

Abstract
Melanoma is the most aggressive type of skin cancer. It is highly metastatic, migrating through lymph nodes to distant sites of the body, especially to lungs, liver and brain. Systemic chemotherapy remains the mainstay of treatment; however, the development of multidrug resistance (MDR) restricts the efficacy of current chemotherapeutic drugs. We synthesized a series of microtubule destabilizers, substituted methoxybenzoylary-thiazole (SMART) compounds, which inhibited tubulin polymerization and effectively circumvented MDR. Due to poor water solubility of SMART compounds, co-solvent delivery is required for their systemic administration, which is usually associated with hepatotoxicity, nephrotoxicity and hemolysis. To solve this problem and also to increase circulation time, we synthesized a new SMART analogue, SMART-OH, and its polymer-drug conjugate, methoxy-poly (ethylene glycol)-block-poly (2-methyl-2-carboxyl-propylene carbonate-graft-SMART-graft-dodecanol) (abbreviated as P-SMART), with 14.3±2.8% drug payload of SMART-OH. Similar to its parent drug, P-SMART showed significant anticancer activity against melanoma cells in cytotoxicity, colony formation, and cell invasion studies. In addition, P-SMART treatment led to cell cycle arrest at G2/M phase and cell accumulation in sub-G1 phase. We established a model of metastatic melanoma to the lung in C57/BL6 albino mice to determine in vivo efficacy of P-SMART and SMART-OH at the dose of 20 mg/kg. P-SMART treatment resulted in significant inhibition of tumor growth and prolonged mouse median survival. In conclusion, P-SMART, a novel polymer-microtubule destabilizer conjugate, has the potential to treat metastatic melanoma.
Keywords: Metastatic melanoma, Microtubule destabilizer, Polymer drug conjugate, Nanomedicine
Graphical abstract
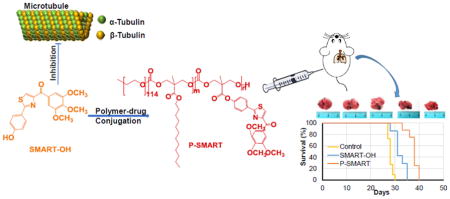
Introduction
Over the past several decades, the development of nanomedicines has been driven by the increasing demands of delivering therapeutic agents to disease sites efficiently. A large amount of pioneering research has highlighted applications of micelles, nanoparticles, liposomes, polymersomes, nanogels and dendrimers as nanocarriers of low molecular-weight drugs, oligonucleotides and genes. Polymer-drug conjugates debuted in 1955 [1], and in the mid-1970s Ringsdorf proposed the idea of conjugating therapeutic agents to water soluble polymers [2]. Since then, the field of polymer-drug conjugates started a new era of drug delivery and has been growing fast. Advantages of conjugates over their corresponding parent drugs include: 1) increased aqueous solubility of hydrophobic drugs; 2) prolonged blood circulation time; 3) enhanced bioavailability; 4) increased protection of drugs from degradation; 5) increased tumor accumulation either due to enhanced permeability and retention (EPR) effect or tunable targeting moieties. Unlike physically drug encapsulation into nanoparticles and micelles, covalent drug conjugation to polymers achieves enhanced drug payload and prevents premature drug release, thereby decreasing undesired toxicities compared to physically drug-encapsulated liposomes, nanoparticles and micelles. The polymer-drug conjugate market is currently becoming well-established with several commercialized products available for a wide range of disease states, such as Adynovate by Baxalta, Movantik™ by AstraZeneca, Oncospar® by Enzon Pharmaceuticals, Plegridy® by Biogen, etc.
Malignant melanoma is the most invasive form of skin cancer with high metastatic propensity, typically metastasizing to the lymph nodes, lungs, liver, brain and heart at late stage of melanoma. The median overall survival time of patients suffering from metastatic melanoma is less than one year, and only about 10% of these patients survive more than 5 years after diagnosis [3]. Unfortunately, the survival of patients with advanced metastatic melanoma has not been significantly improved by current Food and Drug Administration (FDA)-approved systemic chemotherapies [4]. Dacarbazine (DTIC), a widely used chemotherapeutic agent for treatment of metastatic melanoma, shows transient efficacy in most patients, however, only 1–2% of patients achieve a durable long-term response to this therapy [5]. The combination of paclitaxel and carboplatin is used as second-line therapy for patients who suffer from disease progression while receiving DTIC treatment. Clinical benefit of this combination therapy was noted in more than 40% of all patients in the original study [6,7]. Nevertheless, a potential problem when using paclitaxel or other microtubule inhibitors for cancer treatment in the long term is that their anticancer effects could be undermined by intrinsic or acquired drug resistance due to overexpression of drug efflux pumps (P-glycoprotein, MRP and BCRP) in cancer cells [8–10]. To address this problem, we have synthesized a series of novel microtubule destabilizers, substituted methoxybenzoyl-ary-thiazole (SMART) compounds, with nanomolar anticancer activity against melanoma, breast cancer, ovarian cancer, colon cancer and prostate cancer [11]. In addition, their ability to circumvent P-gp mediated drug resistance was confirmed by using prostate cancer cells with P-gp overexpression [12]. However, the clinical translation of SMART compounds is limited due to their poor aqueous solubility as many other anticancer agents. Moreover, these small molecular weight drugs are rapidly eliminated from the circulation, requiring frequent dosing, leading to increased risk of side effects. To address this issue, we formulated SMART-100 in micelles using poly (ethylene)-b-poly(D,L-lactide) (PEG-PLA) in a previous study, but the utility of physical encapsulation is limited by low drug payload. Therefore, in the current study we synthesized a new SMART analogue, SMART-OH and conjugated this compound to the carboxyl pendant groups of poly (ethylene glycol)-block-poly (2-methyl-2-carboxyl-propylene carbonate) (mPEG-b-PCC). The polymeric conjugate consists of three components including biocompatible PEG blocks, a biodegradable polycarbonate backbone and lipid chains of dodecanol (DC). The anticancer effect of SMART-OH and its polymer-drug conjugate mPEG-b-PCC-g-SMART-g-DC (abbreviated as P-SMART) on melanoma cells was determined. Furthermore, a mouse model of metastatic melanoma to the lungs was established to study in vivo efficacy of P-SMART as well as SMART-OH.
