

Abstract
In the ischemic-reperfused (I/R) heart, renin-containing mast cells (MC) release enzymatically active renin, activating a local renin-angiotensin system (RAS), causing excessive norepinephrine release and arrhythmic dysfunction. Activation of Gi-receptors on MC and/or ischemic preconditioning (IPC) prevent renin release, thus providing anti-RAS cardioprotection. We questioned whether sphingosine-1-phosphate (S1P), a sphingolipid produced in the I/R heart, might afford anti-RAS cardioprotection by activating Gi-coupled S1P1 receptors (S1P1R) on MC. We report that activation of Gi-coupled S1P1R in cardiac MC confers IPC-like anti-RAS cardioprotection due to S1P1R-mediated inhibition of I/R-induced cardiac MC degranulation and renin release. This results from an initial translocation of protein kinase C subtype-ε and subsequent activation of aldehyde dehydrogenase type 2 (ALDH2), culminating in the elimination of the MC-degranulating effects of acetaldehyde and other toxic species produced during I/R. Inhibition of toxic aldehydes-induced MC-renin release prevents local RAS activation, reduces infarct size, and alleviates arrhythmias. Notably, these cardioprotective effects are lacking in hearts and MC from gene-targeted knock-in mice (ALDH2*2) in which ALDH2 enzymatic activity is maximally reduced. Thus, ALDH2 appears to play a pivotal role in this protective process. Our findings suggest that MC S1P1R may represent a new pharmacologic and therapeutic target for the direct alleviation of RAS-induced cardiac dysfunctions, including ischemic heart disease and congestive heart failure.
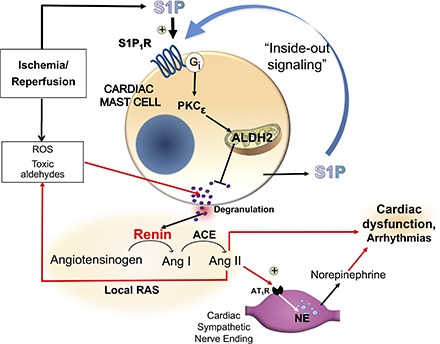
Introduction
Excessive release of norepinephrine (NE) from cardiac sympathetic nerves during ischemia/reperfusion (I/R) is a cause of life-threatening arrhythmias (Airaksinen, 1999; Chen et al., 2001; Selwyn and Braunwald, 2001; Tan and Verrier, 2013). In previous studies we demonstrated that in the I/R heart, renin-containing mast cells (MC) release enzymatically active renin, activating a local renin-angiotensin system (RAS), which culminates in angiotensin-induced NE release from sympathetic nerves and severe arrhythmic dysfunction (Silver et al., 2004; Mackins et al., 2006). We reported that activation of Gi-coupled receptors, such as adenosine A3 and histamine H4, present on the MC surface, prevents MC degranulation and renin release in the I/R heart, thus attenuating angiotensin-induced NE release from sympathetic nerves and alleviating ventricular arrhythmias (Koda et al., 2010; Aldi et al., 2014b). Ischemic preconditioning (IPC) mimics this process via a Gi-mediated translocation/activation of MC protein kinase C subtype-ε (PKCε), followed by phosphorylation/activation of mitochondrial aldehyde dehydrogenase type 2 (ALDH2) (Koda et al., 2010; Aldi et al., 2014b). PKCε translocates from cytosol to mitochondrial membrane and phosphorylates ALDH2 on three phosphorylation sites—threonine 185 (T185), serine 279 (S279), and threonine 412 (T412)—resulting in increased catalytic activity of ALDH2 (Chen et al., 2008). By eliminating toxic aldehydes formed in I/R, and thus preventing MC degranulation and renin release, ALDH2 plays a pivotal anti-RAS cardioprotective role (Koda et al., 2010; Aldi et al., 2014b).
The sphingolipid sphingosine-1-phosphate (S1P) is formed in the ischemic heart, and S1P is thought to be cardioprotective and to mimic IPC via a PKCε-dependent step (Gray et al., 1997; Jin et al., 2004; Vessey et al., 2009). Accordingly, we questioned whether S1P might also afford anti-RAS cardioprotection by activating Gi-coupled S1P1 receptors (S1P1R) on MC, and by PKCε-dependent ALDH2 phosphorylation (Chen et al., 2008).
We questioned whether the stimulation of S1P1R in both cavian and murine hearts mimics the anti-RAS effects of IPC by activating PKCε and ALDH2 in MC and thus, by inhibiting toxic aldehyde-induced MC-renin release. We also investigated whether this process is lacking in the hearts and MC from gene-targeted knock-in mice (ALDH2*2) (Zambelli et al., 2014) in which ALDH2 enzymatic activity is maximally reduced due to a structural point mutation at amino acid position 487 of ALDH2 (a lysine residue replaces a glutamate) (Xiao et al., 1996). We took advantage of the homozygous variant of ALDH2*2 knock-in mice to probe whether the activation of Gi-coupled S1P1R in cardiac MC confers anti-RAS cardioprotection through a PKCε-dependent ALDH2 phosphorylation, prevention of toxic aldehydes-induced MC-renin release, inhibition of local RAS activation, and ultimately antiarrhythmic effects.
Materials and Methods
I/R in Guinea Pig Hearts
Guinea pigs (male Hartley, 300–350 g; Charles River Laboratories, Kingston, NY) were kept in the animal care facility under controlled temperature, humidity, and light/dark cycle, with food and water ad libitum. The animals were anesthetized with CO2 and euthanized by stunning while under general anesthesia. Hearts were rapidly isolated and perfused at constant pressure (55 cm H2O) with oxygenated Ringer’s solution (composition, mM: NaCl 154, KCl 5.61, CaCl2 2.16, NaHCO3 5.95, and dextrose 5.55) at 37°C in a Langendorff apparatus (Radnoti Glass Technology, Monrovia, CA). After equilibration, all hearts were subjected to 20-minute global ischemia, induced by complete cessation of coronary perfusion, followed by 30-minute reperfusion (I/R).
For IPC, hearts were subjected to 2 × 5-minute cycles of ischemia, each followed by 5-minute reperfusion. For the pharmacologic prevention of IPC, S1P1R antagonist was perfused for 20 minutes before and during IPC and then washed out for 15 minutes before I/R. For pharmacologic preconditioning, S1P1R agonist (1 µM, SEW2871: 5-[4-phenyl-5-(trifluoromethyl)thiophen-2-yl]-3-[3-(trifluoromethyl)phenyl]-1,2,4-oxadiazole) was perfused for 2 × 5-minute cycles and then washed out for 5 minutes before I/R.
For prevention of pharmacologic preconditioning, the antagonists W146 1 µM [(3R)-3-amino-4-(3-hexylanilino)-4-oxobutyl]phosphonic acid] and 1 µM selective PKCε inhibitor εV1–2 were perfused for 20 minutes, while the ALDH2 inhibitor 2 µM glyceryl trinitrate (GTN) was perfused for 30 minutes before and during pharmacologic preconditioning and then washed out for 15 minutes before I/R. Coronary flow was measured every 2 minutes; samples were assayed for renin and NE. Two needle electrodes were attached to the surface of the right atrium and left ventricular apex for ECG, which was recorded online (sample frequency of 1 kHz) and analyzed using Powerlab/8SP (AD Instruments, Colorado Springs, CO). Onset and duration of reperfusion arrhythmias were recorded and quantified according to the Lambeth Conventions (Walker et al., 1988; Curtis et al., 2013).
I/R in Mouse Hearts
Hearts were isolated from male C57BL/6 wild-type (WT) and ALDH2*2 mice and perfused as described elsewhere (Mackins et al., 2006). ALDH2*2 knock-in mice were donated by the Mochly-Rosen laboratory, Stanford University (Zambelli et al., 2014). Briefly, 20 minutes after a heparin i.p. injection (100 U) to avoid blood clotting, mice were anesthetized with CO2 vapor and humanely killed by cervical dislocation while under anesthesia. Hearts were quickly excised and cooled in ice-cold Krebs-Henseleit (KH) solution containing pyruvic acid (2 mM) and equilibrated with 95% O2 + 5% CO2. Hearts were then perfused in a Langendorff apparatus (Radnoti, Monrovia, CA) at constant pressure (100 cm H2O) at 37°C with KH buffer. After stabilization, hearts were subjected to 40-minute global ischemia followed by a 30-minute reperfusion with KH buffer (I/R); the reperfusion time was 120 minutes when I/R-induced infarct areas were analyzed.
For IPC, hearts were subjected to 4 × 5-minute cycles of ischemia each followed by a 5-minute perfusion. For pharmacologic prevention of IPC, hearts were pretreated with the selective S1P1R antagonist W146 (3 µM, 20 minutes) and then subjected to IPC (i.e., 4 × 5-minute cycles of ischemia each followed by 5-minute perfusion with W146).
For pharmacologic preconditioning, hearts were perfused with the selective S1P1R agonist SEW2871 (0.1 µM) (Jo et al., 2005) for 4 × 5-minute cycles, each followed by a 5-minute washout before I/R. For prevention of pharmacologic precondition, S1P1R antagonist W146 (3 µM), PKCε inhibitor εV1–2 (1 µM), and ALDH2 desensitizer GTN (2 µM) were perfused for 20 minutes before and during pharmacologic preconditioning and then washed out before I/R. ECG was recorded online (sample frequency of 1 kHz) and analyzed using Powerlab/8SP (AD Instruments).
Onset and duration of reperfusion arrhythmias were recorded and quantified according to the Lambeth Conventions (Walker et al., 1988; Curtis et al., 2013). Coronary flow was measured by timed collections of the effluent every 2 minutes, samples were assayed for renin and NE.
