

Abstract
Within the past several decades, a brigade of dedicated researchers from around the world has provided essential insights into the critical niche of immune-mediated inflammation in the pathogenesis of age-related macular degeneration (AMD). Yet, the question has lingered as to whether disease-initiating events are more or less dependent on isolated immune-related responses, unimpeded inflammation, endogenous pathways of age-related cell senescence and oxidative stress, or any of the other numerous molecular derangements that have been identified in the natural history of AMD. There is now an abundant cache of data signifying immune system activation as an impetus in the pathogenesis of this devastating condition. Furthermore, recent rigorous investigations have revealed multiple inciting factors, including several important complement-activating components, thus creating a new array of disease-modulating targets for the research and development of molecular therapeutic interventions. While the precise in vivo effects of complement activation and inhibition in the progression and treatment of AMD remain to be determined, ongoing clinical trials of the first generation of complement-targeted therapeutics are hoped to yield critical data on the contribution of this pathway to the disease process.
History
The dysregulation of immunologic responses in the pathogenesis of retinal diseases, in particular age-related macular degeneration (AMD), has long been under investigation, but it is only recently that specific molecular mechanisms have been identified [1]. The phalanx of mammalian immunity is controlled by two intercalated pathways: the innate and adaptive (or acquired) systems. Innate immunity is simply defined as the immediate nonspecific host response upon initial encounter with the pathogen. There is mounting evidence that immune complement, a major humoral component of the innate immune system, plays a major role in AMD pathobiology [2]. Immune complements mediate the opsonization and clearance of pathogens through the classical, alternative and lectin pathways, all of which feed into a common terminus (fig. 1). The alternative and lectin pathways differ from the classical pathway in that no specific antibody-antigen interaction is required for complement-mediated cell lysis.
Fig. 1.

There are three pathways to complement activation. To date, complement activation is known to occur through the classical, alternative and lectin pathways. These inter-related molecular cascades share their ability to induce the assembly of C3 convertase, an enzyme that cleaves C3 to its active by products, C3a and C3b. C3 activation channels into a terminal pathway via C5 that leads to the assembly of the membrane attack complement. Classical complement activation is initiated by antibody/antigen recognition, C1 binding and formation of classical C3 convertase, C4bC2a. Conversely, the alternative pathway does not require immunoglobulin and is perpetuated by the binding of C3b to microorganisms and cell surfaces in order to join with cleaved factor B, Bb, and form C3bBb. This complex serves as the alternative form of C3 convertase and regenerates C3b thereby creating a feedback amplification loop. The lectin pathway is regulated by the proteases MASP-1 and -2 which interact via mannose-binding lectin-bound microorganisms to aid in the formation of C3 convertase. All three pathways result in robust host responses to increase opsonization, leukocyte chemotaxis and cell lysis.
Inappropriate activation of complement activation occurs in several immunovascular diseases, including glomerulonephritis and rheumatoid arthritis. This immune-mediated inflammation ultimately serves to repair damaged tissue via cellular remodeling, neovascularization and scar formation. However, when these processes beset the specialized tissues of the eye, they often result in significant vision loss due to hyperplasia of the retinal pigment epithelium (RPE), choroidal neovascularization (CNV), and disciform scarring. Drusen, the hallmark of AMD, actually harbor many of these activated complement factors and other inflammatory mediators that upregulate cytokines critical for CNV [3]. As clinicians and scientists have long suspected, there is now convincing evidence that drusen may not simply precede the pathogenic growth of choroidal blood vessels but actually promote it through specific complement-related mechanisms that are discussed in further detail below.
Key Concepts in Complement Biology
Excessive complement activation through the alternative pathway is proposed to significantly contribute to the progression of AMD [4]. There are several critical complement-mediated interactions which drive the feed-forward amplification of the alternative pathway (fig. 2). Firstly, factor C3b binds to a diverse array of proteins and polysaccharides present on pathogen membranes [5]. C3b is created upon cleavage of C3 in a reaction carried out by C3 convertase. This enzyme may be formed as a complex of C2a and C4b in the classical or lectin pathways or in the alternative pathway as a complex of the factor D cleaved component of factor B (Bb), properdin (factor P) and hydrolyzed C3 [6]. The different forms of C3 convertase channel into a common pathway that supplies the complement cascade with ample C3b, as well as C3a and C5a, both potent anaphylatoxins that mediate leukocyte chemotaxis. It is important to note that the C3a/C5a ligand receptors, C3aR and C5aR, are not only present on circulating proinflammatory leukocytes and mast cells, but also within the retinal nerve fiber and inner plexiform layers [7]. These findings suggest that in situ C3a/C5a generation may directly induce cellular alterations in neural retinal elements independent of leukocyte mediation. In the common terminal pathway, a secondary product of C5 cleavage, C5b, assembles with complement factors 6–9 to form the membrane attack complement which destroys cells to which it binds. An endogenous soluble complement inhibitor, complement factor H (CFH), is also present and capable of inactivating C3b through factor I binding [8] thus preventing complement-mediated host cell death in tissues. CFH is expressed in the RPE and choroid [9], and is hypothesized to damper complement-mediated inflammation.
