

Abstract
Experimental analysis of enzymatic transition-state structures uses kinetic isotope effects (KIEs) to report on bonding and geometry differences between reactants and the transition state. Computational correlation of experimental values with chemical models permits three-dimensional geometric and electrostatic assignment of transition states formed at enzymatic catalytic sites. The combination of experimental and computational access to transition-state information permits (a) the design of transition-state analogs as powerful enzymatic inhibitors, (b) exploration of protein features linked to transition-state structure, (c) analysis of ensemble atomic motions involved in achieving the transition state, (d) transition-state lifetimes, and (e) separation of ground-state (Michaelis complexes) from transition-state effects. Transition-state analogs with picomolar dissociation constants have been achieved for several enzymatic targets. Transition states of closely related isozymes indicate that the protein’s dynamic architecture is linked to transition-state structure. Fast dynamic motions in catalytic sites are linked to transition-state generation. Enzymatic transition states have lifetimes of femtoseconds, the lifetime of bond vibrations. Binding isotope effects (BIEs) reveal relative reactant and transition-state analog binding distortion for comparison with actual transition states.
Keywords: drug design, inhibitor design, isotope effects, protein conformations, protein dynamics
INTRODUCTION
Research in the mechanisms of enzymatic catalysis has redoubled with new efforts in computational chemistry proposing explanations for catalytic rate enhancement and barrier crossing (1–5), in systems biology for annotation of unassigned open reading frames (6, 7), in chemical library screening for new enzyme inhibitors as pharmacology leads (8, 9), in the evolution of enzyme structure and function (10, 11), and in the de novo design of enzymatic activity (12, 13). Catalytic efficiency and the nature of enzymatic transition states are pertinent to each effort. Only by efficient formation of the transition state can enzymes serve their catalytic function. Any interference in catalytic function constitutes an approach to potential pharmacophores. Enzymes remain important pharmaceutical targets as approximately one-third of current drugs act as enzyme inhibitors (14, 15). All biological function is linked to catalytic activity, and increased understanding in genetics invariably generates new questions relative to the catalysts involved in higher-order regulatory processes.
Chemical transition states have short lifetimes of a few femtoseconds, the time required for electron redistribution and the time to convert the restoring mode of a chemical bond to a translational mode (16, 17). Enzymes have functional turnover numbers (kcat) near the millisecond timescale; thus, the transition-state lifetime occupies only about 10−12 of the reaction coordinate cycle (Figure 1). An enzyme fully saturated with reactants and engaged in maximal catalysis will therefore have about 1 part in a trillion occupied by the transition-state complex with the remainder in ground-state complexes. Thus, spectroscopic or static techniques are incapable of characterizing enzymatic transition-state complexes. Although claims have been made for trapping actual transition-state species in crystallographic structures (e.g., References 18 and 19), these claims have not withstood critical analysis (20, 21).
Figure 1.

Relative timescales for motions involved in enzymatic catalysis and interactions of transition-state analogs (adapted from Reference 51 with permission). Note the 1013 timescale difference between kcat for PNP and TS lifetimes (blue). The red bar indicates typical enzymatic kcat ranges. fs, femtosecond; Hb, hemoglobin; μs, microsecond; ms, microsecond; ns, nanosecond; PNP, purine nucleoside phosphorylase; ps, picosecond; TIM, triose phosphate isomerase; TS, transition state.
Indirect methods used to characterize transition states have traditionally been borrowed from physical organic chemistry, including chemical reactivity relationships (22, 23). Kinetic isotope theory and kinetic isotope effects (KIEs) were first developed for isotope fractionation and later used to deduce chemical mechanisms in organic chemistry and transition-state features for solution-based chemical reactions (24–27). The application of isotope effects to enzymatic reactions became more practical with the publication of books summarizing the theory and methods for approaching problems with isotope effects in enzymology (28, 29). With the current applications of KIE experiments exemplified here, technology is now available to understand the structural features of enzymatic transition states.
Enzymes are remarkable for catalytic rate enhancement and for their ability to overcome the single large transition-state energetic barrier of solution reactions by creating multiple steps with smaller barriers. Often, protein conformational changes or rates of reactant release limit the reaction rates and thereby obscure the values of the KIE (intrinsic values) arising from the chemical step. Intrinsic isotope effects are essential for interpretation of KIE information into transition-state structures, and methods to address this problem evolved with the application of transition-state theory to enzymes (30, 31). Multiple KIE approaches were used to map general features of enzymatic transition states before computational approaches were available to fit a family of intrinsic KIEs to a specific transition-state structure (32). Despite the long history of KIE applications to enzymatic reactions, the technology is still evolving. For example, a general assumption has been that binding isotope effects (BIEs) could be ignored. BIEs are now known to be significant in many, and possibly all, enzymatic reactions and often contribute to the calculated intrinsic KIE values (33, 34). BIEs can now be used to provide insights about ground-state (thermodynamic) interactions.
This article provides some examples of the combined experimental-computational approaches to establish transition-state information for specific enzymatic reactions and the use of transition-state structure to design transition-state analogs. The focus is on transition states for a family of N-ribosyltransferases. Despite this focus, the KIE method of transition-state analysis is general and relies only on the ingenuity of the investigator to select appropriate targets, resolve features of the transition state, and synthesize new transition-state analogs. Isotope effects and transition-state analogs also provide research tools to understand catalyst function.
FROM KINETIC ISOTOPE EFFECTS TO TRANSITION-STATE STRUCTURE
KIE measurements compare reaction rates of isotope-labeled and natural abundance reactants. When the reactants compete, the results yield KIEs on kcat/Km and include all steps between reactants free in solution and the first irreversible step in the reaction. These methods have been described (35–38). Recent application of NMR methods allows natural abundance determination of many isotope effects simultaneously or permit accurate analysis by NMR neighbor chemical-shift methods (39–41). Larger isotope effects from solvent 2H2O or for hydride transfer mechanisms can be measured using labeled and unlabeled reactants in separate experiments to obtain distinct values for isotope effects on kcat and Km. Intrinsic KIEs arise from the difference in bond vibrational environment for an atom in the reactant state compared to its environment at the transition state and thus gives an atom-by-atom description of the transition state. Multiple isotope effects, taken from molecular sites that change in bond vibrational environments between reactant and transition states, can be used to recreate a full model of the transition state.
Matching Kinetic Isotope Effects to Transition-State Models
Atom-by-atom KIE values are converted to a specific static model with fixed bond angles and lengths by computational matching to a quantum chemical model of the reaction of interest. A typical approach is to explore the reaction of interest in the Gaussian computational suite using the B3LYP method with a 6-31G* basis set (defining the level of computational theory), although other levels of theory can be used and the method is not strongly dependent on the basis set (42–44). Reactants, transition state, and product geometries are located as the global minima. Transition-state structures are located with a single imaginary frequency, characteristic of true potential energy saddle points. The experimental intrinsic KIEs are then matched in fixed-distance optimizations using a grid of possible transition structures. Fixed bonds are used only along the reaction coordinate (the reaction coordinate is defined as the sites of bond making and bond breaking, for example, the bonds to the leaving group and to the attacking nucleophile in ribosyl transferases). The computed structures are relaxed in other dimensions, and isotope effects are calculated for each transition state using QUIVER or ISOEFF98 software (45, 46). If indicated by structural data and if needed to obtain a KIE match, hydrogen bonds or other interactions from catalytic site residues can be added. Transition-state models matching the intrinsic KIE can be recalculated at the highest practical level (typically, B3LYP/6-31+G**) to obtain the final transition-state model.
Electrostatic Potential Maps
The CUBE subprogram from Gaussian can be used to generate the molecular electrostatic potential surfaces of transition-state structures using the formatted checkpoint files from geometry optimization. The molecular electrostatic potential surfaces can be visualized at the van der Waals surface or closer to the nuclei (iso-values of 0.040 and 0.068, respectively) using GaussView 3.09 (available in Reference 47).
DESIGN OF TRANSITION-STATE ANALOGS
The goal of transition-state analog design is to create stable chemical structures with van der Waals geometry and molecular electrostatic potential surfaces as close as possible to those of the transition state. The original hypothesis for binding of transition-state analogs invoked tight binding of the transition state as the rationale for the tight binding of transition-state mimics (48–50). This interpretation is now debated as the explanation for transition-state formation (discussed below). A dynamic view proposes that enzyme-associated reactants cross the transition-state barrier by stochastic protein vibrational modes without unique equilibrated high-affinity binding features (51, 52). Transition-state analogs convert the dynamic features of the transition state into thermodynamic binding energy and thereby generate tight binding.
Predictive Binding
Before synthetic chemistry efforts are applied to putative transition-state analogs, quantitative measures of similarity to the transition state are useful. One method ranks the similarity of proposed analogs by parameters of geometry and electrostatic potential surface against the same parameters for transition-state analogs (53, 54). As geometry and electrostatics are the dominant parameters for ligand recognition, molecules more similar to the transition state are bound more tightly. For example, using these similarity parameters from 0 (no similarity) to 1.0 (identity to the transition state), the substrate inosine for bovine purine nucleoside phosphorylase (PNP) shares a 0.483 similarity, whereas the analog immucillin-H (ImmH) shares a similarity of 0.723 to the transition state, providing a dissociation constant of 23 picomolar (pM) and a Km/Kd ratio of 739,000 (Figure 2) (55). Values for chemically stable transition-state analogs near 1.0 are not possible because the features of the transition state include nonequilibrium bond lengths and partial charges that cannot be reproduced in stable chemical mimics.
Figure 2.
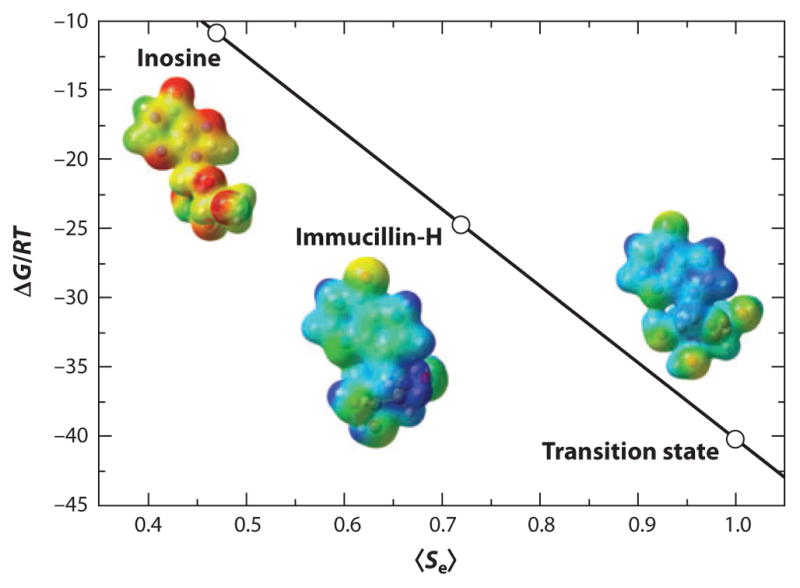
Binding free energy (ΔG/RT) and molecular electrostatic potential surface similarity 〈Se〉 for inosine, immucillin-H, and the transition state for bovine purine nucleoside phosphorylase (modified and reprinted from Reference 55 with permission).
A second predictive affinity method uses neural network similarity measures to rank geometry and electrostatic potential surface parameters from a known set of inhibitors (including some with features of the transition state) and compare these parameters to those from proposed inhibitors prior to chemical synthetic efforts (56). This approach faithfully predicts the binding affinity of a family of unknown inhibitors for the IU-nucleoside hydrolase from Crithidia fasciculata (Figure 3) (57).
Figure 3.

