

Abstract
Cardiac phospholipids, notably cardiolipin, undergo acyl chain remodeling and/or loss of content in aging and cardiovascular diseases, which is postulated to mechanistically impair mitochondrial function. Less is known about how diet-induced obesity influences cardiac phospholipid acyl chain composition and thus mitochondrial responses. Here we first tested if a high fat diet remodeled murine cardiac mitochondrial phospholipid acyl chain composition and consequently disrupted membrane packing, supercomplex formation and respiratory enzyme activity. Mass spectrometry analyses revealed that mice consuming a high fat diet displayed 0.8–3.3 fold changes in cardiac acyl chain remodeling of cardiolipin, phosphatidylcholine, and phosphatidylethanolamine. Biophysical analysis of monolayers constructed from mitochondrial phospholipids of obese mice showed an impairment in the packing properties of the membrane compared to lean mice. However, the high fat diet, relative to the lean controls, had no influence on cardiac mitochondrial supercomplex formation, respiratory enzyme activity, and even respiration. To determine if the effects were tissue specific, we subsequently conducted select studies with liver tissue. Compared to the control diet, the high fat diet remodeled liver mitochondrial phospholipid acyl chain composition by 0.6–5.3 fold with notable increases in n-6 and n-3 polyunsaturation. The remodeling in the liver was accompanied by diminished complex I to III respiratory enzyme activity by 3.5 fold. Finally, qRT-PCR analyses demonstrated an upregulation of liver mRNA levels of tafazzin, which contributes to cardiolipin remodeling. Altogether, these results demonstrate that diet-induced obesity remodels acyl chains in the mitochondrial phospholipidome and exerts tissue specific impairments of respiratory enzyme activity.
Keywords: Cardiolipin, High Fat Diet, Membranes, Mitochondria, Obesity, Polyunsaturated Fatty Acids
1. Introduction
Mitochondria have a central role in a multitude of cellular functions including energy production and apoptosis [1]. Impairments in mitochondrial function, driven through a range of mechanisms, contribute to the etiology of several diseases. One potential set of mechanisms that disrupt mitochondrial respiratory function is through the loss of cardiolipin (CL) content and aberrant remodeling of CL acyl chains in the inner mitochondrial membrane. Specifically, the loss of CL concentration and atypical CL acyl chain composition are associated with impaired enzymatic activity and disrupted respiratory supercomplex assembly [2–4]. For example, in heart failure, mitochondria are unable to produce enough energy to maintain heart homeostasis, which can be partially attributed to the loss of the most abundant CL species, (18:2)4CL [2,5,6]. In turn, the loss of (18:2)4CL is postulated to disrupt supercomplex assembly and this may lead to an increase in reactive oxygen species, which further damages the mitochondria [7,8]. Similarly, in aging, there is a decrease in the amount of (18:2)4CL and an increase in highly polyunsaturated acyl chains (PUFA) such as 20:4 and 22:6 [9], which may contribute to the reduction in supercomplex assembly [10].
Phospholipid acyl chains, particularly those of CL, are important since they can bind a multitude of inner mitochondrial membrane proteins and influence bioenergetics [11]. CL binds to the oxidative phosphorylation machinery including complexes I, III, IV, V, and the mobile electron carrier cytochrome c [12–16]. CL also mediates the formation of respiratory supercomplexes and has been proposed to be the “glue” that holds them together [17,18]. In addition, it is hypothesized that CL acyl chains create distinct microdomains that allow for optimal protein diffusion and clustering [19–21]. There is also some suggestion that CL microdomains serve as platforms for pro-apoptotic signals [20].
CL’s four acyl chains and small headgroup regulate membrane biophysical organization by promoting formation of highly curved membranes, which influences cristae formation [22,23]. CL acyl chain composition, in addition to its content, is also relevant for regulating the packing properties of phospholipids and potentially influencing protein activity [24,25]. Phospholipids such as phosphatidylethanolamine (PE) and phosphatidylcholine (PC) also have an influential role in mitochondrial function, although they are significantly less studied compared to CL. Several studies demonstrate that PE and PC acyl chains are remodeled in diseases such as type 2 diabetes and Barth Syndrome [26,27]. For instance, in human models of Barth syndrome, PC and PE acyl chains are remodeled to fatty acids such as 16:0, 16:1, 18:1, and 20:1 which likely contributes to impaired mitochondrial function [28]. Ischemia-reperfusion injury is also associated with a loss in total PC and PE concentration [29].
The influence of diet-induced obesity on phospholipid acyl chain composition and subsequent mitochondrial function is poorly studied. Obesity contributes to cardiovascular complications by increasing dyslipidemia, hypertension, and glucose dysregulation [30]. These factors and others link obesity to an increased risk of cardiovascular diseases [31–33]. Therefore, the primary objective of this study was to determine if administration of a high fat diet to mice remodeled the cardiac acyl chain profile of key mitochondrial phospholipids and if acyl chain remodeling was associated with impaired membrane packing, formation of respiratory supercomplexes, respiratory enzyme activity, and respiration. We then compared results from cardiac tissue with the liver to determine if the results were tissue specific.
2. Materials and Methods
2.1 Animals and Diets
All experiments were conducted in accordance with guidelines established by the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996) and with prior approval by East Carolina University’s Animal Care and Use Committee. Male C57BL/6 mice (Charles River, Wilmington, MA) were fed a control diet with 10 kcal% lard or a high fat diet with 60 kcal% lard for 20 weeks (Research Diets Inc., New Brunswick, NJ) (Supp. Table S1). The fatty acid composition of the control and high fat diet is provided in Supp. Table S2. Mice were housed on a 12:12 h light-dark cycle with free access to water. Mice were sacrificed via isoflurane inhalation followed by cervical dislocation.
2.2 Isolation of Mitochondria
Mitochondrial isolations were performed on ice and all instruments and buffers were chilled to 4°C before isolation using our established protocols [25]. Hearts and livers were removed and rinsed in mitochondrial isolation medium (MIM) containing 300 mM sucrose, 10 mM Na-HEPES (pH=7.2) and 1 mM EGTA (Sigma-Aldrich, St. Louis, MO). The tissue was minced for 5–6 minutes and diluted in MIM+BSA (1mg/ml BSA) (pH=7.4). Tissue was then subjected to homogenization with a Teflon Potter homogenizer. The homogenate was centrifuged at 800 × g for 10 minutes and the supernatant was centrifuged at 12,000 × g for 15 minutes. The mitochondrial pellet was resuspended in MIM and used immediately for respiration studies or stored at −80°C. Protein content was determined using a BCA protein quantification assay (Thermo Fisher Scientific, Waltham, MA).
2.3 Electrospray Ionization Mass Spectrometry
Phospholipid molecular species were determined in lipid extracts (0.2 mg) of mitochondrial protein by liquid chromatography with electrospray ionization mass spectrometry (LC/MS) and analyzed as previously described [34].
2.4 Lipid Extractions and Phosphorous Quantification Assay
Total lipids were extracted from mitochondria as previously shown [2,25]. Total phosphate of lipids extracted from mitochondrial samples was obtained from UV-Vis spectrum analysis. From the lipid extraction, 1.0 mg of resuspended lipids was dried under nitrogen along with phosphate standards (0.0 μmol – 0.650 μmol). Sulfuric acid (Sigma-Aldrich, St. Louis, MO) was added to all samples and heated to 200°C – 215°C for 25 minutes. Samples were then cooled to room temperature. Hydrogen peroxide (Sigma-Aldrich, St. Louis, MO) was subsequently added to all samples and heated to 200°C – 215°C for 30 minutes. Samples were then cooled to room temperature. Deionized water, ammonium molybdate (VI) tetrahydrate solution, and ascorbic acid solution (Sigma-Aldrich, St. Louis, MO) were added and vortexed. A glass marble was placed on each tube to prevent evaporation of samples while they were heated to 100°C for 7 minutes. Samples and standards were cooled to room temperature a final time. Absorbance (820 nm) of each sample was determined via a UV-Vis Spectrophotometer (Shimadzu, Kyoto, Japan). From the UV-Vis spectrum data, a calibration curve was generated to determine total phosphate in all samples.