Fig. 2.

In the alternative pathway of complement activation, C3 convertase is spontaneously produced by hydrolysis of C3 to form C3(H2O) in a process referred to as ‘tickover’. If CFH is not sufficient to inhibit cell surface binding, then C3(H2O) will bind with Bb to form C3 convertase resulting in the activation of the downstream complement cascade. MAC= Membrane attack complement.
Previously, activation of the classical pathway was not purported to be a major contributory factor in AMD, as little evidence of antibody-mediated complement fixation or localization of specific classical pathway components existed in vivo [10, 11]. However, there are now several studies showing genetic association data with classical pathway factors that are mentioned in the next section. Mediators of the lectin pathway, namely the mannose-binding lectin-associated proteases (MASP-1 and -2), have also been investigated as potential disease modulators of AMD progression and pathologic neovascularization. As modern research strives to unravel the complex interactions of the various complement pathways, the number of potential therapeutic targets for immune-related diseases in the eye and elsewhere will continue to rise. Yet, with only a little more than a decade worth of advanced biomedical and translational research focused on this problem, there are already a multitude of major molecular discoveries that have propelled the development of specific complement factor-targeted interventions currently in clinical trials.
Pathogenic Mechanisms and Metabolism
Our understanding of molecular mechanisms of AMD pathogenesis have undergone a considerable shift with numerous laboratories and pharmaceutical ventures now studying the specific mechanisms, genome-wide associations and effects of excessive complement activation. The pioneering studies linking AMD and complement activation found evidence of premature drusen deposition in patients with membranoproliferative glomerulonephritis type II [12, 13], a disease associated with mutations in the endogenous complement inhibitor, CFH.
CFH quickly advanced to the frontier of AMD biology after a group of independent studies simultaneously reported on a panel of single nucleotide polymorphisms (SNPs) within the human CFH gene that dramatically increase the AMD risk [9, 14–16]. Overall, these studies suggested that CFH accounts for approximately 50% of all cases of AMD. One of the identified mutations, the Y402H variant, was shown to increase the AMD risk nearly 3-fold in patients of European descent. The physiologic effects of the Y402H mutation may be related to a reduction in the heparin binding affinity of CFH on cell membranes thereby suppressing C3b inhibition and fueling alternative complement pathway activation [17]. Y402H has also been linked to other downstream molecular events, including decreased neutralization of the proinflammatory lipid peroxidation product, malondialdehyde, leading to IL-8 and TNF-α induction [18]. C-reactive protein (CRP), an acute-phase protein and commonly employed systemic marker of inflammation that assists in complement-mediated phagocytosis, was elevated in the blood of patients with AMD [19], but further data were contradictory [20] perhaps due to patient variability. CRP was elevated in the choroids of patients that are homozygous for the CFH risk allele, 402HH [21], and protein interaction studies of CFH demonstrate that CRP is a binding partner [22], a function which is significantly reduced with the 402HH gene product [23, 24]. The wide spectrum of biochemical interactions with CFH has continued to grow, with recent evidence demonstrating that a critical short consensus repeat region (SCR7) binds with fibulin-3 (EFEMP-1, 2p16), a protein found in drusen and an established locus for an inherited form of familial drusen [25]. Further data revealed that exposure of human RPE cells harboring pathogenic Cfh mutations to bis-retinoids found in the outer segments of diseased photoreceptors resulted in significantly more complement activation than wild-type Cfh cells [26]. The functional association of CFH with AMD is further complicated by its alternative splicing to FHL-1 (factor H-like 1) which also encodes SCR7 [27] as well as flanking related genes, CFHR1 and CFHR3, which are linked to AMD pathogenesis [28]. Many investigators have utilized mouse models in order to improve our basic understanding of alternative complement activation in retinal homeostasis and in the progression of atrophy and CNV formation.
