clearly pathogenic5 | cDNA sequencing reveals creation of novel acceptor site resulting in inclusion of 8 bp (ACCCCCAG) from 3′ end of intron 8, a frame shift and premature truncation of the peptide—p.(Gly396Aspfs*20).38 | Use of novel splice site |
| 9 | c.1359-31_1359-23delinsCGGCT | Clearly pathogenic5 | mRNA sequencing revealed retention of intron 9 and evidence that two additional transcripts are produced using cryptic splice sites in exon 10. Due to removal of the invariant A at consensus splicing branch site in intron 9.39 | Use of novel splice sites |
| 9 | c.1359-5C>G | Clearly pathogenic5 | mRNA sequencing revealed retention of intron 9, resulting in a frame shift and premature truncation of the peptide—p.(Ser453Argfs*1).34 | Inactivation of wild-type splice site |
| 10 | c.1586+5G>A | Likely to be pathogenic4 | RT-PCR sequencing revealed alternate splicing resulting in two abnormal mRNAs: (a) skipping of exon 10 and (b) inclusion of 22 novel amino acids from the activation of a cryptic splice site.40 | Inactivation of wild-type splice site and use of cryptic splice site |
| 12 | c.1845+11C>G | Likely to be pathogenic4 | RT-PCR revealed that approximately half the transcripts use the novel splice site, resulting in inclusion of 11 bp from 5′ end of intron 12 into the transcript, a frame shift and premature truncation of the peptide—p.(Glu615fs*53).41 | Normal splicing and use of novel splice site |
| 14 | c.2140+5G>A | Unlikely to be pathogenic2 | RT-PCR sequencing reveals normal splicing of intron 14.34 | Normal splicing |
| 14 | c.2140+86C>G | Likely to be pathogenic4 | RT-PCR sequencing revealed creation of a novel splice donor site, resulting in inclusion of 81 bp from 5′ end of intron 14 and a 27aa insertion into peptide p.(Thr713_Glu714ins27). Resulting peptide fails to leave the ER.42 | Use of novel splice site |
| 15 | c.2312-3C>A | Likely to be pathogenic4 | cDNA sequencing reveals skipping of exon 16, predicted to result in an in-frame deletion—p.(Ala771_Ile796del).36 | Inactivation of wild-type splice site |
Only two of the in-frame small rearrangements (c.887_889delinsAGC, c.1659_1661delinsATACTTTCA) were classified as clearly pathogenic as both resulted in the creation of in-frame termination codons. Of the remaining 74 variants in this category, 60 were assigned to class 4 and 13 to class 3 (VUS). Variant c.1120_1121delinsTC was assigned to class 2 because the resultant amino acid change, p.(Gly374Ser), was predicted to be benign according to in silico analysis.
Although the majority (85%) of synonymous substitutions were predicted to be benign (16 class 1, 13 class 2), four were considered to be pathogenic. In silico analyses predicted that c.621C>T, p.(Gly207=), c.1216C>A, p.(Arg406=) and c.1813C>T, p.(Leu605=), would create cryptic splice sites with higher affinities than the wild-type sites, and furthermore, in vitro evidence was available to support the predictions for these variants.43 44 The variant c.1845G>A, p.(Glu615=), which alters the last base of exon 12, was predicted to destroy the exon 12 splice donor site, although because no in vitro analysis was available for this variant it has been classified as class 4.
As mentioned previously, across the database 112 variants (7% of the total) were classified as VUSs (class 3). With the exception of nonsense and frame-shifting small rearrangements, these variants occur in all other variant categories, 43% (n=16) of variants in the promoter region were class 3, as were 17% (n=13) of small non-frame-shifting rearrangements, 9% (n=75) of missense and 4% (n=8) intronic variants. Finally, only one synonymous and one large rearrangement were assigned to class 3.
Discussion
The ACGS practice guidelines for the evaluation of pathogenicity12 designate variants as classes 1 and 2 as clearly not or unlikely to be pathogenic, respectively, 3 as a VUS, and 4 or 5 as likely to be or clearly pathogenic, respectively. In general, we have adhered to these guidelines, but for FH there is such a strong a priori probability that an FH-causing variant will be found in the LDLR gene that we believe less stringent proof of functionality is reasonable. In particular, we believe that showing a novel variant with ‘likely pathogenic’ in silico predictions may be reported as class 4 if it is present in several unrelated patients with FH and is absent or at very low frequency (<MAF=0.0002) in sequence databases such as the 1000 Genomes22 and ExAC database (http://exac.broadinstitute.org/). The finding in this study that in silico predictions matched the in vitro evidence in 63/73 (86%) missense variants lends support to this view.