Infarct Size in WT and ALDH2*2 Mouse Hearts
WT and ALDH2*2 hearts were removed from the Langendorff apparatus after a 120-minute reperfusion. Hearts were cut in 2 mm-thick slices and immersed in an aqueous solution (1% w/v) of 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich, St. Louis, MO) for 20 minutes at 37°C and then in an aqueous solution of formaldehyde (10% v/v). After overnight incubation, heart slices were photographed with a ×16 magnification and analyzed to measure infarct size (expressed as percentage of infarcted versus total left ventricular area).
Cell Lines
HMC-1 Culture.
The human mastocytoma cell line HMC-1 was kindly provided by Dr. J. H. Butterfield (Mayo Clinic, Rochester, MN). Cells were maintained in suspension culture at high density in Iscove’s modified Dulbecco's medium supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin, and kept at 37°C in a 5% CO2 atmosphere (Aldi et al., 2015).
Bone Marrow–Derived Mast Cell Culture.
Bone marrow was collected from femurs and tibias of WT C57BL/6J mice (male, 10 to 12 weeks old; Jackson Laboratory, Bar Harbor, ME) and ALDH2*2 knock-in mice and euthanized by cervical dislocation under light CO2 anesthesia, as approved by the Weill Cornell Medicine Institutional Animal Care and Use Committee. Bone marrow cells were cultured in RPMI 1640 medium (Invitrogen Life Technologies, Carlsbad, CA) containing 1% penicillin/streptomycin, 10% heat-inactivated fetal calf serum, 55 μM 2-mercaptoethanol, and recombinant murine interleukin-3 and stem cell factor (PeproTech, Rocky Hill, NJ), both at 20 ng/ml. Bone marrow cells were counted and placed in culture at a cell density of 5 × 105 cells/ml. The cell medium was changed every 3 to 4 days, and nonadherent cells were transferred to a new flask. Mature murine bone marrow–derived mast cells (BMMC) were obtained after 4 weeks of culture, and stained positive for Toluidine blue; moreover, >90% of cells expressed both c-Kit and FcεRI. All experiments were performed with BMMC cultured for 4–7 weeks (Aldi et al., 2014a).
Immunofluorescence in BMMC
Mature WT- and ALDH2*2-BMMC obtained after 4 weeks of culture were placed on poly-d-lysine–coated coverslips for 2 hours at 37°C to adhere. Cells were then washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 15 minutes at room temperature, washed in PBS, and blocked in 1% bovine serum albumin (BSA) and 0.1% Triton X-100 in PBS for 30 minutes. Then, cells were incubated overnight with mouse anti-S1P1R (cat. no. ab11424; Abcam, Cambridge, MA) and rat anti-c-Kit (cat. no. ab65525; Abcam). The next day, the coverslips were washed with PBS and incubated with secondary Ab Alexa Fluor 488 donkey anti-rabbit IgG (green) and Alexa Fluor 594 rabbit anti-rabbit IgG (red). Nuclei were counterstained with 4ʹ,6-diamidino-2-phenylindole (DAPI) 300 nM and coverslips were mounted on glass slides using VectaMount medium (Vector Laboratories, Burlingame, CA). Immunofluorescence images of cells were captured in a Z-stack with 1-μm steps using a Zeiss Axio Observer.Z1 microscope (Carl Zeiss, Thornwood, NY) followed by deconvolution using the ZEN pro 2012 software (Carl Zeiss). To analyze the image we used ImagePro analyzer 7.0 software (Media Cybernetics, Rockville, MD).
Western Blot Analysis
To ascertain S1P1R expression in HMC-1, WT-BMMC, and ALDH2*2-BMMC cells, we used an anti-S1P1R antibody (rabbit monoclonal anti-EDG1) purchased from Abcam (cat. no. ab125074). Cells were lysed in ice-cold lysis buffer 1X (Cell Signaling Technology, Danvers, MA). Proteins were equalized to the same concentration with the Bradford protein assay kit (Bio-Rad Laboratories, Hercules, CA), separated by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were blocked with 5% BSA dissolved in Tris-buffered saline containing 1% Tween 20 (TBST) for 1 hour at room temperature. After washing with TBST, the membrane was incubated with a 1:10,000 dilution of specific antibody against S1P1R in 5% milk-TBST at 4°C overnight (antibody final concentration was 3.2 ng/ml).
The blots were washed in TBST, and the membranes were incubated in a 1:5,000 dilution of horseradish peroxidase-conjugated IgG secondary antibody (final concentration 13 ng/ml; cat. no. 7074; Cell Signaling Technology) in 5% BSA-TBST at room temperature for 1 hour. Total proteins were evaluated by incubating the PVDF membranes with a 1:50,000 dilution of β-actin horseradish peroxidase (HRP) conjugated (concentration 1 µg/ml; Alpha Diagnostic International, San Antonio, TX) in milk 5% at room temperature. Proteins were detected using the electrochemiluminescence (ECL) system.
PKCε Translocation
After incubation of HMC-1, WT-BMMC, and ALDH2*2-BMMC with phorbol ester myristate acetate (PMA) or SEW2871 for 10 minutes at 37°C, cytosolic and membrane fractions were prepared as described elsewhere (Koda et al., 2010). When necessary, the S1P1R antagonist W146 was preincubated for 20 minutes at 37°C. Na+/K+ ATPase antibody (cat. no. ab185065; Abcam) was used for immunoblotting to verify the purity of the cell membrane fraction. Translocation of PKCε was determined by Western blot analysis using a PKCε antibody (cat. no. sc-214; Santa Cruz Biotechnology, Santa Cruz, CA) at a 1:1,000 dilution in 5% milk-TBST. Blots were washed in TBST, and the membranes were incubated in a 1:5,000 dilution of goat anti-rabbit-IgG HRP secondary antibody (cat. no. 7074; Cell Signaling Technology) in 5% milk-TBST at room temperature for 1 hour. After washing with TBST, blots were developed by an ECL Western Blotting Detection Reagent, and the membranes were exposed to Hyperfilm ECL. Total proteins were evaluated by incubating the PVDF membranes with a 1:50,000 dilution of β-actin HRP conjugated (concentration 1 µg/ml; Alpha Diagnostic International) in milk 5% at room temperature. PKCε translocation was expressed as the ratio between PKCε in the membrane and PKCε in the cytosol. The density of each band was determined with GeneTool software (Syngene USA, Frederick, MD).
ALDH2 Enzymatic Activity Assay
Enzymatic activity of ALDH2 in HMC-1, WT-BMMC, and ALDH2*2-BMMC cells was determined spectrophotometrically by monitoring the reductive reaction of NAD+ to NADH at 340 nm as described elsewhere (Koda et al., 2010). Briefly, cells were treated with SEW2871, Alda-1, or PMA for 10 minutes at 37°C. When indicated, S1P1R antagonist, PKCε, and ALDH2 inhibitors were preincubated in the presence of SEW2871 for 20 minutes at 37°C. The cells were centrifuged at 2,500 rpm for 5 minutes at 4°C, and the pellets resuspended in homogenization buffer (composition: Tris-HCl, pH 8.0, 100 mM; 1,4-dithiothreitol, 10 mM; glycerol 20%; Triton X-100 1%). Assays were performed in 50 mM sodium pyrophosphate buffer, pH 9.0, at 25°C, and 500 μg of cell lysates and 2.5 mM NAD were added to the buffer. To start the reaction, 10 mM acetaldehyde was added, and the accumulation of NADH was recorded for 15 minutes with measurements taken every 20 seconds. The ALDH2 reaction rates were calculated as μmoles of NADH per min per mg of protein and compared with control cells (i.e., treated with Na Ringer [composition, mM: NaCl 154, KCl 5.61, CaCl2 2.16, NaHCO3 5.95, and dextrose 5.55]) and expressed as percentage of increase from control.
β-Hexosaminidase Assay
β-Hexosaminidase (β-HEX) release was measured in HMC-1, WT-BMMC, and ALDH2*2-BMMC. Briefly, cells were washed and resuspended in Ringer buffer. Identical volumes of cells were aliquoted in Eppendorf tubes (HMC-1) or in 96-well plates (BMMC) and incubated with gentle oscillation at 37°C with acetaldehyde (300 and 100 μM for HMC-1 and BMMC, respectively) or 4-hydroxynonenal (4-HNE; 30 and 10 μM, respectively) for 20 minutes (Sigma-Aldrich). When reported, HMC-1 and BMMC cells were treated with the S1P1R agonist SEW2871 at concentrations of 0.1 and 1 μM, respectively, for 10 minutes at 37°C. When required, HMC-1 and BMMC were preincubated with the S1P1R antagonist (1 and 0.1 μM, respectively) and PKCε (1 μM) and ALDH2 (2 μM) inhibitors for 20 minutes at 37°C. At the end of the incubation, the samples were placed in ice and centrifuged at 500 g for 5 minutes.
We placed 20 μl of BMMC or HMC-1 supernatants in 96-well plates, to which 50 μl of p-nitrophenyl-N-acetyl-β-d-glucosaminide (1.3 mg/ml dissolved in 0.1 M citrate buffer at pH 4.5) was added and incubated for 90 minutes at 37°C. Cell pellets were lysed with 0.5% Triton X-100, and the total lysates were used to determine the total β-HEX content. The reaction was stopped by adding 150 μl of 0.2 M glycine (pH 10.7).
Optical density was read at 405 nm on a spectrophotometric plate reader using SoftMax Pro version 4.8 (Molecular Devices, Sunnyvale, CA). β-HEX release was expressed as the percentage of total β-HEX (where total β-HEX denotes the sum of β-HEX in supernatant and total cell lysate) (Aldi et al., 2014a).
