

Abstract
Background
The placenta is the principal organ regulating intrauterine growth and development, performing critical functions on behalf of the developing fetus. The delineation of functional networks and pathways driving placental processes has the potential to provide key insight into intrauterine perturbations that result in adverse birth as well as later life health outcomes.
Results
We generated the transcriptome-wide profile of 200 term human placenta using the Illumina HiSeq 2500 platform and characterized the functional placental gene network using weighted gene coexpression network analysis (WGCNA). We identified 17 placental coexpression network modules that were dominated by functional processes including growth, organ development, gas exchange and immune response. Five network modules, enriched for processes including cellular respiration, amino acid transport, hormone signaling, histone modifications and gene expression, were associated with birth weight; hub genes of all five modules (CREB3, DDX3X, DNAJC14, GRHL1 and C21orf91) were significantly associated with fetal growth restriction, and one hub gene (CREB3) was additionally associated with fetal overgrowth.
Conclusions
In this largest RNA-Seq based transcriptome-wide profiling study of human term placenta conducted to date, we delineated a placental gene network with functional relevance to fetal growth using a network-based approach with superior scale reduction capacity. Our study findings not only implicate potential molecular mechanisms underlying fetal growth but also provide a reference placenta gene network to inform future studies investigating placental dysfunction as a route to future disease endpoints.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3878-0) contains supplementary material, which is available to authorized users.
Keywords: Placenta, RNA-Seq, WGCNA, Birth weight
Background
It is increasingly appreciated that the intrauterine experience impacts the long-term health of the offspring [1–3]. The placenta is the principal organ regulating the intrauterine environment and orchestrating gestational development. Situated at the maternal-fetal interface, the placenta acts as a multi-organ system, performing at times the functions of the lung, liver, gastrointestinal tract, kidney and endocrine organs on behalf of the developing fetus [4]. Through this capacity, the placenta plays an active role in facilitating the transport of nutrients and waste, exchange of gases, immune protection and generation of hormones to carefully regulate fetal growth and organ-specific development throughout gestation [4]. Alterations to the intrauterine environment through the action of various intrinsic and extrinsic stressors can impact placental function, including vascularization, growth, transport activity, metabolism and hormone production, and as a result, impact the physiology of the developing fetus, with potential health consequences across the lifespan [5, 6]. The placenta, therefore, is the primary regulator through which the developmental trajectory of the fetus is altered in response to changes in the intrauterine environment [7]. Despite the centrality of the placenta in the developmental origins of health and disease, this vital organ remains poorly characterized.
Recent developments in high throughput whole-genome sequencing make it feasible to assess the entire transcriptome with the potential to agnostically uncover biological processes driving complex phenotypes. Studies assessing the transcriptome-wide profile of human term placenta are beginning to emerge [8–11], however, the inferences from these findings are limited by the small sample size and the focus on univariate gene expression analyses contrasting normal and adverse phenotypic outcomes. Systems biology methods that apply a networks-based analysis of transcriptome-wide data better capture the complexity of inter-gene relationships and the pathways they participate in to give rise to disease processes, and, therefore, hold the opportunity to better define the co-regulatory patterns that underlie complex phenotypes. Methods such as weighted gene co-expression network analysis (WGCNA) have been successfully applied in a number of tissues in relation to various health outcomes [12–15]. This approach facilitates systems-level characterization of expression changes by clustering highly correlated genes into coexpression modules of conserved biological function [16, 17]. The utility of this approach was demonstrated in a recently published study focusing on highly variable genes (n ~ 3000) in a set of 16 placenta that showcased both conservation and divergence between human and mouse placental networks [18].
Abnormal fetal growth, both undergrowth (fetal growth restriction) and overgrowth (macrosomia), is among the most commonly reported outcomes linked to placental dysfunction. Infants clinically defined as either small (SGA, bottom 10% weight for gestational age) or large (LGA, above 90% weight for gestational age) are of particular concern as they are at elevated risk for postnatal morbidities, including metabolic syndrome and impaired neurobehavioral and cognitive development [19–24], highlighting the public health impetus to identify the molecular mechanisms underlying fetal growth dysregulation. Hence, this outcome provides an exemplary opportunity to demonstrate the importance of placenta gene networks by elucidating fetal-growth related placental processes. In addition, these findings have the potential to be translated into novel approaches for disease intervention or even prevention at an early time-point, when they may be the most effective.
In this study, we comprehensively profiled the transcriptome-wide landscape of the largest birth cohort collection of human placentae to date, implementing a network-based approach to construct a placental gene coexpression network and to delineate a fetal-growth gene signature.
Results
The demographic characteristics of our study population, categorized as small (SGA), large (LGA) and appropriate (AGA) for gestational age infants, are shown in Table 1. Consistent with the literature, a greater proportion of LGA infants were male [25] and born to mothers with elevated pre-pregnancy BMI [26]. A greater proportion of SGA infants were female and born to mothers of non-Caucasian descent [27]. Additionally, LGA infants were more likely to be delivered by cesarean section.
Table 1.
Demographic characteristics of the study population (n = 200)
| Variables | SGAa (n = 33) | AGAb (n = 112) | LGAc (n = 55) | p-valued |
|---|---|---|---|---|
| Mean (SD) | Mean (SD) | Mean (SD) | ||
| Birth weight (g) | 2582.3 (277.3) | 3436.8 (388.9) | 4276.9 (247.2) | <0.01 |
| Gestational age (weeks) | 38.9 (1.2) | 39.1 (0.9) | 39.0 (0.8) | 0.77 |
| Maternal age (years) | 31.9 (5.6) | 31.1 (4.6) | 30.9 (4.1) | 0.59 |
| Maternal BMI (kg/m2) | 25.7 (7.0) | 25.6 (5.8) | 28.4 (6.9) | 0.02 |
| N (%) | N (%) | N (%) | ||
| Infant Gender | 0.05 | |||
| Female | 22 (66.7) | 57 (50.9) | 22 (40.0) | |
| Male | 11 (33.3) | 55 (49.1) | 33 (60.0) | |
| Maternal Ethnicity | <0.01 | |||
| Caucasian | 18 (54.5) | 91 (81.2) | 46 (83.6) | |
| African American | 6 (18.2) | 2 (1.8) | 3 (5.5) | |
| Other | 7 (21.2) | 19 (17.0) | 5 (9.1) | |
| Unknown | 2 (6.0) | 0 (0.0) | 1 (1.8) | |
| Delivery Method | ||||
| Vaginal | 19 (57.6) | 65 (58.0) | 19 (34.5) | 0.01 |
| C-Section | 14 (42.4) | 47 (42.0) | 36 (65.5) |
asmall for gestational age; bappropriate for gestational age; clarge for gestational age; d p-values based on chi-square test (categorical variables) or ANOVA (continuous variables)
The RNA-Seq dataset averaged 44 million reads per sample. Of all annotated NCBI Reference Sequence genes (n = 23,228), 22,518 genes were mapped at least one count per sample, with 52% genes surpassing expression levels above 2 in the log2 scale in a minimum of 30 samples. Among 20 samples run in triplicate, we observed a median R2 value of 0.97, with a range between 0.93 and 0.98. Consistent with previous reports [8, 9], the most abundant placental transcripts observed in our study include placental hormones (CSH1 [28]), placental development factors (PAPPA [29], PAPPA2 [30]), steroidogenesis enzymes (CYP19A1 [31]), tissue remodeling genes (TFPI2 [32], FBLN1 [33], FN1 [11, 34], MALAT1 [35]), ribonucleases (RPPH1 [36]) and signaling molecules (RHOBTB3) [37]. Several of the most abundant transcripts are putative imprinted genes, including TFPI2 [38], RHOBTB3 and PAPPA2 [37] (Additional file 1). For technical validation of the RNAseq results, we additionally examined the agreement between RNASeq and the probe-hybridization based nCounter (NanoString technologies, Seattle, Washington) method for a subset of genes with expression data available on both platforms (n = 80) [39]. A significant, positive correlation was observed in gene rankings based on relative gene expression levels (Spearman rho = 0.76), indicating relative gene expression levels were comparable across the platforms (Additional file 2).