Materials
4-Cyanophenol, L-cysteine, N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU), tert-butyldimethylsilyl chloride (TBDMSCl), n-butyllithium, tetra-n-butylammonium fluoride, 2, 2-bis(hydroxymethyl) propionic acid, methoxy poly(ethylene glycol) (mPEG, Mn =5000), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) 98%, 1-hydroxybenzotriazole (HOBT), Benzyl bromide, Dodecanol (DC), diisopropylethylamine (DIPEA) and cremophor® EL were purchased from Sigma Aldrich (St. Louis, MO). FxCycle™ PI/RNase staining solution was purchased from ThermoFisher Scientific (Waltham, MA). Bovine brain tubulin was purchased from Cytoskeleton (Denver, CO).
Methods
Synthesis and Characterization of SMART-OH
2-(4-Hydroxyphenyl)-4,5-dihydrothiazol-4-yl-3,4,5-trimethoxyphenyl)methanone (abbreviated as SMART-OH) was synthesized as shown in Figure 1A (compound 5). Briefly, 4-cyanophenol (1 equiv.) was mixed with L-cysteine (1 equiv) in a 1:1 solution of MeOH/pH 6.4 PBS. The reaction mixture was heated to 40 °C and stirred for 3 days. The mixture was then filtered to remove the precipitate and MeOH was removed using a rotary evaporator. The remaining solution was then acidified to pH 4 using 1M HCl and CH2Cl2 was added to the solution. The resulting precipitate was filtered to yield a white solid, compound 1. This solid was dried overnight in a vacuum desiccator and then used directly for the next step.
Figure 1. Synthesis and characterization of SMART-OH and its inhibition of tubulin.

A) Synthetic scheme and 1H NMR spectrum of SMART-OH. B) Molecular docking. Docking image showed that SMART-OH bind to the colchicine binding site in tubulin. C) Tubulin polymerization assay. Tubulin (3.33 mg/ml) was exposed to 10 μM of SMART-OH, colchicine or vehicle control (5% DMSO), respectively, and incubated in general tubulin buffer. Absorbance at 340 nm was monitored at 37°C every minute for 15 min. Both SMART-OH and colchicine effectively inhibited polymerization, while robust polymerization was observed in the control group.
Compound 1 (1 equiv.) was dissolved in anhydrous CH2Cl2. HBTU (1.1 equiv) was then added and stirred for 15 minutes. This was followed by addition of DIPEA (2.2 equiv) which was stirred for 2–3 minutes. Finally, HNCH3OCH3 HCl salt (1.1 equiv.) was added and the reaction was stirred at room temperature for 12–18 hours. The reaction mixture was washed once with ddH2O and twice with saturated NaCl solution. The organic layer was then dried over MgSO4. The solvent was removed by a rotary evaporator to yield crude yellow oil. This was then purified by flash chromatography to obtain compound 2.
A solution of compound 2 (1 equiv.) in anhydrous tetrahydrofuran (THF) was kept under argon and cooled to 0 °C. Imidazole (2.5 equiv.) and TBDMSCl (2 equiv) were then added to the solution and the mixture was stirred for 12–18 hours. The solvent was removed by a rotary evaporator and the resulting solid was dissolved in CH2Cl2 and washed once with ddH2O and once with saturated NH4Cl solution. The organic layer was dried over NaSO4 followed by purification using flash chromatography to yield compound 3.
Compound 3 (1 equiv) was dissolved in freshly distilled THF at room temperature while 3,4,5-methoxylphenyl (1.2 equiv) was dissolved in freshly distilled THF in a separate flasks and cooled to −78 °C under argon. n-Butyllithium (1.5 equiv) was then added to the cooled mixture and stirred at −78 °C for 30 minutes. The solution containing compound 3 was then added to the mixture and stirred for 2 hours while returning to room temperature. The reaction was quenched with saturated NH4Cl solution, extracted three times with ethyl acetate, dried over NaSO4, and purified by flash chromatography to yield a bright yellow solid, compound 4.
Compound 4 (1 equiv) was then dissolved in THF and cooled to 0 °C. Tetra-n-butyl ammonium fluoride (2 equiv) was then added and the mixture was stirred for 10 minutes. The reaction was quenched with saturated NH4Cl solution, extracted three times with ethyl acetate, dried over NaSO4, and purified using flash chromatography to yield the final pure product (compound 5, Figure 1A).
Docking
Docking studies were carried out using the crystal structures of the α,β-tubulin dimer in complex with DAMA-colchicine (Protein Data Bank code 1SA0). Schrodinger Molecular Modeling Suite 2016 (Schrodinger Inc., Portland, OR) running on Microsoft Windows 7 platform was used to perform these studies, similar to what we described in previous reports [13–15]. Briefly, the protein-ligand complex was prepared using the Protein Preparation module, and SMART-OH was docked into the colchicine binding site in the structure of 1SA0 using Glide module. Data analyses were performed using the Maestro interface of the software.
Tubulin Polymerization Assay
Bovine brain tubulin (3.33 mg/m) was exposed to 10 μM of SMART-OH, colchicine or vehicle control (5% DMSO), respectively, and incubated in 100 μl of general tubulin buffer (80 mM PIPES, 2.0 mM MgCl2, 0.5 mM EGTA and 1 mM GTP; pH 6.9). Absorbance at 340 nm was monitored at 37°C every minute for 15 min by the SYNERGY 4 Microplate Reader (Bio-Tek Instruments, Winooski, VT).
