

The sequences of 1504 mutagenized lines in the short life cycle (9 weeks) rice variety Kitaake will enable rapid functional genomic studies of this important crop.
Abstract
The availability of a whole-genome sequenced mutant population and the cataloging of mutations of each line at a single-nucleotide resolution facilitate functional genomic analysis. To this end, we generated and sequenced a fast-neutron-induced mutant population in the model rice cultivar Kitaake (Oryza sativa ssp japonica), which completes its life cycle in 9 weeks. We sequenced 1504 mutant lines at 45-fold coverage and identified 91,513 mutations affecting 32,307 genes, i.e., 58% of all rice genes. We detected an average of 61 mutations per line. Mutation types include single-base substitutions, deletions, insertions, inversions, translocations, and tandem duplications. We observed a high proportion of loss-of-function mutations. We identified an inversion affecting a single gene as the causative mutation for the short-grain phenotype in one mutant line. This result reveals the usefulness of the resource for efficient, cost-effective identification of genes conferring specific phenotypes. To facilitate public access to this genetic resource, we established an open access database called KitBase that provides access to sequence data and seed stocks. This population complements other available mutant collections and gene-editing technologies. This work demonstrates how inexpensive next-generation sequencing can be applied to generate a high-density catalog of mutations.
INTRODUCTION
Rice (Oryza sativa) provides food for more than half the world’s population, making it the most important staple crop (Gross and Zhao, 2014). In addition to its critical role in global food security, rice also serves as a model for studies of monocotyledonous species including important cereals and bioenergy crops (Izawa and Shimamoto, 1996). For decades, map-based cloning has been the main strategy for isolating genes conferring agronomically important traits (Peters et al., 2003). In Arabidopsis thaliana and other model plant species (Alonso et al., 2003; Cheng et al., 2014; Li et al., 2016c), indexed mutant collections constitute highly valuable genetic resources for functional genomic studies. In rice, multiple mutant collections have been established in diverse genetic backgrounds including Nipponbare, Dong Jin, Zhonghua 11, and Hwayoung (Wang et al., 2013b; Wei et al., 2013). Rice mutants have been generated through T-DNA insertion (Jeon et al., 2000; Chen et al., 2003; Sallaud et al., 2003; Wu et al., 2003; Hsing et al., 2007), transposon/retrotransposon insertion (Miyao et al., 2003; Kolesnik et al., 2004; van Enckevort et al., 2005; Wang et al., 2013b), RNAi (Wang et al., 2013a), TALEN-based gene editing (Moscou and Bogdanove, 2009; Li et al., 2012),CRISPR/Cas9 genome editing (Jiang et al., 2013; Miao et al., 2013; Xie et al., 2015), chemical induction, such as EMS (Henry et al., 2014), and irradiation (Wang et al., 2013b; Wei et al., 2013). Several databases have been established to facilitate use of the mutant collections (Droc et al., 2006; Zhang et al., 2006; Wang et al., 2013b). These approaches have advanced the characterization of ∼2000 genes (Yamamoto et al., 2012). The usefulness of these rice mutant collections has been hindered by the long life cycle of the genetic backgrounds used (i.e., 6 months) and the lack of sequence information for most of the mutant lines. To address these challenges, we recently established a fast-neutron (FN) mutagenized population in Kitaake, a model rice variety with a short life cycle (9 weeks) (Li et al., 2016b). Here, we report the sequencing of 1504 individual lines. We anticipate that the availability of this mutant population will significantly accelerate rice genetic research.
FN irradiation induces a diversity of mutations that differ in size and copy number, including single-base substitutions (SBSs), deletions, insertions, inversions, translocations, and duplications (Belfield et al., 2012; Bolon et al., 2014; Li et al., 2016b; Dobbels et al., 2017), in contrast to other mutagenesis approaches that mostly generate one type of mutation (Thompson et al., 2013; Wang et al., 2013b). FN irradiation generates a broad spectrum of mutant alleles, including loss-of-function, partial loss-of-function, and gain-of-function alleles that constitute an allelic series, which are highly desirable for functional genomic studies. In addition, FN irradiation induces subtle variations, such as SBSs and in-frame insertions/deletions (Indels), which facilitate the study of protein structure and domain functions (Li et al., 2016b). Finally, FN irradiation induces abundant mutations in noncoding genomic regions that may contain important functional transcription units, such as microRNAs (Lan et al., 2012) and long noncoding RNAs (Ding et al., 2012). The availability of a FN-induced mutant population with these unique characteristics greatly expands the mutation spectrum relative to other collections and provides researchers the opportunity to discover novel genes and functional elements controlling diverse biological pathways.
Whole-genome sequencing (WGS) of a mutant population and pinpointing each mutation at a single-nucleotide resolution using next-generation sequencing technologies is an efficient and cost-effective approach to characterize variants in a mutant collection, in contrast to targeting induced local lesions in genomes (TILLING) collections, for which researchers must scan amplicons from a large set of mutants for each use (McCallum et al., 2000). Another commonly used approach to characterize a genome is whole-exome sequencing (WES) (Krasileva et al., 2017). Though it is relatively low cost, WES does not cover most noncoding regions that potentially contain important functional elements such as microRNAs. Furthermore, WES is unable to identify balanced variants, including inversions and translocations, which are commonly induced by FN irradiation (Biesecker et al., 2011; Li et al., 2016b). Finally, WGS gives more accurate and complete genome-wide variant information than WES, even for the exome (Belkadi et al., 2015). Fully sequenced mutant collections are particularly useful for crops, which have inefficient transformation methods and require more time and space for genetic analyses compared with model organisms (Barampuram and Zhang, 2011). Among major crops, rice has the smallest genome (∼389 Mb) (Michael and Jackson, 2013), making it the most amenable to WGS, especially with the low cost afforded by sample multiplexing.
In this study, taking advantage of the established FN mutant collection in Kitaake (Li et al., 2016b), we whole genome sequenced 1504 lines, identified 91,513 mutations affecting 32,307 genes (58% of all genes in the rice genome), and established a WGS mutant collection in rice. To facilitate the use of this mutant collection, we established an open access resource called KitBase, which integrates multiple bioinformatics tools and enables users to search the mutant collection, visualize mutations, download genome sequences for functional analysis, and order seed stocks.
RESULTS
Genome Sequencing
We sequenced 1504 mutagenized lines, including 1408 M2 lines and 96 M3 lines using Illumina high-throughput sequencing technology, and characterized mutations in these lines. To facilitate downstream analysis, genomic DNA was isolated from a single plant of each line. High-throughput sequencing was performed using the Illumina HiSeq 2500 system, and the resulting sequence reads were mapped to the Nipponbare reference genome using the Burrows-Wheeler Aligner-Maximal Exact Match algorithm (Li, 2013). On average, 183 million paired-end reads (18.6 Gb) were obtained for each line (Table 1; Supplemental Data Set 1), and 170 million high-quality reads (93% of the raw reads) were mapped onto the reference genome, giving an average sequencing depth of 45.3-fold for each line. The high sequencing depth of these rice mutant lines facilitated detection of different types of variants.
Table 1. Genome Sequencing Summary of Mutagenized Rice Plants Used in This Study.
| Summary | Information |
|---|---|
| Total samples | 1504 |
| Mean raw bases (Gb) | 18.6 |
| Mean aligned bases (Gb) | 17.3 |
| Mean sequencing depth (fold)a | 45.3 |
The reference genome size of 374 Mb was used to calculate sequencing depth.
Genomic Variants Detected in the 1504 Mutant Lines
We used an established variant-calling pipeline containing multiple complementary programs to call variants in each rice line, filtering out variants present in the parental line and those found in two or more rice lines (see Methods). A total of 91,513 FN-induced mutations were detected in the 1504 rice lines, including 43,483 SBSs, 31,909 deletions, 7929 insertions, 3691 inversions, 4436 translocations, and 65 tandem duplications (Figure 1; Supplemental Data Set 2). The largest inversion is 36.8 Mb, the largest tandem duplication 4.2 Mb, and the largest deletion 1.7 Mb (Supplemental Figure 1). To assay the false-positive rate, we randomly selected 10 lines and examined all of their mutations (Supplemental Data Set 3). Out of 638 mutation events, we identified 30 false positives (4.7%), indicating that our variant-calling pipeline is robust. Sixty percent of these false positives are either SBSs or small Indels (<30 bp), mostly in the polynucleotide or repetitive regions. Only four false positives out of 638 mutations events (0.6%) are in coding regions, indicating the minimal impact of false positives on mutated genes.
Figure 1.