A molecular electrostatic potential and geometric input for inhibitor recognition by nucleoside hydrolase using a neural training network. The surface (outer sphere of golden squares) is imprinted with molecular electrostatic potentials, where red indicates a partial positive charge, and purple indicates a partial negative charge relative to the average charge at the van der Waals surface. The transition state (left) is similar to the high-affinity inhibitor (middle, Kd = 4 nM) but different from the low-affinity inhibitor (right, Kd = 18 mM). Modified from Reference 57 and reprinted with permission. nM, nanomolar; mM, millimolar.
Selecting Targets
Emphasis on the N-ribosyltransferases in this review is not a random choice. Good target candidates for transition-state analog design should have altered geometry and/or charge on conversion from reactants to the transition state. Glycosytransferases generally share these properties. Thus, most sugar transferases form transition states with cationic charge at the anomeric carbon, and the geometry is altered at this center from sp3 (tetrahedral geometry) in the reactant sugar to sp2 (trigonal planar geometry) at the transition state (58–60). Thus, the chemistry to match these features provides candidates as transition-state analogs (e.g., References 61 and 62). Other reactions that involve displacements at carbon are also candidates for transition-state analysis. Such reactions include a broad range of biological reactions with altered charge and/or geometry at the transition state, including reactions with sp2 reactants and sp3 transition states, for example, the proteases.
TRANSITION-STATE STRUCTURES AND ANALOGS
Examples of transition-state analysis are provided where transition-state information has been obtained from KIEs and computational chemistry. Transition-state analogs have been designed, synthesized, and characterized for most of these targets. Bovine and human PNPs are the most advanced examples, and candidates from the first and second generation transition-state analog inhibitors are currently in advanced clinical trials (see below). Other examples provided here have yielded the most powerful inhibitors known for the targets.
Human and Bovine Purine Nucleoside Phosphorylases
The human genetic deficiency of PNP was linked to T-cell immune deficiency by the early studies of Giblett et al. (63). Subsequent studies revealed that the accumulation of 2′-deoxyguanosine in blood is specifically toxic by causing excess dGTP accumulation in activated T cells (64). Bovine PNP was used as a model for human PNP and assumed to form a similar transition state because of the 87% amino acid sequence identity (65, 66). KIE values and transition-state analysis for the arsenolysis reaction revealed an early dissociative transition-state structure with a significant bond order to the departing hypoxanthine at the transition state (Figure 4). Electrostatic comparison of the transition state and analogs indicated a good match between ImmH and the transition state for bovine PNPs, and indeed, ImmH is a 23-pM inhibitor of bovine PNP and gives a 739,000 Km/Ki value (Figures 3 and 4). As bovine PNP was intended as a surrogate for human PNP, the 56-pM dissociation constant of ImmH for human PNP was a concern because of its lower affinity for the desired target, especially because there is 100% amino acid conservation for inhibitor contacts at the catalytic sites (67). This difference in ImmH binding for the human and bovine PNP isozymes suggested the possibility of distinct transition states.
Figure 4.

Intrinsic kinetic isotope effects (in percentages) for bovine (upper left, in red) and human (upper right) purine nucleoside phosphorylases (PNPs). Intrinsic isotope effects shown in blue for human PNP are significantly different from those for bovine PNP. Reaction coordinate bond lengths (middle) give rise to the molecular electrostatic potentials (bottom) for the transition states (TSs) and transition-state analogs, immucillin-H (ImmH), and 4′-deaza-1′-aza-2′-deoxy-1′-(9-methylene)immucillin-H (DADMe-ImmH). Blue atoms (middle) are sties most changed by the formation of the transition state. pM, picomolar.
Intrinsic KIE values for bovine and human PNPs were different (Figure 4). Because intrinsic KIEs report on the bond vibrational status at the transition state, the primary data set established different transition-state structures for these closely related enzymes (Figure 4). The transition state for human PNP is a fully formed ribocation with the cationic charge located primarily at C1′ (the anomeric carbon), has a fully dissociated bond of 3 Å to the hypoxanthine, and shows no significant participation of the arsenate nucleophile. The physiological substrate for human PNP is 2′-deoxyguanosine; thus, the transition state for human PNP also forms efficiently with 2′-deoxyribosyl sugars. These distinct features of the transition state can be matched with DADMe-ImmH [4′-deaza-1′-aza-2′-deoxy-1′-(9-methylene)immucillin-H] (Figures 4 and 5). DADMe-ImmH shows higher affinity for human than bovine PNP and binds 2,400,000 times tighter than substrate according to the Km/Kd ratio (68).
Figure 5.
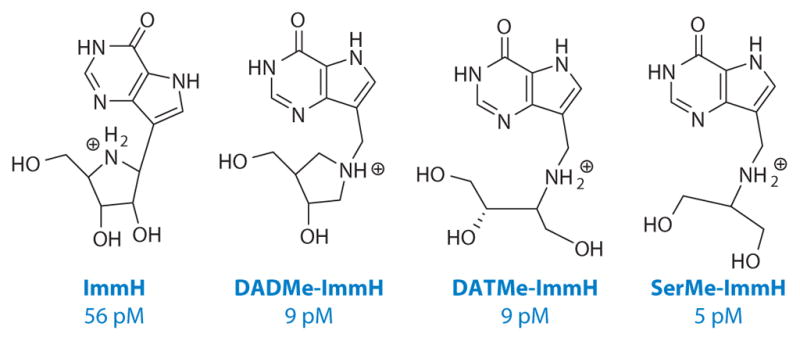
Four generations of transition-state analogs for human purine nucleoside phosphorylases together with their dissociation constants. DADMe-ImmH, 4′-deaza-1′-aza-2′-deoxy-1′-(9-methylene)immucillin-H; ImmH, immucillin-H; pM, picomolar; SerMe-ImmH, the seramide substituent of 9-methylene-9-deazahypoxanthine.
Lessons from comparisons of bovine and human PNPs are that distinct transition-state structures can be formed by closely related enzymes with nearly identical catalytic sites and that transition-state information from KIEs provides a tool for the design of specific transition-state analogs. Distinct transition states from enzymes with near-identical catalytic sites suggest involvement of the protein architecture in transition-state formation. By replacing remote amino acids (>25 Å from the catalytic sites) in human PNP with those of bovine PNP, a chimeric enzyme was formed with altered dynamic properties and a transition state distinct from either parent (69, 70).
ImmH contains four asymmetric carbon centers and reactive nitrogen and oxygen groups that require protection, making the chemical synthesis difficult (71, 72). Use of 2′-deoxynucleosides by human PNP permits design of transition-state analogs with the elimination of one stereocenter, and replacing the C1′-anomeric carbon with nitrogen eliminates a second; thus, DADMe-ImmH has only two stereocenters and can be synthesized using the Mannich reaction without protecting the chemically reactive nitrogen and oxygen atoms (Figure 5) (73). Replacing the hydroxypyrrolidine ribocation mimic of DADMe-ImmH with acyclic groups further simplified these analogs and has generated third- and fourth-generation inhibitors exemplified by DATMe-ImmH and SerMe-ImmH (Figure 5). These inhibitors show similar picomolar binding affinity to human PNP and are increasingly accessible to synthetic chemistry (74, 75). Crystallographic analysis of human PNP with all four generations of the immucillins shows common features linked to their tight binding. Most important is the ability to form an ion pair between the bound N-ribocation mimic and the phosphate anion at the catalytic site. In ImmH, this distance is 3.3 Å and is shorter than with the other analogs (67). The 9-deazahypoxanthine group is common to all inhibitors, but in ImmH, contacts to catalytic site groups are less optimal than for the others. The geometry of ImmH in the dimension of the reaction coordinate (distance from the N4′-cation to N7) is too short to permit optimal contacts to both 9-deazahypoxanthine and the ribocation mimic. Thus, human PNP is resistant to flex beyond the extended geometry of a fully dissociated transition state in this dimension. Inhibitors longer in their reaction coordinate dimension by addition of the methylene bridge more optimally capture the interactions between the inhibitor and the catalytic site.
Transition-state analogs are not useful in drug development unless biological availability permits access to the target. In the case of human PNP, this means access to all tissues because the enzyme is widely distributed. From characterization of human PNP genetic mutants, it is known that >95% inhibition of PNP is needed to cause acumulation of 2′-deoxyguanosine and thereby block T-cell proliferation (76). ImmH enters human cells on the purine nucleoside transporters to provide access to all tissues (77).
Although mechanisms of transport for other immucillins have not been reported, single-dose oral administration of DADMe-ImmH caused rapid inhibition of PNP in mice as indicated by complete inhibition of PNP in mouse blood. Recovery of half-normal PNP activity requires 11.5 days and parallels erythrocyte replacement. Thus, DADMe-ImmH stays on its biological target until cells are replaced (68). Imm-H and DADMe-ImmH show low toxicity in animal studies. ImmH is in clinical trials for T-cell malignancies under the name of Forodesine™, and DADMe-ImmH is in clinical trials for gout under the name BCX4208 (78, 79).
Human 5′-Methylthioadenosine Phosphorylase
The production of 5′-methylthioadenosine (MTA) in humans occurs only in the pathway for conversion of S-adenosylmethionine (SAM) to polyamines (Figure 6). The sole metabolic fate for MTA is conversion to adenine and 5-methylthio-α-D-ribose 1-phosphate by 5′-methylthioadenosine phosphorylase (MTAP). These products of the MTAP reaction are recycled to ATP and methionine, metabolites that can be recycled to SAM (80). MTAP therefore serves as an essential part of adenine, methionine, and SAM salvage pathways. The MTAP reaction is linked to biosynthetic methylation reactions and regulatory methylations of CpG sites in DNA and to protein methylation. Inhibitors of these processes are proposed to function as anticancer agents (81, 82). The polyamine pathway is also an anticancer target. Inhibition of MTAP causes MTA to accumulate and causes product inhibition of polyamine synthetases (82, 83).
Figure 6.