Synthesis and Characterization of mPEG-b-PCC-g-SMART-g-DC (P-SMART)
2-Methyl-2-benzyloxycarbonyl-propylene carbonate (MBC), poly(ethylene glycol)-block-poly(2-methyl-2-benzoxycarbonyl-propylene carbonate) (mPEG114-b-PBC28) and poly(ethylene glycol)-block-poly(2-methyl-2-carboxyl-propylene carbonate) (mPEG114-b-PCC28) were synthesized as described previously [16]. SMART-OH and DC were conjugated to the carboxyl groups of mPEG114-b-PCC28 copolymer using carbodiimide coupling. MPEG-b-PCC (180 mg, 0.019 mmol) was dissolved in anhydrous CH2Cl2 followed by addition of EDC (215 mg, 1.12 mmol), HOBT (101 mg, 0.75 mmol) and the solution was stirred at room temperature. After two hours, N,N-diisopropylethylamine (DIPEA, 98 μl, 0.56 mmol) and SMART-OH (84 mg, 0.23 mmol) were added and the reaction continued for two days. Then, DC (70 mg, 0.37 mmol) was added and the reaction was allowed to proceed for one additional day (Figure 2A). Crude product was purified by precipitation in large excess of diethyl ether and then by dialysis against MeOH using a regenerated cellulose membrane with 3.5 K MWCO.
Figure 2. Synthesis and characterization of mPEG-b-PCC-g-SMART-g-DC (P-SMART).

A) Synthetic scheme of P-SMART. B) 1H NMR (500 MHz, DMSO-d6) spectrum of P-SMART. C) Particle size distribution of P-SMART by dynamic light scattering (DLS). The mean particle size of P-SMART was 71.51±0.47 nm. D) In vitro drug release profile of P-SMART. Concentration of released SMART-OH at each time point was measured by HPLC and drug release profiles were represented as the mean ± SEM (n=3). P-SMART showed a slow but sustained release of SMART-OH at neutral and acidic condition.
1H NMR spectrum was recorded on a Bruker (500 MHz, T = 25 °C) using DMSO-d6 as solvent for P-SMART in a chemical shift range of 0–12 ppm.
Dynamic light scattering (DLS) was used for measuring the particle size distribution of P-SMART. Briefly, 10 mg of P-SMART was dissolved in CH2Cl2 and the solvent was evaporated under reduced pressure. The resulting film was hydrated with 1 ml PBS (pH 7.4) followed by sonication and filtration through 0.22 μm filter. Mean particle size was measured by using a Zetasizer Nano ZS90 (Malvern, Worcestershire, UK) at a scattering angle of 173°. A total of 12 measurements were taken per sample with a time span of 10 s. Particle size distribution was reported as the mean ± SEM. of three independent samples.
Quantification of SMART Payload in P-SMART
To quantify the conjugated drug, alkaline hydrolysis method was used. 1 mg/ml P-SMART was mixed with 1 ml NaOH (1 N) at 37 °C overnight. Samples were then neutralized to pH 7.4 followed by HPLC-PDA analysis. Chromatography was performed on Phenomenex® column (250×4.6 mm; 5 μm) using acetonitrile and water (60:40, v/v) as mobile phase and wavelength of 300 nm. The stability of SMART-OH was also tested under the same alkaline hydrolysis conditions at 37 °C overnight to determine if there was any degradation of SMART-OH. The data was reported as the mean ± SEM of three individual experiments. Payload was calculated using equation 1.
In Vitro Drug Release
Drug release from the P-SMART conjugate was determined at pH 6.5 and 7.4. Briefly, 1 mg of P-SMART was suspended in 1 ml buffer solution (0.1 M acetic acetate, pH 6.5 or 0.1 M PBS, pH 7.4) and diluted with MeOH in a volume ratio of MeOH: aqueous solution (1:4, v/v). All samples were incubated at 37 °C, shaken at 100 rpm for 0, 3, 6, 9, 12, 24, 48, 72, 96, 120 h, and neutralized to pH 7.4 prior to HPLC analysis as described in previous section. All experiments were performed in triplicate and the data reported as the mean ± SEM of three individual experiments.
Cell Culture
Human melanoma cell line A375, mouse melanoma cell lines B16-F10 and B16-F10-luc were cultured in DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin. All cell lines were grown and maintained in a humidified incubator containing 5% CO2 at 37 °C.
Cytotoxicity Assay
A375 and B16-F10 cells were used to determine the cytotoxicity of P-SMART and parent drug. After attached to the bottom of plate, cells were incubated with different concentrations of SMART-OH or P-SMART for another 72 h. Cell viability was determined by MTT assay and the absorbance was measured at 560 nm with subtraction of absorbance at 630 nm. Each group was performed in triplicate and the data reported as the mean ± SEM.
Colony Formation Assay
A375 or B16-F10 cells were seeded at 250 cells/well into 6-well plates and allowed to attach for 24 h. Cells were then treated with SMART-OH or P-SMART at different concentrations. After a 7 day-incubation, colonies in each well were fixed by 10% formalin, stained with 0.5% crystal violet solution and visualized under a microscope. Each group was performed in triplicate.
Transwell Invasion Assay
Cell invasion experiments were carried out using 24-well plates and cell culture inserts with 8 μm pore size (Corning®). The upper sides of the inserts were coated with 40 μL Matrigel® diluted 1:4 (v/v) with serum-free DMEM, were placed in a 24-well plate, and were incubated for 2 h at 37 °C. B16-F10 cells were suspended in serum-free DMEM and placed in the upper chamber of the Transwell insert (1×105 cells/ml) with treatment of SMART-OH or P-SMART at a dose of 0.5 μM. Cell suspension without treatment was used as a control. DMEM containing 10% FBS was added to the corresponding lower chamber. After 24 h, the non-invaded cells in the upper chamber were removed by a cotton swab and the invaded cells were fixed with 10% formaldehyde in PBS and stained with 0.5% crystal violet solution. Each group was conducted in triplicate wells and three 40× imaging areas were randomly selected for each well.