Interestingly, the first mouse models of CFH deficiency did not reveal a robust phenotype and showed limited structural abnormalities which occur in the human condition of AMD [29]. This weak phenotype may be explained by biological differences between total gene deletion versus expression of the mutant Cfh gene as well as divergences in human and mouse complement function. Aged RPE contained less CFH, a process postulated to be related to the photooxidation of photoreceptor outer segments and the generation of reactive oxygen species, both of which are known to occur in AMD [30]. In further studies, aged Cfh-deficient mice (up to 2 years) demonstrated pathologic features such as increased formation of subretinal and mesangial deposits [31] with retinal vasculature significantly attenuated at 1 year [32]. Aged Cfh-deficient mice fed a high glycemic index diet developed AMD-like features, including basal laminar deposits, RPE de-pigmentation and vacuolation [33]. An important observation in another study of aged Cfh null mice showed that concomitant loss of C3 worsened retinal function with enhanced deposition of inflammatory markers [34]. These data suggest that over time unimpeded alternative pathway activation may promote RPE degeneration and pathologic angiogenesis, but that blockage via C3 inhibition may not be beneficial in patients with pathogenic Cfh mutations.
As of 2015, data on the importance of CFH in AMD pathogenesis have been validated in numerous cohorts building a significant case for the potential of therapeutic blockade of complement activation in the treatment of this disease. C3 and CFH SNPs showed significant environmental interactions with odds ratios for current smokers’ rising to nearly 10 with those homozygous of C3 SNP and even higher for Y402H [35]. Recent data highlight the importance of cigarette smoke-induced oxidative stress in RPE cells with accumulation of inflammatory lipid deposits and excessive alternative complement activation in cell culture and animal models [36, 37]. Subsequent studies confirmed the increased prevalence of AMD in carriers of the CFH variants [38–40] and also identified SNPs in the C2, C3, and complement factor B (CFB) and I (CFI) genes that were associated with AMD [41–43]. Cfb expression is increased in aged RPE cells with strong basal localization along with enhanced deposition of C3/C3a [44]. A major question in complement biology is which specific pathways are required and active during AMD progression. Data demonstrating the genetic association of an SNP of C1 inhibitor (also known as SERPING1) with AMD reinvigorated the hypothesis that classical complement activation is at least partially contributory to AMD progression [45]. In support of this theory, experiments with an established model of CNV formation with laser injury in mice revealed that the presence of a functional alternative pathway is essential for promotion of angiogenesis but is not independently sufficient to induce the phenotype [46, 47]. Conditional reconstitution of functional Cfb in a deficient transgenic mouse strain similarly increased CNV size in the laser model. Collectively, these data are interpreted to hypothesize that the alternative complement activation is a critical component in this accelerated animal model of CNV formation [48]. This has led other research groups to rigorously dissect the components of alternative pathway signaling in AMD to identify other potential targets. Complement factor D (CFD), a self-inhibited serine protease involved in alternative pathway activation, was linked to AMD in a single study but has not been independently confirmed [49]. Less-studied regulators of complement activation are being investigated with some encouraging results, including CD59, which exhibited altered expression levels in animal models and human eyes with AMD but without evidence of any SNP associations [50–52]. Most importantly, the clinical significance of this strong genetic signature of complement activation in AMD is now being translated in early and advanced human trials targeting various alternative pathway components with next-generation biologics.
An area of critical scientific work is linking complement activation to specific molecular events in order to determine the pathologic effects of complement activity in AMD. A widely reported hypothesis is that the formation of inflammatory complement byproducts, most notably, C3a and C5a, are responsible of instigating AMD progression. These potent chemoattractant factors were specifically localized to AMD drusen compared to non-AMD peripheral drusen from aged human eyes [53]. In the same study, C3a and C5a upregulated the expression of VEGF-A, a cytokine required for CNV progression. In vitro, primary human RPE cell isolates treated with C3a or C5a for 8 h secreted significantly more VEGF-A in both confluent and subconfluent cultures (correlates of healthy and degenerative RPE states, respectively) [53]. Whereas an immortalized RPE cell line, D407, exhibited similar responses after C3a/C5a stimulation, primary human choroidal endothelial cells did not. In vivo, VEGF-A concentrations in RPE/choroid peaked just 4 h after intravitreous C3a/C5a administration in a dose-dependent fashion demonstrating the tissue-localized and rapid response to these anaphylatoxins. The temporal kinetics of this swift inflammatory reaction suggests that RPE cells, and likely other resident and infiltrating cells, play a major role in VEGF-A production after complement activation (fig. 3). Further investigations demonstrated that exposure of human RPE cells to oxidative stress induced complement activation, subsequent upregulation of matrix metalloproteases (MMP-2/MMP-9) and VEGF-A secretion [54]. While these effects are observed in both cell culture and animal models, several lines of evidence have pointed to the significance of infiltrating cell types in the amplification of cytokine expression with complement activation in vivo. Retinal microglia expressed increased C3 in both phototoxicity and aging models [55, 56], and exposure of RPE to supernatant from activated macrophages hugely elevated CFB and C3 levels [57]. In the mouse model of laser-induced CNV, infiltrating VEGF-A-expressing macrophages were found in areas of pathologic vasculature along with increased deposition of complement components [58]. C3a/C5a levels are selectively increased in the RPE/choroid 12 h after photocoagulation and blockade of C3a and C5a, as in transgenic mice deficient in C3aR and C5aR, VEGF-A production is suppressed both 1 and 3 days after laser treatment. Given that VEGF-A production peaks at 3 days in this animal model, a time point which coincides with maximal macrophage infiltration, implementing a strategy to neutralize C3a/C5a signaling may also significantly inhibit leukocyte trafficking. In corroboration with this hypothesis, C3aR/C5aR null mice exhibited decreased neutrophils and macrophages in RPE/choroid tissues after laser injury resulting in decreased CNV formation. In conclusion, the amplified generation of C3a and C5a fragments during complement activation and the rapid downstream upregulation of VEGF-A may explain the abrupt switch-like progression to neovascular AMD in patients with previously dry fundus examinations. In summary, this expanding cache of data supports the direct response of complement production on proangiogenic cytokine secretion in the retina and a strong paracrine effect between microglia and activated macrophages and RPE further promoting a pharmacologic strategy of complement inhibition in the treatment of AMD.