Of the variants where the results were not in agreement (table 1), five class 2 and three class 3 variants by in silico analyses were shown to be class 4 by in vitro functional studies. The variant c.226G>T, p.(Gly76Trp), resulted in strong pathogenic predictions from PolyPhen2, both SIFT analyses and Mutation Taster, and it might be expected that replacement of the highly conserved small glycine amino acid at position 76 with the larger tryptophan would be detrimental. However, this variant was reported in non-FH family members and displayed normal levels of LDLR expression, LDL-binding and internalisation.26 45 Similarly, c.769C>T, p.(Arg257Trp) (rs200990725), was found in non-FH samples (4 heterozygotes (Htz) in 1000 Genomes and 9 Htz in the ExAC) databases, and although the in silico analyses supported a pathogenic prediction, in vitro analysis revealed that there was no adverse effect on function.29 The two variants c.2389G>A and c.2389G>T, which both result in p.(Val797Met), were predicted to be probably not pathogenic by in silico analyses; however, in vitro studies show that both variants disrupt normal mRNA splicing as they affect the last nucleotide of exon 1633 34 and so were deemed to be class 4. Thus, extra caution must be used with in silico analysis of variants close to intron/exon boundaries as predictions for in silico tools such as PolyPhen2 and SIFT are based on the effects of amino acid changes on the mature peptide rather than differences that the variant DNA may have on normal mRNA processing, and so such variants should also be analysed with the splice predicting programs also. Likewise, three synonymous variants (c.621C>T, p.(Gly207=), c.1216C>A, p.(Arg406=), c.1813C>T, p.(Leu605=)) were shown to disrupt normal splicing by both in silico and in vitro analyses and c.1845G>A, p.(Glu615=) was predicted to do so by in silico analysis alone as it affected the last residue in exon 12 (no in vitro evidence is available for this variant at present). Although the majority of synonymous variants will not be pathogenic, closer examination of their impact is justified if they have not been reported in non-FH subjects, if they are present in unrelated patients with FH and if segregation with FH has been demonstrated.
As mentioned previously, it is generally accepted that intronic variants affecting bases ±1 and 2 from the intron/exon boundary will be pathogenic as these residues are highly conserved.46 The updated UCL LDLR database currently lists 98 such variants of which in vitro evidence was available for 17. The potential significance of variants beyond the immediate intron/exon boundary was examined for those variants with accompanying in vitro data (table 2). Also, 11 variants within the first 12 bases of the intron/exon boundaries were found to be pathogenic by either inactivating the wild-type splice site resulting in exon skipping or by the creation of novel splice sites that were used preferentially. Furthermore, in vitro studies revealed that c.1359-31_1359-23delinsCGGCT resulted in the removal of the invariant adenine at the consensus splicing branch site in intron 9, causing retention of intron 9 and use of cryptic splice sites in exon 10,39 and c.2140+86C>G resulted in the creation and use of a novel splice site in intron 14.42 Thus, there is the potential for variants deep into the introns to be pathogenic, and as with the synonymous variants, some of these intronic variants may warrant further examination.
According to the ACGS guidelines, it is acceptable to predict that any variant that results in a premature termination of the peptide either as a result of a nonsense variant or a reading shift change and will be pathogenic. However, in rearrangements where this is not the case it is more difficult to make a prediction; clearly deletion of whole exons and functional domains would be likely to have deleterious effects. Although changes in the peptide secondary and tertiary structures may result from the addition or removal of small numbers of amino acids, their effect is difficult to predict; furthermore, very few in vitro functional studies have been published for such variants, probably because they would be technically difficult and costly to perform and also because the assumption may be made that these variants are likely to be pathogenic. As advances are made in in vitro functional assays,47 it is hoped that more evidence will be provided to confirm or refute the pathogenicity of non-frame-shifting rearrangements.