Cell Cycle Analysis by Propidium Iodide Staining
A375 and B16-F10 cells were used for cell cycle analysis. Cells were cultured in a 24-well plate to 80% confluence and treated with P-SMART for 48 h. Cells were harvested, fixed in 70% ice-cold ethanol for 1 h and washed by PBS. A cell pellet containing 1×106 cells was then re-suspended in 0.5 mL of FxCycle™ PI/RNase staining solution and incubated for 15 min at room temperature. Cell cycle was measured by a flow cytometer (BD FACSCalibur NJ). Results from 20,000 fluorescent events were obtained for analysis.
In Vivo Study
All animal experiments were performed in accordance with the NIH animal use guidelines and protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Nebraska Medical Center (UNMC), Omaha, NE. A mouse model of metastatic melanoma to the lung was established in 8 week-old female C57BL/6 albino mice by injecting 2 × 105 B16-F10-luc cells suspended in 100 μL PBS into their tail vein. Mice were randomly divided into three groups of five animals per group when the radiance of tumor had reached 105. SMART-OH and P-SMART were administered intravenously to mice once every three days for a total of five times. Group 1 was kept as the control and received normal saline, group 2 received 20 mg/kg SMART-OH in 35 % of cosolvent (50% propylene glycol, 30% Cremophor® EL, and 20% ethanol) and 65 % of dextrose solution, and group 3 received 20 mg/kg P-SMART (equivalent to free SMART-OH). Bioluminescent radiance of tumor was measured every other day using IVIS® Spectrum imaging system (PerkinElmer Inc., MA). At the end of the animal study (i.e., day 24), mice were sacrificed and tumors as well as vital organs (liver, spleen, kidney and heart) were excised.
In a separate survival study, mice were randomly divided into three groups of seven mice for different treatments as described above. Survival observation of mice ceased when death occurred due to uncontrolled tumor growth or the toxicity of treatments. Three representative tumor tissues were collected per group and fixed with 10% buffered formalin for 24h. The fixed samples were embedded in paraffin and thin sections of 4 μm were obtained and immunostained for hematoxylin and eosin (H&E) and cleaved Caspase 3.
Statistical Analysis
Data were represented as the mean ± SEM. The statistical comparisons of the experiments were performed by two-tailed Student’s t test. P <0.05 was considered statistically significant.
RESULTS
Characterization and Molecular Docking of SMART-OH in tubulin
The structures for all synthesized compounds were characterized and confirmed by NMR and high resolution mass spectrometry. The proton NMR for final compound 5 (SMART-OH) was shown in Figure 1A. Docking studies (Figure 1B) indicated excellent binding and interactions between SMART-OH and the tubulin dimer. SMART-OH and the native ligand in the crystal structure of 1SA0 showed good overlap when they bind to the colchicine binding site in tubulin (Figure 1B, enlarged portion). Three hydrogen bonds were formed between SMART-OH and the tubulin dimer, namely oxygen in the 4-methoxy moiety to Cys241; the carbonyl to Asp-251; and the phenol to Val315. These three hydrogen bonds anchor SMART-OH tightly in this colchicine binding pocket, predicting effective disruption of tubulin polymerization.
Inhibition of Tubulin Polymerization
To evaluate the ability of SMART-OH to directly interact with tubulin and confirm its mode of action, we performed a microtubule polymerization assay in vitro. A vehicle and colchicine (10μM), a well-known microtubule destabilizing agent, were used as controls and assayed under the same conditions. Robust polymerization is observed in the control group, while both SMART-OH and colchicine effectively inhibit polymerization (Figure 1C). This result is consistent with the proposed mechanism of action of SMART-OH compound as a potent tubulin polymerization inhibitor.
Characterization and Quantification of P-SMART
P-SMART was synthesized by direct carbodiimide coupling of SMART-OH onto the pendant carboxylic acid groups of the hydrophobic block of mPEG-b-PCC copolymer (Figure 2A). In 1H NMR spectrum of mPEG-b-PCC, protons corresponding to –CH2–CH2–O– of PEG at δ3.4–3.6, –CH2– units of PCC at δ4.2–4.4 and –COOH at δ12–14 were observed and reported earlier by our group. After conjugation of SMART-OH to mPEG-b-PCC, protons corresponding to SMART were all observed in 1H NMR spectrum.1H NMR (500 MHz, DMSO-d6) spectrum of mPEG-b-PCC-g-SMART-g-DC showed peaks corresponding to PEG: (–CH2–CH2–O-) at δ3.5, PCC: (–CO-O-CH2-C-) at δ4.2 (m, 4H), (–CO-O-CH2-C-CH3) at δ1.1–1.3 (t, 3H), Dodecanol: CH3-(CH2)9- at δ0.9 (t, 3H), CH3-(CH2)9- at δ1.0–1.3 (bs, 18H), CH3-(CH2)9-CH2 at δ1.6 (m, 2H), CH3-(CH2)9-CH2-CH2- at δ4.3 (m, 2H), SMART-OH: benzene -CH- at δ8.1 (dt, 2H), δ7.6 (dt, 2H), δ7.2 (t, 2H), thiazole -CH- at δ8.7 (s, 1H), -OCH3 at δ3.83 (s, 6 H) and δ3.71 (s,3 H) (Figure 2B).
DLS showed the mean particle size of P-SMART was 71.51±0.47 nm (PDI: 0.055 ± 0.011) (Figure 2C). Naked SMART-OH was stable under alkaline hydrolysis condition and drug payload of conjugated SMART-OH was determined by HPLC analysis as 14.3±2.8 % (w/w).
pH Dependent Drug Release
In vitro drug release studies were carried out in PBS at pH 7.4 and acetate buffer at pH 6.5 to simulate drug release in blood and tumor environment. P-SMART showed a slow but sustained release of SMART-OH. There was no noticeable initial burst release at the different pH and no significant drug release at neutral conditions afterwards. At pH 6.5, the liberation of SMART-OH from P-SMART was accelerated as expected due to the increased cleavage of ester linkages known to occur in an acidic or basic environment. After five days, more than 25% of SMART-OH was released at pH 6.5, but only 15% of SMART-OH at pH 7.4 (Figure 2D).