Placental gene coexpression network
Network-based analyses provide a means to account for the coordinated expression among genes, thereby reducing the dimensionality of the data-set and affording insight into underlying biological processes. Coexpression network analysis of the placental transcriptome revealed 17 coexpressed gene modules in human placenta (Additional file 3). Module size ranged from 37 to 3073 genes (Fig. 1). Approximately 1000 genes did not load onto any specific module (grey module). To identify biological functions associated with each module, we performed gene ontology (GO) analysis. While there were overlapping processes enriched across several modules, including processes related to chromatin assembly (purple and light cyan), most processes were uniquely enriched in specific modules, including immune response (black module), gas transport (grey60) and cell adhesion (tan). Modules enriched in similar biological processes tended to cluster together using unsupervised hierarchical clustering (Additional file 4). For example, both brown and greenyellow modules are involved in mRNA processing and black and tan modules are involved in extracellular signaling. Next, we investigated whether disease signatures are present within the placental network. For this purpose, we surveyed a curated list of published GWAS, including 23,086 genes and spanning 760 phenotypes, for the enrichment of genes with phenotype-linked variants within our placental network. Overall, 651 GWAS-linked genes encompassing 185 phenotypes were enriched in the placental network (p < 0.05) (Additional file 5). We observed notable trends consistent with the enriched GO-derived biological processes identified in our network modules. For instance, the immune response module (black) was enriched for immune-related disorders, such as systemic lupus erythematosus, inflammatory bowel disease and IgA nephopathy; the vasculature development module (blue) was enriched in vascular endothelial growth factor levels, platelet aggregation and blood pressure; the gas transport module (grey60) was enriched in red blood cell traits, platelet count and mean corpuscular hemoglobin; and the module involved in the secretion of the metabolic hormones gonadotropin/glucagon (salmon) was enriched in low high density lipoprotein cholesterol levels, LDL cholesterol and carotid intima media thickness (Fig. 2).
Fig. 1.
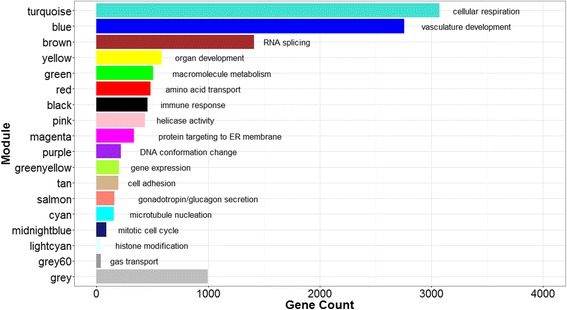
Placental gene coexpression network. We identified 18 network modules ranging in size from 37 to 3073 genes. A total of 998 genes (grey module) did not load onto any specified module. Gene ontology enrichment analysis revealed key growth and developmental processes enriched in each module, including transcriptional activity, cell division and respiration
Fig. 2.
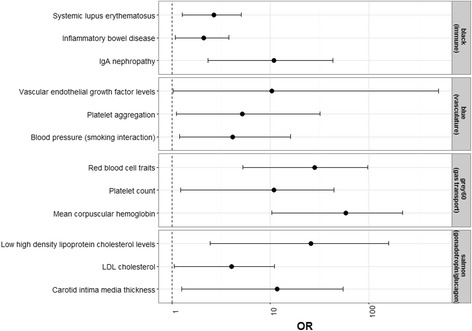
Enrichment of GWAS-associated phenotypes in network modules. Enrichment odds ratios (OR) and 95% confidence intervals are shown for a subset of phenotypes observed to be significantly enriched in the black, blue, grey60 and salmon modules
We defined the 1st principal component of each module, the module eigengene, as a summary measure of each module [17, 40]. Genes most strongly correlated with the eigengene of each module (hub genes) are shown in Table 2. In most instances, the identified hub genes reflect the biological processes enriched in the modules. For example, complement system genes are the hub genes of the immune response module (black), ribosome-coding genes are the hub genes of the protein translation module (magenta) and histone modification genes are the hub genes of the DNA conformation change module (purple).
Table 2.
Placenta gene coexpression network hub genes
| Module | Hub gene |
|---|---|
| Turquoise | HELZ, MLL2, YIF1, C12orf5, GHITM |
| Blue | TCF4, PDGFRB, ETS1, COL6A2, SPTBN1 |
| Brown | RBM33, ATXN2L, MYSM1, MLL4, CBFA2T2 |
| Yellow | ARHGAP23, LAMB1, UTRN, LRP5, MYH10 |
| Green | UBE2W, TMED7, ZNF24, SOCS6, RAP2C |
| Red | FAR1, B4GALT3, POR, PIGH, NISCH |
| Black | C1QC, C1QB, CD68, CTSS, C1QA |
| Pink | MPZL1, SKP2, MCM3, BMP7, MCM |
| Magenta | RPL7A, UBA52, RPS5, RPL18, RPS11 |
| Purple | HIST1H3F, HIST1H2AH, CDK1, FEN1, CDC20 |
| Greenyellow | C21orf91, ZNF721, INO80D, ANKRD12, MBTD1 |
| Tan | NOTUM, HN1, REPS2, B3GNT7, PYCR1 |
| Salmon | PVRL4, NDRG1, GRHL1, PLIN2, HMHA1 |
| Cyan | ZC3H18, UBE2N, PRDX3, CAPZA2, CDK12 |
| Midnight blue | TOP2A, TPX2, LMNB1, CENPF, SLC13A3 |
| Light cyan | DDX3Y, RPS4Y1, KDM5D, ZFY, TTTY15 |
| Grey60 | ALAS2, SLC4A1, HBA1, HBG1, HBA2 |
Correlations between network module eigengenes and continuous demographic variables are shown in Fig. 3. Several interesting patterns are apparent. With increasing gestational age, a negative correlation with drivers of growth and development (purple, midnightblue and yellow) and a positive correlation with immune-related activity (black) was observed. An increase in maternal age was negatively correlated with processes related to development (yellow) and cell replication (pink and midnight blue), while an increase in maternal BMI was correlated with an upregulation of growth-promoting processes (greenyellow). Differences across categorical demographic variables were evaluated based on mean differences in module eigengene values. As seen on Fig. 4, module eigengene values were significantly different with respect to infant gender, maternal race/ethnicity, delivery method and birth weight categories. Differences across infant gender were observed in modules involved in chromatin assembly-related processes (light cyan and purple). An upregulation of growth-promoting processes was observed among white infants compared to non-white infants (blue and pink). Differences in modules involved in growth and development related processes, including respiration (turquoise), amino acid transport (red), metabolic hormone secretion (salmon), gene expression (greenyellow) and histone modification (light cyan), were observed across birth weight categories. Several modules related to birth weight overlapped with modules related to gender (light cyan), maternal BMI (greenyellow) and delivery method (salmon, red, greenyellow).
Fig. 3.
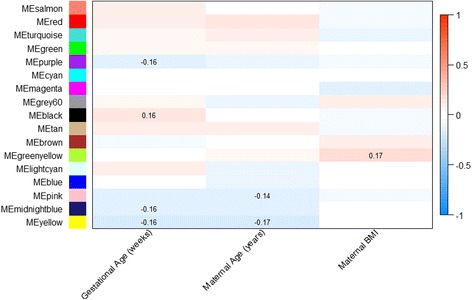
Spearman correlations between placenta network modules and continuous demographic characteristics. The color gradient indicates the direction, positive (red) and negative (blue), and strength of the correlation. Significant correlation coefficients (p < 0.05) are indicated on the plot
Fig. 4.