2.5 Thin-Layer Chromatography (TLC)
TLC was performed as previously described [25]. Briefly, lipid extracts were spotted on a Whatman (10×10 cm, silica gel) TLC plate 2 cm from the bottom. The plate was developed in a glass chamber until the mobile phase consisting of chloroform, methanol, glacial acetic acid and water (100:30:1:4) (v/v/v/v) (Sigma-Aldrich, St. Louis, MO) reached 1 cm from the top of the plate. The plate was air dried, sprayed with a charring solution (4% phosphoric acid and 5% copper sulfate, Sigma-Aldrich, St. Louis, MO) and then placed in an oven at 190°C for 15 minutes. The charred lipid spots were quantified using an Odyssey Infrared Imager (LI-COR Biosciences, Lincoln, NE).
2.6 Generation of Mitochondrial Monolayers for Packing and Elasticity Measurements
Surface pressure-area isotherms (π − A) were obtained on a Mini LB Trough (KSV NIMA, Biolin Scientific, Stockholm, Sweden) using a Wilhelmy plate at a compression rate of 3.0 mm/min [35]. The trough was washed three times with 70% ethanol, Milli-Q water, and subphase (10 mM sodium phosphate buffer, pH 7.4) (Sigma-Aldrich, St. Louis, MO) before each run. Trough barriers were compressed and expanded before spotting lipid, until the surface pressure remained constant (<0.3 mN/m). Extracted mitochondrial lipids (1.09 nmol) were spread onto the subphase, ensuring the surface pressure did not exceed 0.3 mN/m. Compression began 10 minutes after lipid spotting to ensure the evaporation of chloroform. Surface pressure (π) values were measured by the Wilhelmy method and all samples were run in triplicate. The surface pressure-area isotherms were used to calculate the mean molecular area and the surface elasticity modulus at 30mN/m:
where A is the mean molecular area of the lipid mixture at the indicated surface pressure (π). Lower values indicate higher elasticity modulus [36].
2.7 Blue-Native PAGE for Assessing Supercomplex Formation
Mitochondria were centrifuged at 16,873 × g for 10 minutes at 4°C. Pelleted mitochondria were resuspended in Native PAGE Sample Buffer (Life Technologies, Carlsbad, CA). Mitochondria were then solubilized using an 8:1 digitonin (Sigma-Aldrich, St. Louis, MO) to protein ratio for 15 minutes on ice. After solubilization, samples were centrifuged at 16,873 × g for 30 min at 4°C. The supernatants were collected and protein content was determined via a BCA protein quantification assay (Thermo Fisher Scientific, Waltham, MA). Samples were combined with 5% G-250 sample additive (Life Technologies, Carlsbad, CA) and were loaded onto the 3–12% Bis-Tris gel (Life Technologies, Carlsbad, CA). The gel was run on ice at 150 V for 3 hours and had molecular weight markers (NativeMark Unstained Protein Standard, Life Technologies, Carlsbad, CA). Gels were fixed using a solution of 40% methanol and 10% acetic acid (Sigma-Aldrich, St. Louis, MO) and destained in 8% acetic acid overnight or until desired background was reached. Gels were imaged and quantified using the Odyssey Infrared Imager (LI-COR Biosciences, Lincoln, NE). Data were normalized to the total amount of protein in each sample.
2.8 Kinetic Assays
Kinetic assays were done using a UV-Vis Spectrophotometer (Shimadzu, Kyoto, Japan) at 37°C as previously demonstrated [25]. Briefly, complex I activity was measured by the oxidation of 0.8 mM NADH (Sigma-Aldrich, St. Louis, MO) at 340 nm, complex II activity was measured by the reduction of 80 μM dichlorophenolindophenol (DCPIP) (Sigma-Aldrich, St. Louis, MO) at 600 nm, complex III activity was measured by the reduction of 40 μM cytochrome c (Sigma-Aldrich, St. Louis, MO) at 550 nm and complex IV activity was measured by the oxidation of 10 μM reduced cytochrome c (Sigma-Aldrich, St. Louis, MO) at 550 nm. Activity of complexes I+III was measured by the reduction of 40 μM cytochrome c at 550 nm. For complexes II+III, activity was measured by the reduction of 40 μM cytochrome c and measured at 550 nm. Citrate Synthase activity was assayed using 0.1 mM 5,5-dithio-bis-(2-nitobenzoic acid) (DTNB) (Sigma-Aldrich, St. Louis, MO) at 412 nm. Each reaction was performed in triplicate and normalized to citrate synthase activity.
2.9 Mitochondrial Respiration
Mitochondrial respiration was assayed using an Oroboros 2k oxygraph (High-resolution respirometer, Oroboros Instruments, Innsbruck, Austria) using our established techniques [37]. Oxygen consumption was measured using 50–150 μg of freshly isolated mitochondria. Mitochondria were incubated in Buffer Z (105 mM K-MES, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl·6H2O, 1mM EGTA, 0.5 mg/ml BSA, pH=7.1) (Sigma-Aldrich, St. Louis, MO) in the oxygraphic chamber under continuous stirring at 37°C. State 3 (4mM ADP) and state 4 (no ADP) respiration were measured utilizing various substrates as electron donors (10mM/2mM glutamate/malate, 10 mM succinate, 5mM/2mM pyruvate/malate, and 0.018mM/0.02mM/5mM/2mM palmitoylcarnitine/palmitoylCoA/carnitine/malate) (Sigma-Aldrich, St. Louis, MO). State 3 respiration was measured with the addition of 4 mM ADP to the chamber. Oxygen consumption was normalized to protein content. Mitochondrial integrity was ensured by the addition of cytochrome c (10 μM) during state 3 respiration. Mitochondria were considered damaged if there was >10% increase in respiration with the addition of cytochrome c and were not used for functional assays [38].
2.10 Quantitative RT-PCR
Total RNA was isolated from cardiac and liver tissue using the RNeasy Plus Universal Mini Kit (Qiagen, Valencia, CA). RNA was subjected to reverse transcription and qPCR in a one-step reaction using iScript One-Step RT-PCR kit with Syber Green reagents (Biorad, Hercules, CA). Gene products were amplified and detected using a ViiA 7 Real-Time PCR System (Thermo Fisher, Waltham, MA). mRNA quantities were calculated using the 2-ΔΔCt method and Ct values were normalized to GAPDH and β-actin. The following primers were used:
GAPDH 5′-GGTGTGAACGGATTTGGCCGTATT-3′ and 5′-GTCGTTGATGGCAACAATCTCCAC-3′, β-actin 5′-GCAGCCACTGTCGAGTC’3′ and 5′-GCAGCGATATCGTCATCCAT-3′, ALCAT 5′-CCTCAGGGTGGAGAAGATTTG-3′ and 5′-GGCTCTTATCATCCTTCCACTT-3′, TAZ 5′-GGCTGATTGCTGAGTGTCAT-3′ and 5′-GTACTGAAGGGCTTCCCAATC-3′.
2.11 Statistical Analyses
Data are presented as average ± S.E.M. All data are from multiple independent experiments, as indicated in the figure legends. The data were normally distributed and were analyzed using GraphPad Prism 6 software. Statistical significance was established using a student’s t-test. P< 0.05 was considered significant.
3. Results
3.1 Murine cardiac mitochondrial phospholipids are remodeled in response to a high fat diet
We first determined if mice consuming a high fat diet displayed aberrant remodeling of CL acyl chains as previously reported in diabetic and aged animals [9,39,40]. Therefore, mice were administered a control or high fat diet (Supp. Table S1), which resulted in the lean mice having a body weight of 31.8 ± 1.5 grams compared to obese mice with a body weight of 38.6 ± 2.8 grams (p<0.0001). To assay acyl chain composition, LC/MS was performed on mitochondrial lipid extracts. An example of the raw LC/MS data from mice consuming a lean or a high fat diet are presented in Supp. Fig. 1A–B.