Anticancer Activity
We determined the anticancer activity of P-SMART as well as parent drug SMART-OH on A375 and B16-F10 cells for 72 h. Due to the slow release of SMART-OH from the polymer conjugates, the IC50 of P-SMART increased to 0.75 μM in two cell lines while IC50 of SMART-OH was 75 nM in A375 cells and 150 nM in B16-F10 cells. P-SMART effectively killed 80% of melanoma cells at 2 μM parent drug equivalent dose (Figure 3A).
Figure 3. Cytotoxicity and colony formation assay.

A) Cytotoxicity of SMART-OH and P-SMART was determined in A375 and B16-F10 cells for 72 h. The IC50 of P-SMART was 0.75 μM in two cell lines. B) To determine colony formation of melanoma cells, 250 cells per well were seeded to 6-well culture plates. At 24 h, drug formulations were added and at 7 days, cell colonies were fixed, stained and counted. The long-term treatment of P-SMART resulted in significant anti-proliferative effect. Data represented as the mean ± SEM (n=3). *p<0.05, **p<0.01 compared to Control.
Inhibition of Colony Formation
The inhibitory effect of SMART-OH and P-SMART on tumorigenic potential in melanoma cells was determined by colony formation assay. The parent drug SMART-OH greatly reduced colony formation compared with the control group in both cell lines. Meanwhile, long-term treatment of P-SMART allowed much amount of conjugated drug to be released from the conjugate and then expose to melanoma cells, which resulted in significant anti-proliferative effect. Treatment of A375 with P-SMART at a dose of 75 nM reduced colony formation by 94.5% compared to the control and treatment of B16-F10 cells with 200 nM P-SMART reduced colony formation by 79%. The doses of P-SMART in this assay were far below the IC50 of P-SMART in cytotoxicity assay (Figure 3B).
Inhibition of Cell Invasion
We also determined the inhibitory effect of SMART-OH and P-SMART on cell invasion using B16-F10 cells. At a dose of 0.5 μM, both parent drug and prodrug showed effective inhibition of cell invasion. Treatment of SMART-OH suppressed 87% of cell invasion while the ability of P-SMART to prevent cell invasion was slightly less with 73% of cell invasion blocked at 24 h (Figure 4).
Figure 4. Cell invasion assay.

B16-F10 cells were treated with either SMART-OH (0.5 μM) or P-SMART (0.5 μM, equivalent to parent drug) and allowed to invade through Matrigel for 24 h. Results were shown as mean number of invaded cells ± SEM (n=3). Treatment of SMART-OH suppressed 87% of cell invasion and P-SMART blocked 73% of cell invasion. *p<0.05, **p<0.01 compared to Control.
Effect of P-SMART on cell cycle and apoptosis
The effect of P-SMART on cell cycle and apoptosis was determined by PI-staining using A375 and B16-F10 cells. There was observable G2/M phase arrest after treatment of these cells with P-SMART for 48 h and the % of cells in G2/M phase was augmented in a dose dependent manner. Specifically, the number of A375 cells increased from 14.9% in the control group to 19.1% with 1 μM of P-SMART and to 39.0% with 1.5 μM of P-SMART (Figure 5A). Similarly, the % of B16-F10 cells increased from 11.3% in the control group to 22.9% with 1.5 μM of P-SMART and to 53.0% with 2 μM of P-SMART (Figure 5B). In addition, the number of A375 cells in sub-G1 phase elevated from 0.04% in the control group to 24.0% with 1.5 μM of P-SMART (Figure 5A). The number of B16-F10 cells in sub-G1 phase increased from 5.2% in the control group to 28.3% with 2 μM of P-SMART (Figure 5B). The accumulation of cell population in sub-G1 phase indicated that apoptotic cells significantly increased after P-SMART treatment.
Figure 5. Cell cycle analysis and apoptosis of P-SMART.

Cells were treated with P-SMART for 48 h, stained with propidium iodide (PI), and analyzed on a flow cytometer. A) A375. B) B16F10. Results were expressed as the mean ± SEM (n=3). The percent of cells in G2/M phase and sub-G1 phase was augmented in a dose dependent manner after treatment with P-SMART.
In Vivo Efficacy in B16-F10 Lung Metastatic Mouse Model
We successfully established a metastatic melanoma model in 8-week-old female C57BL/6 albino mice by injecting B16-F10-luc cells via tail vein. At day 10, all mice were imaged for luciferase bioluminescence to determine the tumor growth rate. The mice whose bioluminescent radiance reached 105 were randomized into three groups: 1) control, 2) SMART-OH and 3) P-SMART. All treatments were administered intravenously at the equivalent dose of 20 mg/kg SMART-OH. Both parent drug and prodrug groups showed inhibition of tumor growth compared with the control group. Significantly higher tumor growth inhibition was observed in the group treated with P-SMART compared with the group treated with SMART-OH (Figure 6 and 7A). In addition, treatment with P-SMART significantly reduced the number of lung tumor nodules compared to the control and SMART-OH groups (Figure 7B–C). The survival study showed that the median survival was 28 days in the control group and 31 days in the SMART-OH group. The median survival was significantly prolonged (38 days) when mice were treated with P-SMART (Figure 7D).
Figure 6. In vivo representative bioluminescent images at first day and last day of treatments.

Mice (n=5) from Control (saline), SMART-OH and P-SMART groups were taken bioluminescent images every alternate day during the treatment. Images of four mice from each group were shown.
Figure 7. In vivo efficacy of SMART-OH and P-SMART in B16-F10 lung metastatic animal model.

Mice received saline, 20 mg/kg SMART-OH or 20 mg/kg P-SMART intravenously once every three days for a total of five times when the radiance of tumor had reached 105 (day 10 after tumor implantation). A) Radiance intensity plot of all groups was measured from day 10 to day 24. Data represented as the mean ± SEM (n=5). B) Representative tumors of each group were excised after sacrificing the mice at the end of the efficacy study. Significantly higher tumor growth inhibition and less number of lung tumor nodules was observed in the group treated with P-SMART compared to SMART-OH treated group. C) The weight of mouse lungs from each group was measured at the end of the study. D) Survival analysis of control group, SMART-OH group and P-SMART group. The median survival was 31 days in SMART-OH group and 38 days in P-SMART group while in control group median survival was 28 days.