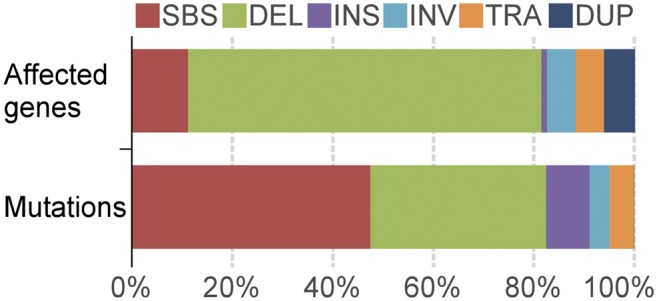
Mutations and Affected Genes in the Kitaake Rice Mutant Population.
DEL, deletions; INS, insertions; INV, inversions; TRA, translocations; DUP, tandem duplications.
Among the 91,513 mutations, SBSs are the most abundant variants, accounting for 48% of mutation events. We identified 48,030 non-SBS mutations, of which deletions account for 66%. Small deletions make up the majority of all deletion events: deletions smaller than 100 bp account for nearly 90% of all deletions (Table 2). There are 7469 single-base deletions, accounting for 23% of all deletion events. The average deletion size is 8.8 kb.
Table 2. Size Distribution of Deletions in the Kitaake Rice Mutant Population.
| Size | Number | Average size | Percentage |
|---|---|---|---|
| 1–10 bp | 21,998 | 3.7 bp | 68.9 |
| 10–100 bp | 6,588 | 21.7 bp | 20.6 |
| 100 bp–10 kb | 1,274 | 2.5 kb | 4.0 |
| 10 kb–1 Mb | 2,029 | 124.3 kb | 6.4 |
| >1 Mb | 20 | 1.2 Mb | 0.1 |
| Total | 31,909 | 8.8 kb | 100.0 |
To analyze the distribution of mutations in the genome, all mutations from the sequenced lines were mapped to the reference genome (Figure 2). We found that the FN-induced mutations are distributed evenly across the genome, except for some repetitive regions with low mapping quality reads or no read coverage caused by the inability to confidently align the reads to the reference. Many translocations were identified in the mutant population, as shown by the connecting lines (Figure 2E). The density of translocations is similar on each chromosome, ranging from 20.4/Mb to 26.8/Mb (Supplemental Table 1). The genome-wide mutation rate of the Kitaake rice mutant population is 245 mutations/Mb. The even distribution of FN-induced mutations is similar to the distribution of mutations generated through chemical mutagenesis of Sorghum bicolor and Caenorhabditis elegans (Thompson et al., 2013; Jiao et al., 2016).
Figure 2.

Genome-Wide Distribution of FN-Induced Mutations in the Kitaake Rice Mutant Population.
(A) The 12 rice chromosomes represented on a megabase scale.
(B) Genome-wide distribution of FN-induced mutations in nonoverlapping 500-kb windows. The highest column equates to 242 mutations/500 kb.
(C) Repetitive sequences in the reference genome in nonoverlapping 500-kb windows. The darker the color, the higher the percentage content of repetitive sequences.
(D) The sequencing depth of the parental line X.Kitaake. The highest column indicates 300-fold.
(E) Translocations are represented with connecting lines in the color of the smaller-numbered chromosome involved in the translocation.
Genes Affected in 1504 Mutant Lines
Genes affected by FN-induced mutation were identified using an established pipeline (see Methods). A total of 32,307 genes, 58% of all 55,986 rice genes (Kawahara et al., 2013), are affected by different types of mutations (Figure 1; Supplemental Data Set 4). On average, two alleles are available for each affected gene, and the maximum number of mutant alleles for a single gene is 17. Deletions affect the greatest number of genes, 27,614, accounting for 70% of the total number of affected genes. SBSs, constituting the most abundant mutation, only affect 4378 genes (11%). Inversions, translocations, and duplications affect 2230, 2218, and 2378 genes, respectively.
To test whether the affected genes are biased with respect to a particular biological process, we used Gene Ontology (GO) analysis to classify all affected genes into major functional categories (Ashburner et al., 2000; Du et al., 2010). The selected biological process categories DNA metabolic process, protein modification process, and transcription have the most hits and show similar percentages to the mutation saturation (58%) (Supplemental Table 2 and Supplemental Figure 2). We observed that the terms DNA metabolic process and cellular component organization show slightly higher percentages within the biological process category, whereas photosynthesis and transcription show much lower percentages (Supplemental Table 2). Core eukaryotic genes are highly conserved and are recalcitrant to modifications (Parra et al., 2009). We analyzed a set of core eukaryotic genes and showed that 40% of these analyzed are affected, mostly by heterozygous mutations (Supplemental Data Set 5). Taken together, these results suggest that, although FN-induced mutations are evenly distributed across the genome in the mutant population, the affected genes are biased against mutations in core gene functions.
FN-Induced Mutations in Each Rice Line
To assess the overall effect of FN irradiation in each sequenced line, the mutation rate and number of genes affected in each line were calculated (Supplemental Data Set 1). On average, each line contains 61 mutations; the average distance between two mutations is 6 Mb. The distribution of the number of mutations per line corresponds to a normal distribution (Figure 3). Of the 1504 lines, 90% have fewer than 83 mutations per line (Figure 3). The average number of genes affected per line is 43 (Supplemental Data Set 1). The variation of affected genes per line is greater than that of mutations per line (Table 3) due to the presence of large mutation events (Supplemental Data Set 4). For example, line FN-259 has the most genes affected (681 genes) in this mutant population, largely due to the 4.2-Mb tandem duplication that affects 667 genes (Supplemental Data Set 4). However, 76% of the mutated lines contain no more than 50 mutated genes per line (Table 3). Only 10% of the mutated lines contain more than 100 affected genes. The relatively low number of mutations per line for most lines in the Kitaake rice mutant population facilitates downstream cosegregation assays.
Figure 3.
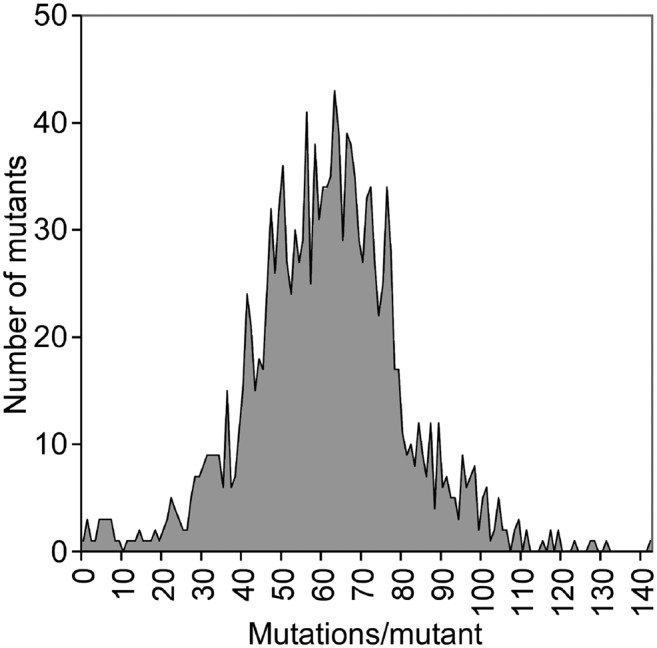
Distribution of the Number of Mutations per Line in the Kitaake Rice Mutant Population.
The x axis represents the number of mutations per line. The y axis indicates the number of mutants containing the indicated number of mutations.
Table 3. Affected Genes per Line in the Kitaake Rice Mutant Population.
| Genes/Mutant | Mutants | Percentage |
|---|---|---|
| <50 | 1,142 | 76 |
| 50–100 | 215 | 14 |
| >100 | 147 | 10 |
| Total | 1,504 | 100 |
Loss-of-Function Mutations
A large number of loss-of-function mutations were identified in this mutant population. Loss-of-function mutations completely disrupt genes. These mutations are of considerable value in functional genomics because they often clearly indicate the function of a gene (MacArthur et al., 2012). To identify loss-of-function mutations from the Kitaake rice mutant population, we adopted the definition as described (MacArthur et al., 2012) with minor modifications: We included mutations affecting start/stop codons and intron splice sites as well as mutations causing frameshifts, gene knockouts, or truncations (see Methods). There are 28,419 genes affected by loss-of-function mutations (Figure 4; Supplemental Data Set 6), accounting for 88% of the genes affected in this mutant population and 51% of all rice genes in the genome. The 340 genes affected by loss-of-function SBSs account for 1% of all genes mutated by all loss-of-function mutations. In contrast, loss-of-function deletions disrupt 26,396 genes, accounting for 85% of genes mutated by loss-of-function mutations. Inversions and translocations disrupt 2230 and 2217 genes, respectively. These results explicitly show that FN irradiation induces a high percentage of loss-of-function mutations and that deletions are the main cause.