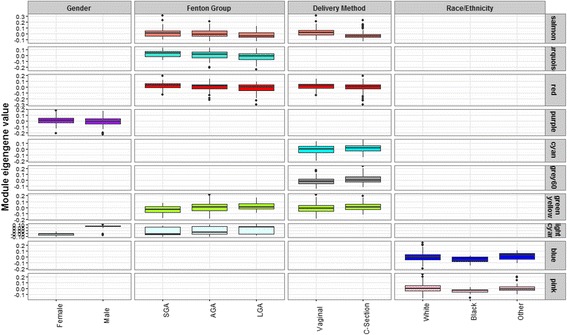
Placenta network module differences across categorical demographic characteristics. Significant differences were determined based on Mann Whitney U tests (2 groups) and Kruskal Wallis tests (>2 groups)
Placental gene network modules associated with birth weight
To demonstrate the functional relevance of the placenta network identified above, we further evaluated module-based differences in fetal-growth related gene pathways. Intramodular hub genes are genes with maximal connections to other genes in their respective modules. Given the importance of these genes in likely determining the behavior of the module-associated biological pathway, we identified the candidate fetal growth-related intramodular hub genes within each of the five fetal growth-related modules, focusing on genes strongly correlated with both Fenton growth curve percentiles and module eigengene values (Additional file 6). Significant associations with deviations in appropriate fetal growth are observed among genes with high module membership (Fig. 5), including CREB3 (turquoise module), DNAJC14 (red module), GRHL1 (salmon module) and C21orf91 (greenyellow module). A log2 unit increase in the expression of CREB3, DNAJC14, DDX3X and GRHL1 is associated with increased odds of SGA status, while a log2 unit increase in the expression of C21orf91 is associated with decreased odds of SGA status. A log2 unit increase in the expression of CREB3 is additionally observed to be protective against LGA status. Comparing the candidate fetal growth-related intramodular hub genes with the overall module hub genes listed in in Table 2, an overlap of two salmon module genes, GRHL1 and PVRL4 as well as one greenyellow module gene, C21orf91, was observed.
Fig. 5.
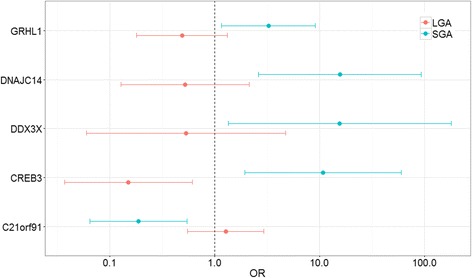
Association between candidate module hub genes and birth weight. Genes shown along y-axis are putative fetal growth-related intramodular hub genes: CREB3 (turquoise), DNAJC14 (red), GRHL1 (salmon), DDX3X (light cyan) and C21orf91 (greenyellow). Each gene’s association with aberrant fetal growth categories is indicated by the odds ratios (OR) and 95% Confidence Intervals (CI) of SGA and LGA infants referenced against AGA infants for a log2 unit increase in expression. Multinomial regression models were adjusted for infant gender and maternal BMI
Conventional univariate analyses across all 12,095 genes indicated 393 genes differentially expressed across the three birth weight categories, primarily between SGA and LGA infants. Genes differentially expressed across the birth weight categories predominately loaded onto modules differentially coexpressed across birth weight categories, with a significant enrichment observed in the turquoise, red and greenyellow modules (Fig. 6).
Fig. 6.
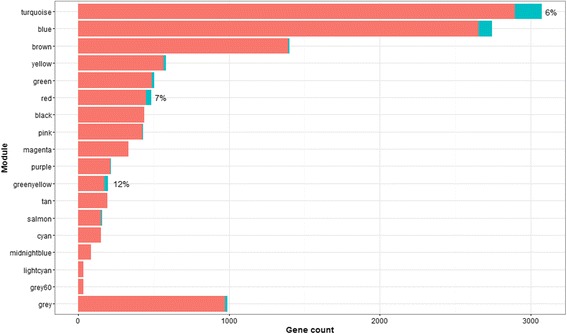
Enrichment of genes differentially expressed by birth weight in network modules. Twelve modules contained genes differentially expressed by birth weight category (n = 393). The proportion of differentially expressed genes within each of the modules is indicated in blue in the stacked bar plot. A significant enrichment of differentially expressed genes based on a fisher’s exact test was observed in the turquoise, red and greenyellow modules
In addition to the global network, we also constructed separate networks for each birth weight category. Fewer modules are observed in the separate networks compared to the global networks, with 15 modules in the SGA network, 12 modules in the LGA network and 13 modules in the AGA network. Next, we compared the network density and connectivity patterns of the SGA and LGA networks to the AGA network to determine whether the topology of the AGA network is preserved in the aberrant fetal growth networks. Network density measures assess whether gene-sets that load onto a common module in the reference network are also adjacent to one another in the test networks, while network connectivity measures assess whether the connection strengths of module gene-pairs in the reference network are preserved among the same gene-pairs in the test networks. While no differences in network densitity measures were observed, as shown in Fig. 7, a significant loss of network connectivity (Z statistic <10) was observed in the salmon module for both the SGA (Z connectivity =7.85) and LGA (Z connectivity = 7.95) networks compared to the AGA network. This loss in network connectivity suggests that a subset of gene interactions in the salmon module are disrupted in the aberrant fetal growth categories, indicative of a breakdown in the associated biological pathway.
Fig. 7.
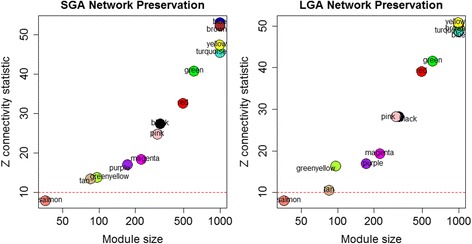
Loss of network conservation in SGA and LGA networks compared to AGA networks. Relative to the AGA network, the salmon module demonstrates loss of connectivity in the SGA (Z = 7.85) and LGA (7.95) networks
An example of module connectivity patterns, indicating both genes differentially expressed across birth weight categories and module hub genes, is shown for the salmon module in Fig. 8. With the exception of INHBA, the differentially expressed genes largely demonstrate weaker connectivity patterns within the module in relation to the hub genes.
Fig. 8.
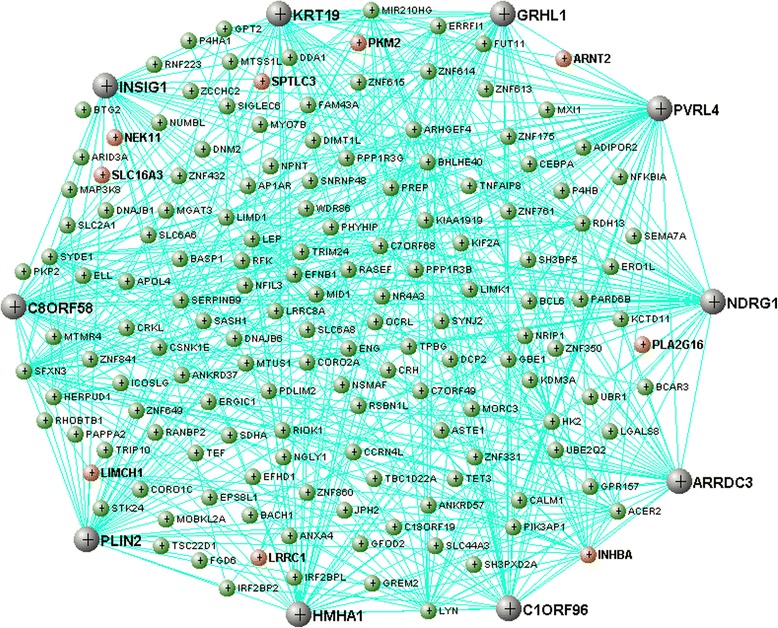
Gene connectivity in the salmon module. Black nodes indicate the most highly connected genes in the module (hub genes). Red nodes indicate genes differentially expressed across birth weight categories
Discussion
In this study, we have taken a systems biology approach to describe the functional gene networks present in the human term placenta. Weighted gene coexpression analysis of the placental transcriptome revealed an enrichment in functional processes related to growth and development, including cellular respiration, transcriptional activity, and signal transduction. Although few descriptions of non-pathological tissue-specific coexpression networks are available in the literature, a few general trends are observable. Placental modules enriched for biological processes involved in transcriptional activity, cellular respiration and immune response were also common to processes observed in other reported tissue networks, including liver [41], skeletal muscle [42] and blood [43]. Compared to these networks, processes uniquely enriched in the placenta include organ and vasculature development, cell replication, cell adhesion, gas transport and hormone secretion. Furthermore, the processes enriched in the placental gene network reflect known critical functions perfomed by the placenta for appropriate fetal development, including establishing a blood supply (vasculature development), trophoblast adherence to maternal decidua (cell adhesion), gas exchange (gas transport), feto-maternal immune tolerance (immune response) and endocrine signaling (hormone secretion). Interestingly, genes with common GWAS-associated biomedical traits also loaded onto our network modules. While the molecular function/process assigned to genes using GO are based on curated in vitro or in silico evidence of participation in biological pathways [44], the GWAS catalog comprises array-based genotyping studies conducted in human population settings [45, 46]. Hence, our observed GWAS-enrichments corroborate the functional processes assigned to the modules based on GO enrichment analyses and also suggest the possible priming of later life health effects during development.