Renin Assay
For the renin assay, human and porcine angiotensinogen was used for HMC-1 and BMMC samples, respectively. Renin activity, in terms of angiotensin I formed, was determined by GammaCoat Plasma Renin Activity [125I] radioimmunoassay (DiaSorin, Stillwater, MN). The cell pellets were lysed with cell lysis buffer 1X (Cell Signaling Technology), and the total lysates were used to determine total protein concentration by the Bradford Protein Assay kit (Bio-Rad Laboratories). Total protein concentration was used to normalize renin release. The results were expressed as the percentage of increase above the basal level.
Statistics
Statistical analysis was performed with GraphPad Prism 6.0 software (San Diego, CA). All data are reported as mean ± S.E.M. Statistical analysis was performed only when a minimum of n = 5 independent samples had been acquired, and was performed with an unpaired t test when comparing two different groups and with one-way analysis of variance followed by Dunnett’s post hoc test when comparing more than two groups of data (Fig. 5). The data and statistical analyses comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The data were considered statistically significant when a value of at least P < 0.05 was achieved.
Fig. 5.

Activation of S1P1R enhances ALDH2 activity in HMC-1 and WT-BMMC but not in ALDH2*2-BMMC. Incubation with the ALDH2 activator Alda-1 (300 μM, 10 minutes, positive control), PMA (300 or 10 nM, 10 minutes), or SEW2871 (1 μM or 0.1 μM, 10 minutes) enhances ALDH2 activity in (A) HMC-1 and (B) WT-BMMC but not in (C) ALDH2*2-BMMC. Pretreatment with W146 (1 or 0.1 μM, 20 minutes), εV1–2 (1 μM, 20 minutes), or GTN (2 μM, 30 minutes) prevents the effects of the S1P1R agonist in HMC-1 (A) and WT-BMMC (B) but not in ALDH2*2-BMMC (C). Bars are mean ± S.E.M. of the percentage of increase from control (HMC-1, WT-BMMC, ALDH2*2-BMMC, respectively: control, n = 12, 17, 17; PMA, n = 9, 8, 7; Alda-1, n = 10, 8, 6; SEW2871, n = 17, 9, 7; SEW2871 + W146, n = 5, 8, 6; SEW2871 + εV1–2, n = 5, 5, 6; SEW2871 + GTN, n = 7, 4, 4). Basal NADH production was 3.5 ± 0.21 μmol/min/mg protein in HMC-1, 0.96 ± 0.12 μmol/min/mg protein in WT-BMMC, and 0.63 ± 0.11 μmol/min/mg protein in ALDH2*2-BMMC. *P < 0.05; **P < 0.01; ***P < 0.001 versus control, by one-way ANOVA analysis of variance followed by Dunnett’s post hoc test.
Study Approval
All animals were kept in the animal care facility under controlled temperature, humidity, and light/dark cycle, with food and water ad libitum. All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health (NRC, 2011), and were approved by the Weill Cornell Medicine Institutional Animal Care and Use Committee.
Results
S1P1R Activation Mimics IPC and Affords Anti-RAS Cardioprotection in Isolated Guinea Pig Hearts.
Because guinea pig hearts share more similarity with human hearts and represent a better and a more relevant clinical model (Choy et al., 2016), and also given our previous experience with other Gi-coupled receptors in this animal model (Koda et al., 2010; Aldi et al., 2014b), our initial experiments were performed in isolated guinea pig hearts. Spontaneously beating Langendorff-perfused guinea pig hearts were subjected to 20-minute global ischemia followed by 30-minute reperfusion (I/R). We had previously shown that I/R results in MC degranulation, demonstrated by a 2-fold increase in β-HEX overflow into the coronary effluent (Koda et al., 2010). I/R also caused large increases in renin and NE overflow (i.e., ∼4- and ∼200-fold, respectively), and severe ventricular arrhythmias (tachycardia and fibrillation, VT/VF) that lasted ∼7 minutes (Fig. 1). We had previously shown that the enhanced NE overflow and arrhythmias caused by I/R result from the activation of a local RAS by renin released from cardiac MC (Mackins et al., 2006).
Fig. 1.

S1P1R activation provides anti-RAS IPC-like cardioprotection in an isolated guinea pig heart model of I/R. S1P1R blockade, PKCε inhibition, and ALDH2 inactivation prevent IPC-like cardioprotection. All guinea pig hearts were subjected to 20 minutes of global ischemia followed by 30 minutes of reperfusion in either the absence (control, n = 8) or presence of W146 (S1P1R antagonist, 1 μM; n = 15) and SEW2871 (S1P1R agonist, 1 μM; n = 10). When necessary, W146 (S1P1R antagonist, 1 μM; n = 15), εV1–2 (PKCε inhibitor, 1 μM; n = 8), or GTN (ALDH2 inactivator, 2 μM; n = 8) were added in the presence of SEW2871 (S1P1R agonist, 1 μM). Other hearts were subjected to I/R preceded by IPC alone (i.e., 2 × 5-minute cycles of ischemia, each followed by 5-minute reperfusion, n = 14) or in the presence of W146 (1 μM; n = 8). Bars indicate mean ± S.E.M. of independent experiments. (A) Duration of reperfusion arrhythmias (VT/VF). (B) Overflow of NE and (C) angiotensin I (Ang I)–forming activity collected over 6 minutes either before ischemia or at the start of reperfusion. *P < 0.05; **P < 0.01; ***P < 0.001, by unpaired t test. (D–F) Representative ECG recordings from guinea pig hearts before ischemia (control, D), at the start of reperfusion (E), and at the end of VT/VF (recovery, F).
When I/R was preceded by a 20-minute perfusion with the selective S1P1R antagonist W146 (1 µM) (Gonzalez-Cabrera et al., 2008) followed by a 10-minute washout, the overflows of renin and NE were each enhanced ∼1.8-fold, and the duration of VT/VF was extended ∼1.4-fold (Fig. 1), clearly indicating that S1P1R blockade exacerbates the effects of I/R-induced activation of a local cardiac RAS and suggesting that S1P1R may play a cardioprotective anti-RAS role.
We had previously shown that IPC affords anti-RAS cardioprotection by stabilizing MC in the heart before I/R (Koda et al., 2010). Because endogenously produced S1P is a mediator of IPC (Vessey et al., 2009), and MC express S1P1R (Jolly et al., 2004), we next assessed whether selective pharmacologic blockade of S1P1R in the heart counteracts the IPC-mediated attenuation of renin release in hearts subjected to I/R. We found that two 5-minute cycles of ischemia, each followed by 5-minute reperfusion (IPC) (Koda et al., 2010), decreased the I/R-induced renin and NE overflow by ∼2.8- and ∼1.6-fold, respectively, and also reduced by one-half the duration of VT/VF (Fig. 1). Notably, selective blockade of S1P1R with W146 (1 µM) prevented these anti-RAS effects of IPC, in that renin and NE overflow and VT/VF duration in IPC hearts with S1P1R blockade did not differ from those in hearts subjected to I/R alone (Fig. 1). Similarly, with S1P1R blockade these parameters did not differ from those in hearts subjected to I/R alone (Fig. 1).
We next perfused hearts with the selective S1P1R agonist SEW2871 (Jo et al., 2005) (1 µM, for two 5-minute cycles each followed by 5-minute washout) before I/R. Preperfusion with SEW2871 mimicked the protective effects of IPC in terms of renin and NE overflow and VT/VF duration, which were reduced as much as by IPC (Fig. 1). Notably, this protective effect of SEW2871 was prevented by selective blockade of S1P1R with compound W146 (1 µM) (Fig. 1). These results indicated that S1P1R activation likely plays a role in the cardioprotective anti-RAS effects of IPC.
PKCε and ALDH2 Mediate the Protective IPC-like Anti-RAS Effects of S1P1R Activation in Isolated Guinea Pig Hearts.
We had previously shown that PKCε activation/translocation is involved in the cardioprotective anti-RAS effects of IPC (Koda et al., 2010). Accordingly, we next investigated the role of PKCε in the cardioprotective anti-RAS effects of S1P1R activation. We found that selective inhibition of PKCε with compound εV1–2 (Johnson et al., 1996) (1 µM) prevented the IPC-like cardioprotective anti-RAS effects of S1P1R activation (i.e., compound εV1-2 abolished the SEW2871-induced inhibition of renin and NE release as well as the alleviation of VT/VF; Fig. 1).
We next assessed whether the anti-RAS effect of S1P1R activation also involves ALDH2 activation. For this, we investigated whether inhibition/inactivation of ALDH2 would abolish the anti-RAS effects of S1P1R activation. We found that nitroglycerine (GTN), perfused for 30 minutes before I/R at a concentration (2 µM), which is known to inactivate ALDH2 (Chen et al., 2008), prevented the IPC-like cardioprotective anti-RAS effects of S1P1R activation (i.e., GTN abolished the SEW2871-induced inhibition of renin and NE release as well as the alleviation of VT/VF; Fig. 1). Thus, activation of both ALDH2 and PKCε appears to be required for the genesis of the IPC-like cardioprotective anti-RAS effects of S1P1R activation.
Lack of IPC- and S1P1R-Mediated Anti-RAS Protection in ALDH2*2 Knock-In Mouse Hearts.