The role of 5-methylthioadenosine phosphorylase (MTAP) in polyamine synthesis, methionine, ATP, and S-adenosylmethionine (SAM) salvage (figure modified and reprinted from Reference 87 with permission).
KIEs for human MTAP and computational modeling (see above) provided a transition-state structure for the arsenolysis of MTA (84). Intrinsic KIEs indicated a dissociative SN1 transition state for MTAP (Figure 7). The anomeric carbon of 5-methylthioribose is converted to a cationic center at the transition state, and bond loss to the adenine leaving group is complete. An unusual feature of the KIEs is the large [9-15N]MTA value of 3.9%, which indicates anionic adenine at the transition state. The primary [1′-14C]MTA KIE of 3.1% indicates a transition state with significant bond order between the anomeric carbon and the attacking nucleophile, an arsenate oxygen (2.0 Å distant from the anomeric carbon). Another unusual feature of this transition state is the polarization or ionization of the ribosyl 3′-OH group. Ionization of the 3′-OH group suggests a zwitterionic ribosyl group with a 3′-endo conformation at the transition state.
Figure 7.

The transition state and transition-state analogs for the arsenolysis reaction of human 5′-methylthioadenosine phosphorylase (MTAP). The intrinsic kinetic isotope effects used in transition-state analysis are shown on the upper right, and the inhibitors are shown below. Inhibitor features that mimic the transition state are in blue. pClPhT- is a 4-chloro-phenylthio group. MT, methylthio; pM, picomolar.
The late SN1 transition state for human MTAP suggested that analogs similar to DADMe-ImmH for human PNP, modified to reflect the substrate specificity of human MTAP, may capture transition-state features. The 9-deazaadenine ring and 5′-methylthio group mimic human MTAP substrates (85). Analogs for the human MTAP transition state were synthesized and provide picomolar inhibitors (84–86). Methylthio-DADMe-immucillin-A (MT-DADMe-ImmA) is a transition-state analog of human MTAP and is a slow-onset, tightly binding inhibitor with a dissociation constant of 86 pM and is orally available in mice (Figure 7) (87).
Screening of human cancer cell lines showed low toxicity for MT-DADMe-ImmA and MTA as individual agents. MT-DADMe-ImmA in combination with MTA causes growth inhibition in some cell lines and apoptosis in others. Human FaDu head and neck cancer cells, when treated with MT-DADMe-ImmA and MTA, revealed inhibition of cellular MTAP, increased cellular MTA concentrations, decreased polyamines, and increased rates of apoptosis. Implanted FaDu tumors in mouse xenografts were treated with MT-DADMe-ImmA, resulting in tumor remission with no apparent toxicity to the host (87). In a related study with human lung cancer cell lines, MT-DADMe-ImmA inhibited growth of primary tumors in mice and decreased metastatic cancers to the lungs (88).
Bacterial Methylthioadenosine/Adenosylhomocysteine Nucleosidase Hydrolases
Induction of luminescence genes in Vibrio harveyi as a function of cell density in culture led to the discovery of quorum-sensing systems in bacteria (89). When bacterial cell populations reach critical levels, cells produce and detect signaling molecules known as autoinducers (AIs) to coordinate gene expression and regulate processes that are presumed to be beneficial for the colony (89). Bacterial multidrug resistance has rapidly appeared with conventional cell-killing antibiotics, and overcoming this problem requires approaches that are nonlethal to bacteria to prevent development of resistance while blocking pathogenicity (90–93). Quorum sensing is linked to virulence but is not essential for growth, making it an attractive target.
One class of quorum-sensing molecules, the autoinducer-1 (AI-1) and autoinducer-2 (AI-2) molecules, is linked to SAM metabolism (Figure 8). 5′-Methylthioadenosine/adenosylhomocysteine nucleosidases (MTANs) hydrolyze both MTA and adenosylhomocysteine, are directly involved in the biosynthesis of autoinducers or in SAM recycling, and are essential for the production of autoinducer molecules (94). AI-1 and AI-2 are synthesized from SAM; thus, MTAN inhibition was proposed to block both AI-1 and AI-2 production, thereby disrupting quorum sensing. Because MTANs are not found in humans, MTAN inhibitors are expected to block quorum sensing in bacteria without effects on human metabolism.
Figure 8.

The role of 5′-methylthioadenosine/adenosylhomocysteine nucleosidase (MTAN) in AI-1 and AI-2 quorum-sensing pathways (reprinted from Reference 94 with permission). AHL, acylhomoserine lactones; AI, autoinducer; MTA, 5′-methylthioadenosine; MTR, methylthioribose; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SRH, S-ribosylhomocysteine.
Transition-state structures of bacterial MTANs were established by quantum chemical analysis using the intrinsic KIE values for MTANs from Escherichia coli, Streptococcus pneumoniae, and Neisseria meningitidis (95–97). A comparison of the intrinsic KIE values is instructive (Figure 9). Intrinsic KIEs for E. coli MTAN-catalyzed hydrolysis of MTA gave large 1′-3H (16%) and small 1′-14C (0.4%) KIEs, indicating that this transition state involves minimal leaving group or attacking nucleophile participation and a transition state with well-developed ribocation character. A transition state matching the intrinsic KIEs was located and indicated the leaving group (N9) was 3.0 Å from the anomeric carbon and a similar distance for the attacking water nucleophile. The relatively small 9-15N KIE indicates that the leaving group is protonated at the the transition state. Ribose pucker at the transition state affects the 2′-3H KIE, and the relatively small value of 4.4% indicated a H1′-C1′-C2′-H2′ dihedral angle of 53° at the transition state, a modest 3′-endo geometry. This transition state predicts that extended transition-state analogs, patterned after the DADMe-ImmH for human PNP, would resemble this transition state, and these compounds are powerful inhibitors (see below).
Figure 9.
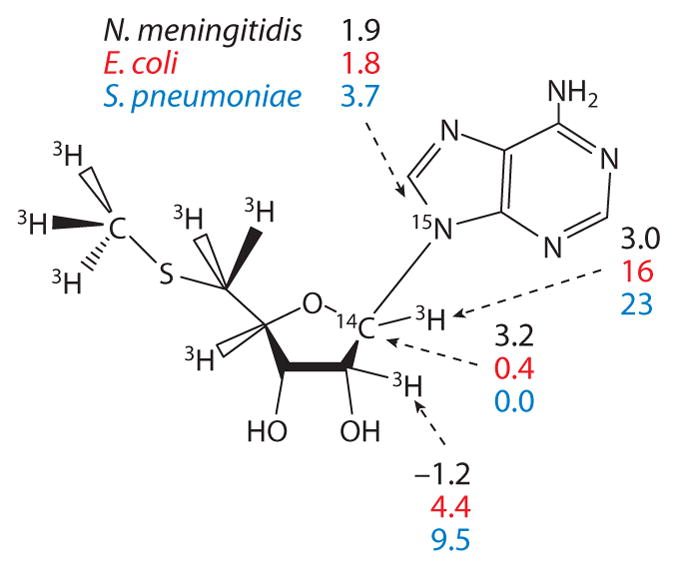
Kinetic isotope effect values (as percentages) used for transition-state analysis of the bacterial 5′-methylthioadenosine/adenosylhomocysteine hydrolases.
The intrinsic KIE values for S. pneumoniae MTAN are similar to those for the E. coli enzyme and also support a dissociative SN1-like transition state with no significant covalent participation of the adenine leaving group or the attacking water nucleophile (96). A quantum chemical model of the transition state as a ribooxacarbenium ion intermediate was found to fit the intrinsic KIEs. A 3′-endo conformation for the ribocation corresponding to H1′-C1′-C2′-H2′ dihedral angle of 70° is consistent with the KIEs. Although both E. coli and S. pneumoniae MTAN transition states exhibit fully developed ribocations, the 9-15N KIEs differ considerably, 1.8% and 3.7%, respectively. The [9-15N]MTA isotope effect reports on the total bond order to N9 at the transition state and is influenced by the protonation state of the leaving group. The value of 3.7% found for the S. pneumoniae MTAN indicates that the adenine leaving group is not protonated at the transition state and therefore is proposed to depart as a catalytic-site-stabilized adenine anion. In this and other transition states, the influence of the virtual solvent (a dielectric constant) was varied as part of the modeling and did not influence calculated KIE values beyond experimental error. S. pneumoniae MTAN differs from most other purine N-ribosyltransferases but resembles human MTAP in the protonation state of the leaving group. Departure of a leaving group from an ion-pair transition state (cationic ribose and anionic adenine) is difficult and is consistent with the 103-fold decrease in the catalytic efficiency of S. pneumoniae MTAN relative to that from E. coli. Because the interaction of transition-state analogs is also related to catalytic efficiency, this transition state is also consistent with weaker binding of transition-state analogs to S. pneumoniae than to E. coli MTANs (see below).
Unlike the well-developed ribocation transition states of S. pneumoniae and E. coli MTANs, the transition state of N. meningitidis MTAN is early in an SN1 reaction path. The 1′-3H KIE is dominated by an out-of-plane mode as the anomeric carbon rehybridizes from sp3 in the reactant to sp2 in dissociative transition states. In E. coli and S. pneumoniae MTANs, the 1′-3H KIEs are 16% and 23%, but for N. meningitidis MTAN, the value is 3%. Thus, sp2 geometry at C1′- is not established at the transition state. Likewise, no isotope effect is seen at 2′-3H, and the isotope effect of 9-15N is 1.9%. These values are consistent with the significant bond order remaining to the adenine leaving group at the transition state. S. pneumoniae MTAN, like bovine PNP, is the second enzyme in the purine N-ribosyltransferase family to exhibit an early transition state. Late dissociative SN1 transition states dominate the N-ribosyltransferases.
Transition-state analogs designed for human MTAP overlap in activity with those designed for bacterial MTANs (Figures 7 and 10). Iminoribitol analogs of MTA mimic early dissociative transition states for MTAN. 5′-Methylthio-immucillin-A (MT-ImmA) was a slow-onset tight-binding inhibitor with a 77-pM dissociation constant for E. coli MTAN, and hydrophobic substituents lowered the dissociation constant to 2 pM. The DADMe-immucillins are more closely related to the transition state, and MT-DADMe-ImmA exhibits a 2-pM dissociation constant. Hydrophobic 5′-substituents in this scaffold gave transition-state analogs with dissociation constants in the range of 10−12 to 10−14 M. The dissociation constant of 47 femtomolar (10−15 M) for 5′-p-Cl-phenylthio-DADMe-immucillin-A (pClPhT-DADMe-ImmA) ranks it among the most powerful noncovalent inhibitors reported for any enzyme. Comparison with substrate interactions by the Km/Kd ratio indicates that this inhibitor binds 91 million times tighter than the substrate.
Figure 10.