Figure 4.
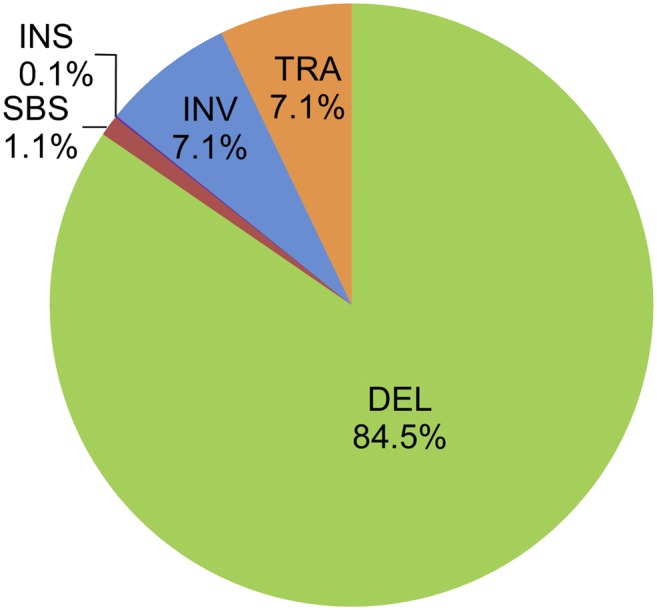
Genes Mutated by Loss-of-Function Mutations in the Kitaake Rice Mutant Population.
The percentage of gene mutated by each type of mutation is shown. DEL, deletions; TRA, translocations; INV, inversions; INS, insertions. Genes affected by tandem duplications, the copy number of which is increased, are not included.
Loss-of-function mutations affecting a single gene allow straightforward functional genomic analysis. We analyzed genes affected by these mutations and cataloged them according to the effect of the mutation and identified 7517 such genes (Table 4; Supplemental Data Set 7). Frameshifts and truncations, mostly a result of deletions, inversions, and translocations, account for 94% of the genes, which indicates the importance of these non-SBS variants.
Table 4. Genes Mutated by Loss-of-Function Mutations Affecting a Single Gene.
| Effect Type | Genes | Percentage |
|---|---|---|
| Start lost | 18 | 0.2 |
| Splice site | 109 | 1.4 |
| Stop gained/lost | 324 | 4.1 |
| Frameshifta | 2,898 | 36.4 |
| Truncationb | 4,622 | 58.0 |
| Totalc | 7,517 | 100.0 |
A frameshift refers to Indels, although it has a truncation effect on the gene.
The breakpoint of the loss-of-function mutation falls in the genic region or the gene is completely deleted due to structural variants.
Only includes unique genes. This number is smaller than the sum of genes affected in each category, as one gene can be affected by different types of mutations.
FN-Induced SBSs
To draw comparisons between the FN-induced and EMS-induced mutant populations, we conducted a detailed analysis of SBSs. There is an average of 29 SBSs per line (Supplemental Figure 3). Ninety percent of our lines contain between 10 and 50 SBSs per line. There are 118 SBSs in mutant FN1423-S, the highest number of SBSs per line in the mutant population. SBSs are evenly distributed in the genome (Supplemental Figure 4), similar to the EMS-induced mutant populations (Thompson et al., 2013; Jiao et al., 2016), and 37.9% of SBSs map within genes and 62.1% to intergenic regions (Supplemental Table 3). Of the genic SBSs, 17.3% are within exons, 17.4% within introns, 3.2% within untranslated regions, and 0.1% at canonical splice sites (GT/AG). Nonsynonymous SBSs, which represent 12.4% of all SBSs, are found in 4378 genes (Supplemental Data Set 4). Of these, 11.5% cause missense mutations, 0.8% cause nonsense mutations, and 0.1% result in read-through mutations (Supplemental Table 3).
The amino acid changes of the three mutant populations were further analyzed using heat maps (Figure 5A). The amino acid changes of the FN-induced Kitaake rice mutant population are relatively evenly distributed compared with the two EMS-induced mutant populations (Figures 5B and 5C). The differences are due to the less biased nucleotide changes of the FN-induced mutant population compared with the two EMS-induced mutant populations (Figure 5D). The frequency of the most common GT>AC nucleotide changes in the FN-induced mutant population is 42.5%, half that in the EMS-induced population (88.3%) (Henry et al., 2014) (Figure 5D). All possible amino acid changes caused by a single nucleotide change are present in the FN-induced mutant population (Figure 5A). Alanine-to-threonine or -valine changes show a much higher frequency, 4.5 and 4.3%, respectively, compared with the average amino acid change frequency of 0.7%. Alanine-to-threonine or -valine changes occur so often because these three amino acids are all encoded by four codons, and a single nucleotide change (GT>AC), the most common nucleotide changes in the mutant population, is enough to change the amino acid (Figure 5E). Similar patterns are found in the two EMS-induced mutant populations (Thompson et al., 2013; Jiao et al., 2016). Some amino acid changes occur infrequently because the occurrence frequency of these amino acids is low in rice (Itoh et al., 2007) and/or a single GT>AC change may not be sufficient to cause the amino acid change. These results demonstrate that FN irradiation induces diverse amino acid changes at higher frequencies than EMS treatment, though both FN-induced and EMS-induced mutations are evenly distributed along the genome, and that FN irradiation can result in amino acid mutations rarely achieved by chemical mutagenesis.
Figure 5.

Amino Acid and Nucleotide Changes in the FN- and Two EMS-Induced Mutant Populations.
(A) Amino acid changes in the FN-induced Kitaake rice mutant population. Single-letter amino acid symbols are shown in heat maps in (A) to (C). Each cell is colored according to the percentage of the specific amino acid change compared with all amino acid changes in the mutant population. The blank cells in (A) represent amino acid changes that require alterations of two or three nucleotides in the codon.
(B) Amino acid changes in the EMS-induced mutant population in Nipponbare rice (Henry et al., 2014).
(C) Amino acid changes in the EMS/ENU-induced mutant population in C. elegans. This population was generated with either EMS, ENU, or a combination of both (Thompson et al., 2013).
(D) Nucleotide changes in the FN-induced Kitaake rice mutant population (left), the EMS-induced mutant population in Nipponbare rice (middle), and the EMS/ENU-induced mutant population in C. elegans (right). Nucleotides are labeled in the heat maps. Each cell is colored according to the percentage of the specific nucleotide change among all missense nucleotide changes in the mutant population.
(E) The most frequent amino acid changes in the three induced mutant populations. The codon changes show that nucleotide changes of alanine (A) to threonine (T) or to valine (V) are induced by the conserved GC>AT changes. Single-letter amino acid symbols are shown in bold, and nucleotides are shown in plain font. N stands for nucleotides A, T, C, and G.
An Inversion in Mutant FN1535 Cosegregates with the Short Grain Phenotype
Grain shape is a key determinant of rice yield (Huang et al., 2013). When growing the mutated lines, we observed that line FN1535 produces significantly shorter grains compared with the parental line (Figure 6). The mutant is also dwarfed and shows a much shorter panicle. In a segregating population, we observed 34 normal plants and 13 short-grain plants, a 3:1 ratio. A goodness-of-fit test based on χ2 analysis of the phenotypic ratio revealed that the observed values are statistically similar to the expected values, indicating that the short-grain phenotype is likely caused by a recessive mutation. Next, we identified all mutations in line FN1535. We identified 76 mutations, including 26 SBSs, 38 deletions, 10 insertions, and 2 inversions (Supplemental Data Set 2). These mutations affect seven non-transposable element genes (Supplemental Table 4). To identify which mutation is responsible for the short-grain phenotype, we prioritized them based on their putative loss-of-function effects and the predicted functions of the affected genes. We prioritized a 37-kb deletion on chromosome 7 that affects five genes, an inversion on chromosome 5 affecting one gene, and a SBS on chromosome 6 that affects one gene. Using the segregating population of 50 plants, we found that the inversion on chromosome 5, not the chromosome 7 deletion or the chromosome 6 SBS, cosegregates with the phenotype (Figure 6D; Supplemental Figure 5). We analyzed the causative inversion in detail. One breakpoint of the inversion is in the fourth exon of gene LOC_Os05g26890, which truncates the gene (Figure 6E). The other breakpoint of the inversion is not in the genic region. This gene, named Dwarf 1/RGA1, was previously isolated using a map-based cloning strategy (Ashikari et al., 1999). Gene Dwarf 1/RGA1 encodes a Gα protein involved in gibberellin signal transduction (Ueguchi-Tanaka et al., 2000). Mutations in gene Dwarf 1/RGA1 cause the dwarf and short-grain phenotypes (Ashikari et al., 1999). Identical phenotypes were observed in line FN1535 (Figure 6). These results demonstrate that we can rapidly pinpoint the genetic lesion and gene conferring a specific phenotype using a small segregating population of the mutant line.