As a negative control, we used a heart model of I/R and I/R preceded by IPC from knock-in mice, in which ALDH2 activity is dramatically reduced (Zambelli et al., 2014). Spontaneously beating Langendorff-perfused WT mouse hearts were subjected to 30-minute normothermic global ischemia followed by 40-minute reperfusion. I/R in the WT ex vivo mouse heart resulted in large increases in renin and NE overflow (i.e., ∼60- and ∼100-fold compared with basal control, respectively) and severe ventricular arrhythmias (VT/VF) that lasted ∼2 minutes (Fig. 2). We had previously shown that the enhanced NE overflow and arrhythmias in the mouse heart exposed to I/R result from the activation of a local RAS initiated by renin released from cardiac MC (Mackins et al., 2006; Aldi et al., 2014b). When mouse hearts were subjected to IPC (i.e., 4 × 5-minute cycles of ischemia each followed by 5-minute reperfusion) before I/R, renin and NE overflow were reduced ∼3.5- and ∼3-fold, respectively, and VT/VF duration was reduced ∼2.5 fold (Fig. 2). Notably, when WT mouse hearts were pretreated with the selective S1P1R antagonist W146 (3 µM, 20 minutes) and then subjected to IPC, the cardioprotective anti-RAS effects of IPC were abolished (Fig. 2).
Fig. 2.

S1P1R activation affords anti-RAS IPC-like cardioprotection in a WT isolated murine heart model of I/R but not in ALDH2*2 hearts. Mouse hearts were subjected to 40 minutes of global ischemia followed by 30 minutes of reperfusion (control, WT, n = 9; ALDH2*2, n = 9). Other hearts were subjected I/R preceded by IPC alone (WT, n = 8; ALDH2*2, n = 15) or in the presence of W146 (3 μM; WT, n = 9). Other hearts were perfused with SEW2871 alone (0.1 μM; WT, n = 13; ALDH2*2, n = 14) or in the presence of W146 (3 μM; WT, n = 8; ALDH2*2, n = 6), εV1–2 (1 μM; WT, n = 6; ALDH2*2, n = 6), or GTN (2 μM; WT, n = 6; ALDH2*2, n = 6). (A) Duration of reperfusion arrhythmias (VT/VF). Overflow of NE (B) and angiotensin I (Ang I)–forming activity (C) collected over 2 minutes before ischemia and 6 minutes at the start of reperfusion. Bars indicate mean ± S.E.M. of independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001, by unpaired t test. (D–F) Representative ECG recordings from mouse hearts before ischemia (control, D), at the start of reperfusion (E), and at the end of VT/VF (recovery, F).
Another group of WT mouse hearts were perfused with the S1P1R agonist SEW2871 (0.1 µM) for 4 × 5-minute cycles, each followed by a 5-minute washout before I/R. Similar to what observed in guinea pig hearts (see Fig. 1), we found that activation of S1P1R with SEW2871 mimicked the cardioprotective anti-RAS effects of IPC (Fig. 2). Notably, this IPC-like anti-RAS effect was prevented in WT mouse hearts pretreated with the selective S1P1R antagonist W146 (3 µM, 20 minutes) (Fig. 2). Moreover, the IPC-like anti-RAS effect was prevented in WT mouse hearts pretreated with the selective PKCε inhibitor compound εV1–2 (1 µM; 20 minutes) or the ALDH2 inactivator GTN (2 µM; 30 minutes) (Fig. 2).
Thus, the anti-RAS cardioprotective effects of IPC and S1P1R activation observed in mouse hearts were similar to those observed in guinea pig hearts, in which ALDH2 activation appeared to play a pivotal role. Hence, to solidify the notion that ALDH2 activation plays a major role in the anti-RAS cardioprotection afforded by IPC and S1P1R, we used the recently developed ALDH2*2 knock-in mouse model, in which ALDH2 enzymatic activity is radically reduced (Zambelli et al., 2014). We found that either IPC or SEW2871 pretreatment (0.1 µM; 4 × 5-minute cycles) failed to protect ALDH2*2 hearts in terms of renin and NE overflow and duration of arrhythmias. In fact, preconditioned and SEW2871-pretreated ALDH2*2 hearts did not differ from ALDH2*2 hearts subjected to I/R alone (Fig. 2). Nor was any difference found when ALDH2*2 hearts were pretreated with the selective S1P1R antagonist, the selective PKCε inhibitor compound εV1–2, or the ALDH2 inactivator GTN (Fig. 2). These findings corroborated the notion that full ALDH2 enzymatic activity is needed to elicit the anti-RAS cardioprotection afforded by either IPC or S1P1R activation.
IPC and S1P1R Activation Fail To Reduce Infarct Size in ALDH2*2 Knock-In Mouse Hearts.
The size of ventricular tissue exhibiting evidence of I/R-induced cell death is widely used as a suitable indicator of I/R-induced injury (Fishbein et al., 1981; Chen et al., 2008; Redfors et al., 2012). Further, angiotensin II formed upon RAS activation is known to generate oxygen radicals, which contribute to ischemic heart damage (Mehta and Griendling, 2007; Wen et al., 2012; Balakumar and Jagadeesh, 2014). Therefore, to test further potential cardioprotective effects of S1P1R activation and their possible dependence on ALDH2 activity, we measured infarct size in Langendorff-perfused mouse hearts preconditioned with IPC or treated with the selective S1P1R agonist (SEW2871, 0.1 µM) in the presence or absence of the S1P1R antagonist (W146, 3 µM). We found that after 40-minute ischemia/2-hour reperfusion, the ventricular infarcted area amounted to ∼40% of the entire area in hearts from both WT and ALDH2*2 mice (Fig. 3, A and B). Notably, either IPC (4 × 5-minute cycles) or S1P1R activation (4 × 5-minute cycles of SEW2871, 0.1 µM) reduced infarct size in WT hearts by ∼75% and ∼60%, respectively. In contrast, no reduction in infarct size was observed when ALDH2*2 hearts were ischemically preconditioned or perfused with the S1P1R agonist, either alone or in the presence of the S1P1R antagonist (Fig. 3, A and B). These findings suggested that ALDH2 plays a role not only in the anti-RAS cardioprotection afforded by either IPC or S1P1R activation but also in the IPC- or S1P1R-induced reduction of infarct size.
Fig. 3.

S1P1R activation reduces the area of I/R-induced ischemic injury in WT but not in ALDH2*2 hearts. (A) Qualitative and (B) quantitative representation of 2,3,5-triphenyltetrazolium chloride (TTC) staining in 2-mm-thick left ventricle slices. WT and ALDH2*2 mouse hearts were subjected to 120 minutes of reperfusion (I/R, WT, n = 5; ALDH2*2, n = 7) with SEW2871 alone (0.1 μM WT, n = 6; ALDH2*2, n = 6) or in the presence of W146 (3 μM; WT, n = 6; ALDH2*2, n = 6). Other hearts were analyzed after IPC (WT, n = 5; ALDH2*2, n = 8). (A) Scale bar = 1 mm. Bars indicate mean ± S.E.M. of independent experiments. Pale areas indicate I/R-injured tissue; healthy tissue is colored in red. *P < 0.05; **P < 0.01; ***P < 0.001, by unpaired t test.
MC Express S1P1R.
Given the pivotal role that local MC play in the activation of RAS in the heart (Mackins et al., 2006), MC are the probable site at which the IPC-like anti-RAS effects of S1P1R activation develop. Thus, we first ascertained by Western immunoblot analysis that HMC-1, WT-BMMC, and ALDH2*2-BMMC all express S1P1R (Fig. 4A). Even though it had already been demonstrated that human LAD2 MC, cord blood–derived human MC (Oskeritzian et al., 2008), WT-BMMC, and RBL-2H3 all express S1P1R (Jolly et al., 2004), here we show for the first time that the S1P1R is expressed in ALDH2*2-BMMC.
Fig. 4.

S1P1R is expressed in HMC-1, WT-BMMC, and ALDH2*2-BMMC, and its activation enhances PKCε translocation. (A) Representative Western blot of total lysate of HMC-1, WT-BMMC, and ALDH2*2-BMMC (50 µg protein/lane) and Jurkat cells (positive control, 15 µg protein/lane) probed with anti-S1P1R antibody. (B) Double immunofluorescence staining of S1P1R (green) and c-Kit (red) in WT- and ALDH2*2-BMMC. (C–E) PKCε translocation in (C) HMC-1, (D) WT-BMMC, and (E) ALDH2*2-BMMC. Incubation of HMC-1 and BMMC WT and ALDH2*2 with S1P1R agonist (SEW2871, 1 µM and 0.1 µM, respectively, 10 minutes) and PMA (10 nM, min, positive control) enhances PKCε translocation from cytosol to membrane in all three cells lines. Bars represent the quantification of PKCε translocated from the cytosol to membrane fraction normalized over β-actin and are mean ± S.E.M. (control, n = 7, 8, 6; PMA, n = 9, 7, 5; SEW2871, n = 10, 5, 5; SEW2871 + W146, n = 5, 4, 6 in HMC-1, WT-BMMC, and ALDH2*2-BMMC, respectively). *P < 0.05; **P < 0.01, ***P < 0.001, by unpaired t test.
To corroborate the presence of S1P1R in MC, we used a triple-staining technique for mature MC (red), S1P1R (green), and nuclei (blue) (Fig. 4B). Clearly, when compared with WT-BMMC, ALDH2*2-BMMC showed neither a lack nor a diminished expression of the receptor, both with immunoblot and immunocytochemistry analyses. This indicates that the lack of S1P1R-induced anti-RAS cardioprotection in ALDH2*2 hearts (see Figs. 2 and 3), as well as the failure of the S1P1R agonist to activate ALDH2 in ALDH2*2-BMMC (see Fig. 5), is not due to a diminished expression of S1P1R in MC.
S1P1R Activation Translocates/Activates PKCε in MC.
Having found that the cardioprotective anti-RAS effects of S1P1R activation are ALDH2-dependent and prevented by PKCε inhibition, and that S1P1R are expressed in MC, we hypothesized that MC are the site at which the IPC-like effects of S1P1R activation are initiated and mediated by PKCε and ALDH2 activation. Thus, we next ascertained that MC PKCε is translocated/activated upon S1P1R activation.