Fig. 3.

Drusen promote CNV via C3a/C5a induction of VEGF-A. Complement factors are abundant in drusen and lead to the activation of numerous inflammatory pathways. C3a and C5a upregulate RPE production of VEGF-A and MMPs which are able to stimulate choroidal endothelial cells to proliferate, migrate and form leaky vasculature beneath Bruch’s membrane, as in occult CNV, or breakthrough to sub-RPE space and neural retina layers, as in classical CNV. Activated macrophages and microglia also express complement components and secrete additional proinflammatory cytokines that promote pathologic angiogenesis.
Implications for Retinal Pharmacotherapy
Even though the field of complement biology within the domain of ophthalmic science is relatively young, a host of new and established pharmaceutical companies are targeting this pathway with novel molecular therapeutics (fig. 4). Already in the past 5 years, there has been a massive shift in the complement pharmaceutical space with numerous consolidations, failed ventures and negative results. Given the provocative genetic association of hypomorphic CFH SNPs and AMD, many drug development efforts focused on strategies to restore CFH or suppress alternative pathway activity. Several early stage companies focused on the synthesis and delivery of recombinant human CFH, yet no clinical trials in AMD have been initiated to date. One drug design (TT30/ALXN1102) consisted of a complement receptor (CR2) fused to a CFH fragment and was in a trial for the treatment of another complement-mediated disease, paroxysmal nocturnal hemoglobinuria, until being recently terminated [59]. There are currently no other active efforts for CFH replacement therapy.
Fig. 4.
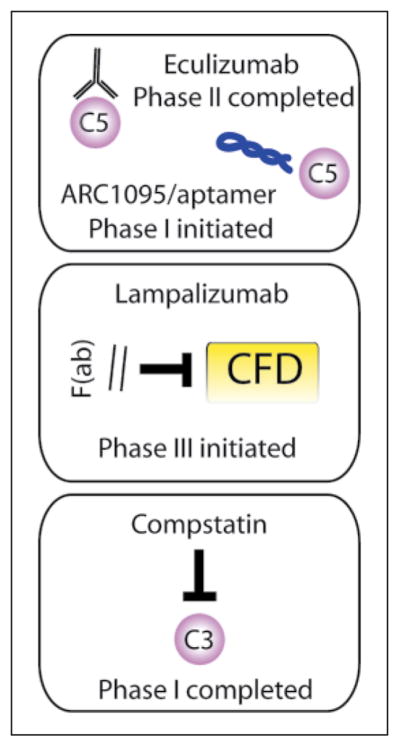
Complement factors are targets for pharmacologic inhibition in retinal disease. Numerous therapeutic targets for complement pathway inhibition are investigated in preclinical and clinical investigations. Alexion engineered a whole IgG humanized antibody (eculizumab) against C5 which was tested in a phase II trial with negative results. Ophthotech developed a pegylated RNA aptamer (ARC1905/Zimura®) that also neutralizes C5 and is currently in a phase I trial for the combination treatment of polypoid choroidal vasculopathy. Potentia revived a previously described small-molecule inhibitor of C3, compstatin, which completed phase I trials for the treatment of wet AMD with no preliminary signature of efficacy. Genentech has advanced the anti-CFD antibody fragment, lampalizumab, to phase III trials for the treatment of geographic atrophy; 1-year results are anticipated in early 2016 for this important trial.