Figure 6.

An Inversion Cosegregates with the Short-Grain Phenotype in Line FN1535.
(A) Seeds of line FN1535 and the nonirradiated parental line X.Kitaake (X.Kit). Bar = 1 cm.
(B) Panicles of line FN1535 and the parental line X.Kit. Bar = 1 cm.
(C) Line FN1535 and the parental line X.Kit at the grain filling stage. Bar = 10 cm.
(D) The inversion on chromosome 5 of line FN1535 cosegregates with the short-grain phenotype. Grain length was measured by lining up 10 mature seeds of each plant as shown in (A), and the average grain length was calculated. The first lane in the top panel represents the parental line X.Kit. The 50 progeny used in the cosegregation analysis, including 15 M2 plants and 35 M3 plants derived from two M2 plants, are represented in the two panels. FN1535Inv indicates the PCR results targeting the inversion on chromosome 5 of line FN1535. A band indicates the presence of at least one parental allele in the plant. Actin primers were used for DNA quality control.
(E) IGV screenshots of the two breakpoints of the inversion on chromosome 5 of line FN1535. Reads of line FN1535 that match the Nipponbare reference genome are shown as gray bars, and reads that do not match the reference genome due to the inversion are show in black in the top panels. The black bars in the bottom panels are random mismatches. Only the left breakpoint affects a gene (LOC_Os05g26890). X.Kit indicates the parental line.
(F) Gene structure of LOC_Os05g26890. The breakpoint of the inversion is marked with a cross symbol. Gray boxes indicate exons, and lines indicate introns. The gene structure diagram is modified from the Nipponbare reference genome.
Access to Mutations, Sequence Data, and Seed Stocks
Publicly available access to high-throughput resources is essential for advancing science (McCouch et al., 2016). To make the mutant collection and associated data available to users, we established an open access web resource named KitBase (http://kitbase.ucdavis.edu/) (Figure 7). KitBase provides mutant collection information, including sequence data, mutation data, and seed information, for each rice line. Users can use different inputs, including gene IDs, mutant IDs, and DNA or protein sequences to search and browse KitBase (Figure 7A). Searches with DNA or protein sequences will be performed with the standalone BLAST tool (Deng et al., 2007). Both MSU LOC gene IDs and RAP-DB gene IDs (Kawahara et al., 2013; Sakai et al., 2013) can be used in searching the database. Mutations are visualized using the web-based interactive JBrowse genome browser, in which different symbols are used to indicate different types of mutations at the corresponding locations. Users interested in a particular region of the genome can browse all of the mutations from KitBase in that region (Figure 7B). This visual approach enables users to identify multiple allelic mutations and elucidate gene function quickly. Mutation information for each line can be downloaded from KitBase. The original sequence data and primary mutation data of lines in KitBase can be accessed through the National Center for Biotechnology Information (NCBI) and the Joint Genome Institute (JGI) (Supplemental Data Set 1). A seed request webpage was set up for seed distribution with a minimal handling fee. The seed distribution (M2 and M3 seeds) was initially subsidized by the Department of Energy via the Joint BioEnergy Institute. The user-friendly genetic resources and tools in this open access platform will facilitate rice functional genomic studies.
Figure 7.

The Navigation Page and Tools in KitBase.
(A) The main navigation page of KitBase. KitBase can be queried using either mutant ID, MSU7 LOC gene ID, or RAP-DB gene ID. Both DNA and protein sequences can be used as the input in BLAST searches.
(B) A JBrowse snapshot of mutations in a genomic region of the mutant population.
DISCUSSION
In this article, we describe a new resource that facilitates functional genomic studies of rice. A key technical feature of our mutant collection is the low level of mutagenesis (Li et al., 2016b). There is an average of 61 mutations per line (Figure 3), which means that only a small segregating population is needed to identify the causative mutation. For example, only 50 plants were needed in our study of the short-grain phenotype. Similar approaches have been used in Arabidopsis and other organisms to clone genes from WGS lines (Schneeberger, 2014; Li et al., 2016a). In contrast, a large segregating population is typically needed to identify the causative mutation using conventional genetic mapping approaches. Another advantage of our population is that it requires at most one round of backcrossing (approximately 6 months) to correlate genotype and phenotype. In contrast, some heavily mutagenized populations (carrying thousands of SBSs) require multiple rounds of time-consuming backcrosses to clean up the genetic background (Jiao et al., 2016). T-DNA insertion lines and Tos17 lines, which have 1.4 and 3.4 copies of the insert per line, respectively, also require assays to assess if the insertion cosegregates with the phenotype. Because we sequenced a single plant instead of pooled samples, users can readily identify segregating populations to pinpoint the mutation responsible for the phenotype, often without carrying out backcrossing. We estimate that 67% of all mutations in the M2 sequenced lines are heterozygous. For these lines, the progeny seeds (M3) available in KitBase can be directly used for cosegregation analysis. For homozygous mutations (33% of detected mutations), the sibling plants of the sequenced lines (M2) or progeny of their sibling plants (M3) that carry the corresponding heterozygous mutations can be used for cosegregation analysis (Figure 6), which significantly expedites genetic analysis. Users can also backcross the mutant to the parental line to create segregating progeny if needed. Compared with other sequence-indexed mutant populations including the T-DNA or Tos17 populations, WGS detects all possible variants, regardless of whether the variant is induced or spontaneous, tagged or not, which avoids the problem of somaclonal variants going undetected, even when the tag is clearly identified in some mutant populations (Wang et al., 2013b). We anticipate that the public availability of the mutant population in the early flowering, photoperiod-insensitive Kitaake variety will lower the threshold for researchers outside the rice community to examine functions of their genes of interest in rice.
FN irradiation induces a high proportion of loss-of-function mutations, which means that a relatively small population is needed to mutate all of the genes in the genome. In 1504 mutated lines, 89.3% of the affected genes are mutated by loss-of-function mutations (Figure 4). In comparison, only 0.2% of the EMS-induced mutations are annotated as loss-of-function mutations in the sequenced sorghum population (Jiao et al., 2016). For T-DNA insertion rice lines with 1.4 copies/line, 80,000 are needed to reach the same mutation saturation level achieved in our population (58%), without taking into account that T-DNA insertions are biased to certain genomic regions (Wang et al., 2013b). In plants, some screens can only be performed when plants are mature, entailing a considerable delay when using a variety with a long life cycle, such as Nipponbare. In contrast, the Kitaake rice mutant population enables researchers to carry out studies and complete screens in a relatively small population in much less time than that required for T-DNA insertion or Tos17 line screens. These features make it easier to use this mutant population (M3) to study complex traits such as yield and stress tolerance (Figure 6), which require much more time and labor. Finally, with FN-induced loss-of-function mutations, researchers can avoid the variation in knockdown efficiency or off-target issues with approaches such as RNAi or CRISPR-Cas9 (Peng et al., 2016).
Structural variants (variants >1 kb) cause many human diseases, such as the well-known Down and Turner syndromes, and are associated with several cancers (Weischenfeldt et al., 2013; Carvalho and Lupski, 2016). Limited studies in plants show that structural variants contribute to important agricultural and biological traits, such as plant height, stress responses, crop domestication, speciation, and genome diversity and evolution (Lowry and Willis, 2010; Huang et al., 2012; Saxena et al., 2014; Żmieńko et al., 2014; Zhang et al., 2015; Zhang et al., 2016). However, the study of structural variants in plants is challenging because they are often identified in different plant varieties/accessions, and the numerous variants between varieties/accessions complicate the study of the function of a specific structural variant (Saxena et al., 2014; Zhang et al., 2016). The Kitaake rice mutant population provides structural variants in the same genetic background, with only a few structural variants per line, significantly facilitating the study of the function and formation of structural variants in plants (Supplemental Data Set 2).