Traditionally LDLR variants have been grouped into one of five classes based on their functional effects (class 1 Null, class 2 Transport defective, ie, retained in the ER, class 3 Binding defective, Class 4 Internalisation defective, Class 5 Recycling deficient).25 However, as more is understood about the different mechanisms that can impact on normal LDLR function, additional classes could usefully be added to this list. In 2001, Koivisto et al48 demonstrated that the cytoplasmic variant c.2531G>A, p.(Gly844Asp) interfered with the basolateral sorting of LDLR with the effect that the peptide was misstargeted to the apical surface of the cell, hence reducing the numbers of receptors on the basolateral surface that would be predicted to reduce LDL-C clearance in vivo. Recently, two in vitro functional studies on transmembrane variants have shown that c.2396T>G, p.(Leu799Arg) and c.2413G>A, p.(Gly805Arg) both result in secretion of variant LDLR peptide from the cell.49 50 It appears that c.2396T>G, p.(Leu799Arg) fails to anchor in the ER membrane, resulting in secretion of mature LDLR peptide, while c.2413G>A, p.(Gly805Arg) undergoes metalloproteinase cleavage, resulting in the secretion of the ectodomain of the variant peptide. Both of these variants would thereby result in reducing the numbers of membrane-bound LDLR molecules for LDL-C clearance.
It is clearly of great importance to be able to assess whether variants identified in clinical settings or as incidental findings in genomics projects are pathogenic or not. Although 93% (n=1595) of LDLR variants in the current upgrade of the database have been assigned to an ACGS pathogenicity category, 7% (n=112) remain as VUS. It is hoped that as more information becomes available from in vitro functional studies, the development of additional in silico tools and from the various genomics studies, it will be possible to determine the pathogenicity of these variants; indeed, the classification of other variants may also change as our knowledge increases.
In conclusion, the LDLR database provides a valuable resource to the research and clinical communities. We would like to encourage the ethos of data sharing and open access to resources, and so we urge researchers and clinicians to submit their variant data to the database via a link on the homepage (http://databases.lovd.nl/shared/genes/LDLR). While every effort has been made to ensure the accuracy of the data in this database, we accept that errors may have occurred and so we would be grateful if you could please inform us of any that you find.
Footnotes
Contributors: SL: principal in collation and analysis of variants, and of manuscript preparation; MF: collation and analysis of variants, contributor in manuscript preparation; RW: collation and analysis of variants; AT-B, MW: clinical contribution of novel variants and knowledge regarding significance of variants to patients; JTdD: responsible for formatting data to be loaded onto the LOVD platform and responsible for maintenance and development of this database platform; SEH: project lead, giving direct guidance, advice and support for the study and assistance in manuscript preparation.
Funding: British Heart Foundation BHF PG08/008.
Competing interests: SEH holds a chair funded by the British Heart Foundation, and SEH and RW are supported by the BHF (PG08/008) and by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. MF is supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Benn M, Watts GF, Tybjærg-Hansen A, Nordestgaard BG. Mutations causative of familial hypercholesterolaemia: screening of 98 098 individuals from the Copenhagen General Population Study estimated a prevalence of 1 in 217. Eur Heart J 2016;37:1384–94. 10.1093/eurheartj/ehw028 [DOI] [PubMed] [Google Scholar]
- 2.Lahtinen AM, Havulinna AS, Jula A, Salomaa V, Kontula K. Prevalence and clinical correlates of familial hypercholesterolemia founder mutations in the general population. Atherosclerosis 2015;238:64–9. 10.1016/j.atherosclerosis.2014.11.015 [DOI] [PubMed] [Google Scholar]
- 3.Marks D, Thorogood M, Farrer JM, Humphries SE. Census of clinics providing specialist lipid services in the United Kingdom. J Public Health 2004;26:353–4. 10.1093/pubmed/fdh176 [DOI] [PubMed] [Google Scholar]