Given that most complement activity channels through C3 and C5, these factors have provided two major targets for intervention. In 2007, the first FDA-approved complement inhibitor reached the market, an anti-C5 humanized monoclonal antibody (eculizumab, Solaris™) developed by Alexion for the treatment of paroxysmal nocturnal hemoglobinuria. In 2014, data were presented from a phase II trial comparing the efficacy of eculizumab with placebo in patients with advanced dry AMD or geographic atrophy. Thirty eyes were enrolled in a 2: 1 randomization to high and low doses of C5 antibody treatment, but there were no significant changes in expansion of atrophy at the 6-month endpoint [60]. Ophthotech is in phase I trials of its anti-C5 aptamer, ARC1905/Zimura®, to study its safety profile and efficacy when used in combination with anti-VEGF-A antibodies for the treatment of idiopathic polypoid choroidal vasculopathy, a variant of neovascular AMD that is also associated with multiple SNPs in the complement alternative pathway [61, 62]. Most efforts to translate complement inhibition have strategized in favor of targeting C5, as there are no known endogenous C5 inhibitors and activation may occur independent of C3. A single dose-escalation study of a rediscovered small molecule C3 inhibitor, compstatin (POT-4), was studied in a phase I clinical trial in patients with neo-vascular AMD without any preliminary signs of efficacy and no evidence of toxicity in the midrange doses [63]. This compound was not studied further although a second generation C3 inhibitor derived from compstatin (APL-2) may be studied in a follow-up trial.
Other approaches to alternative pathway inhibition have included targeting CFB using a humanized monoclonal antibody, but there are no clinical trials planned at this stage. CFD SNPs were identified in patients with AMD, but this association was mostly found in females and was not observed in additional cohorts [64, 65]. Yet, animal models revealed that CFD inhibition prevented photoreceptor cell death [66] supporting the rationale for ongoing clinical studies of an anti-CFD monoclonal antibody fragment (FCFD4514S/lampalizumab; Genentech). Intra-vitreous injection of lampalizumab showed no retinal toxicity [67], and in a completed phase II study (MAHALO), expansion of RPE atrophy in advanced dry AMD is slowed by 20% with monthly lampalizumab (10 mg) and by over 40% in patients with an associated SNP in Cfi at 18 months [68]. Phase III studies of lampalizumab are now underway comparing sham versus treatment every 4 or 6 weeks in Cfi biomarker-positive and -negative patients with the primary endpoint of aggregate geographic atrophy size measured at 1 year. While there are likely many more startups and lead compounds under investigation at larger pharmaceutical companies, determining the efficacy of complement inhibition in the treatment of neovascular and advanced dry AMD in large clinical trials will be the next major step in improving the molecular and clinical implications of this complex biological pathway within the context of this blinding disease.
Conclusion
Major scientific breakthroughs within the past decade revealed that complement dysregulation may provide a critical component to the inflammatory siege in the pathogenesis of AMD. Many of the specific complement factors responsible for these deleterious effects were identified through genome-wide association and functional studies in vivo. CFH, C2, C3 and CFB have all been genetically tied to AMD; however, the association data for CFH remain the most startling with mutations in this endogenous complement inhibitor accounting for approximately 50% of this disease. C3a and C5a, which are generated as byproducts of the common complement pathway and localized to AMD drusen, induce the expression of VEGF-A which increased the formation of CNV. Pharmacologic inhibition of the alternative complement pathway either via introduction of recombinant complement inhibitors or neutralization of specific complement factors is a valuable strategy to halting this disease. Significant advancements in clinical trials have translated this approach into a potential therapeutic for the treatment of geographic atrophy. In the coming years, fundamental knowledge will be acquired about the role of complement inhibition and the efficacy of the specific inhibitors as more clinical trials are conducted.