Transitions state analogs of E. coli 5′-methylthioadenosine/adenosylhomocysteine hydrolase. Dissociation constants are shown, and Ki* indicates the observation of slow-onset, tight-binding inhibition. The prefix indicates the 5′-substituents to the parent DADMe-ImmA, thus, BnO-DADMe-ImmA, benzyl-O-DADMe-ImmA; BnT-DADMe-ImmA, benzylthio-DADMe-ImmA; BnT-Pz-DADMe-ImmA, benzylthio-pyrazolo-DADMe-ImmA; BuT-DADMe-ImmA, butylthio-DADMe-ImmA; cHxT-DADMe-ImmA, cyclohexylthio-DADMe-ImmA; EtT-DADMe-ImmA, ethylthio-DADMe-ImmA; fM, femtomolar; Homocys-DADMe-ImmA, homocysteinyl-DADMe-ImmA; mClPhT-DADMe-ImmA, meta-chlorophenylthio-DADMe-ImmA; MT-DADMe-ImmA, methylthio-DADMe-immucillin-A; pClPhT-DADMe-ImmA, para-chlorol-phenylthio-DADMe-immucillin-A; pFPhT-DADMe-ImmA, para-fluorophenylthio-DADMe-ImmA; PhT-DADMe-ImmA, phenylthio-DADMe-ImmA; PrT-DADMe-ImmA, propylthio-DADMe-ImmA; 4-PyrT-DADMe-ImmA, 4-pyridylthio-DADMe-ImmA; 5′-dEt-DADMe-ImmA, 5′-deoxyethyl-DADMe-ImmA.
The MTAN inhibitors were tested for the ability to disrupt quorum sensing in E. coli and Vibrio cholerae. MT-DADMe-ImmA, EtT-DADMe-ImmA, and BuT-DADMe-ImmA (see Figures 10 and 11) bind to V. cholerae MTAN, with dissociation constants of 73, 70, and 208 pM, respectively. Enzymatic assays of MTAN in intact V. cholerae cells demonstrated cell permeability and potent MTAN inhibition with 50% inhibitory concentration (IC50) values of 6 to 27 nM. To cause quorum-sensing disruption without induction of resistance, MTAN inhibitors should not disrupt bacterial growth. No growth inhibition was observed at concentrations 1,000 times the IC50values, and autoinducer production was blocked at nanomolar concentrations (94). Similar results were obtained with enterohemorrhagic E. coli O157:H7. Inhibitor studies conducted in parallel with bacterial cells genetically deleted in MTAN affirmed the role of this pathway in quorum sensing and its potential as a target for bacterial quorum sensing (94).
Figure 11.
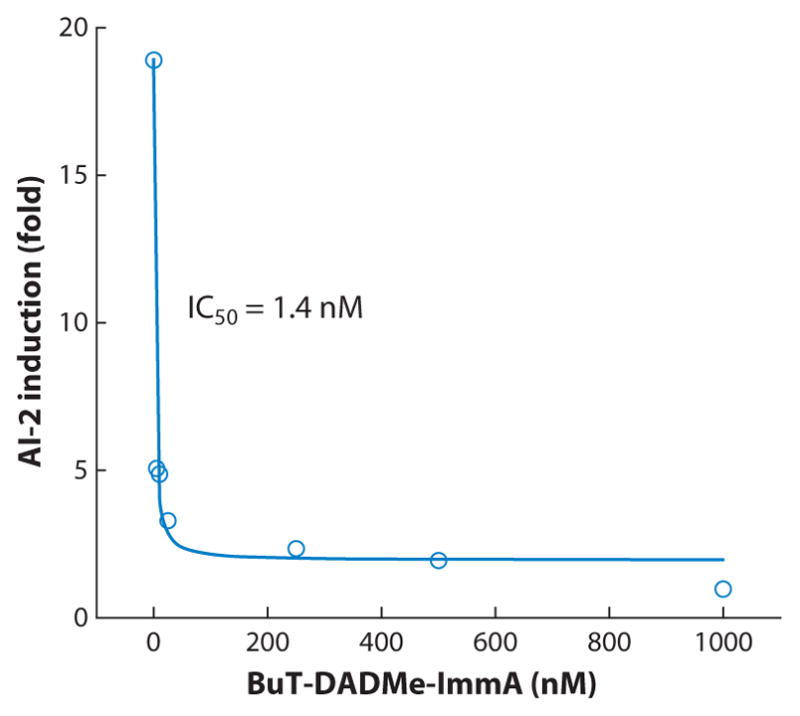
Inhibition of autoinducer-2 (AI-2) formation in Vibrio cholerae by BuT-DADMe-ImmA (see Figure 10) (reprinted from Reference 94 with permission). IC50 refers to the concentration of BuT-DADMe-ImmA that causes 50% inhibition of AI-2 formation.
With the three MTAN transition states described above, the transition states for other members of the bacterial MTAN family can be predicted from MTAN affinity to transition-state analogs. Thus, transition-state analogs with short distances between the ribocation mimic and leaving group mimic will bind better to the MTANs that form early transition states. Transition-state analogs with extended distances between the ribocation mimic and leaving group mimic will bind better to MTANs with fully dissociated transition states. This approach was tested on six MTANs, the three described above with known transition states and three MTANs with uncharacterized transition states. Using this approach, the transition states of N. meningitidis and Helicobacter pylori MTANs appear similar, and both are early dissociative. By contrast, E. coli, Staphylococcus aureus, S. pneumoniae, and Klebsiella pneumoniae MTANs all bind more tightly to extended transition-state analogs, resembling fully dissociated ribocation transition states. Intrinsic KIE values were used to confirm this prediction. Characteristic large [1′-3H]MTA and unity [1′-14C]MTA KIEs were observed for the MTANs predicted to have fully dissociative transition states, whereas MTANs with early dissociative states showed small [1′-3H]MTA and significant [1′-14C]MTA KIEs (98). A caveat to analysis of transition-state structure by transition-state analog specificity is the requirement for a family of well-characterized transition-state analogs for the enzymes of interest, a rarity in the current development of transition-state analogs.
Ricin A Chain and Saporin
Ricin toxin A chain (RTA) and saporin are plant ribosome-inactivating proteins (RIPs) with catalytic activity for depurination of adenine residues from RNA, making them N-ribosylhydrolases (99). Biomedical interest in the RIPs arises from their use as anticancer agents when linked to monoclonal antibodies or other molecules designed to recognize cancer cells. Off-target toxicity causes vascular leak syndrome, an unacceptable side effect. Transition-state analogs that inhibit RIP activity following therapy are being explored to provide an approach to limit the side effects.
Ricin A chain shows high substrate specificity for a single adenine in the elongation factor–binding site of 28S ribosomal RNA located in a 5′-GAGA-3′ tetraloop where the 5′-A is hydrolyzed. The transition state for depurination of stem-loop RNA by RTA was solved by KIEs using stem-loop RNA (5′-GGCGAGAGCC-3′) with isotopically labeled adenosine at the depurination site (100). The KIEs of 16.3% for [1′-3H], −1.9% for [7-15N], 1.6% for [9-15N], −0.7% for [1′-14C], and 1.2% for [2′-3H] demonstrated formation of an RNA ribooxocarbenium ion and adenine in equilibrium with the Michaelis complex (101). The ribosyl ring pucker at the transition state was found to be 3′-endo, giving rise to the small [2′-3H] KIE. The unusual dihedral angle of approximately 50° between C2′-H2′ and the vacant p orbital of the anomeric carbon are consistent with the relatively rigid geometry of the stem-loop RNA backbone (Figure 12). An irreversible nonchemical step initiates water attack. RTA has a stepwise DN*AN mechanism, and the cationic intermediate has a finite lifetime. This study with RTA was the first to establish that KIEs and transition-state analysis could be extended to the reactions of nucleic acid chemistry.
Figure 12.
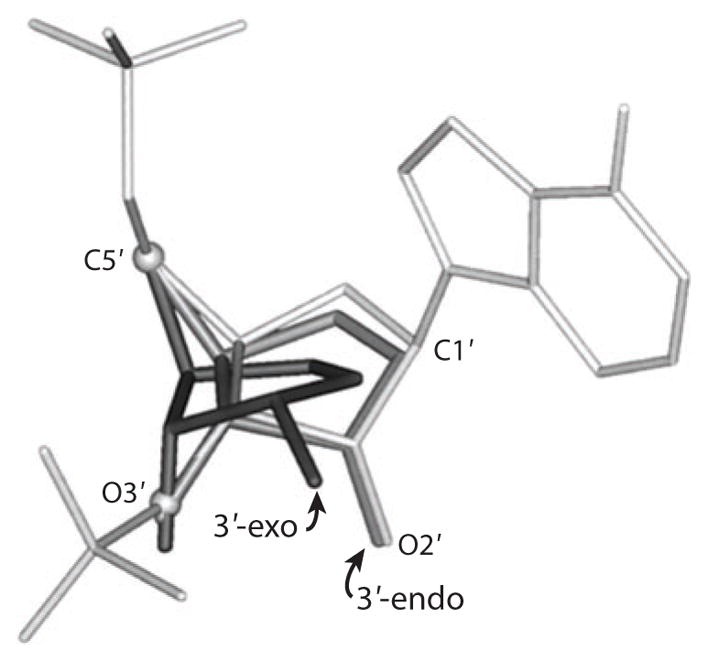
Transition-state geometry for ricin A chain with stem-loop RNA (dark gray) and with stem-loop DNA (medium gray) superimposed on the crystal structure of stem-loop RNA geometry (light gray) (reprinted from Reference 102 with permission). Ribosyl carbons C1′ and C5′ as well as ribosyl oxygens O2′ and O3′ are shown for the two geometries of the transition state, 3′-exo for the transition state with RNA and 3′-endo for the transition state with DNA. 3′-Exo and 3′-endo ribosyl geometry refer to the 3′C being below or above the plane of the ribosyl ring formed by the other four atoms.
Transition-state analysis for oligonucleotides was extended to DNA in a KIE and transition-state analysis study using RTA with stem-loop DNA as a slow substrate. Although the transition state for stem-loop RNA is constrained by the rigid RNA scaffold, stem-loop DNA is more flexible and permits formation of the most chemically stabilized geometry for the deoxyribocation at the transition state (Figure 12) (102).
On the basis of transition-state structures of RTA with stem-loop RNA and DNA, ribocation characteristics with N7-protonated leaving groups are implicated as transition-state mimics. Because RTA is catalytically efficient only on stem-loop RNAs, loop mimics of the stem-loop structures were constructed and tested as inhibitors using small stem-loop RNAs as substrates (103, 104). Nanomolar inhibitors were achieved with an N-benzyl-hydroxypyrrolidine (N-Bn) transition-state analog at the RTA depurination site in a circular GAGA motif. A cyclic G(N-Bn)GA provided a new RTA inhibitory scaffold and the most powerful RTA inhibitor (Kd = 70 nM) known. The transition-state analysis and inhibitor design for RTA was done at pH 4.0, required for RTA activity on small stem-loop substrates. These inhibitors did not protect mammalian ribosomes from RTA at physiological pH values. Transition-state inhibitor design was therefore extended to saporin, a RIP from the soapwort plant with the assumption that it forms a similar transition state.
Saporin-L1 from Saponaria officinalis (soapwort) leaves is highly active against small nucleic acid substrates and mammalian ribosomes and removes multiple adenines from RNA, including small linear sequences (105). Linear, cyclic, and stem-loop oligonucleotide inhibitors containing 9-deazaadenine-9-methylene-N-hydroxypyrrolidine and based on the 5′-GAGA-3′ sarcin-ricin tetraloop gave Kd values in the range of 2 to 9 nM (Figure 13). Under physiological pH values, these analogs bind up to 40,000-fold tighter than similar RNA substrates. Importantly, the destruction of ribosomes by saporin-L1 in rabbit reticulocyte translation was protected by these inhibitors (106).
Figure 13.