Using Western analysis in cytosolic and membrane fractions of HMC-1 and BMMC cells, we found that the phorbol ester PMA (10 nM; positive control) markedly increased the translocation of PKCε from cytosol to membrane (i.e., a hallmark of PKCε activation) (Fig. 4, C and D). Incubation of HMC-1 and BMMC cells with the S1P1R agonist (SEW2871, 1 and 0.1 µM, respectively, for 10 minutes) also enhanced PKCε translocation from cytosol to membrane, an effect which was blocked by preincubation with the S1P1R antagonist W146 (1 and 0.1 µM, respectively, for 20 minutes) (Fig. 4, C and D). Similarly, PMA and S1P1R activation each enhanced PKCε translocation in BMMC from ALDH2*2 mice (Fig. 4E), a process that was prevented by S1P1R blockade. This indicates that S1P1R-induced PKCε translocation/activation in MC is independent of ALDH2.
S1P1R Activation Enhances ALDH2 Activity in MC.
Our findings in guinea pig and mouse hearts ex vivo suggested that ALDH2 mediates the anti-RAS, IPC-like effects of S1P1R activation (see Figs. 1 and 2); thus, we next assessed whether S1P1R activation in MC elicits an increase in ALDH2 activity. For this, we measured ALDH2 activity in HMC-1 and BMMC cells in response to the S1P1R agonist SEW2871.
We found that in both HMC-1 and WT-BMMC, SEW2871 (1 and 0.1 μM, respectively, 10 minutes) elicited a ∼30% increase in ALDH2 enzymatic activity (i.e., NADH production), comparable to that induced by the specific ALDH2 activator Alda-1 (300 µM, 10 minutes) (Budas et al., 2009) used as the positive control (Fig. 5, A and B). Notably, the S1P1R-induced increase in ALDH2 activity was prevented by the S1P1R antagonist W146 (1 and 0.1 μM, respectively, 20 minutes), by the selective ALDH2 inactivator GTN (2 μM, 25 minutes) (Koda et al., 2010), and by the PKCε inhibitor compound εV1–2 (1 μM, 20 minutes) (Fig. 5, A and B). This suggested that activation of PKCε is necessary for the S1P1R-induced increase in ALDH2 activity. In fact, the general PKC activator PMA (300 and 10 nM, 10 minutes) also significantly increased NADH production in HMC-1 and WT-BMMC (Fig. 5, A and B).
As a negative control, we investigated the effects of Alda-1, PMA, and SEW2871 in BMMC obtained from ALDH2*2 mice. We found that none of these compounds elicited a significant change in ALDH2 activity, nor was ALDH2 activity changed when the S1P1R agonist was used in the presence of its antagonist, of the PKCε inhibitor, or of the ALDH2 inactivator (Fig. 4C).
S1P1R Activation Inhibits MC Degranulation and Renin Release Elicited by Acetaldehyde: Dependence on ALDH2.
We next investigated the role of ALDH2 in MC degranulation and renin release elicited by acetaldehyde, a prototypic toxic compound that accumulates during I/R (Chen et al., 2008; Koda et al., 2010). First, by comparing WT-BMMC with ALDH2*2-BMMC, we correlated the level of ALDH2 activity with the extent of inhibition of acetaldehyde-induced MC degranulation and renin release. We found that the selective ALDH2 activator Alda-1 (300 µM) enhanced ALDH2 activity by 60% in WT-BMMC, but it failed to affect it in ALDH2*2-BMMC (Fig. 6, A and B). At the same time, Alda-1 (20 µM) markedly inhibited acetaldehyde-induced degranulation and renin release in WT-BMMC, but it failed to affect it in ALDH2*2-BMMC (Fig. 6, C and D). This indicates that a fully functioning ALDH2 is a prerequisite for the inhibition of acetaldehyde-induced MC degranulation and associated renin release.
Fig. 6.

Alda-1 increases ALDH2 activity and reduces acetaldehyde-induced degranulation and renin release in WT-BMMC but not in ALDH2*2-BMMC cells. (A) Incubation of WT-BMMC and ALDH2*2-BMMC with the selective ALDH2 activator Alda-1 (300 μM, 10 minutes) elicits an increase in ALDH2 activity in WT-BMMC (n = 7) but not in ALDH2*2-BMMC cells (n = 5). Basal NADH production was 0.96 ± 0.12 μmol/min/mg protein in WT-BMMC (n = 17) and 0.63 ± 0.11 μmol/min/mg protein in ALDH2*2-BMMC (n = 17). (B) Enzymatic activity of ALDH2 in WT-BMMC and ALDH2*2-BMMC cells. Curves start at 5 minutes, the time at which the activity began in all samples. (C, D) Incubation with acetaldehyde (100 µM, 20 minutes, 37°C) induces release of β-HEX and renin release in WT-BMMC (n = 20 and 12, respectively) and ALDH2*2-BMMC cells (n = 20 and 8, respectively). Preincubation with Alda-1 (20 µM, 10 minutes, 37°C) inhibits degranulation and renin release in WT-BMMC (n = 6 and 5, respectively) but not in ADH2*2-BMMC (n = 9 and 5, respectively). Bars are mean ± S.E.M. β-HEX and renin were measured in the supernatants at the end of acetaldehyde incubation. * P < 0.05; **P < 0.01; ***P < 0.001 by unpaired t test.
We next assessed whether S1P1R activation inhibits acetaldehyde-induced MC degranulation and renin release, and we also investigated the role played by PKCε and ALDH2 in this action. We found that S1P1R activation with SEW2871 (1 and 0.1 µM, respectively) inhibited acetaldehyde-induced degranulation and renin release in HMC-1 and WT-BMMC; this effect was prevented by the S1P1R antagonist W146 (1 and 0.1 µM, respectively) (Fig. 7, A and B). In contrast, S1P1R activation did not affect acetaldehyde-induced degranulation and renin release in ALDH2*2-BMMC (Fig. 7C), indicating the indispensable role of ALDH2 in the S1P1R-induced prevention of degranulation and renin release elicited by acetaldehyde. Notably, the selective PKCε inhibitor, εV1–2, and the ALDH2 inactivator, GTN, each prevented the SEW2871-induced abolition of acetaldehyde-induced MC degranulation and renin release in HMC-1, WT-BMMC, and ALDH2*2-BMMC cells (Fig. 7). Collectively, these findings indicate that activation of S1P1R on the MC membrane leads sequentially to PKCε translocation and ALDH2 activation, which prevents the degranulating effects of acetaldehyde, known to be produced in I/R.
Fig. 7.

S1P1R activation attenuates acetaldehyde-induced degranulation and renin release in HMC-1 and WT-BMMC but not in ALDH2*2-BMMC. Incubation with SEW2871 (0.1 and 1 µM, 10 minutes) diminishes degranulation and renin release evoked by acetaldehyde (100–300 µM, 20 minutes) in (A) HMC-1 and (B) WT-BMMC but not in (C) ALDH2*2 cells. Pretreatment with W146 (0.1 and 1 µM, 20 minutes), εV1–2 (1 μM, 20 minutes), or GTN (2 μM, 30 minutes) prevents the effect of the S1P1R agonist in (A) HMC-1, (B) WT-BMMC, and (C) ALDH2*2-BMMC cells. Bars are mean ± S.E.M. of the percentage of increase above control (β-HEX and renin in HMC-1, WT-BMMC, ALDH2*2-BMMC, respectively: acetaldehyde, n = 20, 22, 19 and n = 9, 13, 8; SEW2871, n = 9, 10, 7 and 9, 10, 7; SEW2871 + W146, n = 17, 14, 13 and n = 7, 10, 6; SEW2871 + εV1–2, n = 5, 5, 5 and n = 5, 5, 5; and SEW2871 + GTN, n = 7, 5, 5 and n = 5, 5, 5). β-HEX and renin content was measured in the supernatants at the end of acetaldehyde incubation. Basal β-HEX in HMC-1, WT-BMMC, and ALDH2*2-BMMC was 3.14 ± 0.41 (n = 14), 2.87 ± 0.27 (n = 23), and 2.92 ± 0.31 (n = 21), the percentage of total, respectively; basal renin release (Ang-I formed) for HMC-1 was 18.26 ± 4.12 (n = 17) ng/h/mg protein, and for WT-BMMC and ALDH2*2-BMMC was 0.25 ± 0.04 (n = 29) and 0.49 ± 0.15 (n = 22) µg/h/mg protein, respectively. *P < 0.05; **P < 0.01; ***P < 0.001 by unpaired t test.
Similar to acetaldehyde, 4-HNE is another toxic compound produced in large quantities during oxidative stress (Esterbauer et al., 1991; Eaton et al., 1999; Kaludercic et al., 2014). 4-HNE also elicited BMMC degranulation and renin release that were markedly reduced by the S1P1R agonist SEW2871; this effect was abolished by the S1P1R antagonist W146 (Fig. 8). Notably, SEW2871 failed to inhibit BMMC degranulation and renin release in BMMC from ALDH2*2 animals (Fig. 8).
Fig. 8.