LC/MS analyses revealed that cardiac CL acyl chains were remodeled with the high fat diet compared to the lean control. The CL species (18:2)3(16:1), (18:2)3(16:0), (18:2)3(18:1), (18:2)3(20:4), (16:1)(18:1)(18:2)(22:6), (16:0)(18:2)2(22:6), (16:1)(18:2)(20:3)(20:4), and (16:0)(18:2)(20:4)2 were significantly lowered with the high fat diet relative to the lean controls by 0.76 to 3.32 fold (Fig. 1A). There was also an increase in the CL species (18:2)2(18:1)(20:4) in obese mice compared to lean controls by 0.75 fold (Fig. 1A). There were no alterations in the highly abundant (18:2)4CL. With cardiac PC, (18:2)(18:2) and (18:0)(20:5) increased in the obese mice compared to the lean controls by 0.87 and 1.40 fold respectively (Fig. 1B). In addition, (16:0)(18:1)PC was decreased in the high fat diet relative to the lean controls by 0.73 fold (Fig. 1B). With cardiac PE, (16:0)(18:1) was reduced in the obese animals compared to the lean controls by 2.0 fold (Fig 1C). In addition, (18:2)(20:4)PE was elevated in obese mice compared to lean controls by 0.93 fold (Fig. 1C). TLC analysis revealed the total amount of each phospholipid remained unchanged between lean controls and the high fat fed animals (Fig. 1D–E).
Figure 1. Cardiac mitochondrial phospholipid acyl chains are remodeled in response to a high fat diet.
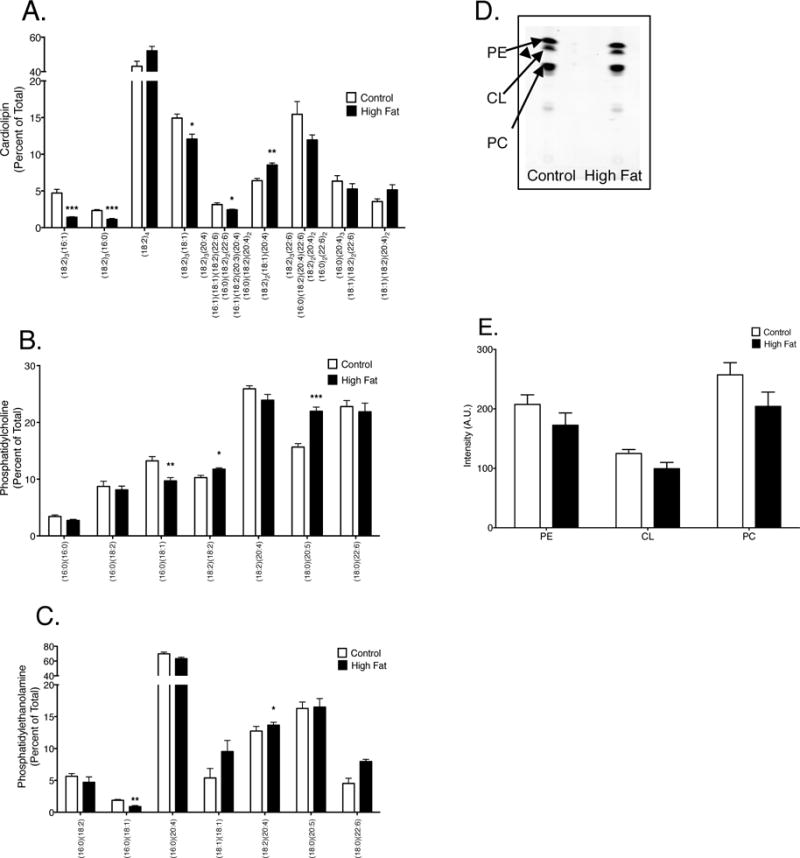
Acyl chain composition of (A) CL, (B) PC, and (B) PE in cardiac mitochondria. (D) Representative TLC plate of phospholipids assayed from cardiac mitochondria of lean and obese mice. (E) Quantification of total CL, PC, and PE in lean controls versus obese animals. Data are the average ± S.E.M. from 4 independent experiments. The asterisks indicate significance from control (*p < 0.05, ** p < 0.01, ***p < 0.001).
3.2 Cardiac mitochondrial monolayer packing is impaired with mice consuming a high fat diet
We next determined if the structural organization of mitochondrial phospholipids was altered in response to remodeling of CL, PC, and PE acyl chains. Monolayers were constructed from extracted mitochondrial phospholipids. Pressure-area isotherm analysis of monolayers (Fig. 2A) provided quantitative values for area per molecule (a measure of lipid packing) and the elasticity modulus of the monolayers. Data were quantified at the physiological surface pressure of 30mN/m [41,42]. There was a 10% increase in the area per molecule of monolayers composed of phospholipids isolated from high fat fed animals compared to those constructed from lean animals (Fig. 2B). This increase suggested that each phospholipid was occupying a larger area of the monolayer and was therefore less packed. The high fat diet had no influence on the elasticity modulus of the monolayer (Fig. 2C).
Figure 2. Mitochondrial monolayer packing is impaired in response to a high fat diet.
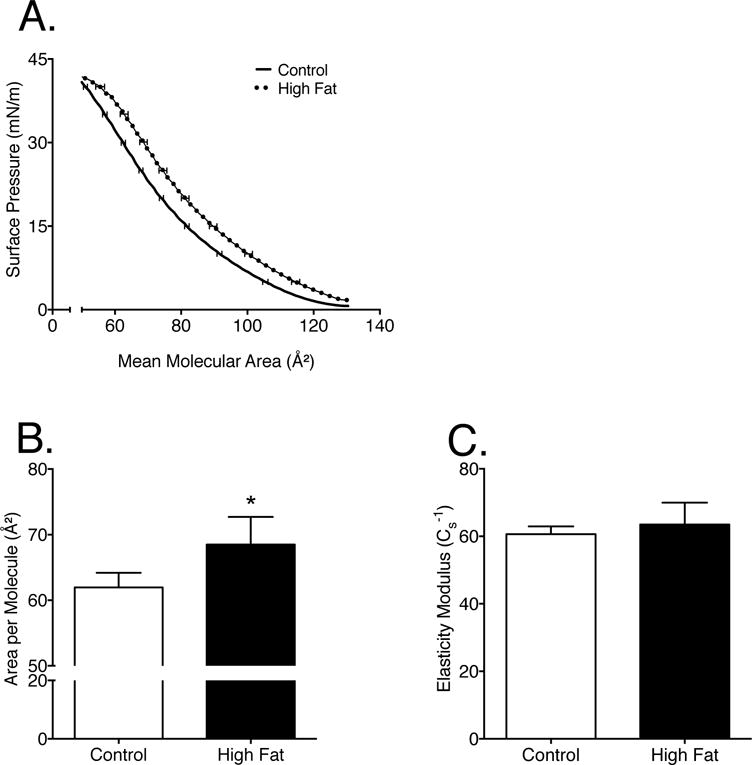
(A) Sample pressure-area isotherms constructed from extracted mitochondrial lipids from mice consuming a control or high fat diet. (B) Area per molecule, and (C) elasticity modulus were calculated from the pressure-area isotherms as described in the methods. Data are the average ± S.E.M. from 4 independent experiments. The asterisk indicates significance from control (*p < 0.05).
3.3 Cardiac mitochondrial supercomplex formation and oxidative phosphorylation enzyme activities are maintained in mice consuming a high fat diet
We next tested if the aforementioned changes were mechanistically associated with impaired functionality of the mitochondrial membrane. BN-PAGE analysis revealed that the extent of mitochondrial respiratory complex or supercomplex formation in high fat fed animals was unchanged when compared to lean controls (Fig. 3A–B). Analysis of the specific activities for single oxidative phosphorylation enzymes (Fig. 3C) and the combined activities of CI to CIII and CII to CIII (Fig. 3D) showed no differences between high fat fed animals and lean controls. We further measured state 3 and state 4 respiration with a variety of substrates. Mice consuming the control and high fat diets responded similarly to each substrate, indicating no differences in respiration (Supp. Fig. 2).