Inhibitors for saporin L-1 and their dissociation constants. A-10 RNA is a substrate with 440 min−1 kcat and 95 μM Km (reprinted from Reference 106 with permission). A-10, the 10-base RNA stem-loop shown in the figure; DADMeA (9-DA), DADMe-immucillin-A, a transition state analog mimic for adenine-hydrolyzing ribosome inactivating proteins when incorporated into RNA constructs; Ki*, the dissociation constant for inhibitors with saporin L-1; the asterisk indicates slow-onset inhibition is observed.
The availability of powerful inhibitors for ricin and saporin permitted the first crystal structures of RTA and saporin with oligonucleotides bound in the catalytic sites. The inhibitors resemble both the sarcin-ricin recognition sequence and the ribocation transition state established for RTA. RTA and saporin share unique purine-binding geometry at their catalytic sites with quadruple pi-stacking interactions between adjacent adenine and guanine bases and two conserved tyrosines (107). An arginine at one end of the pi stack provides cationic polarization and is proposed to enhance the adenine leaving group ability (Figure 14). Adenine leaving group activation dominates the reaction mechanism with no apparent features to assist in ribocation formation. Conserved glutamates activate the water nucleophile. Ricin A chain and saporin-L1 exemplify the ability of transition-state analogs to organize catalytic sites and permit mechanistically meaningful crystal structures.
Figure 14.

(a) The inhibitor G(9DA)GA 2′-OMe is shown at the catalytic sites of ricin A chain and (b) saporin L-1. (c) In both cases, a catalytic site tyrosine [Tyr80 or (d) Tyr73] is base stacked between the transition-state analog and the adjacent guanosine. The second tyrosine (Tyr123 in both cases) is base stacked on the other side of the transition-state analog. The nucleophilic water is in contact with a catalytic site glutamic acid (Glu177 or Glu174) (reprinted from Reference 107 with permission).
Orotate Phosphoribosyltransferases
Orotate phosphoribosyltransferases from Plasmodium falciparum and human sources (PfOPRT and HsOPRT) use the pyrimidine nucleoside orotidine as a slow substrate for pyrophosphorolysis. Intrinsic KIEs were measured to provide the first enzymatic transition state using pyrophosphate as the nucleophile. PfOPRT and HsOPRT are characterized by late transition states with fully dissociated orotate, well-developed ribocations, and weakly bonded PPi nucleophiles (108). The N-ribosidic bond to the leaving orotate is 2.8 Å, whereas weak participation of the PPi nucleophile gave a bond distance of approximately 2.3 Å (Figure 15). These phosphoribosyltransferase transition states are similar to but occur earlier in the reaction coordinate than transition states for PfOPRT and HsOPRT previously determined with orotidine 5′-monophosphate and phosphonoacetic acid as substrates (109) and occur much later in the reaction coordinate than a transition-state structure reported for a bacterial OPRT using orotidine 5′-monophosphate and phosphonoacetic acid (110). Similar transition states for PfOPRT and HsOPRT with different substrate analogs support similar transition-state structures imposed by the enzymes. Geometric constraints imposed by the catalytic sites may imprint these transition states.
Figure 15.