References
- 1.Ambati J, Ambati BK, Yoo SH, et al. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003;48:257–293. doi: 10.1016/s0039-6257(03)00030-4. [DOI] [PubMed] [Google Scholar]
- 2.Nussenblatt RB, Ferris F., 3rd Age-related macular degeneration and the immune response: implications for therapy. Am J Ophthalmol. 2007;144:618–626. doi: 10.1016/j.ajo.2007.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donoso LA, Kim D, Frost A, et al. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2006;51:137–152. doi: 10.1016/j.survophthal.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration – emerging pathogenetic and therapeutic concepts. Ann Med. 2006;38:450–471. doi: 10.1080/07853890600946724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sahu A, Lambris JD. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol Rev. 2001;180:35–48. doi: 10.1034/j.1600-065x.2001.1800103.x. [DOI] [PubMed] [Google Scholar]
- 6.Janeway C. Immunobiology. New York: Garland Science; 2005. [Google Scholar]
- 7.Vogt SD, Barnum SR, Curcio CA, Read RW. Distribution of complement anaphylatoxin receptors and membrane-bound regulators in normal human retina. Exp Eye Res. 2006;83:834–840. doi: 10.1016/j.exer.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Józsi M, Oppermann M, Lambris JD, Zipfel PF. The C-terminus of complement factor H is essential for host cell protection. Mol Immunol. 2007;44:2697–2706. doi: 10.1016/j.molimm.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in drusen formation and age related macular degeneration. Exp Eye Res. 2001;73:887–896. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- 11.Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–431. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- 12.Mullins RF, Aptsiauri N, Hageman GS. Structure and composition of drusen associated with glomerulonephritis: implications for the role of complement activation in drusen biogenesis. Eye. 2001;15:390–395. doi: 10.1038/eye.2001.142. [DOI] [PubMed] [Google Scholar]
- 13.Duvall-Young J, MacDonald MK, McK-echnie NM. Fundus changes in (type II) mesangiocapillary glomerulonephritis simulating drusen: a histopathological report. Br J Ophthalmol. 1989;73:297–302. doi: 10.1136/bjo.73.4.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 16.Edwards AO, Ritter R, 3rd, Abel KJ, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 17.Clark SJ, Higman VA, Mulloy B, et al. His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. J Biol Chem. 2006;281:24713–24720. doi: 10.1074/jbc.M605083200. [DOI] [PubMed] [Google Scholar]
- 18.Weismann D, Hartvigsen K, Lauer N, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478:76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seddon JM, Gensler G, Milton RC, et al. Association between C-reactive protein and age-related macular degeneration. JAMA. 2004;291:704–710. doi: 10.1001/jama.291.6.704. [DOI] [PubMed] [Google Scholar]
- 20.McGwin G, Hall TA, Xie A, Owsley C. The relation between C reactive protein and age related macular degeneration in the Cardiovascular Health Study. Br J Ophthalmol. 2005;89:1166–1170. doi: 10.1136/bjo.2005.067397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson PT, Betts KE, Radeke MJ, et al. Individuals homozygous for the age-related macular degeneration risk-conferring variant of complement factor H have elevated levels of CRP in the choroid. Proc Natl Acad Sci U S A. 2006;103:17456–17461. doi: 10.1073/pnas.0606234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giannakis E, Jokiranta TS, Male DA, et al. A common site within factor H SCR 7 responsible for binding heparin, C-reactive protein and streptococcal M protein. Eur J Immunol. 2003;33:962–969. doi: 10.1002/eji.200323541. [DOI] [PubMed] [Google Scholar]
- 23.Laine M, Jarva H, Seitsonen S, et al. Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. J Immunol. 2007;178:3831–3836. doi: 10.4049/jimmunol.178.6.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ormsby RJ, Ranganathan S, Tong JC, et al. The complement factor H Y402H polymorphism associated with age-related macular degeneration affects interactions with multiple ligands. Invest Ophthalmol Vis Sci. 2008;49:1763–1770. doi: 10.1167/iovs.07-1297. [DOI] [PubMed] [Google Scholar]
- 25.Wyatt MK, Tsai JY, Mishra S, et al. Interaction of complement factor H and fibulin3 in age-related macular degeneration. PLoS One. 2013;8:e68088. doi: 10.1371/journal.pone.0068088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Radu RA, Hu J, Jiang Z, Bok D. Bisretinoid-mediated complement activation on retinal pigment epithelial cells is dependent on complement factor H haplotype. J Biol Chem. 2014;289:9113–9120. doi: 10.1074/jbc.M114.548669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Estaller C, Schwaeble W, Dierich M, Weiss EH. Human complement factor H: two factor H proteins are derived from alternatively spliced transcripts. Eur J Immunol. 1991;21:799–802. doi: 10.1002/eji.1830210337. [DOI] [PubMed] [Google Scholar]