Figure 3. Mice consuming a high fat diet have appropriately assembled cardiac mitochondrial supercomplexes and no alterations in specific respiratory enzyme activities.
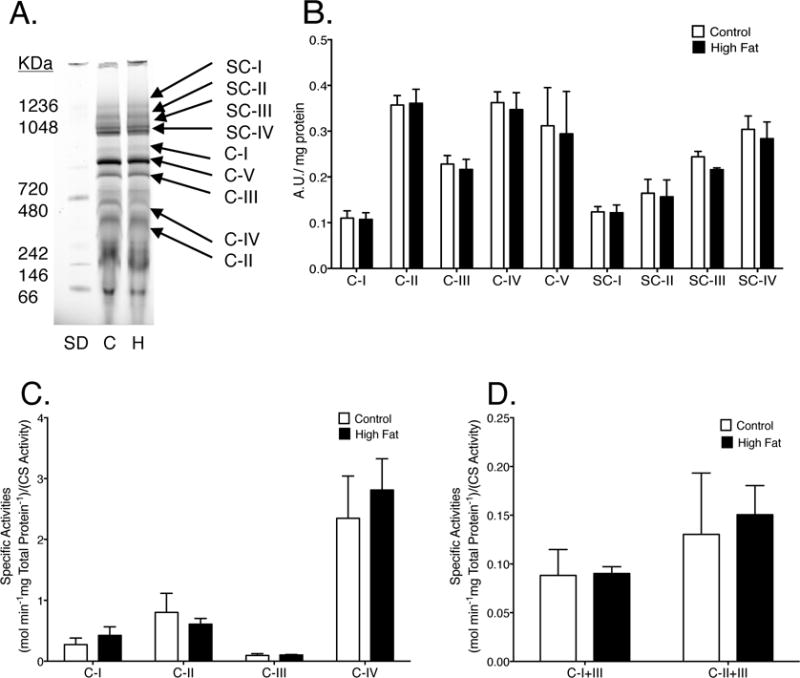
(A) Representative BN-PAGE gel of complexes (C-I-V) and supercomplexes (SC-I-IV) from lean and obese mice. “SD” is the molecular weight marks, “C” is for lean control, and “H” is for high fat diet. (B) Quantification of BN-PAGE. (C) Specific enzymatic activities of individual complexes and (D) NADH oxidation or succinate oxidation coupled to cytochrome c reduction (CI or CII to CIII). Activities were determined relative to total protein content and then normalized to citrate synthase (CS) activity. Data are the average ± S.E.M. from 4 independent experiments.
3.4 Murine liver mitochondrial phospholipids are remodeled in response to a high fat diet
Given that we observed no major change in cardiac mitochondrial enzyme activity, we next determined if the effects were tissue specific. We focused on the liver since it is a major metabolic tissue and there is precedence in the literature to suggest that obesity impairs mitochondrial function although underlying mechanisms are not completely established [43–46]. LC/MS analyses revealed that liver CL acyl chains were remodeled with the high fat diet compared to the lean control. The CL species (18:2)3(16:1), (18:2)3(16:0), (18:2)2(18:1)(20:4), (18:2)3(22:6), (16:0)(18:2)(20:4)(22:6), (18:2)2(20:4)2, and (16:0)2(22:6)2 were all significantly reduced by 0.73–5.25 fold in the high fat fed animals compared to the lean controls (Fig. 4A). There was also an increase in the (18:2)4CL and (18:2)3(20:2)CL by 1.6 and 0.7 fold respectively in the obese mice compared to lean controls (Fig. 4A). The liver PC species (16:0)(16:0), (16:0)(18:1), and (18:2)(18:2) were decreased by 0.6, 2.02, and 0.74 fold respectively in the high fat diet animals compared to the lean controls (Fig. 4B). In contrast, liver (18:0)(20:5)PC and (18:0)(22:6)PC were significantly increased in obese animals compared to lean controls by 0.8 and 0.72 fold (Fig. 4B). With PE, (16:0)(18:1) was reduced in the obese animals compared to the lean controls by 2.10 fold and liver (18:0)(22:6)PE was elevated in obese mice compared to lean controls by 1.76 fold (Fig. 2C). Finally, TLC analysis revealed the total amount of each phospholipid remained unchanged between lean controls and the high fat fed animals (Fig. 4D–E). Although there were significant alterations in the phospholipid profile of liver mitochondria, there were no changes in membrane packing (data not shown).
Figure 4. Liver mitochondrial phospholipid acyl chains are remodeled in response to a high fat diet.
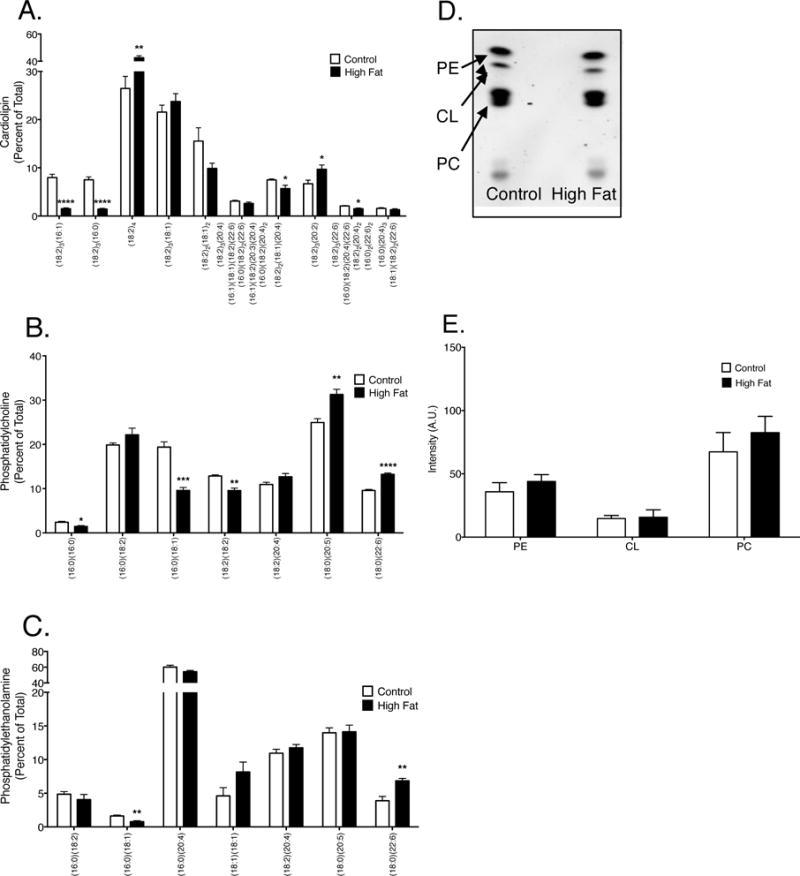
Acyl chain composition of (A) CL, (B) PC, and (B) PE in liver mitochondria. (D) Representative TLC plate of phospholipids assayed from liver mitochondria. (E) Quantification of total CL, PC, and PE in lean controls versus obese animals. Data are the average ± S.E.M. from 4 independent experiments. The asterisks indicate significance from control (*p < 0.05, ** p < 0.01, ***p < 0.001, ****p < 0.0001).
3.5 Complex I to III activity in mitochondria isolated from the liver is impaired with mice consuming a high fat diet
Supercomplex formation and oxidative phosphorylation enzymatic activities were measured in liver mitochondria isolated from mice consuming a high fat or control diet. There was no apparent impairment in supercomplex formation in the liver (Fig. 5A–B). In addition, individual complex enzymatic activities were unchanged between mice consuming a high fat diet compared to control mice (Fig. 5C). However, there was a 3.5 fold reduction in the rate of NADH oxidation coupled to cytochrome c reduction (complex I to III activity) in obese mice compared with lean controls (Fig. 5D).
Figure 5. Mice consuming a high fat diet have impaired liver complex I to III enzyme activity.