The Kitaake rice mutant population is complementary to other mutant populations and will significantly facilitate rice functional genomics; however, there are still some factors to consider when using this population. The Kitaake rice mutant population contains the Xa21 transgene driven by the maize ubiquitin promoter in the Kitaake genetic background (Supplemental Figure 6) (Park et al., 2010). For researchers studying innate immunity, the presence of the Xa21 transgene, which confers resistance to the bacterial pathogen Xanthomonas oryzae pv oryzae (Xoo) (Niño-Liu et al., 2006), facilitates research on the X.Kitaake-Xoo pathosystem, an excellent model for studies of infectious diseases (Ronald and Beutler, 2010; Pruitt et al., 2015). For example, the Ronald laboratory has performed forward genetic screens of this population to identify genes required for the XA21-mediated immune response. One drawback of the presence of the transgene is that, although transgenic rice lines, including T-DNA insertion lines and Xa21 lines, have been widely distributed and used in a variety of research projects, the planting of transgenic lines is restricted in some locales. In the United States, researchers routinely apply and are granted permits for interstate transport and field trials of transgenic plants, but this is not the case in all countries. In cases where the presence of a transgene is not desired, the Xa21 transgene can be segregated out by crossing the mutant with the nontransgenic parental line Kitaake.
One limitation of this Kitaake rice mutant population is that large deletions cause loss of function of many genes at once. Although such large deletions are important in achieving saturation of the genome and are valuable in screens, they pose challenges. A large deletion is likely homozygous lethal, and lethality makes it hard to study genes within the deletion. In addition, if a large deletion is identified as the causative mutation, determining which gene causes the phenotype requires multiple complementation tests (Wei et al., 2013; Chern et al., 2016). However, as more mutagenized rice lines are collected, multiple lines carrying independent mutations of the same gene will allow researchers to quickly identify the gene associated with the phenotype (Henry et al., 2014).
Another disadvantage of the current mutant population is the lack of enough mutant alleles in core eukaryotic genes and genes involved in photosynthesis and developmental process (Supplemental Table 2 and Supplemental Data Set 5), which is likely due to the lethality of these genes and the high portion of loss-of-function mutations induced by FN irradiation. Other rice mutant collections, for example, the EMS-induced mutant populations, would be complementary on this aspect by providing alleles with less severe effects on these genes (Krishnan et al., 2009; Henry et al., 2014).
Though we have sequenced the rice lines at a high depth (45-fold), it is still challenging to accurately call dispersed duplications that might result from unbalanced translocations; therefore, we include only tandem duplications. Because of the algorithms used, the genotype (homozygosity/heterozygosity) of large structural variants is not included. However, users can use tools such as Integrative Genomic Viewer (IGV; Robinson et al., 2011) to obtain genotype information with available mutant files from KitBase (Figure 7).
In the current pipeline, we used the Nipponbare reference genome because the X.Kitaake genome was not yet available. X.Kitaake is highly similar to Nipponbare, and more than 97% of the Nipponbare genome is covered by the X.Kitaake reads. Thus, variants in the Kitaake-specific regions that we failed to detect are likely minimal (∼3%) (Li et al., 2016b). To facilitate identification of all mutations in the mutant population, we are assembling the X.Kitaake genome, which we anticipate releasing in late 2017. The availability of the X.Kitaake genome will also improve mutation-calling efficiency and reduce costs.
Cost is another factor to consider when using WGS in profiling variants in a population, though this consideration is not specific to the Kitaake mutant population. Establishing a WGS population still requires a considerable initial investment, but the price of sequencing has dropped dramatically due to technological improvements (Goodwin et al., 2016). One approach to alleviate the financial challenge is through community collaboration, as a WGS population greatly benefits every researcher in that community.
The availability of the Kitaake rice mutant population complements the use of gene-editing technologies such as CRISPR/Cas9, which can generate SBSs and small Indels at target sites (Weeks et al., 2016). One drawback to gene editing in rice is that the process requires plant tissue culture and transformation, which is time-consuming and can induce somaclonal variation (Ma et al., 2016). Furthermore, gene-editing efficiency varies significantly from gene to gene (Feng et al., 2013), and off-target mutations can arise. For these reasons, the current gene-editing technologies in rice are useful for studying individual genes but are challenging for use in high-throughput studies.
A systematically phenotyped WGS mutant population is highly desirable for functional genomic studies and can rapidly bridge the genotype-to-phenotype knowledge gap. The Kitaake rice mutant population we describe in this study paves the way toward the use of a genomics-phenomics approach for functional genomics. Recently developed high-throughput phenotyping platforms make it feasible to conduct large-scale phenotyping in rice (Yang et al., 2014). We anticipate that adding systematic phenotypic data to the Kitaake WGS lines will significantly boost the utilization of the mutant collection in this model rice variety. Pairing our genomics resource with a high-throughput phenomics platform will greatly expand the capacity of researchers in rice functional genomic studies.
This study provides a cost-efficient and time-saving open access resource to gene discovery in a short life cycle rice variety by integrating physical mutagenesis, WGS, and a publicly available online database. With the WGS approach, crops are advantageous compared with some mammalian systems because a sufficiently large mutagenized population can be easily generated and maintained as seed stocks at a low cost, and the mutagenized lines can be directly planted and screened on a large scale in the field. Furthermore, as physical mutagenesis is not considered a transgenic approach, mutants with elite traits from the screens can be directly used in breeding. Given the close phylogenetic relations of rice to other grasses (Devos and Gale, 2000), this resource will also facilitate the functional studies of other grasses, such as cereals and candidate bioenergy crops (Yuan et al., 2008).
METHODS
Plant Materials and Growth Conditions
The mutagenized rice (Oryza sativa) lines used in this study were generated using FN irradiation as described previously (Li et al., 2016b). Briefly, 10,000 rice seeds of the parental line X.Kitaake, a line of the japonica cv Kitaake carrying the XA21 gene under the control of the maize (Zea mays) ubiquitin promoter, were mutagenized at 20 grays of irradiation (Li et al., 2016b). Over 7300 fertile M1 lines constitute the mutant population. The sequenced plants are mainly derived from the M2 generation and some from the M3 generation (Supplemental Data Set 1). The seeds from each line were dried and stored. To collect leaf tissues for DNA isolation, seeds were soaked in water in Petri dishes at 28°C in a growth chamber for 1 week and then transplanted to an environmentally controlled greenhouse at the University of California, Davis. In the greenhouse, light intensity across the spectrum from 400 to 700 nm was ∼250 μmol m−2s−1 and the temperature was set to 28 to 30°C and humidity to 75 to 85%. During November to April, artificial lights were supplemented to maintain the light intensity and the day/night period to 14/10 (Schwessinger et al., 2015).
DNA Sequencing and Read Mapping
DNA isolation and sequencing were done as described previously (Li et al., 2016b). Briefly, the young leaf tissue from a 3-week-old plant of each line was frozen in liquid nitrogen and stored in the −80°C freezer for DNA isolation. High-quality genomic DNA was isolated from young leaves using the cetyltrimethyl ammonium bromide (CTAB) method (Xu et al., 2011). DNA was quantified using a NanoDrop (Thermo Scientific) and fluorometer (Tecan) with the PicoGreen dsDNA assay kit (Life Technologies). The integrity of DNA samples was assayed by running the samples through a 0.7% agarose gel. Only high-quality DNA was used in sequencing. Sequencing was performed on the HiSeq 2500 sequencing system (Illumina) at the JGI following the manufacturer’s instructions. Sequencing was targeted to a minimum sequencing depth of 25-fold for each rice line to facilitate the downstream variant detection. The 2 × 100-bp paired-end sequence reads were mapped to the Nipponbare genome version 7 (Kawahara et al., 2013) using the mapping tool Burrows-Wheeler Aligner-MEM (BWA version 0.7.10) with default parameters (Li, 2013). The 41 mutant lines published in the pilot study were also included (Li et al., 2016b).
Genomic Variant Detection
Genomic variant detection was conducted as described (Li et al., 2016b) with minor modifications. Samples were analyzed in groups of no more than 50 mutant lines including the nonirradiated control line, given the high computational requirement of handling such a large data set. Genomic variants were called using a set of complementary tools, including SAMtools (Li and Durbin, 2009), BreakDancer (Chen et al., 2009), Pindel (Ye et al., 2009), CNVnator (Abyzov et al., 2011), and DELLY (Rausch et al., 2012). For the results from each tool, we removed all variants detected in the parental genome and those found in two or more samples in that group. We then merged results from each tool by filtering out redundant records. SAMtools and Pindel were used to call SBSs and small Indels (<30 bp). The minimum phred scaled quality score of variants called by SAMtools was set to 100. Pindel (version 0.2.4) was run using BreakDancer results as the input. Small Indel results from Pindel were filtered with three criteria: (1) the variant site had at least 10 reads, (2) no less than 30% of the reads supported the variant, and (3) the control line had at least 50 reads as described (Li et al., 2016b). Large variants (≥30 bp) were called using BreakDancer, Pindel, CNVnator, and DELLY as described (Li et al., 2016b). For large variants, Pindel results were filtered using the criteria listed above. Pindel sometimes reports the same common variant at multiple close positions in different samples. Therefore, we merged the same type of event if they are <10 bp from each other. We used a bin size of 1 kb for CNVnator to detect large deletions (≥30 bp). Inversion and translocation results were used from DELLY. Due to the nature of variant calls made by the algorithms (Ye et al., 2009), our results only included tandem duplications but not dispersed duplications. Only tandem duplications from Pindel were used and further filtered based on read depth variance. The false positive rate was calculated by manually examining all mutations in silico using IGV (Robinson et al., 2011) from 10 randomly selected samples. Snapshots of mutations were generated using IGV unless stated otherwise. The mutation density was calculated by adding up all mutations from the mutant population in every nonoverlapping 500-kb window for each chromosome. The genome-wide distribution of mutations was generated using Circos version 0.66 (Krzywinski et al., 2009).