- 4.Neil A, Cooper J, Betteridge J, Capps N, McDowell I, Durrington P, Seed M, Humphries SE. Reductions in all-cause, cancer, and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolaemia: a prospective registry study. Eur Heart J 2008;29:2625–33. 10.1093/eurheartj/ehn422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Borén J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjærg-Hansen A, European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 2013;34:3478–90a. 10.1093/eurheartj/eht273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Versmissen J, Oosterveer DM, Yazdanpanah M, Defesche JC, Basart DCG, Liem AH, Heeringa J, Witteman JC, Lansberg PJ, Kastelein JJ, Sijbrands EJ. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ 2008;337:a2423 10.1136/bmj.a2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huijgen R, Hutten BA, Kindt I, Vissers MN, Kastelein JJP. Discriminative ability of LDL-cholesterol to identify patients with familial hypercholesterolemia: a cross-sectional study in 26,406 individuals tested for genetic FH. Circ Cardiovasc Genet 2012;5:354–9. 10.1161/CIRCGENETICS.111.962456 [DOI] [PubMed] [Google Scholar]
- 8.Wilson DJ, Gahan M, Haddad L, Heath K, Whittall RA, Williams RR, Humphries SE, Day IN. A World Wide Web site for low-density lipoprotein receptor gene mutations in familial hypercholesterolemia: sequence-based, tabular, and direct submission data handling. Am J Cardiol 1998;81:1509–11. 10.1016/S0002-9149(98)00215-X [DOI] [PubMed] [Google Scholar]
- 9.Heath KE, Gahan M, Whittall RA, Humphries SE. Low-density lipoprotein receptor gene (LDLR) world-wide website in familial hypercholesterolaemia: Update, new features and mutation analysis. Atherosclerosis 2001;154:243–6. 10.1016/S0021-9150(00)00647-X [DOI] [PubMed] [Google Scholar]
- 10.Leigh SEA, Foster AH, Whittall RA, Hubbart CS, Humphries SE. Update and analysis of the university college London low density lipoprotein receptor familial hypercholesterolemia database. Ann Hum Genet 2008;72:485–98. [DOI] [PubMed] [Google Scholar]
- 11.Usifo E, Leigh SEA, Whittall RA, Lench N, Taylor A, Yeats C, Orengo CA, Martin AC, Celli J, Humphries SE. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Ann Hum Genet 2012;76:387–401. 10.1111/j.1469-1809.2012.00724.x [DOI] [PubMed] [Google Scholar]
- 12.Wallis Y, Payne S, McAnulty C, Bodmer D, Sistermans E, Robertson E, Moore D, Abbs S, Zandra Deans AD. Practice Guidelines for the Evaluation of Pathogenicity and the Reporting of Sequence Variants in Clinical Molecular Genetics. http://www.acgs.uk.com/media/774853/evaluation_and_reporting_of_sequence_variants_bpgs_june_2013_-_finalpdf.pdf (accessed Oct 2016).
- 13.Wildeman M, Van Ophuizen E, Den Dunnen JT, Taschner PEM. Improving sequence variant descriptions in mutation databases and literature using the mutalyzer sequence variation nomenclature checker. Hum Mutat 2008;29:6–13. 10.1002/humu.20654 [DOI] [PubMed] [Google Scholar]
- 14.Den Dunnen JT, Antonarakis E. Nomenclature for the description of human sequence variations. Hum Genet 2001;109:121–4. 10.1007/s004390100505 [DOI] [PubMed] [Google Scholar]
- 15.Fokkema IFAC, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat 2011;32:557–63. 10.1002/humu.21438 [DOI] [PubMed] [Google Scholar]
- 16.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS SS. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–81. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- 18.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11:361–2. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- 19.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol 1997;4:311–23. 10.1089/cmb.1997.4.311 [DOI] [PubMed] [Google Scholar]