Transition-state structures of Plasmodium falciparum (PfOPRT) and human (HsOPRT) for the pyrophosphorolysis of orotidine. The reaction coordinate bond distances are shown above, and the electrostatic potential maps are shown below (red is electron rich, purple is electron deficient) (reprinted from Reference 108 with permission). TS, transition state.
GENERAL APPLICABILITY OF ENZYMATIC TRANSITION-STATE ANALYSIS
The examples provided above focus on N-ribosyltransferases. However, the method can be applied to any enzymatic reaction (see side-bar Beyond N-Ribosyltransferases).
TRANSITION-STATE VERSUS GROUND-STATE COMPLEXES
Glycosyltransferases are known to pass through cationic transition states, and the resulting glycocations are often considered to be intermediates with significant lifetimes (111). In some cases, experimental evidence exists for intermediate status; for example, in the case of ricin, a ribocationic intermediate is equilibrated with the Michaelis complex (101, 102). A chemical model for N-ribosyl hydrolysis has been provided by the acid-catalyzed solvolysis of dAMP in 0.1 N HCl, where a deoxyribocation lifetime of 10−11 to 10−10 s was estimated based on the 8.5 to 1 ratio of α- to β-methyl-deoxyribose 5-phosphate formed in the presence of 50% methanol as the nucleophile (112). The DNA repair enzyme MutY, which hydrolyzes adenine from 8-oxo-G:A mismatches, is also reported to form a deoxyribocation intermediate with a lifetime of less than 10−10 s (113). As human PNP is known to form a fully developed ribocation at the transition state, computational studies have been employed to investigate the lifetime of the ribocation and the accompanying reaction coordinate motion. As competitive, intrinsic KIEs are a two-state comparison (reactants free in solution are compared to the transition state), temporal and reaction coordinate information is not provided, and computational chemistry approaches are needed to provide additional insights.
Purine Nucleoside Phosphorylase Transition-State Motion and Lifetime
Transition path sampling is an unbiased computational method used to locate transition states in complex systems (114, 115). Recently, it has been applied to the chemical steps of lactate dehydrogenase and human PNP (116). Starting conditions are provided by the crystal structure of PNP with transition-state analogs at the catalytic sites. That this model is a reliable estimate of the geometry near the transition state is supported by the high success rate of unbiased barrier crossings using this method (17). Results of 220 barrier crossings show some common features associated with the transition state. Early loss of the ribosidic bond forms a ribocation transition state, followed by dynamic local motion of the enzyme and the ribosyl group, causing migration of the anomeric carbon toward bound phosphate to form ribose 1-phosphate. Common features identified by transition path sampling are (a) compression of the O4′…O5′ vibrational motion linked to ribocation formation, (b) optimized leaving group interactions, and (c) activation of the phosphate nucleophile as the reaction proceeds through the transition-state region. This complex reaction coordinate is complete in approximately 70 femtoseconds (fs), with the transition-state lifetime being only 10 fs (Figure 16). An important conclusion of this study is that simultaneous optimization of the effects needed to reach the transition state occur on the femtosecond timescale and require coincident interactions for simultaneous ribocation formation and leaving group activation.
Figure 16.

(a–d) Catalytic site contacts during catalysis in human purine nucleoside phosphorylase. (a) The reaction involves compression of three oxygens from the 5′-O, 4′-O and phosphate groups, (b) loss of the N-riboside bond, (c) ribosyl migration, and (d) formation of the new C-O bond to phosphate. (e) The reaction coordinate lifetime is approximately 70 fs (50 fs shown here), and the transition state (0.5 probability of product formation) is approximately 10 fs (reprinted from Reference 5 with permission). The limits of zero (0) and unity (1.0) in (e) represent stable reactants and fully formed products, respectively. The inset in (e) provides a more detailed plot of the probability of product formation at 55 to 70 fs.
Test of the Reactive Intermediate Hypothesis for Human Purine Nucleoside Phosphorylase
The 10 fs transition-state lifetime (10−14 s) for human PNP is on the timescale of a single bond vibration and cannot be considered an intermediate. Because the diffusion of water is approximately 1 Å per picosecond (ps) (10−12 s), water would be unlikely to react with an enzyme-generated ribocation when phosphate is in the vicinity if the computational lifetime of 10 fs is valid. If the lifetime of the ribocation is longer, for example, 10−10 s (100 ps), water diffusion could easily capture the ribocation provided that solvent has access to the catalytic site. Human PNP, like many enzymes, has hydrophobic groups that cover the catalytic site as a result of the loop motion associated with substrate binding. One interpretation of burying substrates into enzymatic catalytic sites is to shield reactive intermediates from solvent. This hypothesis was tested for human PNP by systematically removing four amino acid residues that sequester bound inosine in the catalytic site from solvent water (117). Solvent leaks were introduced into the catalytic site by replacing individual catalytic site–sequestering amino acids with glycine. Reactivity of the ribocation transition state was tested for capture by water relative to phosphate in the glycine mutants. NMR was used to follow ribose and ribose 1-phosphate formation for approximately 106 catalytic cycles. Without phosphate, the phosphate-binding site fills with water, and inosine is hydrolyzed by native PNP as well as by the “leaky” Y88G, F159G, H257G, and F200G enzymes. Hydrolysis did not occur in any of the leaky mutants when phosphate is bound despite the solvents’ access to the catalytic site. The result supports a ribocation lifetime too short to permit water diffusion into the catalytic site, a time estimated to be <5 ps (117).
An unprecedented N9-to-N3 isomerization of inosine is catalyzed by H257G and F200G in the presence of phosphate and by all PNPs in the absence of phosphate. Expansion of the catalytic site permits ribocation formation with relaxed geometric constraints, permitting nucleophilic rebound and N3-inosine isomerization (Figure 17).
Figure 17.

Experimental test of water access to the ribocation transition state of human purine nucleoside phosphorylase (PNP). The catalytic site of human PNP is covered by F200, H257, F159, and Y88. Replacement of each by glycine introduces leaks into the catalytic site (a). After approximately one million catalytic turnovers, 13C-NMR (b) demonstrates that ribose is not formed; thus, water cannot react because the carbocation lifetime is too short to permit water diffusion. These chemical reactions, including the unprecedented formation of N3-inosine, are summarized in (c) (derived from Reference 117).
Thermodynamics of Transition-State Analog Binding
Thermodynamic views of enzymatic transition-state theory suggest that the limiting affinity for binding transition-state analogs is the substrate affinity enhanced by magnitude of the enzymatic rate enhancement (118, 119). For PNP, the limiting dissociation constant is estimated to be 6 × 10−18 M based on the rate of spontaneous cleavage of the glycosidic bond of adenosine (120), equivalent to a ΔG binding of −23.6 kcal/mol. This value assumes a perfect mimic of the transition state, a physical impossibility because the transition state has nonequilibrium bond lengths and angles.
Transition-state analog binding to human PNP has picomolar affinity, and the thermodynamics of binding to the first catalytic site have been analyzed by isocalorimetry (121). Inhibitor binding exhibits negative cooperativity, and inhibition of PNP catalytic activity is complete when the inhibitor binds to the first of the three subunits. Binding of the structurally rigid first-generation ImmH (Kd = 56 pM) is driven by negative enthalpy of ΔH = −21.2 kcal/mol, teasingly close to the limit for a perfect transition-state analog, a ΔG binding of −23.6 kcal/mol. The ΔH of − 21.2 kcal/mol is significantly greater than the ΔH of 18.6 kcal/mol or ΔG of 14.3 kcal/mol measured for kcat during the chemical step of PNP (122). However, an entropic (−TΔS) penalty of 7.1 kcal/mol for ImmH binding provides a ΔG binding of −14.1 kcal/mol, almost exactly matching the ΔG for kcat.
DADMe-ImmH (Kd = 9 pM) has increased conformational flexibility compared to ImmH, and it binds with a reduced entropic penalty of 4.3 kcal/mol but is dominated by the ΔH of −18.6 kcal/mol (Figure 18). The thermodynamic binding profile for DADMe-ImmH is a near perfect inverse reflection of the catalytic energy barrier. Entropic penalties arise from protein and/or inhibitor conformational loss and changes in solvent organization, but such penalties are small compared to enthalpic energy, emphasizing the large electrostatic contributions to both catalysis and inhibitor binding. H/D exchange and sedimentation velocity confirmed that ImmH, with a large entropy penalty, forms the least dynamic and structurally tightest complex with human PNP (123). DATMe-ImmH, a highly flexible inhibitor, shows a smaller entropic penalty of 2.3 kcal/mol, less than that for ImmH or DADMe-ImmH (123). The results have implications for catalysis by suggesting that enzyme-inhibitor conformational flexibility can assist in tight binding and that tight binding of transition-state analogs is compatible with dynamic protein flexibility. Computational molecular dynamics have provided a detailed look at the conformational differences for PNP in substrate and different transition-state analog complexes (124). The favorable contribution of entropy to barrier crossing also suggests dynamic assistance in barrier crossing.
Figure 18.

Thermodynamics of transition-state barrier crossing compared to transition-state analog binding for DADMe-ImmA and human purine nucleoside phosphorylase (PNP). The upper portion of the plot (red) reports the thermodynamics for the chemical step (single-turnover kinetics), and the lower portion (blue) reports the energetic signature of transition-state analog binding to the first catalytic site of human PNP-PO4. Numerical values are in kcal/mol. E, PNP; A, guanosine; ΔG, Gibbs free energy; ΔH, enthalpy; TΔS, entropy; I, DADMe-ImmA.
SUBSTRATE AND TRANSITION-STATE BINDING ISOTOPE EFFECTS
The bond vibrational environment of individual atoms at enzymatic transition states is probed by KIE experiments, whereas those for binding interactions are probed by equilibrium BIEs (33, 34). By comparing KIE and BIE experiments, distortion of inosine in the Michaelis complex can be compared to distortion of inosine at the transition state of human PNP. Binding of [5′-3H]inosine to human PNP gives a BIE of 1.5%, but transition-state formation causes an intrinsic KIE (from transition-state distortions) of 4.7%. These values reflect atomic vibrational distortions in the 5′-C-H bonds at the Michaelis complex and the transition state. The degree of atomic distortion for catalysis was compared to that for binding of transition-state analogs. [5′-3H]immucillin-H and [5′-3H]DADMe-immucillin-H gave large 5′-3H BIEs of 12.6% and 29.2%, respectively (Figure 19). This unprecedented result revealed weak C5′-H distortion at the transition state and strong distortion in complexes with transition-state analogs. A dynamic model of catalysis without the need for tight binding at the transiton state is consistent with these results (125).
Figure 19.