Functional Annotation of Mutations
SnpEff (Yang et al., 2015) was used to annotate functional effects of the mutation based on the reference genome version 7 (Kawahara et al., 2013). Genes affected by each type of mutation were further analyzed using specific approaches as described (Li et al., 2016b). Briefly, we only include missense mutations and SBSs affecting the start/stop codon or the canonical GT/AG intron splicing sites for SBSs. Deletions or insertions overlapping with exons taken from the Gff3 file from the reference genome were counted (Kawahara et al., 2013). Genes disrupted by the breakpoint of inversions or translocations were counted for these two types of variants. Genes in the duplicated regions were counted for each tandem duplication event. We performed GO analysis on the affected genes using agriGO (http://bioinfo.cau.edu.cn/agriGO/) (Du et al., 2010). In the GO analysis, we used the biological process category.
Loss-of-Function Mutations
The definition of loss-of-function mutations was adapted from MacArthur et al. (2012) with minor modifications. SBSs/Indels (<30 bp) causing changes in the canonical GT/AG intron splicing sites or loss of the start codon or gains/losses of the stop codon and Indels (<30 bp) causing frameshifts are designated as loss-of-function mutations. Structural variants, including deletions/insertions (≥30 bp) overlapping genes, and inversions and translocations whose breakpoints fall in genic regions are also designated as loss-of-function mutations. Tandem duplications were not considered as loss-of-function mutations in this study.
Heat Maps
To compare the amino acid changes caused by FN irradiation to those caused by chemical mutagens, such as EMS, we selected one EMS-induced mutant population in rice (Henry et al., 2014) and one EMS/N-ethyl-N-nitrosourea (EMS/ENU)-induced mutant population in Caenorhabditis elegans (Thompson et al., 2013), the most comprehensive whole-genome sequenced population of its type in animals. The EMS/ENU-induced C. elegans population was created predominantly with either EMS (37% of strains), ENU (13% of strains), or a combination of both (50% of strains) in the published C. elegans population (Thompson et al., 2013). We analyzed the nucleotide changes of missense mutations and the resulting amino acid changes of these three FN- or EMS/ENU-induced mutant populations. The analyzed results were incorporated into a matrix format that was used in drawing the heat maps using the R/qplots package (https://www.R-project.org/).
Cosegregation Assays of the Short Grain Phenotype in Mutant FN1535
A segregating population, including the M2 and M3 plants derived from FN1535, was used in the cosegregation assay. Fifty plants were used in the assays. Individual plants were phenotyped by measuring grain length when seeds were mature. Average seed length was calculated by measuring 10 representative seeds in a row. χ2 analyzes were conducted to assay the goodness of fit between the observed the expected values of the segregation ratio. Genomic DNA was isolated from the plants using the CTAB method (see above). Mutation-specific primers Inv/F (5′-TTCCGTTGCTTTGGAACTTT-3′) and Inv/R (5′-CACAGCAGTTTTGCACCCTA-3′) were designed from the flanking sequences of the breakpoint of the inversion on chromosome 5 so that PCR will amplify products from the wild-type plant and plants heterozygous at the mutation sites, but not from plants homozygous at the inversion site. Primers targeting the 37-kb deletion region on chromosome 7 are Del/F (5′-CATCCTCACGGCTATACCAA-3′) and Del/R (5′-GGTGACGACGAGCGAGAG-3′). The actin primers ActF (5′-ATCCTTGTATGCTAGCGGTCGA-3′) and ActR (5′-ATCCAACCGGAGGATAGCATG-3′) were used for DNA quality control. Snapshots of the breakpoints of the inversion on chromosome 5 were taken using IGV (Robinson et al., 2011). The diagram of the structure of the mutated gene was modified from the reference genome (Kawahara et al., 2013). PCR was performed with the DreamTaq enzyme (Thermo Scientific).
KitBase
The open access resource named KitBase (http://kitbase.ucdavis.edu/) integrates genomic data, mutation data, and seed information of the Kitaake rice mutant population. Open source software and tools were used for the development of KitBase. The mutation data of each line were stored in the relational database using MySQL (https://www.mysql.com/). We used the PHP: Hypertext Preprocessor (PHP) scripting language (http://php.net/) to create the web interface and to make the data accessible. Variant Call Format files were generated for each type of mutation and embedded in the JBrowse genome browser (Skinner et al., 2009) to visualize the mutations. Standalone BLAST was incorporated into KitBase to facilitate DNA and protein sequence searching (Deng et al., 2007). Both MSU7 LOC gene IDs (http://rice.plantbiology.msu.edu/) and RAP-DB gene IDs (http://rapdb.dna.affrc.go.jp/) were incorporated into KitBase; users can use either when searching KitBase. The seed request webpage facilitates seed distribution. The KitBase server is hosted by the University of California, Davis.
Accession Numbers
All sequencing data have been deposited into NCBI’s Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra). The BioProject ID for the whole study is PRJNA385509. Accessions for individual lines are listed in Supplemental Data Set 1. Sequencing data are also available from the JGI website (http://genome.jgi.doe.gov/). Seed stocks of the Kitaake rice mutant lines used in this study are available at KitBase (http://kitbase.ucdavis.edu/kitbase/seed-order).
Supplemental Data
Supplemental Figure 1. The Largest Inversion, Tandem Duplication, and Deletion Events Detected in the Kitaake Rice Mutant Population.
Supplemental Figure 2. Gene Ontology Analysis of Affected Genes in the Kitaake Rice Mutant Population.
Supplemental Figure 3. Distribution of the Number of Single Base Substitutions per Line in the Kitaake Rice Mutant Population.
Supplemental Figure 4. Genome-Wide Distribution of Single Base Substitutions in the Kitaake Rice Mutant Population.
Supplemental Figure 5. Neither the 37-kb Deletion on Chromosome 7 nor the Single Base Substitution on Chromosome 6 of Line FN1535 Cosegregates with the Short-Grain Phenotype.
Supplemental Figure 6. Integrative Genomics Viewer Screenshot of the Insertion of the Ubi:Xa21 Transgene in the X.Kitaake Genome.
Supplemental Table 1. Translocation Density per Chromosome.
Supplemental Table 2. GO Analysis of Mutated Genes in the Kitaake Rice Mutant Population.
Supplemental Table 3. Functional Impacts of Single Base Substitutions in the Kitaake Rice Mutant Population.
Supplemental Table 4. Non-TE Genes Mutated in Line FN1535.
Supplemental Data Set 1. Genome Sequencing Summary of Rice Plants Used in This Study.
Supplemental Data Set 2. Mutations Identified in the Kitaake Rice Mutant Population.
Supplemental Data Set 3. Mutations Selected for Validation.
Supplemental Data Set 4. Genes Affected in the Kitaake Rice Mutant Population.
Supplemental Data Set 5. Core Eukaryotic Genes Affected in the Kitaake Rice Mutant Population.
Supplemental Data Set 6. Genes Mutated by Loss-of-Function Mutations.
Supplemental Data Set 7. Genes Mutated by Loss-of-Function Mutations Affecting a Single Gene.
Acknowledgments
We thank Patrick E. Canlas, Shuwen Xu, Li Pan, Kira H. Lin, Rick A. Rios, Anton D. Rotter-Sieren, Saghi Nojoomi, Hans A. Vasquez-Gross, Maria E. Hernandez, Furong Liu, Anna Joe, and Natasha Brown for assistance in genomic DNA isolation and submission, seed organization, and data processing. We thank Catherine R. Nelson for her extensive, high-quality editing and Jenny C. Mortimer, Brittany Anderton, and Oliver X. Dong for critical reading of the manuscript. We also thank Chongyun Fu, Jiandi Xu, and other Ronald lab members for insightful discussions. This work was part of the DOE Joint BioEnergy Institute (http://www.jbei.org) supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the U.S. Department of Energy. The work conducted by the U.S. Department of Energy JGI was supported by the Office of Science of the U.S. Department of Energy under Contract DE-AC02-05CH11231. This work was also supported by NIH (GM59962) and NSF (IOS-1237975) to P.C.R.