- 20.Dogan RI, Getoor L, Wilbur WJ, Mount SM. SplicePort-An interactive splice-site analysis tool. Nucleic Acids Res 2007;35(Suppl. 2):W285–91. 10.1093/nar/gkm407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khamis A, Palmen J, Lench N, Taylor A, Badmus E, Leigh S, Humphries SE. Functional analysis of four LDLR 5′UTR and promoter variants in patients with familial hypercholesterolaemia. Eur J Hum Genet 2015;23:790–5. 10.1038/ejhg.2014.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR, 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015;526:68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG. Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Futema M, Whittall RA, Kiley A, Steel LK, Cooper JA, Badmus E, Leigh SE, Karpe F, Neil HA, Simon Broome Register Group, Humphries SE. Analysis of the frequency and spectrum of mutations recognised to cause familial hypercholesterolaemia in routine clinical practice in a UK specialist hospital lipid clinic. Atherosclerosis 2013;229:161–8. 10.1016/j.atherosclerosis.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat 1992;1:445–66. 10.1002/humu.1380010602 [DOI] [PubMed] [Google Scholar]
- 26.Benito-Vicente A, Alves AC, Etxebarria A, Medeiros AM, Martin C, Bourbon M. The importance of an integrated analysis of clinical, molecular, and functional data for the genetic diagnosis of familial hypercholesterolemia. Genet Med 2015;17:980–8. 10.1038/gim.2015.14 [DOI] [PubMed] [Google Scholar]
- 27.Chang JH, Pan JP, Tai DY, Huang A-C, Li P, Ho H-L, Hsieh HL, Chou SC, Lin WL, Lo E, Chang CY, Tseng J, Su MT, Lee-Chen GJ. Identification and characterization of LDL receptor gene mutations in hyperlipidemic Chinese. J Lipid Res 2003;44:1850–8. 10.1194/jlr.M200470-JLR200 [DOI] [PubMed] [Google Scholar]
- 28.Koivisto UM, Gylling H, Miettinen TA, Kontula K. Familial moderate hypercholesterolemia caused by Asp235—>Glu mutation of the LDL receptor gene and co-occurrence of a de novo deletion of the LDL receptor gene in the same family. Arterioscler Thromb Vasc Biol 1997;17:1392–9. 10.1161/01.ATV.17.7.1392 [DOI] [PubMed] [Google Scholar]
- 29.Etxebarria A, Benito-Vicente A, Stef M, Ostolaza H, Palacios L, Martin C. Activity-associated effect of LDL receptor missense variants located in the cysteine-rich repeats. Atherosclerosis 2015;238:304–12. 10.1016/j.atherosclerosis.2014.12.026 [DOI] [PubMed] [Google Scholar]
- 30.Etxebarria A, Palacios L, Stef M, Tejedor D, Uribe KB, Oleaga A, Irigoyen L, Torres B, Ostolaza H, Martin C. Functional characterization of splicing and ligand-binding domain variants in the LDL receptor. Hum Mutat 2012;33:232–43. 10.1002/humu.21630 [DOI] [PubMed] [Google Scholar]
- 31.Silva S, Alves AC, Patel D, Malhó R, Soutar AK, Bourbon M. In vitro functional characterization of missense mutations in the LDLR gene. Atherosclerosis 2012;225:128–34. 10.1016/j.atherosclerosis.2012.08.017 [DOI] [PubMed] [Google Scholar]
- 32.Etxebarria A, Benito-Vicente A, Palacios L, Stef M, Cenarro A, Civeira F, Ostolaza H, Martin C. Functional characterization and classification of frequent low-density lipoprotein receptor variants. Hum Mutat 2015;36:129–41. 10.1002/humu.22721 [DOI] [PubMed] [Google Scholar]
- 33.Mak YT, Pang CP, Tomlinson B, Zhang J, Chan Y, Mak WL, Masarei JR. Mutations in the low-density lipoprotein receptor gene in Chinese familial hypercholesterolemia patients. Arterioscler Thromb Vasc Biol 1998;18:1600–5. 10.1161/01.ATV.18.10.1600 [DOI] [PubMed] [Google Scholar]
- 34.Bourbon M, Duarte MA, Alves AC, Medeiros AM, Marques L, Soutar AK. Genetic diagnosis of familial hypercholesterolaemia: the importance of functional analysis of potential splice-site mutations. J Med Genet 2009;46:352–7. 10.1136/jmg.2007.057000 [DOI] [PubMed] [Google Scholar]
- 35.Medeiros AM, Alves AC, Francisco V, Bourbon M. Update of the Portuguese familial hypercholesterolaemia study. Atherosclerosis 2010;212:553–8. 10.1016/j.atherosclerosis.2010.07.012 [DOI] [PubMed] [Google Scholar]
- 36.Liguori R, Bianco AM, Argiriou A, Pauciullo P, Giannino A, Rubba P, De Simone V. LDL receptor cDNA sequence analysis in familial hypercholesterolemia patients: 5 novel mutations with high prevalence in families originating from southern Italy. Hum Mutat 2001;17:433 10.1002/humu.1122 [DOI] [PubMed] [Google Scholar]