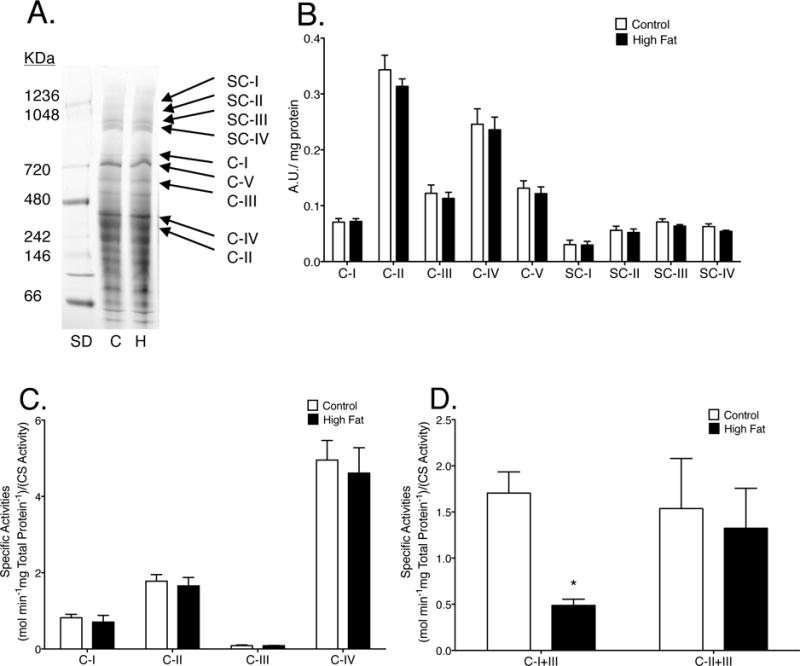
(A) Representative BN-PAGE gel, complexes (C-I-V) and supercomplexes (SC) from mice consuming a lean or high fat diet. “SD” is the molecular weight markers, “C” is lean control, and “H” is high fat diet. (B) Quantification of BN-PAGE. (C) Specific enzymatic activities of individual complexes and (D) NADH oxidation or succinate oxidation coupled to cytochrome c reduction (CI or CII to CIII). Activities were determined relative to total protein content and then normalized to citrate synthase (CS) activity. Data are the averages ± S.E.M. from 4 independent experiments. The asterisk indicates significance from control (*p < 0.05).
3.6 Tafazzin mRNA levels in the liver are increased with mice consuming a high fat diet
Acyltransferases catalyze CL acyl chain remodeling in the mitochondria [47]. Therefore, we determined if acyltransferases were upregulated given that PE, PC, and particularly CL had aberrantly remodeled acyl chains with the high fat in the heart and the liver. We specifically assayed for lysocardiolipin acyltransferase (ALCAT1), which remodels CL to more highly oxidizable acyl chains and is upregulated in select murine obesity models [48]. In addition, we determined if there were any differences in tafazzin (TAZ), a well-known acyltansferase [49]. qRT-PCR analysis showed no increase in ALCAT1 or TAZ mRNA levels in high fat fed animals compared to lean controls in cardiac tissue (Fig. 6, left panel). In liver, ALCAT1 mRNA levels were similar between control and high fat fed mice (Fig. 6, right panel). However, TAZ mRNA levels in the liver increased by 2.4 fold in obese mice compared with lean controls (Fig. 6, right panel).
Figure 6. Mice consuming a high fat diet have increased liver tafazzin mRNA levels.
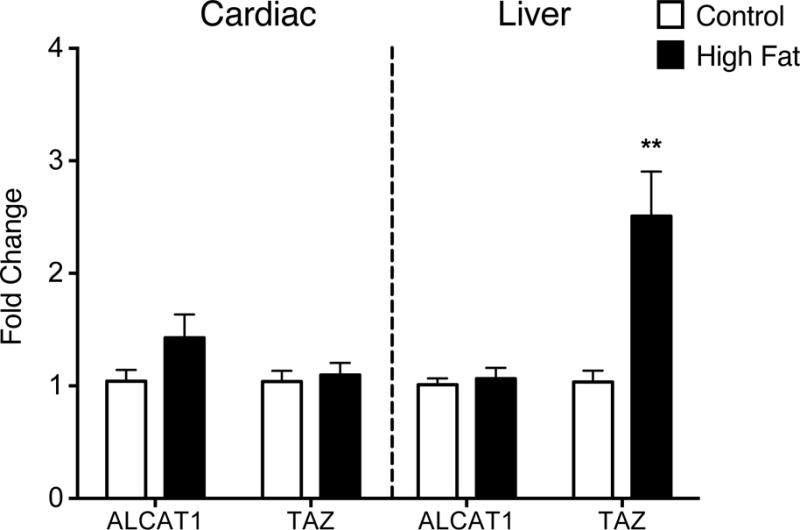
ALCAT1 and TAZ mRNA levels were measured in cardiac and liver tissues isolated from lean and obese mice using qRT-PCR. The fold change is relative to GAPDH and β-actin. Data are the average ± S.E.M. from 8 independent experiments. The asterisk indicates significance from control (*p < 0.05).
4. Discussion
The objectives of this study were to determine if diet-induced obesity remodeled myocardial mitochondrial phospholipid acyl chains and if the remodeling was mechanistically associated with impaired mitochondrial membrane structure and protein function. The rationale for the study was based on data that show alterations in mitochondrial phospholipid acyl chain remodeling, particularly with CL, are associated with a multitude of diseases including Barth Syndrome, cardiovascular diseases, diabetes, and aging. These diseases generally display decreased mitochondrial supercomplex formation, decreased respiration, and increased oxidative stress [2,5,7–10,40,50–53]. Therefore, we specifically hypothesized that remodeling of phospholipid acyl chains in response to a high fat diet would be associated with impaired mitochondrial membrane structure and enzyme activity.
4.1 High fat diet remodels cardiac and liver mitochondrial phospholipids with differential effects on levels of n-6 and n-3 polyunsaturated fatty acids
The data establish that a high fat diet remodels the phospholipidome of cardiac and liver mitochondria. We speculate that the remodeling is driven by fatty acids directly obtained from the diet and due to changes in lipid metabolism as a consequence of the obese phenotype [54,55]. To exemplify, there were no detectable levels of 20:5 in the high fat diet, yet in cardiac and liver tissue (18:0)(20:5)PC levels were increased in obese mice. In contrast, 20:4 levels, which were elevated in the high fat diet, were increased in the phospholipidome of cardiac tissue of obese mice. Thus, future studies will need to tease out how the diet and obesity-associated changes in lipid metabolism contribute toward phospholipid acyl chain remodeling.
There were clear differences in the acyl chain composition of cardiac and liver tissue in response to the high fat diet. There were losses in several shorter CL acyl chains as well as in increase in 20:4 in cardiac mitochondria. In liver mitochondria, CL acyl chain remodeling followed a similar pattern to cardiac phospholipid remodeling but had significant decreases in acyl chains with 20:4 and an elevation in (18:2)4. Furthermore, in cardiac mitochondria, there were increases in 18:2(n-6) and 20:5 whereas liver mitochondria showed a reduction in 18:2(n-6) and increases in 20:5 and 22:6.
We speculate that the change in membrane packing in cardiac tissue may have been driven by changes in polyunsaturation, consistent with data in rodent models and biomimetic membranes that show that PUFAs regulates membrane packing [56–58]. To exemplify, the increase in the relatively abundant (18:0)(20:5)PC may have promoted a significant decrease in packing due to the presence of 20:5, which has a highly disordered structure [59]. Surprisingly, there was no change in membrane packing in liver mitochondria (data not shown) despite elevated levels of 20:5, 22:6, and even 18:2. More studies are needed in this area given that the role of n-3 and n-6 PUFAs on liver mitochondrial structure are not well understood.