- 28.Ansari M, McKeigue PM, Skerka C, et al. Genetic influences on plasma CFH and CFHR1 concentrations and their role in susceptibility to age-related macular degeneration. Hum Mol Genet. 2013;22:4857–4869. doi: 10.1093/hmg/ddt336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coffey PJ, Gias C, McDermott CJ, et al. Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc Natl Acad Sci U S A. 2007;104:16651–16656. doi: 10.1073/pnas.0705079104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen M, Forrester JV, Xu H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp Eye Res. 2007;84:635–645. doi: 10.1016/j.exer.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 31.Ding JD, Kelly U, Landowski M, et al. Expression of human complement factor H prevents age-related macular degeneration-like retina damage and kidney abnormalities in aged CFH knockout mice. Am J Pathol. 2015;185:29–42. doi: 10.1016/j.ajpath.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lundh von Leithner P, Kam JH, Bain-bridge J, et al. Complement factor H is critical in the maintenance of retinal perfusion. Am J Pathol. 2009;175:412–421. doi: 10.2353/ajpath.2009.080927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rowan S, Weikel K, Chang ML, et al. CFH genotype interacts with dietary glycemic index to modulate age-related macular degeneration-like features in mice. Invest Ophthalmol Vis Sci. 2014;55:492–501. doi: 10.1167/iovs.13-12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoh Kam J, Lenassi E, Malik TH, et al. Complement component C3 plays a critical role in protecting the aging retina in a murine model of age-related macular degeneration. Am J Pathol. 2013;183:480–492. doi: 10.1016/j.ajpath.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 35.Seddon JM, Reynolds R, Maller J, et al. Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Invest Ophthalmol Vis Sci. 2009;50:2044–2053. doi: 10.1167/iovs.08-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kunchithapautham K, Atkinson C, Rohrer B. Smoke exposure causes endoplasmic reticulum stress and lipid accumulation in retinal pigment epithelium through oxidative stress and complement activation. J Biol Chem. 2014;289:14534–14546. doi: 10.1074/jbc.M114.564674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, Kondo N, Cano M, et al. Nrf2 signaling modulates cigarette smoke-induced complement activation in retinal pigmented epithelial cells. Free Radic Biol Med. 2014;70:155–166. doi: 10.1016/j.freeradbiomed.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Francis PJ, Schultz DW, Hamon S, et al. Haplotypes in the complement factor H (CFH) gene: associations with drusen and advanced age-related macular degeneration. PLoS One. 2007;2:e1197. doi: 10.1371/journal.pone.0001197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim NR, Kang JH, Kwon OW, et al. Association between complement factor H gene polymorphisms and neovascular age-related macular degeneration in Koreans. Invest Ophthalmol Vis Sci. 2008;49:2071–2076. doi: 10.1167/iovs.07-1195. [DOI] [PubMed] [Google Scholar]
- 40.Souied EH, Leveziel N, Richard F, et al. Y402H complement factor H polymorphism associated with exudative age-related macular degeneration in the French population. Mol Vis. 2005;11:1135–1140. [PubMed] [Google Scholar]
- 41.Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van de Ven JP, Nilsson SC, Tan PL, et al. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat Genet. 2013;45:813–817. doi: 10.1038/ng.2640. [DOI] [PubMed] [Google Scholar]
- 43.Yates JR, Sepp T, Matharu BK, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–561. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 44.Chen M, Muckersie E, Robertson M, et al. Up-regulation of complement factor B in retinal pigment epithelial cells is accompanied by complement activation in the aged retina. Exp Eye Res. 2008;87:543–550. doi: 10.1016/j.exer.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 45.Ennis S, Jomary C, Mullins R, et al. Association between the SERPING1 gene and age-related macular degeneration: a two-stage case-control study. Lancet. 2008;372:1828–1834. doi: 10.1016/S0140-6736(08)61348-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rohrer B, Coughlin B, Kunchithapautham K, et al. The alternative pathway is required, but not alone sufficient, for retinal pathology in mouse laser-induced choroidal neovascularization. Mol Immunol. 2011;48:e1–e8. doi: 10.1016/j.molimm.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bora NS, Kaliappan S, Jha P, et al. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: role of factor B and factor H. J Immunol. 2006;177:1872–1878. doi: 10.4049/jimmunol.177.3.1872. [DOI] [PubMed] [Google Scholar]