Several network modules were observed to be related to maternal-infant demographic variables, suggesting that the activity of these modules reflect gestational characteristics. For example, the observed downregulation in growth-related processes with increasing gestational age may indicate a shift in placental processes to ready the fetus for postnatal life. Similarly the gestational age associated upregulation of immune-related processes is consistent with the onset of parturition [47, 48]. Given the narrow range in gestational age in our study population, however, our observations, while consistent with known trajectories in feto-placental development, would require further in-depth physiologic analysis to establish biologic relevance. Several modules were associated with multiple demographic characteristics. For example, an upregulation of the histone modification (lightcyan) module was observed both among male infants and LGA infants, consistent with known trends in gender differences based on birth size. The upregulation of the gene expression (greenyellow) module with increasing maternal BMI and birth weight is also consistent with a posititve relationship between maternal BMI and birth size. Similarly, changes in modules by delivery status tracked closely with patterns of modules distinguishing LGA and AGA infants, consistent with observations in our study that LGA infants were more likely to be delivered by cesarean section.
To evaluate the utility of the derived placental gene network in elucidating the molecular underpinnings of placental dysfunction, we examined fetal growth-related perturbations to the network. The network-based analysis reduced the dimensionality of the data and accounted for the interdependent structure among the transcripts, enabling us to derive functional insight into the biological processes affected by fetal growth-related dysregulation of coexpression. In our study, we identified 5 network modules with differential coexpression across birth weight categories that are functionally enriched for cellular respiration (turquoise), amino acid transport (red), gonadotropin/glucagon secretion (salmon), gene expression (greenyellow) and histone modification (light cyan). Several of these modules, ones related to cellular respiration, amino acid transport and gene expression, were also significantly enriched with genes differentially expressed across birth weight categories.
A loss in overall network connectivity in SGA and LGA infants was observed for the gonadotropin/glucagon secretion (salmon) module, highlighting a breakdown in key gene interactions within this module among the aberrant growth categories. A closer look at the salmon module highlights the added value gained by using a networks-based approach. This module includes differentially expressed genes previously shown to be relevant to fetal growth and development, i.e., PKM2 [49], SPTLC3 [37] and INHBA [50]. Additionally, this module includes genes that were not identified as differentially expressed in the current study but have been previously shown to be relevant to fetal growth and pregnancy complications in independent studies, primarily LEP [51], as well as the hub genes PVRL4 and PLIN2 [52]. The fact that these genes, which were previously independently implicated with fetal growth and development, load onto a common module in the current study suggests that the mechanism through which these genes exert their effect may converge on a common biological pathway.
While the genes we observed to be differentially expressed were enriched in modules where we observed differential coexpression, it is of interest to note that none of the differentially expressed genes represent hub genes of fetal growth related modules. In fact, differentially expressed genes tended to be relatively weakly connected within network modules. This highlights a key distinction between differential expression and network-based coexpression analyses. Coexpression networks follow a small world architecture, whereby the presence of highly connected hub genes facilitates information flow through the network in relatively few hops [53]. Due to this small world structure of coexpression networks, there is redundancy in the network, allowing for tolerance in perturbations among the majority of nodes. Large-scale perturbations among the handful of highly connected nodes, however, would result in network failure [53, 54]. As differential expression analysis identifies genes with the largest fold changes, these genes are likely situated at the periphery of the network, whereas genes central to fetal growth-related biological processes may be missed. Additionally, the fact that the majority of genes in modules observed to be relevant to fetal growth were not observed to be differentially expressed also highlights that disrupted coexpression in a module can occur in the absence of net differential expression among the individual genes within the module. This suggests that network-based methods may be more robust at uncovering key determinants of affected biological processes and potentially more relevant interventional targets than differential expression analysis.
Several limitations in our study design warrant further discussion. The placenta is a complex organ, consisting of various cell sub-types with transcriptional profiles that likely vary. Nevertheless, our placental samples were collected from four quadrants exclusively on the fetal membrane side and within 2 cm from the cord insertion site, a region identified as the least variable in the placenta [55]. Furthermore, as our study represents a single assessment at term, a causal link between gene expression and birth weight cannot be drawn. As such, follow up studies are warranted to determine whether placental gene signatures bear relevance on postnatal health, extending into childhood as well as adulthood. Such studies also have the potential to additionally provide further mechanistic insight into the roots and consequences of abnormal fetal growth.
Conclusions
The current study is the largest comprehensive human placental profiling study to date. The generated profile stems from non-pathological, term placenta, providing a characterization of the baseline placental transcriptional landscape and the role that normal variation might play in defining patterns of intrauterine development in the general population. Importantly, we implemented a systems biology-based approach to characterize the placental gene networks, and we demonstrated the utility of this approach by delineating a fetal growth-related placental gene signature. This signature highlights the potential for leveraging the generated placental gene network to uncover novel insight into the molecular underpinnings of placental dysfunction. This includes the identification of both established and novel genes related to fetal growth, and importantly, the interrelationship among these genes in common deregulated biological pathways. As such, these findings highlight the placenta, a temporal organ that is commonly discarded, as a valuable resource for identifying biomarkers relevant to fetal development and postnatal health effects.
Methods
Placenta collection
Placenta tissues were collected as part of the Rhode Island Child Health Study (RICHS), a birth cohort representing the populations of Rhode Island and Southeastern Massachusetts, USA [56]. This population consists of singleton, term infants (≥37 weeks gestation) born without congenital or chromosomal abnormalities and born to women without life-threatening pregnancy complications. The cohort includes large for gestational age (LGA, >90% 2013 Fenton Growth Curve), small for gestational age (SGA, <10% 2013 Fenton Growth Curve) and adequate for gestational age (AGA) infants. Given an a priori interest to study fetal growth, this population was oversampled for both LGA and SGA infants. Of the entire RICHS cohort (n = 841), a subset of 200 subjects, representative of the full cohort (Table 1), were selected for RNA sequencing and subsequent analyses. All subjects provided written informed consent approved by the Institutional Review Boards at Women and Infants Hospital and Emory University.
Placental biopsies free of maternal decidua were excised from four quadrants within 2 cm of the cord insertion site, placed in RNALater at 4 °C within 2 h of delivery and at least 72 h later were removed from RNALater, pooled, snap-frozen, homogenized to powder, and stored at −80 °C.