Binding isotope effects (BIEs) compared to the kinetic isotope effect (KIE) for tritium at the 5′-position of human purine nucleoside phosphorylase (PNP). The KIE is from bond changes between the Michaelis complex and the transition state of purine nucleoside phosphorylase, and the BIEs are from equilibrated thermodynamic complexes between unbound inosine (with bound SO4 as a phosphate mimic) and the Michaelis complex. For inhibitors, the equilibrium is between unbound and bound complexes with PNP-PO4 (reprinted from Reference 125 with permission). DADMe-ImmH, DADMe-immucillin-H; ImmH, immucillin-H.
CONCLUSIONS AND THE FUTURE
Enzymatic transition states are accessible with the combined use of intrinsic KIEs and computational chemistry. Molecular electrostatic potential maps of reactants at enzymatic transition states provide blueprints for the design of transition-state analogs. Several inhibitors designed by these methods are in clinical trials. These early successes in drug design support future contributions of transition-state analysis and inhibitor design to drug development. Access to transition-state analogs has also provided structural access to difficult targets and insights to thermodynamic contributions for binding and catalysis.
BEYOND N-RIBOSYLTRANSFERASES.
The examples of enzymatic transition-state structures and transition-state analogs discussed here are N-ribosyltransferases. Are KIE approaches to enzymatic transition-state structure limited to certain classes of reactions? No, KIE analysis can be applied to any chemical reaction, enzymatic or nonenzymatic, using approaches similar to those presented here. Are there advantages to focusing on one class of reactions? Yes, a major effort in transition-state analysis is the synthesis of substrates with individual isotopic labels. An advantage of the N-ribosyltransferases is the ability to use chemoenzymatic synthesis to convert labeled glucose precursors into 5-phosphoribosyl-α-D-1-pyrophosphate and then to ATP. Additional enzymatic reactions are available to convert the labeled ATP molecules into most other purine and pyrimidine nucleosides and nucleotides. Other glycosyltransferases are prime targets for transition-state analysis because many sugars can be converted from labeled glucose or are available with isotopic labels. Primary, α-secondary and β-secondary isotope effects give information on the extent of bond breaking at the transition state, the degree of rehybridization at the anomeric carbon, and the geometry of the sugar ring at the transition state. Similar logic can be directly extended to all biological reactions involving substitutions at carbon. New NMR and mass spectrometry techniques are being developed to permit accurate analysis of natural abundance isotope effects. These methods will expand the experimental access to transition-state analysis. Expansion of enzymatic transition-state knowledge will contribute to the next generation of drug discovery efforts.
SUMMARY POINTS.
Closely related enzymes with near-identical catalytic sites can form distinct transition states.
Transition-state analysis from kinetic isotope effects (KIEs) and computational chemistry permits the design of isozyme-specific transition-state analogs.
Inhibitors designed from the first principles of transition-state theory are among the most powerful noncovalent enzymatic inhibitors.
Transition-state analog inhibitors designed to bind to specific enzymatic targets are finding applications in clinical trials.
Transition-state analogs can demonstrate binding enthalpies greater than the ΔG‡ barrier for catalysis.
The purine nucleoside phosphorylase transition state has a lifetime of 10 fs (10−14 s), whereas enzymatic complexes with transition-state analogs have lifetimes of 1 h (103 to 104 s).
Binding of transition-state analogs causes remarkable distortion of analogs together with the organization and stabilization of the enzymatic protein.
FUTURE ISSUES.
How specific are transition-state analogs? Because analogs capture unique catalytic features of an enzyme, they may target either single or a few enzymes in the human catalytic repertoire.
Biological lifetimes of transition-state analogs are often longer than predicted by the dissociation rates of enzyme-inhibitor complexes. The mechanisms for analog recycling are not well known, do not follow the normal rules of pharmacokinetics, and will require parallel in vitro and in vivo studies.
Time constants for the range of protein dynamics span over 10 orders of magnitude. Which of these are most closely linked with transition-state formation, binding events, and interactions of transition-state analogs including slow-onset binding and inhibitor dissociation?
Transition-state analogs commonly demonstrate slow-onset, tight-binding inhibition. Experimental approaches are needed to distinguish initial and final complex structures.
Acknowledgments
V.L.S. is supported by National Institutes of Health research grants GM41916, CA072444, AI049512, and CA135405, as well as by National Institutes of Health program project P01-GM068036.
Glossary
- Kinetic isotope effect (KIE)
a ratio of reaction rates for labeled (klabel) and unlabeled reactants (kunlabeled); KIE = (kunlabeled)/(klabel)
- Binding isotope effect (BIE)
a ratio of dissociation constants for labeled and unlabeled ligands; BIE = (Kdunlabeled)/(Kdlabel)
- Transition-state analysis
(a) measuring intrinsic KIEs for atomic centers and (b) matching a computational transition state to KIEs
- Molecular electrostatic potential
electron force applied to a positive charge in a defined position relative to a molecule
- Purine nucleoside phosphorylase (PNP)
catalyzes the phosphorolysis of 6-oxypurine nucleosides and 6-oxypurine 2′-deoxynucleosides to purine bases and α-D-ribose 1-phosphate or 2-deoxy-α-D-ribose 1-phosphate
- Immucillin
a chemically stable analog of the transition states for 6-oxypurine N-ribosyltransferases with early transition states
- 5′-Methylthioadenosine phosphorylase (MTAP)
an enzyme that converts 5′-methylthioadenosine (MTA) and phosphate to adenine and 5-methylthio-α-D-ribose 1-phosphate
- 5′-Methylthioadenosine/adenosylhomocysteine nucleosidase (MTAN)
an enzyme that catalyzes the hydrolysis of 5′-methylthioadenosine (MTA) and adenosylhomocysteine to adenine and 5-methylthio-α-D-ribose or ribosylhomocysteine
- DADMe-immucillin
a chemically stable analog of the transition states for purine N-ribosyltransferases with late transition states
- Ribosome-inactivating protein (RIP)
an enzyme that catalyzes the functional inactivation of ribosomes, often with high specificity and often by depurination of adenine
Footnotes
DISCLOSURE STATEMENT
The author is a consultant to pharmaceutical companies developing transition-state analog inhibitors for clinical applications, an employee of Albert Einstein College of Medicine, and the holder of multiple patents based on transition-state analogs. Many of these patents have been licensed by the college to the pharmaceutical industry. The author receives royalty income from the development of these patents.
LITERATURE CITED
- 1.Garcia-Viloca M, Gao J, Karplus M, Truhlar DG. How enzymes work: analysis by modern rate theory and computer simulations. Science. 2004;303:186–95. doi: 10.1126/science.1088172. [DOI] [PubMed] [Google Scholar]
- 2.Kamerlin SC, Mavri J, Warshel A. Examining the case for the effect of barrier compression on tunneling, vibrationally enhanced catalysis, catalytic entropy and related issues. FEBS Lett. 2010;584:2759–66. doi: 10.1016/j.febslet.2010.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagel ZD, Klinman JP. A 21st century revisionist’s view at a turning point in enzymology. Nat Chem Biol. 2009;5:543–50. doi: 10.1038/nchembio.204. [DOI] [PubMed] [Google Scholar]
- 4.Schramm VL. Enzymatic transition states: thermodynamics, dynamics and analogue design. Arch Biochem Biophys. 2005;433:13–26. doi: 10.1016/j.abb.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz SD, Schramm VL. Enzymatic transition states and dynamic motion in barrier crossing. Nat Chem Biol. 2009;5:551–58. doi: 10.1038/nchembio.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerlt JA, Babbitt PC. Enzyme (re)design: lessons from natural evolution and computation. Curr Opin Chem Biol. 2009;13:10–18. doi: 10.1016/j.cbpa.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon GM, Cravatt BF. Activity-based proteomics of enzyme superfamilies: serine hydrolases as a case study. J Biol Chem. 2010;285:11051–55. doi: 10.1074/jbc.R109.097600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ciulli A, Abell C. Fragment-based approaches to enzyme inhibition. Curr Opin Biotechnol. 2007;18:489–96. doi: 10.1016/j.copbio.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nielsen TE, Schreiber SL. Towards the optimal screening collection: a synthesis strategy. Angew Chem Int Ed Engl. 2008;47:48–56. doi: 10.1002/anie.200703073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khersonsky O, Tawfik DS. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu Rev Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- 11.Lassila JK, Herschlag D. Promiscuous sulfatase activity and thio-effects in a phosphodiesterase of the alkaline phosphatase superfamily. Biochemistry. 2008;47:12853–59. doi: 10.1021/bi801488c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thyme SB, Jarjour J, Takeuchi R, Havranek JJ, Ashworth J, et al. Exploitation of binding energy for catalysis and design. Nature. 2009;461:1300–4. doi: 10.1038/nature08508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Y, Yeung N, Sieracki N, Marshall NM. Design of functional metalloproteins. Nature. 2009;460:855–62. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robertson JG. Enzymes as a special class of therapeutic target: clinical drugs and modes of action. Curr Opin Struct Biol. 2007;17:674–79. doi: 10.1016/j.sbi.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5:821–34. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- 16.Quaytman SL, Schwartz SD. Comparison studies of the human heart and Bacillus stearothermophilus lactate dehydrogreanse by transition path sampling. J Phys Chem A. 2009;113:1892–97. doi: 10.1021/jp804874p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saen-Oon S, Quaytman-Machleder S, Schramm VL, Schwartz SD. Atomic detail of chemical transformation at the transition state of an enzymatic reaction. Proc Natl Acad Sci USA. 2008;105:16543–48. doi: 10.1073/pnas.0808413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN. The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction. Science. 2003;299:2067–71. doi: 10.1126/science.1082710. [DOI] [PubMed] [Google Scholar]