4.2 Cardiac supercomplex formation and enzyme activity are not impaired with a high fat diet
Despite the alterations in cardiac phospholipid acyl chains and membrane packing with the high fat diet, there were no abnormalities with either supercomplex formation or respiratory enzymatic activity, contrary to our hypothesis. In addition, we analyzed supercomplex formation and oxidative phosphorylation enzyme activity in the liver and indeed found that liver complex I to III activity was lowered in mice consuming a high fat diet relative to lean controls but supercomplex formation was not impaired. Perhaps liver mitochondria have significantly less supercomplex formation than cardiac mitochondria and therefore may rely more heavily on the diffusion and movement of the electron carriers. Future studies need to assay for differences in supercomplex formation between the heart and liver, which is critical since supercomplexes stabilize electron transport complexes and improve electron transport efficiency [60,61].
Our functional data with the high fat diet model provide new information not seen in previous studies. In ob/ob mice, there is a reduction in mitochondrial oxidative capacity that may contribute to cardiac dysfunction. This reduction in oxidative capacity correlated with a significant loss of oxidative phosphorylation machinery, including complexes I, II, III, and V [62]. Ob/ob mice also have significant aberrant CL remodeling including acyl chains that are redistributed from (18:2)4 to highly unsaturated acyl chains such as 22:6 [40]. The lack of effect between our study and the previous work with ob/ob mice may reflect differences in model systems. Ob/ob mice gain weight at a much more rapid rate compared to our model system of diet-induced obesity. Furthermore, ob/ob mice are genetically different than the C57BL/6 mice used in this study.
In another study using an obese model, ALCAT1 was upregulated, which remodels CL with species that are highly oxidizable, leading to mitochondrial damage [40,63,64]. However, in our high fat diet model we observed no change in ALCAT1 mRNA levels compared with lean controls in both cardiac and liver tissues. The failure to upregulate ALCAT1 mRNA in this study may contribute to the lack of respiratory dysfunction and oxidative stress, at least in cardiac mitochondria. This may be a kinetic effect and perhaps ALCAT1 mRNA levels change with time and we did not capture this change. Similarly, we measured mRNA levels of TAZ, a non-specific acyltransferase, known to incorporate 18:2 acyl chains into CL [49]. Although there was a no reduction in TAZ mRNA levels in cardiac tissue, there was a significant increase in TAZ mRNA in liver of high fat fed mice compared to lean controls (Fig 6). This increase in TAZ mRNA may explain the significant increase in (18:2)4CL observed in liver mitochondria of obese mice. Subsequent studies will need to address if ALCAT1 and TAZ mRNA levels change with time in response to a high fat diet. We fed mice for 20 weeks to ensure a strong difference in fat mass between the control and obese mice. We acknowledge that additional feeding over time may have resulted in stronger effects on the phospholipidome and thereby mitochondrial function.
4.3 Importance of acyl chain remodeling versus loss of phospholipid content
The results from this study showed that total CL levels were not lowered in response to a high fat diet. In most reported diseases with impaired CL remodeling, there is also a loss in overall phospholipid concentration. For example, Barth Syndrome and other models of aberrantly remodeled CL such as the knockdown of ALCAT1, result in a loss of total CL content [48,50]. Therefore, models with both remodeled CL and a loss of total concentration make it difficult to distinguish the importance of only remodeled CL on the endpoints of mitochondrial function. Thus, an advancement from this study was that the high fat diet is not promoting a loss of phospholipid content but only the remodeling of associated acyl chains in cardiac and liver tissue.
These results support an emerging view suggesting that mitochondrial acyl chain remodeling is not a major driver of disrupting mitochondrial function in cardiac tissue. In agreement, it was recently suggested that nascent CL does not have a negative impact on the function of the mitochondria. Using a yeast mutant that only affected CL remodeling and not concentration, it was found that nascent CL had no effect on multiple mitochondrial functions including respiration and supercomplex formation [65]. It was concluded that nascent CL is equally able to maintain mitochondrial morphology and respiration, which disagrees with the prevailing model that CL remodeling is critical for optimal mitochondrial function [2–4,24,25]. In addition, it is important to note that the high fat diet fed animals did not display dramatic acyl chain remodeling in CL, PE, and PC. More substantial pathological remodeling and/or a loss of total CL may lead to dysfunctional mitochondria in cardiac tissue.
Our results were also in agreement with work focused on cardiac metabolism and the delta-6 desaturase, the rate-limiting enzyme of long chain polyunsaturated fatty acid biosynthesis [66,67]. To exemplify, inhibiting the delta-6 desaturase with a pharmacological inhibitor in aged mice prevented the age-associated increase in several PUFAs such as 22:6 in CL [67]. However, inhibition of the enzyme did not influence state 3 respiratory capacity although select measures of cardiac function such as hypertrophy were improved. Thus, our results in parallel with others provide additional evidence that CL remodeling is not mechanistically driving an impairment in select measures of respiratory function.
4.4 Potential mechanisms for cardioprotection in an obesity model
Multiple studies have shown that an increase in PUFAs is beneficial to heart function [68,69]. Although the mechanism of protection is still widely unknown, there is a link between increased PUFAs and cardioprotection. One hypothesis is PUFAs incorporate into mitochondrial membrane phospholipids and allow for efficient cardiac function. We discovered a significant reduction in shorter saturated acyl chains, thereby increasing the relative levels of PUFAs in cardiac tissue. This increase in the levels of PUFA acyl chains could potentially be contributing to the maintenance of mitochondrial function we observed during this study. In addition, some studies show that a high fat diet can potentially be cardioprotective [70–72]. Lastly, the heart is a metabolically flexible organ and it is possible that cardiac mitochondria, even with the onset of obesity, adapt to the substrate supplied by the diet and continue to function normally. In contrast, liver enzymatic activity may be impaired due to an imbalance in the appropriate levels of n-3 and n-6 PUFAs [73].
4.5 Conclusions
The data suggest that a chronic high fat diet that promotes mitochondrial phospholipid remodeling may not be detrimental to cardiac mitochondrial function but could contribute to mitochondrial dysfunction in liver. Furthermore, the results open the door to investigating how acyl chain remodeling enzymes are regulated in response to dietary changes.
Supplementary Material
Highlights.
Diet-induced obesity remodels mitochondrial acyl chains in heart and liver tissue.
Membrane packing is modified in cardiac tissue of obese mice.
Supercomplex formation is maintained in heart and liver mitochondria of obese mice.
Diet-induced obesity impairs liver but not heart respiratory enzyme activity.
TAZ mRNA levels are upregulated in the liver of obese mice.