S1P1R activation attenuates 4-HNE–induced degranulation and renin release in HMC-1 and WT-BMMC but not in ALDH2*2-BMMC. Incubation with SEW2871 (0.1 and 1 µM, 10 minutes) diminishes degranulation (i.e., β-HEX) and renin release evoked by 4-HNE (30 µM, 20 minutes) in (A) HMC-1 and (B) WT-BMMC but not in (C) ALDH2*2 cells. Pretreatment with W146 (0.1 and 1 µM, 20 minutes) prevents the effect of the S1P1R agonist in (A) HMC-1, (B) WT-BMMC, and (C) ALDH2*2-BMMC cells. Bars are mean ± S.E.M. of the percentage of increase above control (β-HEX and renin in HMC-1, WT-BMMC, ALDH2*2-BMMC, respectively: 4-HNE, n = 5, 7, 7 and n = 5, 5, 5; SEW2871, n = 5, 6, 5 and 5, 5, 5; and SEW2871 + W146, n = 5, 6, 5 and 5, 5, 5). β-HEX and renin content was measured in the supernatants at the end of the 4-HNE incubation. Basal β-HEX in HMC-1, WT-BMMC, and ALDH2*2-BMMC was 4.61 ± 0.17 (n = 5), 3.08 ± 0.58 (n = 7), and 2.17 ± 0.25 (n = 7), the percentage of total, respectively; basal renin release (Ang-I formed) for HMC-1 was 5.25 ± 0.42 (n = 6) ng/h/mg protein, and for WT-BMMC and ALDH2*2-BMMC was 0.36 ± 0.12 (n = 6) and 0.20 ± 0.08 (n = 5) µg/h/mg protein, respectively. *P < 0.05; **P < 0.01; ***P < 0.001 by unpaired t test.
Discussion
Our results clearly show that activation of S1P1R on the MC membrane during I/R affords cardioprotective anti-RAS effects, which—similar to IPC—include a reduction of renin and NE release, and alleviation of reperfusion arrhythmias. Activation of ALDH2 in MC plays a pivotal role in this cardioprotection.
MC number increases in ischemic canine and human hearts (Frangogiannis et al., 1998; Patella et al., 1998). Cardiac MC synthesizes renin, which is released in enzymatically active form in I/R and promotes the activation of a local RAS, culminating in enhanced NE release and VT/VF (Silver et al., 2004; Mackins et al., 2006). Notably, in the absence of cardiac MC (i.e., c-Kit knockout mice), renin immunoreactivity disappears, and I/R fails to activate a local RAS and to elicit VT/VF (Mackins et al., 2006). In previous studies we reported that cardiac MC degranulation by toxic aldehydes produced during I/R was inhibited by ALDH2 activation, either directly via selective ALDH2 activators (i.e., Alda-1) or via stimulation of Gi-coupled A3 and H4 receptors with adenosine and histamine, respectively. This process affords a MC-mediated anti-RAS cardioprotection (Koda et al., 2010; Aldi et al., 2014b). Accordingly, we questioned whether the stimulation of other Gi-coupled receptors expressed on MC, and the consequent activation of mitochondrial ALDH2, might provide similar protective IPC-like effects in I/R.
We focused on S1P1R because these receptors are Gi-coupled (Means and Brown, 2009) and are found to be present in rat and mouse hearts (Egom et al., 2010; Keul et al., 2016) and in RBL-2H3 MC lines (Jolly et al., 2004). MC S1P1R are plausibly activatable by their natural endogenous ligand (i.e., S1P), known to be made in the I/R heart (Means and Brown, 2009) and in MC themselves (Obinata and Hla, 2012). In fact, MC-derived S1P might well elicit “inside-out” effects (Takabe et al., 2008).
In support of the hypothesis that S1P1R might provide protective IPC-like anti-RAS effects in I/R, we found that pharmacologic blockade of S1P1R with the selective antagonist W146 (Gonzalez-Cabrera et al., 2008) prevents the protective effects of IPC in cavian and murine hearts, indicating that functional S1P1R are indispensable for the development of the anti-RAS effects of IPC. This notion is corroborated by the discovery that pharmacologic activation of S1P1R with the selective agonist SEW2871 (Jo et al., 2005) mimics the anti-RAS effects of IPC in both cavian and murine hearts, as shown by a marked reduction of renin and NE release and alleviation of arrhythmias. A definitive MC involvement was proven by the finding that pharmacologic activation of S1P1R in cultured human and murine MC prevented degranulation and renin release elicited by acetaldehyde and 4-HNE, two characteristic endogenous products of I/R (Chen et al., 2008). It is noteworthy that this protective effect was prevented by pharmacologic S1P1R blockade.
Moreover, we found that not only IPC but also the administration of the S1P1R agonist SEW2871 markedly reduced the I/R-induced left ventricular infarct size in a murine ex vivo model. In contrast, infarct size increased when treatment with SEW2871 was preceded by the administration of the selective S1P1R antagonist W146. Blockade of IPC-induced protection against ischemic injury had been previously observed with a S1P1R antagonist in the I/R rat heart (Vessey et al., 2009), while deletion of S1P1R in mouse myocytes was found to be associated with ischemic injury due to an impaired phosphorylation of the protein kinase B (Akt) survival kinase pathway, disruption of intracellular Ca2+ homeostasis, and Na+/H+ exchange (NHE) activity (Keul et al., 2016). In addition, Akt phosphorylation, as a mechanism of preconditioning, did not occur in a sphingosine kinase 1–deficient mice (Sphk1−/−) (Jin et al., 2004), suggesting that S1P1R activation exerts cardioprotective effects through Akt. Thus, S1P1R stimulation appears to protect the heart not only from the severe arrhythmias due to RAS activation but also from ischemic injury, probably due to different mechanisms possibly initiated by the activation of S1P1R on cardiomyocytes.
Yet, angiotensin II generated upon RAS activation can lead to the formation of reactive oxygen species, which are likely contributors to ischemic heart damage (Mehta and Griendling, 2007; Wen et al., 2012; Balakumar and Jagadeesh, 2014). Hence the S1P1R-induced reduction of infarct size can still be part of the overall anti-RAS cardioprotection. Recently, activation of S1P receptor subtype 3 (S1P3R) was found to protect against I/R (Yung et al., 2017), suggesting that therapeutic benefits in ischemic heart disease could be provided by activating both S1P1R and S1P3R.
Intrigued by these S1P1R-mediated anti-RAS protective effects, we next investigated their signaling mechanisms. Given that Gi-coupled receptors are known to translocate/activate PKCε (Inagaki et al., 2006), probably via phospholipase C-diacylglycerol activation (Takai et al., 1979), we asked whether PKCε inhibition would preclude the effects of S1P1R activation. We found that PKCε inhibition with compound εV1–2 (Johnson et al., 1996) not only abolished the IPC-like anti-RAS effects of S1P1R activation with SEW2871 (Jo et al., 2005) in cavian and murine hearts, but also prevented SEW2871 from attenuating the degranulating and renin-releasing effects of acetaldehyde and 4-HNE in cultured human and murine MC, clearly indicating an involvement of PKCε in the anti-RAS effects of SEW2871.
Indeed, SEW2871 translocated/activated PKCε from cytosol to membrane in HMC-1 and BMMC, confirming that the cardioprotective effects of S1P1R activation depend on the translocation/activation of PKCε. PKCε is known to phosphorylate mitochondrial ALDH2 in cardiac myocytes (Chen et al., 2008), while in MC ALDH2 phosphorylation has been shown to prevent the MC-degranulating and renin-releasing effects of reactive oxygen species and toxic aldehydes (Koda et al., 2010; Aldi et al., 2014b). Hence, to uncover the relevance of ALDH2, we preempted its action with an excess of GTN (Koda et al., 2010). Similar to PKCε blockade, ALDH2 inhibition abolished the protective anti-RAS effects of IPC in ex vivo hearts and prevented SEW2871 from attenuating the degranulating and renin-releasing effects of acetaldehyde in both HMC-1 and BMMC. It is noteworthy that PKCε blockade prevented ALDH2 activation by SEW2871.
Most importantly, either IPC or S1P1R activation failed to elicit anti-RAS cardioprotection in ex vivo hearts excised from ALDH2*2 mice, engineered to practically lack ALDH2 activity (Zambelli et al., 2014). In in vitro experiments on BMMC cultured from ALDH2*2 mice, we found that S1P1R is equally expressed on MC membranes from ALDH2*2 and WT mice. Moreover, we showed that the selective ALDH2 activator Alda-1 (Chen et al., 2008) did not activate ALDH2 in ALDH2*2-BMMC and that SEW2871 also failed to activate ALDH2 in the same cells. Nevertheless, SEW2871 was capable of translocating/activating PKCε in ALDH2*2-BMMC, indicating that this is a necessary step for ALDH2 activation because it is preserved even when ALDH2 is nonfunctioning. Notably, in ALDH2*2-BMMC, as opposed to WT-BMMC, ALDH2 activation with Alda-1 did not prevent acetaldehyde-induced degranulation and renin release. Similarly, S1P1R activation markedly reduced 4-HNE-induced degranulation and renin release in WT-BMMC but not in ALDH2*2-BMMC; furthermore, infarct size in ex vivo ALDH2*2 hearts was not reduced by S1P1R activation, demonstrating the pivotal role of MC ALDH2 in anti-RAS cardioprotection.
Collectively, these findings indicate that the IPC-like S1P1R-mediated inhibition of I/R-induced cardiac MC degranulation and renin release results from an initial translocation of PKCε and subsequent phosphorylation of ALDH2, culminating in the elimination of the MC degranulating effects of acetaldehyde and other toxic species produced during I/R (see Fig. 9). Although we did not measure translocation of PKCε to cardiac mitochondria, work by the Mochly-Rosen laboratory has demonstrated that PKCε activates the intramitochondrial enzyme ALDH2 in an ex vivo model of myocardial infarction. This protection coincides with the translocation of PKCε to cardiac mitochondria, where it associates with ALDH2 (Chen et al., 2008; Churchill et al., 2009). Importantly, as mentioned previously, we found that in hearts and BMMC from ALDH2*2 mice no protective anti-RAS effects of IPC occurred, nor did S1P1R-mediated ALDH2 activation, even though PKCε was still activatable with PMA and/or with the S1P1R agonist.