- 37.Marduel M, Carrié A, Sassolas A, Devillers M, Carreau V, Di Filippo M, Erlich D, Abifadel M, Marques-Pinheiro A, Munnich A, Junien C, French ADH Research Network, Boileau C, Varret M, Rabès JP. Molecular spectrum of autosomal dominant hypercholesterolemia in France. Hum Mutat 2010;31:E1811–24. 10.1002/humu.21348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun LY, Zhang YB, Jiang L, Wan N, Wu W-F, Pan X-D, Yu J, Zhang F, Wang L-Y. Identification of the gene defect responsible for severe hypercholesterolaemia using whole-exome sequencing. Sci Rep 2015;5:11380 10.1038/srep11380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Webb JC, Patel DD, Shoulders CC, Knight BL, Soutar AK. Genetic variation at a splicing branch point in intron 9 of the low density lipoprotein (LDL)-receptor gene: a rare mutation that disrupts mRNA splicing in a patient with familial hypercholesterolaemia and a common polymorphism. Hum Mol Genet 1996;5:1325–31. 10.1093/hmg/5.9.1325 [DOI] [PubMed] [Google Scholar]
- 40.Jensen HK, Jensen LG, Holst HU, Andreasen PH, Hansen PS, Larsen ML, Kølvraa S, Bolund L, Gregersen N, Faergeman O. Normolipidemia and hypercholesterolemia in persons heterozygous for the same 1592+5GA splice site mutation in the low-density lipoprotein receptor gene. Clin Genet 1999;56:379–89. 10.1034/j.1399-0004.1999.560506.x [DOI] [PubMed] [Google Scholar]
- 41.Graham CA, McIlhatton BP, Kirk CW, Beattie ED, Lyttle K, Hart P, Neely RD, Young IS, Nicholls DP. Genetic screening protocol for familial hypercholesterolemia which includes splicing defects gives an improved mutation detection rate. Atherosclerosis 2005;182:331–40. 10.1016/j.atherosclerosis.2005.02.016 [DOI] [PubMed] [Google Scholar]
- 42.Kulseth MA, Berge KE, Bogsrud MP, Leren TP. Analysis of LDLR mRNA in patients with familial hypercholesterolemia revealed a novel mutation in intron 14, which activates a cryptic splice site. J Hum Genet 2010;55:676–80. 10.1038/jhg.2010.87 [DOI] [PubMed] [Google Scholar]
- 43.Defesche JC, Schuurman EJM, Klaaijsen LN, Khoo KL, Wiegman A, Stalenhoef AFH. Silent exonic mutations in the low-density lipoprotein receptor gene that cause familial hypercholesterolemia by affecting mRNA splicing. Clin Genet 2008;73:573–8. 10.1111/j.1399-0004.2008.00999.x [DOI] [PubMed] [Google Scholar]
- 44.Ho CKM, Musa FR, Bell C, Walker SW. LDLR gene synonymous mutation c.1813C>T results in mRNA splicing variation in a kindred with familial hypercholesterolaemia. Ann Clin Biochem 2015;52(Pt 6):680–4. 10.1177/0004563215572702 [DOI] [PubMed] [Google Scholar]
- 45.Bourbon M, Alves AC, Medeiros AM, Silva S, Soutar AK. Familial hypercholesterolaemia in Portugal. Atherosclerosis 2008;196:633–42. 10.1016/j.atherosclerosis.2007.07.019 [DOI] [PubMed] [Google Scholar]
- 46.Stephens RM, Schneider TD. Features of spliceosome evolution and function inferred from an analysis of the information at human splice sites. J Mol Biol 1992;228:1124–36. 10.1016/0022-2836(92)90320-J [DOI] [PubMed] [Google Scholar]
- 47.Di Taranto MD, D'Agostino MN, Fortunato G. Functional characterization of mutant genes associated with autosomal dominant familial hypercholesterolemia: integration and evolution of genetic diagnosis. Nutr Metab Cardiovasc Dis 2015;25:979–87. 10.1016/j.numecd.2015.06.007 [DOI] [PubMed] [Google Scholar]
- 48.Koivisto U-M, Hubbard AL, Mellman I. A novel cellular phenotype for familial hypercholesterolemia due to a defect in polarized targeting of LDL receptor. Cell 2001;105:575–85. 10.1016/S0092-8674(01)00371-3 [DOI] [PubMed] [Google Scholar]
- 49.Strøm TB, Tveten K, Laerdahl JK, Leren TP. Mutation G805R in the transmembrane domain of the LDL receptor gene causes familial hypercholesterolemia by inducing ectodomain cleavage of the LDL receptor in the endoplasmic reticulum. FEBS Open Biol 2014;4:321–7. 10.1016/j.fob.2014.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Strøm TB, Laerdahl JK, Leren TP. Mutation p.L799R in the LDLR, which affects the transmembrane domain of the LDLR, prevents membrane insertion and causes secretion of the mutant LDLR. Hum Mol Genet 2015;24: 5836–44. 10.1093/hmg/ddv304 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Accession numbers of peptide sequences used to generate LDLR refined SIFT analysis
jmedgenet-2016-104054supp001.pdf (346.1KB, pdf)
Refined SIFT Analasis of Human LDLR
jmedgenet-2016-104054supp002.pdf (1.8MB, pdf)
LDLR Structural Conservation Scores
jmedgenet-2016-104054supp003.pdf (558.6KB, pdf)