AUTHOR CONTRIBUTIONS
G.L., M.C., and P.C.R. participated in the design of the project, coordination of the project, and data interpretation. G.L., R.J., and P.C.R. drafted and revised the manuscript. M.C. developed and maintained the mutagenized population. G.L., R.J., N.T.P., M.C., J.A.M., T.W., W.S.S., A.M.L., K.C.J., L.J., P.Q.D., D.R., D.B., Y.P., K.W.B., and J.S. performed the sample preparation and sequencing and participated in in-house script development and statistical analyses. All authors read and approved the final manuscript.
Glossary
- FN
fast-neutron
- SBS
single-base substitution
- WGS
whole-genome sequencing
- WES
whole-exome sequencing
- GO
Gene Ontology
- IGV
Integrative Genomic Viewer
- ENU
N-ethyl-N-nitrosourea
References
- Abyzov A., Urban A.E., Snyder M., Gerstein M. (2011). CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21: 974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso J.M., et al. (2003). Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301: 653–657. [DOI] [PubMed] [Google Scholar]
- Ashburner M., et al.; The Gene Ontology Consortium (2000). Gene Ontology: tool for the unification of biology. Nat. Genet. 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashikari M., Wu J., Yano M., Sasaki T., Yoshimura A. (1999). Rice gibberellin-insensitive dwarf mutant gene Dwarf 1 encodes the alpha-subunit of GTP-binding protein. Proc. Natl. Acad. Sci. USA 96: 10284–10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barampuram S., Zhang Z.J. (2011). Recent advances in plant transformation. Methods Mol. Biol. 701: 1–35. [DOI] [PubMed] [Google Scholar]
- Belfield E.J., et al. (2012). Genome-wide analysis of mutations in mutant lineages selected following fast-neutron irradiation mutagenesis of Arabidopsis thaliana. Genome Res. 22: 1306–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkadi A., Bolze A., Itan Y., Cobat A., Vincent Q.B., Antipenko A., Shang L., Boisson B., Casanova J.L., Abel L. (2015). Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 112: 5473–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker L.G., Shianna K.V., Mullikin J.C. (2011). Exome sequencing: the expert view. Genome Biol. 12: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolon Y.T., et al. (2014). Genome resilience and prevalence of segmental duplications following fast neutron irradiation of soybean. Genetics 198: 967–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho C.M., Lupski J.R. (2016). Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 17: 224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., et al. (2009). BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6: 677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Jin W., Wang M., Zhang F., Zhou J., Jia Q., Wu Y., Liu F., Wu P. (2003). Distribution and characterization of over 1000 T-DNA tags in rice genome. Plant J. 36: 105–113. [DOI] [PubMed] [Google Scholar]
- Cheng X., Wang M., Lee H.-K., Tadege M., Ratet P., Udvardi M., Mysore K.S., Wen J. (2014). An efficient reverse genetics platform in the model legume Medicago truncatula. New Phytol. 201: 1065–1076. [DOI] [PubMed] [Google Scholar]
- Chern M., Xu Q., Bart R.S., Bai W., Ruan D., Sze-To W.H., Canlas P.E., Jain R., Chen X., Ronald P.C. (2016). A genetic screen identifies a requirement for cysteine-rich-receptor-like kinases in rice NH1 (OsNPR1)-mediated immunity. PLoS Genet. 12: e1006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W., Nickle D.C., Learn G.H., Maust B., Mullins J.I. (2007). ViroBLAST: a stand-alone BLAST web server for flexible queries of multiple databases and user’s datasets. Bioinformatics 23: 2334–2336. [DOI] [PubMed] [Google Scholar]
- Devos K.M., Gale M.D. (2000). Genome relationships: the grass model in current research. Plant Cell 12: 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J., Lu Q., Ouyang Y., Mao H., Zhang P., Yao J., Xu C., Li X., Xiao J., Zhang Q. (2012). A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 109: 2654–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbels A.A., Michno J.M., Campbell B.W., Virdi K.S., Stec A.O., Muehlbauer G.J., Naeve S.L., Stupar R.M. (2017). An induced chromosomal translocation in soybean disrupts a KASI ortholog and is associated with a high-sucrose and low-oil seed phenotype. G3 (Bethesda) 7: 1215–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droc G., Ruiz M., Larmande P., Pereira A., Piffanelli P., Morel J.B., Dievart A., Courtois B., Guiderdoni E., Périn C. (2006). OryGenesDB: a database for rice reverse genetics. Nucleic Acids Res. 34: D736–D740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z., Zhou X., Ling Y., Zhang Z., Su Z. (2010). agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38: W64–W70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z., Zhang B., Ding W., Liu X., Yang D.L., Wei P., Cao F., Zhu S., Zhang F., Mao Y., Zhu J.K. (2013). Efficient genome editing in plants using a CRISPR/Cas system. Cell Res. 23: 1229–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin S., McPherson J.D., McCombie W.R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17: 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross B.L., Zhao Z. (2014). Archaeological and genetic insights into the origins of domesticated rice. Proc. Natl. Acad. Sci. USA 111: 6190–6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry I.M., Nagalakshmi U., Lieberman M.C., Ngo K.J., Krasileva K.V., Vasquez-Gross H., Akhunova A., Akhunov E., Dubcovsky J., Tai T.H., Comai L. (2014). Efficient genome-wide detection and cataloging of EMS-induced mutations using exome capture and next-generation sequencing. Plant Cell 26: 1382–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsing Y.I., et al. (2007). A rice gene activation/knockout mutant resource for high throughput functional genomics. Plant Mol. Biol. 63: 351–364. [DOI] [PubMed] [Google Scholar]
- Huang R., Jiang L., Zheng J., Wang T., Wang H., Huang Y., Hong Z. (2013). Genetic bases of rice grain shape: so many genes, so little known. Trends Plant Sci. 18: 218–226. [DOI] [PubMed] [Google Scholar]
- Huang X., et al. (2012). A map of rice genome variation reveals the origin of cultivated rice. Nature 490: 497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T., et al.; Rice Annotation Project (2007). Curated genome annotation of Oryza sativa ssp. japonica and comparative genome analysis with Arabidopsis thaliana. Genome Res. 17: 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa T., Shimamoto K. (1996). Becoming a model plant: The importance of rice to plant science. Trends Plant Sci. 1: 95–99. [Google Scholar]
- Jeon J.S., et al. (2000). T-DNA insertional mutagenesis for functional genomics in rice. Plant J. 22: 561–570. [DOI] [PubMed] [Google Scholar]
- Jiang W., Zhou H., Bi H., Fromm M., Yang B., Weeks D.P. (2013). Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 41: e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y., Burke J., Chopra R., Burow G., Chen J., Wang B., Hayes C., Emendack Y., Ware D., Xin Z. (2016). A sorghum mutant resource as an efficient platform for gene discovery in grasses. Plant Cell 28: 1551–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y., et al. (2013). Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice (N. Y.) 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnik T., Szeverenyi I., Bachmann D., Kumar C.S., Jiang S., Ramamoorthy R., Cai M., Ma Z.G., Sundaresan V., Ramachandran S. (2004). Establishing an efficient Ac/Ds tagging system in rice: large-scale analysis of Ds flanking sequences. Plant J. 37: 301–314. [DOI] [PubMed] [Google Scholar]
- Krasileva K.V., et al. (2017). Uncovering hidden variation in polyploid wheat. Proc. Natl. Acad. Sci. USA 114: E913–E921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan A., et al. (2009). Mutant resources in rice for functional genomics of the grasses. Plant Physiol. 149: 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y., et al. (2012). Identification of novel miRNAs and miRNA expression profiling during grain development in indica rice. BMC Genomics 13: 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.L., Santhanam B., Webb A.N., Zupan B., Shaulsky G. (2016a). Gene discovery by chemical mutagenesis and whole-genome sequencing in Dictyostelium. Genome Res. 26: 1268–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Chern M., Jain R., Martin J.A., Schackwitz W.S., Jiang L., Vega-Sánchez M.E., Lipzen A.M., Barry K.W., Schmutz J., Ronald P.C. (2016b). Genome-wide sequencing of 41 rice (Oryza sativa L.) mutated lines reveals diverse mutations induced by fast-neutron irradiation. Mol. Plant 9: 1078–1081. [DOI] [PubMed] [Google Scholar]
- Li H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, https://arxiv.org/pdf/1303.3997.pdf.