Hematoxylin and eosin (H&E) stain of lung tissues confirmed the extensive metastasis throughout the lung lobe in the control group and the inhibition of metastasis and proliferation of tumor cells in the treated groups. As compared with the control and parent drug groups, the lung samples from P-SMART treated group exhibited alveolar lumen with limited mass of metastatic cells (Figure 8A). Furthermore, cleaved caspase-3 stain indicated the induction of significant apoptosis by treatment of P-SMART compared to the treatment of SMART-OH (Figure 8B). Additionally, the chronic toxicities of these treatments were also evaluated by histological analysis of the major organs (Figure 8C). No obvious histological changes were observed in the livers, spleens, kidneys and hearts from all the treated groups, which suggested that mice tolerated all treatments well.
Figure 8. Analysis of lung samples by hematoxylin and eosin (H&E), Caspase 3 stain and analysis of major organs by H&E stain.

Lung samples from control, SMART-OH and P-SMART treated groups were excised, fixed and immunostained for A) H&E and B) Caspase 3. In control group the metastasis was throughout the lung lobe but in the treated groups the metastasis and proliferation of tumor cells was inhibited. Cleaved caspase-3 stain indicated significant the induction of apoptosis by P-SMART. C) Organ samples from three groups were excised, fixed and stained for H&E. No obvious histological changes were observed in the livers, spleens, kidneys and hearts from all the treated groups.
Discussion
Conventional therapies of melanoma such as dacarbazine (DTIC) and combination of paclitaxel and carboplatin are small molecular weight drugs, which are rapidly eliminated from the circulation, requiring frequent dosing, leading to increased risk of side effects. From a clinical standpoint, many anticancer agents that are hydrophobic require appropriate drug delivery system to help them reach tumors after systemic administration. To solve this problem, hydrophobic drugs are encapsulated into liposomes, nanoparticles and micelles. However, physical encapsulation into these nanoparticulate systems usually results in fast drug release with burst effect. This means higher initial drug loading is needed because most drugs should ideally be released at the disease site to reduce their adverse effects. In contrast, chemical conjugation of drugs to polymers or lipids prevents immediate drug release during the transportation of polymer-drug conjugates and provides long-term sustained drug release (Figure 2D). Therefore, conjugation of a drug to the polymer prolongs drug circulation and enhances therapeutic efficacy. So far polymer-drug conjugates with linear backbone have undergone clinical evaluation, such as poly(ethylene glycol) (PEG), poly[N-(2-hydroxypropyl)methacrylamide] (PHPMA) copolymers, dextran and poly(glutamic acid) (PGA) [17–19].
We previously physically encapsulated SMART-100 into poly(ethylene glycol)-b-poly(D,L-lactide) (PEG-PLA) micelles by film dispersion.[12] As expected, drug loading in these physically drug encapsulated micelles was 1.5%. To increase drug loading, in previous studies we conjugated gemcitabine or paclitaxel to mPEG-b-PCC, and in the current study we conjugated a novel microtubule inhibitor, SMART-OH, to the copolymer.
PEG was used as a hydrophilic block for synthesizing methoxy-poly (ethylene glycol)-block-poly (2-methyl-2-carboxyl-propylene carbonate-graft-dodecanol) copolymer. To form micelles, we need to maintain a delicate balance between hydrophilic and lipophilic components of the copolymer. Since the molecular weight of our polymer is in the range of 10,000–11,000 Da, we chose PEG of 5000 Da for synthesizing this polymer before conjugating our drug SMART-OH. If we use PEG of 2,000 Da, we could not have conjugated SMART-OH as much as what we have done in this work, otherwise no micelles could be formed. This PEG length helps us maintain higher HLB easily and allows us conjugating large amount of SMART-OH. The attachment of dodecanol (DC) also enhanced requisite hydrophobicity to form micelles of polymer-drug conjugate. Our polymer-drug conjugation system offers the following distinct advantages: a) PEG corona on the polymer imparts stealth property; b) conjugation ensures in vivo stability and no premature drug release in the circulation; c) small size of this conjugate facilitates the EPR effect to maximize drug delivery to the tumor. Therefore, these polymer-drug conjugate showed increased stability and antitumor effect compared to parent drugs.
Microtubule targeting agents that alter microtubule dynamics have been developed as anticancer drugs for more than several decades, and they have achieved exceptional clinical success acting as essential roles in combination therapy and adjuvant therapy [20–22]. These compounds are currently classified as microtubule stabilizers and destabilizers based on their function on microtubule mass at high concentrations. Stabilizing agents such as taxanes, epothilones, discodermolide, laulimalide, peloruside A, etc., enhance tubulin polymerization and block microtubule dynamics [23]. On the other hand, microtubule-destabilizing agents suppress tubulin polymerization and can be further characterized into two groups: vinca-domain binders and colchicine-domain binders. Although the wide application of microtubule inhibitors is observed, there is urgent need to overcome several emerging challenges including drug-resistance and neurotoxicity [24,25].
In this study, we synthesized a novel microtubule destabilizer, SMART-OH, with a hydroxyl group for conjugation with the copolymer (Figure 1A). Molecular modeling suggested strong interactions between SMART-OH and the tubulin dimer, with the phenolic moiety forming a strong hydrogen bond interaction to Val315 in tubulin, in addition to the other two hydrogen bonds and hydrophobic interactions between SMART-OH and tubulin dimer (Figure 1B). Further, in vitro tubulin polymerization assay confirmed experimentally that SMART-OH effectively disrupted tubulin polymerization, serving as a potent microtubule-targeting agent (Figure 1C). It is also known that microtubule-targeting agents suppress microtubule dynamics leading to cell cycle arrest at the mitotic phase. In cell cycle analysis, cells were arrested in G2/M phase after treatment with P-SMART (Figure 5), which confirmed that the mechanism of action of P-SMART was through destabilization of microtubules. Treatment of P-SMART also resulted in cell accumulation in sub-G1 phase indicating cell apoptosis and DNA damage was induced by P-SMART treatment.