Acknowledgments
Research was supported by NIH R01HL123647 (S.R.S. and D.A.B.) and NIH R15HL12292201 (S.R.S. and D.A.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10(9):369–377. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- 2.Sparagna GC, Chicco AJ, Murphy RC, Bristow MR, Johnson CA, Rees ML, et al. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J Lipid Res. 2007;48(7):1559–1570. doi: 10.1194/jlr.M600551-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res. 2004;94(1):53–59. doi: 10.1161/01.RES.0000109416.56608.64. [DOI] [PubMed] [Google Scholar]
- 4.Petrosillo G, Ruggiero FM, Di Venosa N, Paradies G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. FASEB J. 2003;17(6):714–716. doi: 10.1096/fj.02-0729fje. [DOI] [PubMed] [Google Scholar]
- 5.Petrosillo G, Di Venosa N, Ruggiero FM, Pistolese M, D’Agostino D, Tiravanti E, et al. Mitochondrial dysfunction associated with cardiac ischemia/reperfusion can be attenuated by oxygen tension control. Role of oxygen-free radicals and cardiolipin. Biochim Biophys Acta. 2005;1710(2–3):78–86. doi: 10.1016/j.bbabio.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Taegtmeyer H. Cardiac metabolism as a target for the treatment of heart failure. Circulation. 2004;110(8):894–896. doi: 10.1161/01.CIR.0000139340.88769.D5. [DOI] [PubMed] [Google Scholar]
- 7.Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80(1):30–39. doi: 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Genova ML, Lenaz G. The Interplay Between Respiratory Supercomplexes and ROS in Aging. Antioxid Redox Signal. 2015;23(3):208–238. doi: 10.1089/ars.2014.6214. [DOI] [PubMed] [Google Scholar]
- 9.Lee HJ, Mayette J, Rapoport SI, Bazinet RP. Selective remodeling of cardiolipin fatty acids in the aged rat heart. Lipids Health Dis. 2006;5:2. doi: 10.1186/1476-511X-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez LA, Monette JS, Chavez JD, Maier CS, Hagen TM. Supercomplexes of the mitochondrial electron transport chain decline in the aging rat heart. Arch Biochem Biophys. 2009;490(1):30–35. doi: 10.1016/j.abb.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Planas-Iglesias J, Dwarakanath H, Mohammadyani D, Yanamala N, Kagan VE, Klein-Seetharaman J. Cardiolipin Interactions with Proteins. Biophys J. 2015;109(6):1282–1294. doi: 10.1016/j.bpj.2015.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene. 2002;286(1):135–141. doi: 10.1016/s0378-1119(01)00814-9. [DOI] [PubMed] [Google Scholar]
- 13.Arnarez C, Marrink SJ, Periole X. Identification of cardiolipin binding sites on cytochrome c oxidase at the entrance of proton channels. Sci Rep. 2013;3:1263. doi: 10.1038/srep01263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnarez C, Mazat JP, Elezgaray J, Marrink SJ, Periole X. Evidence for cardiolipin binding sites on the membrane-exposed surface of the cytochrome bc1. J Am Chem Soc. 2013;135(8):3112–3120. doi: 10.1021/ja310577u. [DOI] [PubMed] [Google Scholar]
- 15.Eble KS, Coleman WB, Hantgan RR, Cunningham CC. Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J Biol Chem. 1990;265(32):19434–19440. [PubMed] [Google Scholar]
- 16.Iverson SL, Orrenius S. The cardiolipin-cytochrome c interaction and the mitochondrial regulation of apoptosis. Arch Biochem Biophys. 2004;423(1):37–46. doi: 10.1016/j.abb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Zhang M, Mileykovskaya E, Dowhan W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem. 2002;277(46):43553–43556. doi: 10.1074/jbc.C200551200. [DOI] [PubMed] [Google Scholar]
- 18.Zhang M, Mileykovskaya E, Dowhan W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J Biol Chem. 2005;280(33):29403–29408. doi: 10.1074/jbc.M504955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Renner LD, Weibel DB. Cardiolipin microdomains localize to negatively curved regions of Escherichia coli membranes. Proc Natl Acad Sci U S A. 2011;108(15):6264–6269. doi: 10.1073/pnas.1015757108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sorice M, Manganelli V, Matarrese P, Tinari A, Misasi R, Malorni W, et al. Cardiolipin-enriched raft-like microdomains are essential activating platforms for apoptotic signals on mitochondria. FEBS Lett. 2009;583(15):2447–2450. doi: 10.1016/j.febslet.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 21.Brown DA, Sabbah HN, Shaikh SR. Mitochondrial inner membrane lipids and proteins as targets for decreasing cardiac ischemia/reperfusion injury. Pharmacol Ther. 2013;140(3):258–266. doi: 10.1016/j.pharmthera.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 22.Unsay JD, Cosentino K, Subburaj Y, Garcia-Saez AJ. Cardiolipin effects on membrane structure and dynamics. Langmuir. 2013;29(51):15878–15887. doi: 10.1021/la402669z. [DOI] [PubMed] [Google Scholar]
- 23.Wilkens V, Kohl W, Busch K. Restricted diffusion of OXPHOS complexes in dynamic mitochondria delays their exchange between cristae and engenders a transitory mosaic distribution. J Cell Sci. 2013;126(Pt 1):103–116. doi: 10.1242/jcs.108852. [DOI] [PubMed] [Google Scholar]
- 24.Zeczycki TN, Whelan J, Hayden WT, Brown DA, Shaikh SR. Increasing levels of cardiolipin differentially influence packing of phospholipids found in the mitochondrial inner membrane. Biochem Biophys Res Commun. 2014;450(1):366–371. doi: 10.1016/j.bbrc.2014.05.133. [DOI] [PubMed] [Google Scholar]
- 25.Shaikh SR, Sullivan EM, Alleman RJ, Brown DA, Zeczycki TN. Increasing mitochondrial membrane phospholipid content lowers the enzymatic activity of electron transport complexes. Biochemistry. 2014;53(35):5589–5591. doi: 10.1021/bi500868g. [DOI] [PubMed] [Google Scholar]
- 26.Vecchini A, Del Rosso F, Binaglia L, Dhalla NS, Panagia V. Molecular defects in sarcolemmal glycerophospholipid subclasses in diabetic cardiomyopathy. J Mol Cell Cardiol. 2000;32(6):1061–1074. doi: 10.1006/jmcc.2000.1140. [DOI] [PubMed] [Google Scholar]
- 27.Schlame M, Kelley RI, Feigenbaum A, Towbin JA, Heerdt PM, Schieble T, et al. Phospholipid abnormalities in children with Barth syndrome. J Am Coll Cardiol. 2003;42(11):1994–1999. doi: 10.1016/j.jacc.2003.06.015. [DOI] [PubMed] [Google Scholar]
- 28.Xu Y, Sutachan J, Plesken H, Kelley RI, Schlame M. Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab Invest. 2005;85(6):823–830. doi: 10.1038/labinvest.3700274. [DOI] [PubMed] [Google Scholar]
- 29.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Serena D, Ruggiero FM. Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free Radic Biol Med. 1999;27(1–2):42–50. doi: 10.1016/s0891-5849(99)00032-5. [DOI] [PubMed] [Google Scholar]
- 30.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444(7121):875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 31.DeBoer MD. Obesity, systemic inflammation, and increased risk for cardiovascular disease and diabetes among adolescents: a need for screening tools to target interventions. Nutrition. 2013;29(2):379–386. doi: 10.1016/j.nut.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ginsberg HN, MacCallum PR. The obesity, metabolic syndrome, and type 2 diabetes mellitus pandemic: Part I. Increased cardiovascular disease risk and the importance of atherogenic dyslipidemia in persons with the metabolic syndrome and type 2 diabetes mellitus. J Cardiometab Syndr. 2009;4(2):113–119. doi: 10.1111/j.1559-4572.2008.00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stringer DM, Sellers EA, Burr LL, Taylor CG. Altered plasma adipokines and markers of oxidative stress suggest increased risk of cardiovascular disease in First Nation youth with obesity or type 2 diabetes mellitus. Pediatr Diabetes. 2009;10(4):269–277. doi: 10.1111/j.1399-5448.2008.00473.x. [DOI] [PubMed] [Google Scholar]
- 34.Sparagna GC, Johnson CA, McCune SA, Moore RL, Murphy RC. Quantitation of cardiolipin molecular species in spontaneously hypertensive heart failure rats using electrospray ionization mass spectrometry. J Lipid Res. 2005;46(6):1196–1204. doi: 10.1194/jlr.M500031-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.Pennington ER, Fix A, Sullivan EM, Brown DA, Kennedy A, Shaikh SR. Distinct membrane properties are differentially influenced by cardiolipin content and acyl chain composition in biomimetic membranes. Biochim Biophys Acta. 2016 doi: 10.1016/j.bbamem.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaikh SR, Dumaual AC, Jenski LJ, Stillwell W. Lipid phase separation in phospholipid bilayers and monolayers modeling the plasma membrane. Biochim Biophys Acta. 2001;1512(2):317–328. doi: 10.1016/s0005-2736(01)00335-2. [DOI] [PubMed] [Google Scholar]