- Li H., Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Liu B., Spalding M.H., Weeks D.P., Yang B. (2012). High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat. Biotechnol. 30: 390–392. [DOI] [PubMed] [Google Scholar]
- Li X., Zhang R., Patena W., Gang S.S., Blum S.R., Ivanova N., Yue R., Robertson J.M., Lefebvre P.A., Fitz-Gibbon S.T., Grossman A.R., Jonikas M.C. (2016c). An indexed, mapped mutant library enables reverse genetics studies of biological processes in Chlamydomonas reinhardtii. Plant Cell 28: 367–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry D.B., Willis J.H. (2010). A widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biol. 8: e1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X., Zhu Q., Chen Y., Liu Y.-G. (2016). CRISPR/Cas9 platforms for genome editing in plants: developments and applications. Mol. Plant 9: 961–974. [DOI] [PubMed] [Google Scholar]
- MacArthur D.G., et al.; 1000 Genomes Project Consortium (2012). A systematic survey of loss-of-function variants in human protein-coding genes. Science 335: 823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum C.M., Comai L., Greene E.A., Henikoff S. (2000). Targeting induced local lesions IN genomes (TILLING) for plant functional genomics. Plant Physiol. 123: 439–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCouch S.R., et al. (2016). Open access resources for genome-wide association mapping in rice. Nat. Commun. 7: 10532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J., Guo D., Zhang J., Huang Q., Qin G., Zhang X., Wan J., Gu H., Qu L.J. (2013). Targeted mutagenesis in rice using CRISPR-Cas system. Cell Res. 23: 1233–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael T.P., Jackson S. (2013). The first 50 plant genomes. Plant Genome 6: 10.3835/plantgenome2013.03.0001in. [DOI] [Google Scholar]
- Miyao A., Tanaka K., Murata K., Sawaki H., Takeda S., Abe K., Shinozuka Y., Onosato K., Hirochika H. (2003). Target site specificity of the Tos17 retrotransposon shows a preference for insertion within genes and against insertion in retrotransposon-rich regions of the genome. Plant Cell 15: 1771–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscou M.J., Bogdanove A.J. (2009). A simple cipher governs DNA recognition by TAL effectors. Science 326: 1501. [DOI] [PubMed] [Google Scholar]
- Niño-Liu D.O., Ronald P.C., Bogdanove A.J. (2006). Xanthomonas oryzae pathovars: model pathogens of a model crop. Mol. Plant Pathol. 7: 303–324. [DOI] [PubMed] [Google Scholar]
- Park C.J., Bart R., Chern M., Canlas P.E., Bai W., Ronald P.C. (2010). Overexpression of the endoplasmic reticulum chaperone BiP3 regulates XA21-mediated innate immunity in rice. PLoS One 5: e9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G., Bradnam K., Ning Z., Keane T., Korf I. (2009). Assessing the gene space in draft genomes. Nucleic Acids Res. 37: 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng R., Lin G., Li J. (2016). Potential pitfalls of CRISPR/Cas9-mediated genome editing. FEBS J. 283: 1218–1231. [DOI] [PubMed] [Google Scholar]
- Peters J.L., Cnudde F., Gerats T. (2003). Forward genetics and map-based cloning approaches. Trends Plant Sci. 8: 484–491. [DOI] [PubMed] [Google Scholar]
- Pruitt R.N., et al. (2015). The rice immune receptor XA21 recognizes a tyrosine-sulfated protein from a Gram-negative bacterium. Sci. Adv. 1: e1500245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T., Zichner T., Schlattl A., Stütz A.M., Benes V., Korbel J.O. (2012). DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28: i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. (2011). Integrative genomics viewer. Nat. Biotechnol. 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald P.C., Beutler B. (2010). Plant and animal sensors of conserved microbial signatures. Science 330: 1061–1064. [DOI] [PubMed] [Google Scholar]
- Sakai H., et al. (2013). Rice Annotation Project Database (RAP-DB): an integrative and interactive database for rice genomics. Plant Cell Physiol. 54: e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallaud C., et al. (2003). Highly efficient production and characterization of T-DNA plants for rice (Oryza sativa L.) functional genomics. Theor. Appl. Genet. 106: 1396–1408. [DOI] [PubMed] [Google Scholar]
- Saxena R.K., Edwards D., Varshney R.K. (2014). Structural variations in plant genomes. Brief. Funct. Genomics 13: 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger K. (2014). Using next-generation sequencing to isolate mutant genes from forward genetic screens. Nat. Rev. Genet. 15: 662–676. [DOI] [PubMed] [Google Scholar]
- Schwessinger B., et al. (2015). Transgenic expression of the dicotyledonous pattern recognition receptor EFR in rice leads to ligand-dependent activation of defense responses. PLoS Pathog. 11: e1004809. Erratum. PLoS Pathog. 11: e1004872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.E., Uzilov A.V., Stein L.D., Mungall C.J., Holmes I.H. (2009). JBrowse: a next-generation genome browser. Genome Res. 19: 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson O., et al. (2013). The million mutation project: a new approach to genetics in Caenorhabditis elegans. Genome Res. 23: 1749–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueguchi-Tanaka M., Fujisawa Y., Kobayashi M., Ashikari M., Iwasaki Y., Kitano H., Matsuoka M. (2000). Rice dwarf mutant d1, which is defective in the alpha subunit of the heterotrimeric G protein, affects gibberellin signal transduction. Proc. Natl. Acad. Sci. USA 97: 11638–11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Enckevort L.J., et al. (2005). EU-OSTID: a collection of transposon insertional mutants for functional genomics in rice. Plant Mol. Biol. 59: 99–110. [DOI] [PubMed] [Google Scholar]
- Wang N., Long T., Yao W., Xiong L., Zhang Q., Wu C. (2013b). Mutant resources for the functional analysis of the rice genome. Mol. Plant 6: 596–604. [DOI] [PubMed] [Google Scholar]
- Wang L., Zheng J., Luo Y., Xu T., Zhang Q., Zhang L., Xu M., Wan J., Wang M.B., Zhang C., Fan Y. (2013a). Construction of a genomewide RNAi mutant library in rice. Plant Biotechnol. J. 11: 997–1005. [DOI] [PubMed] [Google Scholar]
- Weeks D.P., Spalding M.H., Yang B. (2016). Use of designer nucleases for targeted gene and genome editing in plants. Plant Biotechnol. J. 14: 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F.J., Droc G., Guiderdoni E., Hsing Y.I.C. (2013). International consortium of rice mutagenesis: resources and beyond. Rice (N. Y.) 6: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J., Symmons O., Spitz F., Korbel J.O. (2013). Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 14: 125–138. [DOI] [PubMed] [Google Scholar]
- Wu C., Li X., Yuan W., Chen G., Kilian A., Li J., Xu C., Li X., Zhou D.X., Wang S., Zhang Q. (2003). Development of enhancer trap lines for functional analysis of the rice genome. Plant J. 35: 418–427. [DOI] [PubMed] [Google Scholar]
- Xie K., Minkenberg B., Yang Y. (2015). Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc. Natl. Acad. Sci. USA 112: 3570–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., et al. (2011). Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 30: 105–111. [DOI] [PubMed] [Google Scholar]
- Yamamoto E., Yonemaru J., Yamamoto T., Yano M. (2012). OGRO: The overview of functionally characterized genes in rice online database. Rice (N.Y.) 5: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S., Wang L., Huang J., Zhang X., Yuan Y., Chen J.Q., Hurst L.D., Tian D. (2015). Parent-progeny sequencing indicates higher mutation rates in heterozygotes. Nature 523: 463–467. [DOI] [PubMed] [Google Scholar]
- Yang W., et al. (2014). Combining high-throughput phenotyping and genome-wide association studies to reveal natural genetic variation in rice. Nat. Commun. 5: 5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K., Schulz M.H., Long Q., Apweiler R., Ning Z. (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25: 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J.S., Tiller K.H., Al-Ahmad H., Stewart N.R., Stewart C.N. Jr. (2008). Plants to power: bioenergy to fuel the future. Trends Plant Sci. 13: 421–429. [DOI] [PubMed] [Google Scholar]
- Zhang J., Li C., Wu C., Xiong L., Chen G., Zhang Q., Wang S. (2006). RMD: a rice mutant database for functional analysis of the rice genome. Nucleic Acids Res. 34: D745–D748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., et al. (2016). Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. Proc. Natl. Acad. Sci. USA 113: E5163–E5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., et al. (2015). Genome-wide mapping of structural variations reveals a copy number variant that determines reproductive morphology in cucumber. Plant Cell 27: 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Żmieńko A., Samelak A., Kozłowski P., Figlerowicz M. (2014). Copy number polymorphism in plant genomes. Theor. Appl. Genet. 127: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]