RNA sequencing
Total RNA was isolated from homogenized placental tissue using the RNeasy Mini Kit (Qiagen, Valencia, CA) and stored in RNAse-free water at −80 °C. The yield was quantified using a Quibit Fluorometer (Thermo Scientific, Waltham, MA) and the integrity was assessed using an Agilent Bioanalyzer (Agilent, Santa Clara, CA). Ribosomal RNA was removed using a Ribo-Zero Kit [57]. RNA was converted to cDNA using random hexamers (Thermo Scientific, Waltham, MA). Transcriptome-wide 50 bp single-end RNA sequencing was conducted using the HiSeq 2500 platform (Illumina, San Diego, CA) [58]. Samples were run in three sequencing batches, with 10% of the samples run in triplicate within each batch.
Statistical analysis
QC filtering and normalization
The raw RNA sequencing data (fastq files) were assessed for quality control, including read length and GC content, using the FastQC software. Reads that passed the quality control metrics were mapped to the human reference genome (hg19) in a splice-aware manner using the Spliced Transcripts Alignment to a Reference (STAR) aligner [59], with common SNPs in the reference genome masked prior to alignment. Genes with counts per million <1 in greater than 30 samples (the sample size of the smallest phenotypic group in this study) were considered unexpressed and removed. Read counts were adjusted for GC content using the EDASeq R package [60], followed by TMM correction for library size differences across samples using the calcNormFactors function in edgeR R package [61]. The data was then transformed into logCPM values accounting for the mean-variance relationship in the data using the voom function of the limma R package [62]. Following assessements of Pearson correlations in gene expression among the triplicate samples, duplicated repeat samples were removed from the analysis. Finally, the normalized log2 counts per million (logCPM) reads were filtered to genes with expression levels above 2 in the log2 scale in a minimum of 30 samples. The final filtered, normalized data-set included 12,135 genes. Eight percent of the variability in our data-set was attributable to batch as determined by the implementation of the pvca R package [63, 64]. Expression level differences based on demographic characteristics were assessed based on Mann Whitney U test for variables with two categories, Kruskal-Wallis test for variables with >2 categories, and Spearman correlations for continuous variables.
Weighted gene coexpression network
Prior to constructing the co-expression network, the gene expression data was adjusted to remove potential confounding due to batch effect using the ComBat function in the R package sva [65]. The gene co-expression network was generated using the WCGNA R package [17]. Briefly, a similarity matrix was generated from the normalized RNA-Seq data using absolute values of Pearson correlation coefficients among all gene pairs {sij = |cor(xi,xj)|}. The similarity matrix was transformed into an adjacency matrix using an adjacency function based on a weighted soft threshold (β =6) {aij = sij β}. The selected β parameter value satisfied the minimum value required to generate a scale-free topology network (linear regression R2 ≥ 0.8). The components of the resulting adjacency matrix indicate connection strengths among gene pairs, with connections among strongly correlated genes emphasized and weakly correlated genes suppressed.
To delineate modules, the relative interconnectedness between each node pair was calculated as a topological overlap similarity measure. Here, the topological overlap for each node pair was defined as the proportion of node connections that are shared between the two nodes out of the total number of node connections of the node with fewer connections, thereby, capturing the similarity in the coexpression relationship with all other genes in the network. The reciprocal topological dissimilarity matrix was used as input for hierarchical clustering, and gene modules were defined based on hierarchical clustering guided by topological overlap, using a dynamic tree cut algorithm to establish modules [66]. Finally, highly correlated modules were merged based on a merging threshold set at a height cut-off of 0.25. In the resulting network, as neighbors in a cluster share high topological overlap, the resulting modules likely indicate a common functional class. As a summary measure of the gene modules, each module was defined by the first principal component of each module (module eigengene) to represent the weighted average gene expression profile of the module. Module membership of genes within each module was determined based on the correlation between individual gene expression values and the module eigengene value. Top five genes based on module membership (correlation with module eigengene) were classified as hub genes. Network modules were further assessed for enrichment of biological processes across Gene Ontology (GO) categories based on Fisher’s exact test. Similarly, Fisher’s exact test was applied to assess network module enrichment of genes linked to specific phenotypes (diseases/traits) based on a curated catalog of genome wide association studies (GWAS) [46, 67]. Phenotypes included in the analysis were restricted to those associated with a minimum of 10 reported gene variants. Fisher’s exact test was applied to trait/module gene membership contingency tables with non-zero cell counts. Spearman correlations were calculated between module eigengenes and continuous mother-infant demographic and gestational variables. Significant differences (p < 0.05) in module eigengene values across categorical mother-infant demographic and gestational variables were determined using a Mann-Whitney U test (2 categories) or a Kruskal Wallis test (>2 categories).
Network-based differential gene coexpression analysis
Module hub genes related to fetal growth were identified as genes highly correlated with the Fenton growth curve percentile r > |0.2| and the module eigengene (r > |0.8|). To assess the association between the expression of these candidate hub genes and fetal growth, we conducted multinomial regression models using the nnet R package [68], setting SGA and LGA status as the outcomes referenced against AGA status. Models were adjusted for infant gender, delivery method and maternal prepregnancy body mass index (BMI). These variables were selected based on observed differences in eigengene values of modules related to fetal growth (infant gender and lightcyan module; delivery method and salmon, red and greenyellow modules; maternal prepregnancy BMI and greenyellow module).
To assess whether module topology patterns are altered among the adverse growth phenotypes, separate networks were generated for each birth weight category as described for the global network. Module conservation between the reference AGA network and the SGA and LGA networks was evaluated based on four network connectivity and four network density measurements. Network density measurements assess whether genes that are highly connected in the reference network (assigned to a common module) are also highly connected in test networks. Density preservation was assessed based on the correlations between module genes in the reference network and the corresponding genes in the test network on measures of mean correlation, mean adjacency, mean module membership, and proportion of variance explained by the module eigengene. Network connectivity measurements assess whether connection strengths among module gene-pairs are conserved in reference and test networks. Connectivity preservation was assessed based on the correlations between module genes in the reference network and the corresponding genes in the test network on measures of intramodular connectivity (conservation of hubs; rowsum of adjacency), adjacency matrix, correlation between each gene and the module eigengene (module membership), and correlation among pairwise gene expression correlations. The significance of the observed preservation statistics was determined using 200 permutation tests where gene labels are randomly assigned in the test network to estimate the mean and standard deviation under the null hypothesis of no preservation. The resulting Z-scores provide a measure of the whether the observed gene connectivity pattern of the test network is significantly more conserved than random. The median of the four Connectivity Z-scores and the median of the four density Z-scores are averaged to generate a summary Z-score [69].
Differential gene expression analysis
Differential gene expression across the three birth weight categories was assessed using the limma R package [62]. Briefly, limma implements an empirical Bayes method to generate gene-wise moderated t-statistics across contrasts of interests. Observed associations were considered significant at FDR < 0.05. Enrichment for differentially expressed genes in network modules was determined using Fisher’s exact test.
Figures depicting enrichment of GO terms and differentially expressed genes in network modules, differential expression of modules across birth weight categories and the forest plot of the association between hub gene expression and aberrant fetal growth were generated using the ggplot2 R package [70]. The figure highlighting differentially expressed genes and hub genes in the salmon module was generated using visANT 5.0 [71, 72]. Figures showing hierarchical clustering of network modules and network preservation measures were generated using the WGCNA R package [17]. The remaining figures were generated using R base plotting functions. All analysis was conducted using R 3.3.1 [73].