Fig. 9.

Schematic representation of our findings and the pathway downstream of S1P1R activation as a novel anti-RAS cardioprotective mechanism.
Interestingly, I/R leads to monoamine oxidase (MAO) activation (Kaludercic et al., 2011). MAO is also expressed in MC (Vitalis et al., 2003) and contributes to the generation of H2O2 and toxic aldehydes (Bianchi et al., 2005). Moreover, it was recently demonstrated that inhibition of ALDH2 by small interfering RNA in combination with MAO activation leads to mitochondrial dysfunction and cell death in heart failure (Kaludercic et al., 2010; Kaludercic et al., 2014). Accordingly, S1P1R-induced ALDH2 activation could play an additional cardioprotective role by decreasing MAO-produced toxic aldehydes and preventing myocardial damage.
Conclusion
Our collective evidence delineates a novel cardioprotective role for S1P1R on the MC membrane. Their direct pharmacologic activation by selective agonists and/or in an autocrine mode by MC-derived S1P (i.e., by “inside-out” signaling) (Takabe et al., 2008) leads sequentially to PKCε and ALDH2 activation, reduction of toxic aldehyde-induced MC degranulation, decreased renin release, prevention of RAS activation, reduction of NE release, and ultimately alleviation of reperfusion arrhythmias (see Fig. 9). Moreover, S1P1R activation mimics IPC in reducing infarct size in hearts isolated from WT but not from ALDH2*2 mice.
Aside from the physiologic and pathophysiologic relevance of this newly discovered protective pathway, our findings suggest that MC S1P1R may represent a new pharmacologic and therapeutic target for the direct alleviation of RAS-induced cardiac dysfunctions, including ischemic heart disease and congestive heart failure. It is noteworthy that S1P1R could also grant cardioprotection by additional mechanisms initiated by the activation of S1P1R on cardiomyocytes (Jin et al., 2004; Keul et al., 2016) and sympathetic nerve endings. Indeed, PC12 cells, a known model of sympathetic nerve endings (Greene and Tischler, 1976), express S1P1R (Safarian et al., 2015). Being Gi-coupled (Means and Brown, 2009), S1P1R could likely inhibit NE release from cardiac sympathetic nerves as other Gi-coupled receptors do (Levi et al., 2007). This effect could complement the reduction of NE release ultimately resulting from S1P1R-mediated actions at the MC level. Yet, we had found that when hearts of MC-deleted mice are exposed to I/R, renin release and VT/VF are abolished (Mackins et al., 2006). This supports the notion that MC and MC-expressed S1P1R are essential for the anti-RAS cardioprotective effects of S1P1R activation.
Lastly, in addition to the heart, S1P1R-mediated protective mechanisms may impact other organs (e.g., brain, liver, and kidney) that have renin-containing MC (Reid et al., 2007; Biran et al., 2008; Veerappan et al., 2008), can suffer ischemic episodes (Kaneko et al., 1998; Guo et al., 2004; Chen et al., 2009), and have been shown to be protected by IPC (Nandagopal et al., 2001; Adachi et al., 2006; Koda et al., 2010; Kukreja, 2012).
Acknowledgments
The authors thank Dr. Annarita Di Lorenzo for expert assistance and advice.
Abbreviations
- Akt
protein kinase B
- ALDH2
aldehyde dehydrogenase type 2
- ALDH2*2
a knock-in mouse model with radically reduced ALDH2 enzymatic activity
- BMMC
murine bone marrow-derived mast cells
- BSA
bovine serum albumin
- ECL
electrochemiluminescence
- GTN
glyceryl trinitrate, nitroglycerin
- β-HEX
β-hexosaminidase
- HMC-1
human mastocytoma cell line
- 4-HNE
4-hydroxynonenal
- IPC
ischemic preconditioning
- I/R
ischemia/reperfusion
- HRP
horseradish peroxidase
- KH
Krebs-Henseleit
- MAO
monoamine oxidase
- MC
mast cells
- NE
norepinephrine
- PBS
phosphate-buffered saline
- PKCε
protein kinase C subtype-ε
- PMA
phorbol ester myristate acetate
- PVDF
polyvinylidene fluoride
- RAS
renin-angiotensin system
- SEW2871
5-[4-phenyl-5-(trifluoromethyl)thiophen-2-yl]-3-[3-(trifluoromethyl)phenyl]-1,2,4-oxadiazole
- S1P
sphingosine-1-phosphate
- S1P1R
S1P1 receptors
- TBST
Tris-buffered saline containing 1% Tween 20
- εV1–2
selective PKCε inhibitor
- VT/VF
ventricular tachycardia/ventricular fibrillation
- WT
wild type
- W146
[(3R)-3-amino-4-(3-hexylanilino)-4-oxobutyl]phosphonic acid
Authorship Contributions
Participated in research design: Marino, Robador, Levi.
Conducted experiments: Marino, Sakamoto, Robador, Tomita, Levi.
Performed data analysis: Marino, Sakamoto, Robador, Tomita, Levi.
Wrote or contributed to the writing of the manuscript: Levi, Marino.
Footnotes
This work was supported by grants from the National Institutes of Health National Heart, Lung, and Blood Institute [Grant HL034215] and the American Heart Association [Grant-in-Aid].
References
- Adachi N, Liu K, Motoki A, Nishibori M, Arai T. (2006) Suppression of ischemia/reperfusion liver injury by histamine H4 receptor stimulation in rats. Eur J Pharmacol 544:181–187. [DOI] [PubMed] [Google Scholar]
- Airaksinen KE. (1999) Autonomic mechanisms and sudden death after abrupt coronary occlusion. Ann Med 31:240–245. [DOI] [PubMed] [Google Scholar]
- Aldi S, Marino A, Tomita K, Corti F, Anand R, Olson KE, Marcus AJ, Levi R. (2015) E-NTPDase1/CD39 modulates renin release from heart mast cells during ischemia/reperfusion: a novel cardioprotective role. FASEB J 29:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldi S, Robador PA, Tomita K, Di Lorenzo A, Levi R. (2014a) IgE receptor-mediated mast-cell renin release. Am J Pathol 184:376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldi S, Takano K, Tomita K, Koda K, Chan NY, Marino A, Salazar-Rodriguez M, Thurmond RL, Levi R. (2014b) Histamine H4-receptors inhibit mast cell renin release in ischemia/reperfusion via protein kinase C ε-dependent aldehyde dehydrogenase type-2 activation. J Pharmacol Exp Ther 349:508–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakumar P, Jagadeesh G. (2014) A century old renin-angiotensin system still grows with endless possibilities: AT1 receptor signaling cascades in cardiovascular physiopathology. Cell Signal 26:2147–2160. [DOI] [PubMed] [Google Scholar]
- Bianchi P, Kunduzova O, Masini E, Cambon C, Bani D, Raimondi L, Seguelas MH, Nistri S, Colucci W, Leducq N, et al. (2005) Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation 112:3297–3305. [DOI] [PubMed] [Google Scholar]
- Biran V, Cochois V, Karroubi A, Arrang JM, Charriaut-Marlangue C, Héron A. (2008) Stroke induces histamine accumulation and mast cell degranulation in the neonatal rat brain. Brain Pathol 18:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budas GR, Disatnik MH, Mochly-Rosen D. (2009) Aldehyde dehydrogenase 2 in cardiac protection: a new therapeutic target? Trends Cardiovasc Med 19:158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. (2008) Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 321:1493–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PS, Chen LS, Cao JM, Sharifi B, Karagueuzian HS, Fishbein MC. (2001) Sympathetic nerve sprouting, electrical remodeling and the mechanisms of sudden cardiac death. Cardiovasc Res 50:409–416. [DOI] [PubMed] [Google Scholar]
- Chen S, Li G, Zhang W, Wang J, Sigmund CD, Olson JE, Chen Y. (2009) Ischemia-induced brain damage is enhanced in human renin and angiotensinogen double-transgenic mice. Am J Physiol Regul Integr Comp Physiol 297:R1526–R1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy L, Yeo JM, Tse V, Chan SP, Tse G. (2016) Cardiac disease and arrhythmogenesis: mechanistic insights from mouse models. Int J Cardiol Heart Vasc 12:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill EN, Disatnik MH, Mochly-Rosen D. (2009) Time-dependent and ethanol-induced cardiac protection from ischemia mediated by mitochondrial translocation of varεPKC and activation of aldehyde dehydrogenase 2. J Mol Cell Cardiol 46:278–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA, Gilchrist A, Hoyer D, Insel PA, Izzo AA, et al. (2015) Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172:3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Hancox JC, Farkas A, Wainwright CL, Stables CL, Saint DA, Clements-Jewery H, Lambiase PD, Billman GE, Janse MJ, et al. (2013) The Lambeth Conventions (II): guidelines for the study of animal and human ventricular and supraventricular arrhythmias. Pharmacol Ther 139:213–248. [DOI] [PubMed] [Google Scholar]
- Eaton P, Li JM, Hearse DJ, Shattock MJ. (1999) Formation of 4-hydroxy-2-nonenal-modified proteins in ischemic rat heart. Am J Physiol 276:H935–H943. [DOI] [PubMed] [Google Scholar]
- Egom EE, Ke Y, Musa H, Mohamed TM, Wang T, Cartwright E, Solaro RJ, Lei M. (2010) FTY720 prevents ischemia/reperfusion injury-associated arrhythmias in an ex vivo rat heart model via activation of Pak1/Akt signaling. J Mol Cell Cardiol 48:406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H. (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11:81–128. [DOI] [PubMed] [Google Scholar]