In addition to its effects on microtubules, P-SMART demonstrated other anticancer activities in cytotoxicity, colony formation and cell invasion assays. As expected, P-SMART resulted in lower toxicity in A375 and B16-F10 cells when compared with the parent drug SMART-OH in a dose dependent manner. The IC50 of P-SMART was 0.75 μM in both cell lines which was about 10-fold higher than SMART-OH in A375 cells and 4-fold higher in B16-F10 cells (Figure 3A). This finding is in good agreement with previous reports suggesting that conjugated anticancer drugs have higher IC50 than their corresponding parent drugs due to slow drug release kinetics [26,27]. Unlike cytotoxicity assay, SMART-OH and P-SMART showed nearly equivalent activity in the colony formation assays (Figure 3B) likely due to the 7-day incubation period, which provided P-SMART with more time for cellular uptake and drug release. Therefore, the difference in anticancer effect between free drug and conjugated drug was reduced and this result demonstrated that the potency of SMART-OH was maintained after conjugation. As discussed above, melanoma has high metastatic propensity and easily metastasizes to other organs. Thus, we did a transwell invasion assay to determine whether SMART-OH and P-SMART can impede the migratory potential and the invasive property of melanoma cells. Both SMART-OH and P-SMART showed significant cell invasion inhibition (Figure 4), which confirmed this drug had good anti-metastatic properties in vitro.
To investigate in vivo efficacy of SMART-OH and P-SMART, B16-F10-luc cells were selected to establish a metastatic model in C57BL/6 albino mice. We chose 20 mg/kg as the dose of SMART-OH and P-SMART based on our previous studies. We plan to do dose escalation studies and will report in our future publication. Both treated groups exhibited promising tumor inhibitory results. However, P-SMART treatment showed further enhanced reduction in tumor growth rate and tumor size compared with SMART-OH treatment (Figures 6–7). Apart from tumor growth suppression, P-SMART also extended mouse survival compared to other groups. In addition, a significantly lower burden of metastatic cells and increased level of apoptotic cells were found in P-SMART group. H&E stain of vital organs demonstrated our formulation carrying P-SMART was well tolerated, as other healthy organs did not show obvious histological changes after treatments (Figure 8). This is in agreement with our recent report indicating that our biodegradable copolymer mPEG-b-PCC as the backbone of delivery system has less toxicity and good safety [28].
Conclusion
We have synthesized a novel microtubule destabilizer SMART-OH and its corresponding polymer-drug conjugate P-SMART. Our results demonstrate that SMART-OH binds to microtubules and suppresses tubulin polymerization. Both SMART-OH and P-SMART inhibit in vitro proliferation and invasion of melanoma cells. When tested in vivo, P-SMART treatment shows increased anticancer efficacy in a melanoma model with lung metastases compared to the control and SMART-OH treatment. Future work to fight with metastatic melanoma will focus on improving the potency of novel microtubule inhibitors and optimization of our delivery system by including targeting moieties.
Acknowledgments
This work was supported by the National Institutes of Health (R01CA148706 to WL and DDM) and the Faculty Start-up fund from the University of Nebraska Medical Center to RIM.
We would like to acknowledge the National Institutes of Health (NIH) (R01CA148706 to WL and DDM) and the Faculty Start-up fund from the University of Nebraska Medical Center to RIM for financial support.
Footnotes
Conflict of interest: none
Author contributions
Ruinan Yang, Goutam Mondal, Vaibhav Mundra and Ram Mahato designed the study. Ruinan Yang carried out all experimental work with assistance from Goutam Mondal, Rachel A. Ness, and Kinsie Arnst, and under guidance from Ram Mahato, Wei Li, Duane D. Miller and Vaibhav Mundra. Ruinan Yang, Ram Mahato, Rachel A. Ness and Wei Li wrote the manuscript. Ram Mahato is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jatzkewitz H. Peptamin (glycyl-L-leucyl-mescaline) bound to blood plasma expander (polyvinylpyrrolidone) as a new depot form of a biologically active primary amine (mescaline) Z Naturforsch. 1955;10:27–31. [Google Scholar]
- 2.Ringsdorf H. Structure and properties of pharmacologically active polymers. 1975;51:135–153. [Google Scholar]
- 3.Garbe C, Eigentler TK, Keilholz U, Hauschild A, Kirkwood JM. Systematic review of medical treatment in melanoma: current status and future prospects. Oncologist. 2011;16:5–24. doi: 10.1634/theoncologist.2010-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coit DG, Andtbacka R, Bichakjian CK, Dilawari RA, Dimaio D, Guild V, Halpern AC, Hodi FS, Kashani-Sabet M, Lange JR, Lind A, Martin L, Martini MC, Pruitt SK, Ross MI, Sener SF, Swetter SM, Tanabe KK, Thompson JA, Trisal V, Urist MM, Weber J, Wong MK. NCCN Melanoma Panel, Melanoma. J Natl Compr Canc Netw. 2009;7:250–275. doi: 10.6004/jnccn.2009.0020. [DOI] [PubMed] [Google Scholar]
- 5.Eigentler TK, Caroli UM, Radny P, Garbe C. Palliative therapy of disseminated malignant melanoma: a systematic review of 41 randomised clinical trials. Lancet Oncol. 2003;4:748–759. doi: 10.1016/s1470-2045(03)01280-4. [DOI] [PubMed] [Google Scholar]
- 6.Rao RD, Holtan SG, Ingle JN, Croghan GA, Kottschade LA, Creagan ET, Kaur JS, Pitot HC, Markovic SN. Combination of paclitaxel and carboplatin as second-line therapy for patients with metastatic melanoma. Cancer. 2006;106:375–382. doi: 10.1002/cncr.21611. [DOI] [PubMed] [Google Scholar]