Additional files
Distribution in expression level (logCPM) among top expressed genes in placenta. Shown in grey is the distribution in expression level across all genes (n = 12,135). (TIFF 1487 kb)
Agreement in gene expression rankings between RNAseq and NanoString (n = 197). Gene expression across 80 genes was assessed by both the RNAseq and NanoString platforms. Genes were ranked by expression level (lowest rank signifies highest expression across samples). Gene ranks across the platforms were significantly correlated (Spearman rho = 0.76, p < 0.01). (TIFF 1666 kb)
Gene loadings in placental gene coexpression network. Genes loading into separate modules are listed. Additionally the correlation between genes and module eigengenes are indicated. (CSV 2815 kb)
Hierarchical clustering of network modules. (TIFF 1474 kb)
Mapping of GWAS-linked genes in placental gene coexpression network. Genes linked to GWAS-associated traits that are enriched in the placental gene coexpression network are listed alongside assigned modules and GWAS-linked traits. (CSV 58 kb)
Candidate module hub genes relevant to birth weight. Plots indicate the correlation between gene expression and module eigengene values (x-axis) and the correlation between gene expression and birth weight category (y-axis). Genes that demonstrate both module importance and birth weight relevance are indicated for each module. Genes of interest include MKL2, PSMD4, ADRM1, AC3H15 and CREB3 in the turquoise module, LAD1, KAT5, DNAJC14 and BECN1 in the red module, GRHL1, INHBA, PVRL4, LEP and C8orf58 in the salmon module, DDX3X in the lightcyan module, and ZNF460, C21orf91 and PAN3 in the greenyellow module. (TIFF 169 kb)
Acknowledgements
Not applicable.
Funding
This work is supported by NIH-NIMH R01MH094609, NIH-NIEHS R01ES022223 and NIH-NIEHS R01ES022223-03S1.
Availability of data and materials
The datasets generated and analysed during the current study are available in the Sequence Read Archive [SRA] repository under accession number.
SRP095910.
Authors’ contributions
MD analyzed and interpreted the RNASeq data and was the primary author responsible for manuscript preparation. SP preprocessed the raw RNASeq data and provided an analysis pipeline to generate the placental gene network. KH oversaw the analytic pipeline of the RNASeq analysis. LL, CM and JC were instrumental in the design of the study and contributed to the interpretation and writing of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All subjects enrolled in the RICHS cohort provided written informed consent approved by the Institutional Review Boards at Women and Infants Hospital and Emory University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3878-0) contains supplementary material, which is available to authorized users.
Contributor Information
Maya A. Deyssenroth, Email: maya.kappil@mssm.edu
Shouneng Peng, Email: Shouneng.Peng@mssm.edu.
Ke Hao, Email: ke.hao@mssm.edu.
Luca Lambertini, Email: luca.lambertini@mssm.edu.
Carmen J. Marsit, Email: carmen.j.marsit@emory.edu
Jia Chen, Email: jia.chen@mssm.edu.
References
- 1.Barker DJ. The Wellcome Foundation Lecture, 1994. The fetal origins of adult disease. Proc Biol Sci. 1995 [cited 2015 Dec 9];262:37–43. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7479990. [DOI] [PubMed]
- 2.de Boo HA, Harding JE. The developmental origins of adult disease (Barker) hypothesis. Aust N Z J Obstet Gynaecol. 2006 [cited 2015 Nov 7];46:4–14. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16441686. [DOI] [PubMed]
- 3.Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science (80-. ). 2004 [cited 2015 Sep 29];305:1733–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15375258. [DOI] [PubMed]
- 4.Burton GJ, Jauniaux E. What is the placenta? Am J Obstet Gynecol. 2015;213(4 Suppl):S6.e1–S6.e4. doi: 10.1016/j.ajog.2015.07.050. [DOI] [PubMed] [Google Scholar]
- 5.Jansson T, Powell TL. Role of the placenta in fetal programming: underlying mechanisms and potential interventional approaches. Clin Sci (Lond). 2007 [cited 2016 Aug 16];113:1–13. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17536998. [DOI] [PubMed]
- 6.Burton GJ, Fowden AL, Thornburg KL. Placental origins of chronic disease. Physiol Rev. 2016;96(4):1509–1565. doi: 10.1152/physrev.00029.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandovici I, Hoelle K, Angiolini E, Constância M. Placental adaptations to the maternal-fetal environment: implications for fetal growth and developmental programming. Reprod Biomed Online. 2012 [cited 2015 Aug 19];25:68–89. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22560117. [DOI] [PubMed]
- 8.Sõber S, Reiman M, Kikas T, Rull K, Inno R, Vaas P, et al. Extensive shift in placental transcriptome profile in preeclampsia and placental origin of adverse pregnancy outcomes. Sci Rep. Nature Publishing Group; 2015 [cited 2016 Nov 25];5:13336. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26268791. [DOI] [PMC free article] [PubMed]
- 9.Saben J, Zhong Y, McKelvey S, Dajani NK, Andres A, Badger TM, et al. A comprehensive analysis of the human placenta transcriptome. Placenta. NIH Public Access; 2014 [cited 2016 Aug 9];35:125–31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24333048. [DOI] [PMC free article] [PubMed]
- 10.Kim J, Zhao K, Jiang P, Lu Z, Wang J, Murray JC, et al. Transcriptome landscape of the human placenta. BMC Genomics. BioMed Central; 2012 [cited 2016 Nov 25];13:115. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22448651. [DOI] [PMC free article] [PubMed]
- 11.Altmäe S, Segura MT, Esteban FJ, Bartel S, Brandi P, Irmler M, et al. Maternal pre-pregnancy obesity is associated with altered placental transcriptome. Torrens C, editor. PLoS One. 2017 [cited 2017 May 22];12:e0169223 Available from: http://dx.plos.org/10.1371/journal.pone.0169223 [DOI] [PMC free article] [PubMed]
- 12.Maschietto M, Tahira AC, Puga R, Lima L, Mariani D, Paulsen B da S, et al. Co-expression network of neural-differentiation genes shows specific pattern in schizophrenia. BMC Med Genomics. 2015 [cited 2016 Sep 16];8:23. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25981335. [DOI] [PMC free article] [PubMed]
- 13.Giulietti M, Occhipinti G, Principato G, Piva F. Weighted gene co-expression network analysis reveals key genes involved in pancreatic ductal adenocarcinoma development. Cell Oncol (Dordr). 2016 [cited 2016 Sep 16];39:379–88. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27240826. [DOI] [PubMed]
- 14.Huang J-Y, Tian Y, Wang H-J, Shen H, Wang H, Long S, et al. Functional genomic analyses identify pathways dysregulated in animal model of autism. CNS Neurosci Ther. 2016 [cited 2016 Sep 16]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/27321591. [DOI] [PMC free article] [PubMed]
- 15.Sundarrajan S, Arumugam M. Weighted gene co-expression based biomarker discovery for psoriasis detection. Gene. 2016 [cited 2016 Sep 16]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/27523473. [DOI] [PubMed]
- 16.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005 [cited 2014 Jul 18];4:Article17. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16646834. [DOI] [PubMed]
- 17.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008 [cited 2014 Jul 11];9:559. Available from: http://www.biomedcentral.com/1471-2105/9/559 [DOI] [PMC free article] [PubMed]
- 18.Buckberry S, Bianco-Miotto T, Bent SJ, Clifton V, Shoubridge C, Shankar K, et al. Placental transcriptome co-expression analysis reveals conserved regulatory programs across gestation. BMC Genomics. 2017 [cited 2017 Feb 7];18:10. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28049421. [DOI] [PMC free article] [PubMed]
- 19.Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005 [cited 2015 Jan 22];115:e290–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15741354. [DOI] [PubMed]
- 20.Van Lieshout RJ, Boyle MH. Canadian youth born large or small for gestational age and externalizing and internalizing problems. Can J Psychiatry. 2011 [cited 2015 Feb 4];56:227–34. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21507279. [DOI] [PubMed]