- Fishbein MC, Meerbaum S, Rit J, Lando U, Kanmatsuse K, Mercier JC, Corday E, Ganz W. (1981) Early phase acute myocardial infarct size quantification: validation of the triphenyl tetrazolium chloride tissue enzyme staining technique. Am Heart J 101:593–600. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG, Perrard JL, Mendoza LH, Burns AR, Lindsey ML, Ballantyne CM, Michael LH, Smith CW, Entman ML. (1998) Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation 98:687–698. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Cabrera PJ, Jo E, Sanna MG, Brown S, Leaf N, Marsolais D, Schaeffer MT, Chapman J, Cameron M, Guerrero M, et al. (2008) Full pharmacological efficacy of a novel S1P1 agonist that does not require S1P-like headgroup interactions. Mol Pharmacol 74:1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MO, Karliner JS, Mochly-Rosen D. (1997) A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J Biol Chem 272:30945–30951. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA 73:2424–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Richardson KS, Tucker LM, Doll MA, Hein DW, Arteel GE. (2004) Role of the renin-angiotensin system in hepatic ischemia reperfusion injury in rats. Hepatology 40:583–589. [DOI] [PubMed] [Google Scholar]
- Inagaki K, Churchill E, Mochly-Rosen D. (2006) Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc Res 70:222–230. [DOI] [PubMed] [Google Scholar]
- Jin ZQ, Goetzl EJ, Karliner JS. (2004) Sphingosine kinase activation mediates ischemic preconditioning in murine heart. Circulation 110:1980–1989. [DOI] [PubMed] [Google Scholar]
- Jo E, Sanna MG, Gonzalez-Cabrera PJ, Thangada S, Tigyi G, Osborne DA, Hla T, Parrill AL, Rosen H. (2005) S1P1-selective in vivo-active agonists from high-throughput screening: off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chem Biol 12:703–715. [DOI] [PubMed] [Google Scholar]
- Johnson JA, Gray MO, Chen CH, Mochly-Rosen D. (1996) A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. J Biol Chem 271:24962–24966. [DOI] [PubMed] [Google Scholar]
- Jolly PS, Bektas M, Olivera A, Gonzalez-Espinosa C, Proia RL, Rivera J, Milstien S, Spiegel S. (2004) Transactivation of sphingosine-1-phosphate receptors by FcepsilonRI triggering is required for normal mast cell degranulation and chemotaxis. J Exp Med 199:959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaludercic N, Carpi A, Menabò R, Di Lisa F, Paolocci N. (2011) Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim Biophys Acta 1813:1323–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaludercic N, Carpi A, Nagayama T, Sivakumaran V, Zhu G, Lai EW, Bedja D, De Mario A, Chen K, Gabrielson KL, et al. (2014) Monoamine oxidase B prompts mitochondrial and cardiac dysfunction in pressure overloaded hearts. Antioxid Redox Signal 20:267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, Chen K, Gabrielson KL, Blakely RD, Shih JC, et al. (2010) Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res 106:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko H, Koshi S, Hiraoka T, Miyauchi Y, Kitamura N, Inoue M. (1998) Inhibition of post-ischemic reperfusion injury of the kidney by diamine oxidase. Biochim Biophys Acta 1407:193–199. [DOI] [PubMed] [Google Scholar]
- Keul P, van Borren MM, Ghanem A, Müller FU, Baartscheer A, Verkerk AO, Stümpel F, Schulte JS, Hamdani N, Linke WA, et al. (2016) Sphingosine-1-phosphate receptor 1 regulates cardiac function by modulating Ca2+ sensitivity and Na+/H+ exchange and mediates protection by ischemic preconditioning. J Am Heart Assoc 5:e003393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koda K, Salazar-Rodriguez M, Corti F, Chan NY-K, Estephan R, Silver RB, Mochly-Rosen D, Levi R. (2010) Aldehyde dehydrogenase activation prevents reperfusion arrhythmias by inhibiting local renin release from cardiac mast cells. Circulation 122:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukreja RC. (2012) Phosphodiesterase-5 and retargeting of subcellular cGMP signaling during pathological hypertrophy. Circulation 126:916–919. [DOI] [PubMed] [Google Scholar]
- Levi R, Seyedi N, Schaefer U, Estephan R, Mackins CJ, Tyler E, Silver RB. (2007) Histamine H3-receptor signaling in cardiac sympathetic nerves: identification of a novel MAPK-PLA2-COX-PGE2-EP3R pathway. Biochem Pharmacol 73:1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackins CJ, Kano S, Seyedi N, Schäfer U, Reid AC, Machida T, Silver RB, Levi R. (2006) Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J Clin Invest 116:1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means CK, Brown JH. (2009) Sphingosine-1-phosphate receptor signalling in the heart. Cardiovasc Res 82:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta PK, Griendling KK. (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292:C82–C97. [DOI] [PubMed] [Google Scholar]
- Nandagopal K, Dawson TM, Dawson VL. (2001) Critical role for nitric oxide signaling in cardiac and neuronal ischemic preconditioning and tolerance. J Pharmacol Exp Ther 297:474–478. [PubMed] [Google Scholar]
- National Research Council (NRC) (2011) Guide for the Care and Use of Laboratory Animals. 8th ed National Academies Press, Washington, DC, http://grants.nih.gov/grants/olaw/Guide-for-the-care-and-use-of-laboratory-animals.pdf. [Google Scholar]
- Obinata H, Hla T. (2012) Sphingosine 1-phosphate in coagulation and inflammation. Semin Immunopathol 34:73–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskeritzian CA, Alvarez SE, Hait NC, Price MM, Milstien S, Spiegel S. (2008) Distinct roles of sphingosine kinases 1 and 2 in human mast-cell functions. Blood 111:4193–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patella V, Marinò I, Arbustini E, Lamparter-Schummert B, Verga L, Adt M, Marone G. (1998) Stem cell factor in mast cells and increased mast cell density in idiopathic and ischemic cardiomyopathy. Circulation 97:971–978. [DOI] [PubMed] [Google Scholar]
- Redfors B, Shao Y, Omerovic E. (2012) Myocardial infarct size and area at risk assessment in mice. Exp Clin Cardiol 17:268–272. [PMC free article] [PubMed] [Google Scholar]
- Reid AC, Silver RB, Levi R. (2007) Renin: at the heart of the mast cell. Immunol Rev 217:123–140. [DOI] [PubMed] [Google Scholar]
- Safarian F, Khallaghi B, Ahmadiani A, Dargahi L. (2015) Activation of S1P1 receptor regulates PI3K/Akt/FoxO3a pathway in response to oxidative stress in PC12 cells. J Mol Neurosci 56:177–187. [DOI] [PubMed] [Google Scholar]
- Selwyn AP, Braunwald E. (2001) Ischemic heart disease, in Harrison’s Principles of Internal Medicine (Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, Jameson JL, eds) pp 1399–1410, McGraw-Hill, New York. [Google Scholar]
- Silver RB, Reid AC, Mackins CJ, Askwith T, Schaefer U, Herzlinger D, Levi R. (2004) Mast cells: a unique source of renin. Proc Natl Acad Sci USA 101:13607–13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takabe K, Paugh SW, Milstien S, Spiegel S. (2008) “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev 60:181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai Y, Kishimoto A, Kikkawa U, Mori T, Nishizuka Y. (1979) Unsaturated diacylglycerol as a possible messenger for the activation of calcium-activated, phospholipid-dependent protein kinase system. Biochem Biophys Res Commun 91:1218–1224. [DOI] [PubMed] [Google Scholar]
- Tan AY, Verrier RL. (2013) The role of the autonomic nervous system in cardiac arrhythmias. Handb Clin Neurol 117:135–145. [DOI] [PubMed] [Google Scholar]
- Veerappan A, Reid AC, Estephan R, O’Connor N, Thadani-Mulero M, Salazar-Rodriguez M, Levi R, Silver RB. (2008) Mast cell renin and a local renin-angiotensin system in the airway: role in bronchoconstriction. Proc Natl Acad Sci USA 105:1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessey DA, Li L, Honbo N, Karliner JS. (2009) Sphingosine 1-phosphate is an important endogenous cardioprotectant released by ischemic pre- and postconditioning. Am J Physiol Heart Circ Physiol 297:H1429–H1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalis T, Alvarez C, Chen K, Shih JC, Gaspar P, Cases O. (2003) Developmental expression pattern of monoamine oxidases in sensory organs and neural crest derivatives. J Comp Neurol 464:392–403. [DOI] [PubMed] [Google Scholar]
- Walker MJA, Curtis MJ, Hearse DJ, Campbell RW, Janse MJ, Yellon DM, Cobbe SM, Coker SJ, Harness JB, Harron DW, et al. (1988) The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia infarction, and reperfusion. Cardiovasc Res 22:447–455. [DOI] [PubMed] [Google Scholar]
- Wen H, Gwathmey JK, Xie LH. (2012) Oxidative stress-mediated effects of angiotensin II in the cardiovascular system. World J Hypertens 2:34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Weiner H, Crabb DW. (1996) The mutation in the mitochondrial aldehyde dehydrogenase (ALDH2) gene responsible for alcohol-induced flushing increases turnover of the enzyme tetramers in a dominant fashion. J Clin Invest 98:2027–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung BS, Brand CS, Xiang SY, Gray CB, Means CK, Rosen H, Chun J, Purcell NH, Brown JH, Miyamoto S. (2017) Selective coupling of the S1P3 receptor subtype to S1P-mediated RhoA activation and cardioprotection. J Mol Cell Cardiol 103:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambelli VO, Gross ER, Chen CH, Gutierrez VP, Cury Y, Mochly-Rosen D. (2014) Aldehyde dehydrogenase-2 regulates nociception in rodent models of acute inflammatory pain. Sci Transl Med 6:251ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]