- 7.Hauschild A, Agarwala SS, Trefzer U, Hogg D, Robert C, Hersey P, Eggermont A, Grabbe S, Gonzalez R, Gille J, Peschel C, Schadendorf D, Garbe C, O’Day S, Daud A, White JM, Xia C, Patel K, Kirkwood JM, Keilholz U. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J Clin Oncol. 2009;27:2823–2830. doi: 10.1200/JCO.2007.15.7636. [DOI] [PubMed] [Google Scholar]
- 8.Chen ZS, Hopper-Borge E, Belinsky MG, Shchaveleva I, Kotova E, Kruh GD. Characterization of the transport properties of human multidrug resistance protein 7 (MRP7, ABCC10) Mol Pharmacol. 2003;63:351–358. doi: 10.1124/mol.63.2.351. [DOI] [PubMed] [Google Scholar]
- 9.Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8:411–424. doi: 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- 10.Risinger AL, Jackson EM, Polin LA, Helms GL, LeBoeuf DA, Joe PA, Hopper-Borge E, Luduena RF, Kruh GD, Mooberry SL. The taccalonolides: microtubule stabilizers that circumvent clinically relevant taxane resistance mechanisms. Cancer Res. 2008;68:8881–8888. doi: 10.1158/0008-5472.CAN-08-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu Y, Li CM, Wang Z, Ross CR, 2nd, Chen J, Dalton JT, Li W, Miller DD. Discovery of 4-substituted methoxybenzoyl-aryl-thiazole as novel anticancer agents: synthesis, biological evaluation, and structure-activity relationships. J Med Chem. 2009;52:1701–1711. doi: 10.1021/jm801449a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li F, Lu Y, Li W, Miller DD, Mahato RI. Synthesis, formulation and in vitro evaluation of a novel microtubule destabilizer, SMART-100. J Control Release. 2010;143:151–158. doi: 10.1016/j.jconrel.2009.12.028. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Ahn S, Wang J, Lu Y, Dalton JT, Miller DD, Li W. Discovery of novel 2-aryl-4-benzoyl-imidazole (ABI-III) analogues targeting tubulin polymerization as antiproliferative agents. J Med Chem. 2012;55:7285–7289. doi: 10.1021/jm300564b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang DJ, Wang J, Li W, Miller DD. Structural Optimization of Indole Derivatives Acting at Colchicine Binding Site as Potential Anticancer Agents. ACS Med Chem Lett. 2015;6:993–997. doi: 10.1021/acsmedchemlett.5b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao M, Ahn S, Wang J, Chen J, Miller DD, Dalton JT, Li W. Discovery of 4-Aryl-2-benzoyl-imidazoles as tubulin polymerization inhibitor with potent antiproliferative properties. J Med Chem. 2013;56:3318–3329. doi: 10.1021/jm4001117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li F, Danquah M, Mahato RI. Synthesis and characterization of amphiphilic lipopolymers for micellar drug delivery. Biomacromolecules. 2010;11:2610–2620. doi: 10.1021/bm100561v. [DOI] [PubMed] [Google Scholar]
- 17.Hu X, Jing X. Biodegradable amphiphilic polymer-drug conjugate micelles. Expert Opin Drug Deliv. 2009;6:1079–1090. doi: 10.1517/17425240903158917. [DOI] [PubMed] [Google Scholar]
- 18.Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 19.Greco F, Vicent MJ. Combination therapy: opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv Drug Deliv Rev. 2009;61:1203–1213. doi: 10.1016/j.addr.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 20.Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Risinger AL, Giles FJ, Mooberry SL. Microtubule dynamics as a target in oncology. Cancer Treat Rev. 2009;35:255–261. doi: 10.1016/j.ctrv.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stanton RA, Gernert KM, Nettles JH, Aneja R. Drugs that target dynamic microtubules: a new molecular perspective. Med Res Rev. 2011;31:443–481. doi: 10.1002/med.20242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buey RM, Barasoain I, Jackson E, Meyer A, Giannakakou P, Paterson I, Mooberry S, Andreu JM, Diaz JF. Microtubule interactions with chemically diverse stabilizing agents: thermodynamics of binding to the paclitaxel site predicts cytotoxicity. Chem Biol. 2005;12:1269–1279. doi: 10.1016/j.chembiol.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 24.Chandrasekaran G, Tátrai P, Gergely F. Hitting the brakes: targeting microtubule motors in cancer. Br J Cancer. 2015;113:693–698. doi: 10.1038/bjc.2015.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fanale D, Bronte G, Passiglia F, Calo V, Castiglia M, Di Piazza F, Barraco N, Cangemi A, Catarella MT, Insalaco L, Listi A, Maragliano R, Massihnia D, Perez A, Toia F, Cicero G, Bazan V. Stabilizing versus destabilizing the microtubules: a double-edge sword for an effective cancer treatment option? Anal Cell Pathol (Amst) 2015;2015:690916. doi: 10.1155/2015/690916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larson N, Yang J, Ray A, Cheney DL, Ghandehari H, Kopecek J. Biodegradable multiblock poly(N-2-hydroxypropyl)methacrylamide gemcitabine and paclitaxel conjugates for ovarian cancer cell combination treatment. Int J Pharm. 2013;454:435–443. doi: 10.1016/j.ijpharm.2013.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang L, Lin SW, Dai W, Lu JK, Yang TY, Xiang Y, Zhang Y, Li RT, Zhang Q. Novel cathepsin B-sensitive paclitaxel conjugate: Higher water solubility, better efficacy and lower toxicity. J Control Release. 2012;160:618–629. doi: 10.1016/j.jconrel.2012.02.020. [DOI] [PubMed] [Google Scholar]
- 28.Yang R, Mondal G, Wen D, Mahato RI. Combination therapy of paclitaxel and cyclopamine polymer-drug conjugates to treat advanced prostate cancer. Nanomedicine. 2016 doi: 10.1016/j.nano.2016.07.017. [DOI] [PubMed] [Google Scholar]