- 21.Moore GS, Kneitel AW, Walker CK, Gilbert WM, Xing G. Autism risk in small- and large-for-gestational-age infants. Am J Obstet Gynecol. 2012 [cited 2015 Feb 4];206:314.e1–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22464070. [DOI] [PMC free article] [PubMed]
- 22.Colman I, Ataullahjan A, Naicker K, Van Lieshout RJ. Birth weight, stress, and symptoms of depression in adolescence: evidence of fetal programming in a national Canadian cohort. Can J Psychiatry. 2012 [cited 2015 Feb 4];57:422–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22762297. [DOI] [PubMed]
- 23.Lundgren EM, Cnattingius S, Jonsson B, Tuvemo T. Intellectual and psychological performance in males born small for gestational age with and without catch-up growth. Pediatr Res. 2001 [cited 2017 Mar 22];50:91–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11420424. [DOI] [PubMed]
- 24.Grissom NM, Reyes TM. Gestational overgrowth and undergrowth affect neurodevelopment: similarities and differences from behavior to epigenetics. Int. J. Dev. Neurosci. [Internet]. 2013 [cited 2017 Mar 22];31:406–14. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0736574812005977 [DOI] [PMC free article] [PubMed]
- 25.Di Renzo GC, Rosati A, Sarti RD, Cruciani L, Cutuli AM. Does fetal sex affect pregnancy outcome? Gend Med. 2007;4:19–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17584623. [DOI] [PubMed]
- 26.Chiavaroli V, Castorani V, Guidone P, Derraik JGB, Liberati M, Chiarelli F, et al. Incidence of infants born small- and large-for-gestational-age in an Italian cohort over a 20-year period and associated risk factors. Ital J Pediatr. 2016 [cited 2016 Aug 9];42:42. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27117061. [DOI] [PMC free article] [PubMed]
- 27.Khalil A, Rezende J, Akolekar R, Syngelaki A, Nicolaides KH. Maternal racial origin and adverse pregnancy outcome: a cohort study. Ultrasound Obstet Gynecol. 2013 [cited 2016 Aug 9];41:278–85. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23023978. [DOI] [PubMed]
- 28.Männik J, Vaas P, Rull K, Teesalu P, Laan M. Differential placental expression profile of human growth hormone/chorionic somatomammotropin genes in pregnancies with pre-eclampsia and gestational diabetes mellitus. Mol Cell Endocrinol. 2012;355:180–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22387044. [DOI] [PMC free article] [PubMed]
- 29.Tornehave D, Chemnitz J, Teisner B, Folkersen J, Westergaard JG. Immunohistochemical demonstration of pregnancy-associated plasma protein A (PAPP-A) in the syncytiotrophoblast of the normal placenta at different gestational ages. Placenta. W.B. Saunders; 1984 [cited 2016 Aug 9];5:427–31. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0143400484800235 [DOI] [PubMed]
- 30.Wang J, Qiu Q, Haider M, Bell M, Gruslin A, Christians JK. Expression of pregnancy-associated plasma protein A2 during pregnancy in human and mouse. J Endocrinol. BioScientifica; 2009 [cited 2016 Aug 9];202:337–45. Available from: http://joe.endocrinology-journals.org/cgi/doi/10.1677/JOE-09-0136 [DOI] [PubMed]
- 31.Perez-Sepulveda A, Monteiro LJ, Dobierzewska A, España-Perrot PP, Venegas-Araneda P, Guzmán-Rojas AM, et al. Placental aromatase is deficient in placental ischemia and preeclampsia. PLoS One. Public Library of Science; 2015 [cited 2016 Aug 9];10:e0139682. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26444006. [DOI] [PMC free article] [PubMed]
- 32.Udagawa K, Miyagi Y, Hirahara F, Miyagi E, Nagashima Y, Minaguchi H, et al. Specific expression of PP5/TFPI2 mRNA by syncytiotrophoblasts in human placenta as revealed by in situ hybridization. Placenta [Internet]. [cited 2016 Aug 9];19:217–23. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9548189. [DOI] [PubMed]
- 33.Singh U, Sun T, Larsson T, Elliott RW, Kostka G, Fundele RH. Expression and functional analysis of Fibulin-1 (Fbln1) during normal and abnormal placental development of the mouse. Placenta. 2006;27:1014–1021. doi: 10.1016/j.placenta.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 34.He P, Shao D, Ye M, Zhang G. Analysis of gene expression identifies candidate markers and pathways in pre-eclampsia. J Obstet Gynaecol (Lahore). Taylor & Francis; 2015 [cited 2016 Aug 9];35:578–84 Available from: http://www.tandfonline.com/doi/full/10.3109/01443615.2014.990430 [DOI] [PubMed]
- 35.Zhang Y, Wang T, Huang H-Q, Li W, Cheng X-L, Yang J. Human MALAT-1 long non-coding RNA is overexpressed in cervical cancer metastasis and promotes cell proliferation, invasion and migration. J BUON. [cited 2016 Aug 9];20:1497–503. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26854446. [PubMed]
- 36.Esakova O, Krasilnikov AS. Of proteins and RNA: the RNase P/MRP family. RNA. Cold Spring Harbor Laboratory Press; 2010 [cited 2016 Aug 9];16:1725–47. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20627997. [DOI] [PMC free article] [PubMed]
- 37.Metsalu T, Viltrop T, Tiirats A, Rajashekar B, Reimann E, Kõks S, et al. Using RNA sequencing for identifying gene imprinting and random monoallelic expression in human placenta. Epigenetics. Landes Bioscience; 2014 [cited 2016 Aug 9];9:1397–409. Available from: http://www.tandfonline.com/doi/full/10.4161/15592294.2014.970052 [DOI] [PMC free article] [PubMed]
- 38.Monk D, Wagschal A, Arnaud P, Müller P-S, Parker-Katiraee L, Bourc’his D, et al. Comparative analysis of human chromosome 7q21 and mouse proximal chromosome 6 reveals a placental-specific imprinted gene, TFPI2/Tfpi2, which requires EHMT2 and EED for allelic-silencing. Genome Res. 2008 [cited 2016 Aug 9];18:1270–81. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18480470. [DOI] [PMC free article] [PubMed]
- 39.Kappil MA, Green BB, Armstrong DA, Sharp AJ, Lambertini L, Marsit CJ, et al. Placental Expression Profile of Imprinted Genes Impacts Birth Weight. Epigenetics [Internet]. 2015 [cited 2015 Jul 28]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/26186239. [DOI] [PMC free article] [PubMed]
- 40.Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007 [cited 2016 Dec 15];1:54. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18031580. [DOI] [PMC free article] [PubMed]
- 41.Liu W, Ye H. Co-expression network analysis identifies transcriptional modules in the mouse liver. Mol Genet Genomics. Springer Berlin Heidelberg; 2014 [cited 2016 Aug 18];289:847–53. Available from: http://link.springer.com/10.1007/s00438-014-0859-8 [DOI] [PubMed]
- 42.Liu X, Du Y, Trakooljul N, Brand B, Muráni E, Krischek C, et al. Muscle Transcriptional Profile Based on Muscle Fiber, Mitochondrial Respiratory Activity, and Metabolic Enzymes. Int J Biol Sci. 2015 [cited 2016 Aug 18];11:1348–62. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26681915. [DOI] [PMC free article] [PubMed]
- 43.van Eijk KR, de Jong S, Boks MPM, Langeveld T, Colas F, Veldink JH, et al. Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMC Genomics. BioMed Central; 2012 [cited 2016 Aug 18];13:636. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23157493. [DOI] [PMC free article] [PubMed]
- 44.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000 [cited 2017 mar 23];25:25–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10802651. [DOI] [PMC free article] [PubMed]
- 45.Malone J, Holloway E, Adamusiak T, Kapushesky M, Zheng J, Kolesnikov N, et al. Modeling sample variables with an experimental factor ontology. Bioinformatics. Oxford University Press; 2010 [cited 2017 Mar 23];26:1112–8 Available from: https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btq099 [DOI] [PMC free article] [PubMed]
- 46.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. Oxford University Press; 2014 [cited 2017 Feb 3];42:D1001–6 Available from: https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkt1229 [DOI] [PMC free article] [PubMed]
- 47.Tang M-X, Hu X-H, Liu Z-Z, Kwak-Kim J, Liao A-H. What are the roles of macrophages and monocytes in human pregnancy? J Reprod Immunol. 2015 [cited 2016 Dec 15];112:73–80 Available from: http://linkinghub.elsevier.com/retrieve/pii/S0165037815300085 [DOI] [PubMed]