- 37.Alleman RJ, Tsang AM, Ryan TE, Patteson DJ, McClung JM, Spangenburg EE, et al. Exercise-induced protection against reperfusion arrhythmia involves stabilization of mitochondrial energetics. Am J Physiol Heart Circ Physiol. 2016;310(10):H1360–70. doi: 10.1152/ajpheart.00858.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perry CG, Kane DA, Lanza IR, Neufer PD. Methods for assessing mitochondrial function in diabetes. Diabetes. 2013;62(4):1041–1053. doi: 10.2337/db12-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han X, Yang J, Cheng H, Yang K, Abendschein DR, Gross RW. Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry. 2005;44(50):16684–16694. doi: 10.1021/bi051908a. [DOI] [PubMed] [Google Scholar]
- 40.Han X, Yang J, Yang K, Zhao Z, Abendschein DR, Gross RW. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: a shotgun lipidomics study. Biochemistry. 2007;46(21):6417–6428. doi: 10.1021/bi7004015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blume A. A comparative study of the phase transitions of phospholipid bilayers and monolayers. Biochim Biophys Acta. 1979;557(1):32–44. doi: 10.1016/0005-2736(79)90087-7. [DOI] [PubMed] [Google Scholar]
- 42.Marsh D. Lateral pressure in membranes. Biochim Biophys Acta. 1996;1286(3):183–223. doi: 10.1016/s0304-4157(96)00009-3. [DOI] [PubMed] [Google Scholar]
- 43.Mantena SK, Vaughn DP, Andringa KK, Eccleston HB, King AL, Abrams GA, et al. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem J. 2009;417(1):183–193. doi: 10.1042/BJ20080868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brady LJ, Hoppel CL. Hepatic mitochondrial function in lean and obese Zucker rats. Am J Physiol. 1983;245(3):E239–45. doi: 10.1152/ajpendo.1983.245.3.E239. [DOI] [PubMed] [Google Scholar]
- 45.Chavin KD, Yang S, Lin HZ, Chatham J, Chacko VP, Hoek JB, et al. Obesity induces expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. J Biol Chem. 1999;274(9):5692–5700. doi: 10.1074/jbc.274.9.5692. [DOI] [PubMed] [Google Scholar]
- 46.Perez-Carreras M, Del Hoyo P, Martin MA, Rubio JC, Martin A, Castellano G, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38(4):999–1007. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- 47.Ye C, Shen Z, Greenberg ML. Cardiolipin remodeling: a regulatory hub for modulating cardiolipin metabolism and function. J Bioenerg Biomembr. 2016;48(2):113–123. doi: 10.1007/s10863-014-9591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J, Romestaing C, Han X, Li Y, Hao X, Wu Y, et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010;12(2):154–165. doi: 10.1016/j.cmet.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schlame M. Cardiolipin remodeling and the function of tafazzin. Biochim Biophys Acta. 2013;1831(3):582–588. doi: 10.1016/j.bbalip.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 50.Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJ, et al. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun. 2000;279(2):378–382. doi: 10.1006/bbrc.2000.3952. [DOI] [PubMed] [Google Scholar]
- 51.Schlame M, Towbin JA, Heerdt PM, Jehle R, DiMauro S, Blanck TJ. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann Neurol. 2002;51(5):634–637. doi: 10.1002/ana.10176. [DOI] [PubMed] [Google Scholar]
- 52.McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol. 2006;361(3):462–469. doi: 10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 53.Kiebish MA, Yang K, Liu X, Mancuso DJ, Guan S, Zhao Z, et al. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J Lipid Res. 2013;54(5):1312–1325. doi: 10.1194/jlr.M034728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raffaella C, Francesca B, Italia F, Marina P, Giovanna L, Susanna I. Alterations in hepatic mitochondrial compartment in a model of obesity and insulin resistance. Obesity. 2008;16(5):958–964. doi: 10.1038/oby.2008.10. [DOI] [PubMed] [Google Scholar]
- 55.Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000;279(5):E1104–13. doi: 10.1152/ajpendo.2000.279.5.E1104. [DOI] [PubMed] [Google Scholar]
- 56.Shaikh SR. Biophysical and biochemical mechanisms by which dietary N-3 polyunsaturated fatty acids from fish oil disrupt membrane lipid rafts. J Nutr Biochem. 2012;23(2):101–105. doi: 10.1016/j.jnutbio.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shaikh SR, Edidin M. Polyunsaturated fatty acids, membrane organization, T cells, and antigen presentation. Am J Clin Nutr. 2006;84(6):1277–1289. doi: 10.1093/ajcn/84.6.1277. [DOI] [PubMed] [Google Scholar]
- 58.Fan YY, McMurray DN, Ly LH, Chapkin RS. Dietary (n-3) polyunsaturated fatty acids remodel mouse T-cell lipid rafts. J Nutr. 2003;133(6):1913–1920. doi: 10.1093/jn/133.6.1913. [DOI] [PubMed] [Google Scholar]
- 59.Williams JA, Batten SE, Harris M, Rockett BD, Shaikh SR, Stillwell W, et al. Docosahexaenoic and eicosapentaenoic acids segregate differently between raft and nonraft domains. Biophys J. 2012;103(2):228–237. doi: 10.1016/j.bpj.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, Enriquez JA. Respiratory active mitochondrial supercomplexes. Mol Cell. 2008;32(4):529–539. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 61.Genova ML, Lenaz G. Functional role of mitochondrial respiratory supercomplexes. Biochim Biophys Acta. 2014;1837(4):427–443. doi: 10.1016/j.bbabio.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 62.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112(17):2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 63.Taylor WA, Hatch GM. Identification of the human mitochondrial linoleoylcoenzyme A monolysocardiolipin acyltransferase (MLCL AT-1) J Biol Chem. 2009;284(44):30360–30371. doi: 10.1074/jbc.M109.048322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cao J, Liu Y, Lockwood J, Burn P, Shi Y. A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J Biol Chem. 2004;279(30):31727–31734. doi: 10.1074/jbc.M402930200. [DOI] [PubMed] [Google Scholar]
- 65.Baile MG, Sathappa M, Lu YW, Pryce E, Whited K, McCaffery JM, et al. Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. J Biol Chem. 2014;289(3):1768–1778. doi: 10.1074/jbc.M113.525733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Le CH, Mulligan CM, Routh MA, Bouma GJ, Frye MA, Jeckel KM, et al. Delta-6-desaturase links polyunsaturated fatty acid metabolism with phospholipid remodeling and disease progression in heart failure. Circ Heart Fail. 2014;7(1):172–183. doi: 10.1161/CIRCHEARTFAILURE.113.000744. [DOI] [PubMed] [Google Scholar]
- 67.Mulligan CM, Le CH, deMooy AB, Nelson CB, Chicco AJ. Inhibition of delta-6 desaturase reverses cardiolipin remodeling and prevents contractile dysfunction in the aged mouse heart without altering mitochondrial respiratory function. J Gerontol A Biol Sci Med Sci. 2014;69(7):799–809. doi: 10.1093/gerona/glt209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kris-Etherton PM, Harris WS, Appel LJ. American Heart Association. Nutrition Committee. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation. 2002;106(21):2747–2757. doi: 10.1161/01.cir.0000038493.65177.94. [DOI] [PubMed] [Google Scholar]
- 69.Lavie CJ, Milani RV, Mehra MR, Ventura HO. Omega-3 polyunsaturated fatty acids and cardiovascular diseases. J Am Coll Cardiol. 2009;54(7):585–594. doi: 10.1016/j.jacc.2009.02.084. [DOI] [PubMed] [Google Scholar]
- 70.Yu L, Fink BD, Herlein JA, Oltman CL, Lamping KG, Sivitz WI. Dietary fat, fatty acid saturation and mitochondrial bioenergetics. J Bioenerg Biomembr. 2014;46(1):33–44. doi: 10.1007/s10863-013-9530-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brainard RE, Watson LJ, Demartino AM, Brittian KR, Readnower RD, Boakye AA, et al. High fat feeding in mice is insufficient to induce cardiac dysfunction and does not exacerbate heart failure. PLoS One. 2013;8(12):e83174. doi: 10.1371/journal.pone.0083174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stanley WC, Dabkowski ER, Ribeiro RF, Jr, O’Connell KA. Dietary fat and heart failure: moving from lipotoxicity to lipoprotection. Circ Res. 2012;110(5):764–776. doi: 10.1161/CIRCRESAHA.111.253104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hagopian K, Weber KL, Hwee DT, Van Eenennaam AL, Lopez-Lluch G, Villalba JM, et al. Complex I-associated hydrogen peroxide production is decreased and electron transport chain enzyme activities are altered in n-3 enriched fat-1 mice. PLoS One. 2010;5(9):e12696. doi: 10.1371/journal.pone.0012696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.