- 48.Schnabolk G, Coughlin B, Joseph K, et al. Local production of the alternative pathway component factor B is sufficient to promote laser-induced choroidal neovascularization. Invest Ophthalmol Vis Sci. 2014;56:1850–1863. doi: 10.1167/iovs.14-15910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stanton CM, Yates JR, den Hollander AI, et al. Complement factor D in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52:8828–8834. doi: 10.1167/iovs.11-7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bora NS, Kaliappan S, Jha P, et al. CD59, a complement regulatory protein, controls choroidal neovascularization in a mouse model of wet-type age-related macular degeneration. J Immunol. 2007;178:1783–1790. doi: 10.4049/jimmunol.178.3.1783. [DOI] [PubMed] [Google Scholar]
- 51.Cipriani V, Matharu BK, Khan JC, et al. Genetic variation in complement regulators and susceptibility to age-related macular degeneration. Immunobiology. 2012;217:158–161. doi: 10.1016/j.imbio.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ebrahimi KB, Fijalkowski N, Cano M, Handa JT. Decreased membrane complement regulators in the retinal pigmented epithelium contributes to age-related macular degeneration. J Pathol. 2013;229:729–742. doi: 10.1002/path.4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006;103:2328–2333. doi: 10.1073/pnas.0408835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bandyopadhyay M, Rohrer B. Matrix metalloproteinase activity creates proangiogenic environment in primary human retinal pigment epithelial cells exposed to complement. Invest Ophthalmol Vis Sci. 2012;53:1953–1961. doi: 10.1167/iovs.11-8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rutar M, Valter K, Natoli R, Provis JM. Synthesis and propagation of complement C3 by microglia/monocytes in the aging retina. PLoS One. 2014;9:e93343. doi: 10.1371/journal.pone.0093343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rutar M, Natoli R, Kozulin P, et al. Analysis of complement expression in light-induced retinal degeneration: synthesis and deposition of C3 by microglia/macrophages is associated with focal photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2011;52:5347–5358. doi: 10.1167/iovs.10-7119. [DOI] [PubMed] [Google Scholar]
- 57.Luo C, Zhao J, Madden A, et al. Complement expression in retinal pigment epithelial cells is modulated by activated macrophages. Exp Eye Res. 2013;112:93–101. doi: 10.1016/j.exer.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 58.Liu J, Copland DA, Horie S, et al. Myeloid cells expressing VEGF and arginase-1 following uptake of damaged retinal pigment epithelium suggests potential mechanism that drives the onset of choroidal angiogenesis in mice. PLoS One. 2013;8:e72935. doi: 10.1371/journal.pone.0072935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fridkis-Hareli M, Storek M, Mazsaroff I, et al. Design and development of TT30, a novel C3d-targeted C3/C5 convertase inhibitor for treatment of human complement alternative pathway-mediated diseases. Blood. 2011;118:4705–4713. doi: 10.1182/blood-2011-06-359646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yehoshua Z, de Amorim Garcia Filho CA, Nunes RP, et al. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: the COMPLETE study. Ophthalmology. 2014;121:693–701. doi: 10.1016/j.ophtha.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang L, Li Y, Guo S, et al. Different hereditary contribution of the CFH gene between polypoidal choroidal vasculopathy and age-related macular degeneration in Chinese Han people. Invest Ophthalmol Vis Sci. 2014;55:2534–2538. doi: 10.1167/iovs.13-13437. [DOI] [PubMed] [Google Scholar]
- 62.Hayashi H, Yamashiro K, Gotoh N, et al. CFH and ARMS2 variations in age-related macular degeneration, polypoidal choroidal vasculopathy, and retinal angiomatous proliferation. Invest Ophthalmol Vis Sci. 2010;51:5914–5919. doi: 10.1167/iovs.10-5554. [DOI] [PubMed] [Google Scholar]
- 63.Kaushal S, Grossi F, Francois C, et al. Complement C3 inhibitor POT-4: clinical safety of intravitreal administration. Invest Ophthalmol Vis Sci. 2009;50:5010. [Google Scholar]
- 64.Maloney SC, Antecka E, Orellana ME, et al. Choroidal neovascular membranes express toll-like receptor 3. Ophthalmic Res. 2010;44:237–241. doi: 10.1159/000313989. [DOI] [PubMed] [Google Scholar]
- 65.Haines JL, Schnetz-Boutaud N, Schmidt S, et al. Functional candidate genes in age-related macular degeneration: significant association with VEGF, VLDLR, and LRP6. Invest Ophthalmol Vis Sci. 2006;47:329–335. doi: 10.1167/iovs.05-0116. [DOI] [PubMed] [Google Scholar]
- 66.Rohrer B, Guo Y, Kunchithapautham K, Gilkeson GS. Eliminating complement factor D reduces photoreceptor susceptibility to light-induced damage. Invest Ophthalmol Vis Sci. 2007;48:5282–5289. doi: 10.1167/iovs.07-0282. [DOI] [PubMed] [Google Scholar]
- 67.Do DV, Pieramici DJ, van Lookeren Campagne M, et al. A phase Ia dose-escalation study of the anti-factor D monoclonal antibody fragment FCFD4514S in patients with geographic atrophy. Retina. 2014;34:313–320. doi: 10.1097/IAE.0b013e3182979ddd. [DOI] [PubMed] [Google Scholar]
- 68.Regillo CD. Lampalizumab (anti-factor D) in patients with geographic atrophy: the MAHALO phase II results in 2013. Annual Meeting of the American Academy of Ophthalmology; New Orleans. 2013. [Google Scholar]