- 48.Gleicher N. Does the immune system induce labor? Lessons from preterm deliveries in women with autoimmune diseases. Clin Rev Allergy Immunol. 2010 [cited 2016 Dec 15];39:194–206. Available from: http://link.springer.com/10.1007/s12016-009-8180-8 [DOI] [PubMed]
- 49.Bahr BL, Price MD, Merrill D, Mejia C, Call L, Bearss D, et al. Different expression of placental pyruvate kinase in normal, preeclamptic and intrauterine growth restriction pregnancies. Placenta. 2014 [cited 2016 Dec 2];35:883–90. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25260566. [DOI] [PubMed]
- 50.McMinn J, Wei M, Schupf N, Cusmai J, Johnson EB, Smith AC, et al. Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta. [cited 2016 Sep 13];27:540–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16125225. [DOI] [PubMed]
- 51.Madeleneau D, Buffat C, Mondon F, Grimault H, Rigourd V, Tsatsaris V, et al. Transcriptomic analysis of human placenta in intrauterine growth restriction. Pediatr Res. 2015 [cited 2016 Dec 2];77:799–807. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25734244. [DOI] [PubMed]
- 52.Kaartokallio T, Cervera A, Kyllönen A, Laivuori K, FINNPEC Core Investigator Group. Gene expression profiling of pre-eclamptic placentae by RNA sequencing. Sci Rep. 2015 [cited 2016 sep 12];5:14107. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26388242. [DOI] [PMC free article] [PubMed]
- 53.Watts DJ, Strogatz SH. Collective dynamics of “small-world” networks. Nature. 1998 [cited 2016 Sep 27];393:440–2. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9623998. [DOI] [PubMed]
- 54.Gaiteri C, Ding Y, French B, Tseng GC, Sibille E. Beyond modules and hubs: the potential of gene coexpression networks for investigating molecular mechanisms of complex brain disorders. Genes Brain Behav. Blackwell Publishing Ltd; 2014 [cited 2016 Sep 27];13:13–24. Available from: http://doi.wiley.com/10.1111/gbb.12106. [DOI] [PMC free article] [PubMed]
- 55.Wyatt SM, Kraus FT, Roh C-R, Elchalal U, Nelson DM, Sadovsky Y. The correlation between sampling site and gene expression in the term human placenta. Placenta. 2005 [cited 2016 Dec 1];26:372–9. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0143400404001869. [DOI] [PubMed]
- 56.Paquette AG, Lester BM, Koestler DC, Lesseur C, Armstrong DA, Marsit CJ. Placental FKBP5 genetic and epigenetic variation is associated with infant neurobehavioral outcomes in the RICHS cohort. Potash JB, editor. PLoS One [Internet]. 2014 [cited 2015 Sep 23];9:e104913. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4130612&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed]
- 57.Huang R, Jaritz M, Guenzl P, Vlatkovic I, Sommer A, Tamir IM, et al. An RNA-Seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs. PLoS One. 2011 [cited 2015 Jun 29];6:e27288 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3213133&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed]
- 58.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. Macmillan Publishers Limited. All rights reserved; 2008 [cited 2014 Jul 9];456:53–9 Available from: 10.1038/nature07517 [DOI] [PMC free article] [PubMed]
- 59.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013 [cited 2014 Jul 13];29:15–21 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3530905&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed]
- 60.Risso D, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-Seq data. BMC Bioinformatics. 2011 [cited 2015 Apr 30];12:480. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22177264. [DOI] [PMC free article] [PubMed]
- 61.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010 [cited 2016 Jul 25];26:139–40. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19910308. [DOI] [PMC free article] [PubMed]
- 62.Smyth GK. LIMMA: linear models for microarray data. In: Gentleman R, Carey V, Dudoit R, Irizarry W, Huber W, editors. Bioinforma. Comput. Biol. Solut. Using {R} Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 63.Li J, Bushel PR, Chu T-M, Wolfinger RD. Principal variance components analysis: estimating batch effects in microarray gene expression data. Batch Eff. Noise Microarray Exp. Chichester, UK: John Wiley & Sons, Ltd; [cited 2016 Aug 17]. p. 141–54. Available from: http://doi.wiley.com/10.1002/9780470685983.ch12
- 64.Bushel P. pvca: Principal Variance Component Analysis (PVCA) 2013. [Google Scholar]
- 65.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007 [cited 2014 Jul 10];8:118–27. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16632515. [DOI] [PubMed]
- 66.Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics [Internet]. Oxford University Press; 2008 [cited 2017 May 24];24:719–20 Available from: https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btm563 [DOI] [PubMed]
- 67.Burdett T, Hall P, Hastings E, Hindorff L, Junkins H, Klemm A, et al. The NHGRI-EBI Catalog of published genome-wide association studies [Internet]. Available from: www.ebi.ac.uk/gwas. [DOI] [PMC free article] [PubMed]
- 68.Venables WN, Ripley BD. Modern Applied Statistics with S. Fourth Edition. New York: Springer; 2002. Available from: http://www.stats.ox.ac.uk/pub/MASS4.
- 69.Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011 [cited 2016 Jul 20];7:e1001057. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21283776. [DOI] [PMC free article] [PubMed]
- 70.Wickham H. ggplot2: elegant graphics for data analysis [Internet]. New York: Springer; 2009. Available from: http://www.springer.com/br/book/9780387981413.
- 71.Hu Z. Using VisANT to analyze networks. Curr Protoc Bioinformatics. 2014 [cited 2016 Oct 10];45:8.8.1–39. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25422679. [DOI] [PMC free article] [PubMed]
- 72.Hu Z, Mellor J, Wu J, DeLisi C. VisANT: an online visualization and analysis tool for biological interaction data. BMC Bioinformatics. 2004 [cited 2016 Oct 10];5:17. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15028117. [DOI] [PMC free article] [PubMed]
- 73.R Core Team. R: A language and environment for statistical computing [Internet]. Vienna, Austria: R Foundation for Statistical Computing; 2015. Available from: http://www.r-project.org/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distribution in expression level (logCPM) among top expressed genes in placenta. Shown in grey is the distribution in expression level across all genes (n = 12,135). (TIFF 1487 kb)
Agreement in gene expression rankings between RNAseq and NanoString (n = 197). Gene expression across 80 genes was assessed by both the RNAseq and NanoString platforms. Genes were ranked by expression level (lowest rank signifies highest expression across samples). Gene ranks across the platforms were significantly correlated (Spearman rho = 0.76, p < 0.01). (TIFF 1666 kb)
Gene loadings in placental gene coexpression network. Genes loading into separate modules are listed. Additionally the correlation between genes and module eigengenes are indicated. (CSV 2815 kb)
Hierarchical clustering of network modules. (TIFF 1474 kb)
Mapping of GWAS-linked genes in placental gene coexpression network. Genes linked to GWAS-associated traits that are enriched in the placental gene coexpression network are listed alongside assigned modules and GWAS-linked traits. (CSV 58 kb)
Candidate module hub genes relevant to birth weight. Plots indicate the correlation between gene expression and module eigengene values (x-axis) and the correlation between gene expression and birth weight category (y-axis). Genes that demonstrate both module importance and birth weight relevance are indicated for each module. Genes of interest include MKL2, PSMD4, ADRM1, AC3H15 and CREB3 in the turquoise module, LAD1, KAT5, DNAJC14 and BECN1 in the red module, GRHL1, INHBA, PVRL4, LEP and C8orf58 in the salmon module, DDX3X in the lightcyan module, and ZNF460, C21orf91 and PAN3 in the greenyellow module. (TIFF 169 kb)
Data Availability Statement
The datasets generated and analysed during the current study are available in the Sequence Read Archive [SRA] repository under accession number.
SRP095910.