- 19.Tremblay LW, Zhang G, Dai J, Dunaway-Mariano D, Allen KN. Chemical confirmation of a pentavalent phosphorane in complex with beta-phosphoglucomufase. J Am Chem Soc. 2005;127:5298–99. doi: 10.1021/ja0509073. [DOI] [PubMed] [Google Scholar]
- 20.Baxter NJ, Hounslow AM, Bowler MW, Williams NH, Blackburn GM, Waltho JP. MgF3− and α-galactose 1-phosphate in the active site of β-phosphoglucomutase form a transition state analogue of phosphoryl transfer. J Am Chem Soc. 2009;131:16334–35. doi: 10.1021/ja905972m. [DOI] [PubMed] [Google Scholar]
- 21.Baxter NJ, Bowler MW, Alizadeh T, Cliff MJ, Hounslow AM, et al. Atomic details of near-transition state conformers for enzyme phosphoryl transfer revealed by MgF-3 rather than by phosphoranes. Proc Natl Acad Sci USA. 2010;107:4555–60. doi: 10.1073/pnas.0910333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leffler JE. Parameters for the description of transition states. Science. 1953;117:340–41. doi: 10.1126/science.117.3039.340. [DOI] [PubMed] [Google Scholar]
- 23.Hammond GS. A correlation of reaction rates. J Am Chem Soc. 1955;77:334–38. [Google Scholar]
- 24.Bigeleisen J, Mayer MG. Calculations of equilibrium constants for isotope exchange reactions. J Chem Phys. 1947;15:261–67. [Google Scholar]
- 25.Bigeleisen J, Wolfsberg M. Theoretical and experimental aspects of isotope effects in chemical kinetics. Adv Chem Phys. 1958;1:15–76. [Google Scholar]
- 26.Sims LB, Fry A, Netherton LT, Wilson JC, Reppond KD, Crook SW. Variations of heavy-atom kinetic isotope effects in SN2 displacement reactions. J Am Chem Soc. 1972;94:1364–73. [Google Scholar]
- 27.Sims LB, Lewis DE. Bond order methods for calculating isotope effects in organic reactions. In: Buncel E, Lee CC, editors. Chemistry. Vol. 6: Isotopes in Organic Chemistry. New York: Elsevier; 1984. pp. 161–259. [Google Scholar]
- 28.Cleland WW, O’Leary MH, Northrop DB, editors. Isotope Effects on Enzyme-Catalyzed Reactions. Baltimore, MD: Univ. Park; 1977. [Google Scholar]
- 29.Gandour RD, Schowen RL, editors. Transition States of Biochemical Processes. New York: Plenum; 1978. [Google Scholar]
- 30.Cleland WW. Isotope effects: determination of enzyme transition state structure. Methods Enzymol. 1995;249:341–73. doi: 10.1016/0076-6879(95)49041-8. [DOI] [PubMed] [Google Scholar]
- 31.Cook PF, editor. Enzyme Mechanism from Isotope Effects. Boca Raton, FL: CRC Press; 1991. [Google Scholar]
- 32.Hegazi MF, Borchard RT, Schowen RL. Letter: SN2-like transition state for methyl transfer catalyzed by catechol-O-methyl-transferase. J Am Chem Soc. 1976;98:3048–49. doi: 10.1021/ja00426a079. [DOI] [PubMed] [Google Scholar]
- 33.Lewis BE, Schramm VL. Binding equilibrium isotope effects for glucose at the catalytic domain of human brain hexokinase. J Am Chem Soc. 2003;125:4785–98. doi: 10.1021/ja0298242. [DOI] [PubMed] [Google Scholar]
- 34.Lewis BE, Schramm VL. Enzymatic binding isotope effects and the interaction of glucose with hexokinase. In: Kohen A, Limbach H-H, editors. Isotope Effects in Chemistry and Biology. Boca Raton, FL: CRC Press; 2006. pp. 1019–53. [Google Scholar]
- 35.Northrop DB. The expression of isotope effects on enzyme-catalyzed reactions. Annu Rev Biochem. 1981;50:103–31. doi: 10.1146/annurev.bi.50.070181.000535. [DOI] [PubMed] [Google Scholar]
- 36.Cleland WW. Use of isotope effects to elucidate enzyme mechanisms. CRC Crit Rev Biochem. 1982;13:385–428. doi: 10.3109/10409238209108715. [DOI] [PubMed] [Google Scholar]
- 37.Parkin DW. Methods for the determination of competitive and noncompetitive kinetic isotope effects. 1991:269–90. See Ref. 31. [Google Scholar]
- 38.Paneth P. Chlorine kinetic isotope effects on enzymatic dehalogenations. Acc Chem Res. 2003;36:120–26. doi: 10.1021/ar010101h. [DOI] [PubMed] [Google Scholar]
- 39.Singleton DA, Thomas J. High-precision simultaneous determination of multiple small kinetic isotope effects at natural abundance. J Am Chem Soc. 1995;117:9357–58. [Google Scholar]
- 40.Lee JK, Bain AD, Berti PJ. Probing the transition state of four glucoside hydrolyses with 13C kinetic isotope effects measured at natural abundance by NMR spectroscopy. J Am Chem Soc. 2004;126:3769–76. doi: 10.1021/ja0394028. [DOI] [PubMed] [Google Scholar]
- 41.Chan J, Lewis AR, Gilbert M, Karwaski MF, Bennet AJ. A direct NMR method for the measurement of competitive kinetic isotope effects. Nat Chem Biol. 2010;6:405–7. doi: 10.1038/nchembio.352. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Luo M, Schramm VL. Transition states of Plasmodium falciparum and human orotate phosphoribosyltransferases. J Am Chem Soc. 2009;131:4685–94. doi: 10.1021/ja808346y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Schramm VL. Pyrophosphate interactions at the transition states of Plasmodium falciparum and human orotate phosphoribosyltransferases. J Am Chem Soc. 2010;132:8787–94. doi: 10.1021/ja102849w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirschi JS, Takeya T, Hang C, Singleton DA. Transition-state geometry measurements from 13C isotope effects. The experimental transition state for the epoxidation of alkenes with oxaziridines. J Am Chem Soc. 2009;131:2397–403. doi: 10.1021/ja8088636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saunders M, Laidig EE, Wolfsberg M. Theoretical calculations of equilibrium isotope effects using ab initio force constants: application to NMR isotopic perturbation studies. J Am Chem Soc. 1989;11:8989–94. [Google Scholar]
- 46.Anisimov V, Paneth P. ISOEFF98. A program for studies of isotope effects using Hessian modifications. J Math Chem. 1999;26:75–86. [Google Scholar]
- 47.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al. Gaussian 03, Revis. C02. Wallingford, CT: Gaussian; 2004. http://www.gaussian.com/ [Google Scholar]
- 48.Lienhard GE. Enzymatic catalysis and transition-state theory. Science. 1973;180:149–54. doi: 10.1126/science.180.4082.149. [DOI] [PubMed] [Google Scholar]
- 49.Wolfenden R. Transition state analogues for enzyme catalysis. Nature. 1969;223:704–5. doi: 10.1038/223704a0. [DOI] [PubMed] [Google Scholar]
- 50.Wolfenden R. Analog approaches to the structure of the transition state in enzyme reactions. Acc Chem Res. 1972;5:10–18. [Google Scholar]
- 51.Schramm VL. Enzymatic transition states and transition state analogues. Curr Opin Struct Biol. 2005;15:604–13. doi: 10.1016/j.sbi.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 52.Machleder SQ, Pineda ET, Schwartz SD. On the origin of the chemical barrier and tunneling in enzymes. J Phys Org Chem. 2010;23:690–95. doi: 10.1002/poc.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bagdassarian CK, Schramm VL, Schwartz SD. Molecular electrostatic potential analysis for enzymatic substrates, competitive inhibitors and transition-state inhibitors. J Am Chem Soc. 1996;118:8825–36. [Google Scholar]
- 54.Bagdassarian CK, Braunheim BB, Schramm VL, Schwartz SD. Quantitative measures of molecular similarity: methods to analyze transition-state analogs for enzymatic reactions. Int J Quant Chem: Quant Biol Symp. 1996;23:1797–804. [Google Scholar]
- 55.Miles RW, Tyler PC, Furneaux RH, Bagdassarian CK, Schramm VL. One-third-the-sites transition-state inhibitors for purine nucleoside phosphorylase. Biochemistry. 1998;37:8615–21. doi: 10.1021/bi980658d. [DOI] [PubMed] [Google Scholar]
- 56.Braunheim BB, Bagdassarian CK, Schramm VL, Schwartz SD. Quantum neural networks can predict binding free energies for enzymatic inhibitors. Int J Quantum Chem. 2000;78:195–204. [Google Scholar]
- 57.Braunheim BB, Miles RW, Schramm VL, Schwartz SD. Prediction of inhibitor binding free energies by quantum neural networks. Nucleoside analogues binding to trypanosomal nucleoside hydrolase. Biochemistry. 1999;38:16076–83. doi: 10.1021/bi990830t. [DOI] [PubMed] [Google Scholar]
- 58.Sinnott ML. Catalytic mechanisms of enzymic glycosyl transfer. Chem Rev. 1990;90:1171–202. [Google Scholar]
- 59.Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–55. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 60.Schramm VL. Enzymatic transition state poise and transition state analogues. Acc Chem Res. 2003;36:588–96. doi: 10.1021/ar0200495. [DOI] [PubMed] [Google Scholar]
- 61.Gloster TM, Davies GJ. Glycosidase inhibition: assessing mimicry of the transition state. Org Biomol Chem. 2010;8:305–20. doi: 10.1039/b915870g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wong C-H, Dumas DP, Ichikawa Y, Koseki K, Danishefsky SJ, et al. Specificity, inhibition, and synthetic utility of a recombinant human α-1,3-fucosyltransferase. J Am Chem Soc. 1992;114:7321–22. [Google Scholar]
- 63.Giblett ER, Ammann AJ, Wara DW, Sandman R, Diamond LK. Nucleoside-phosphorylase deficiency in a child with severely defective T-cell immunity and normal B-cell immunity. Lancet. 1975;1:1010–13. doi: 10.1016/s0140-6736(75)91950-9. [DOI] [PubMed] [Google Scholar]
- 64.Ullman C, Gudas LJ, Clift SM, Martin DW. Isolation and characterization of purine-nucleoside phosphorylase-deficient T-lymphoma cells and secondary mutants with altered ribonucleotide reductase: genetic model for immunodeficiency disease. Proc Natl Acad Sci USA. 1979;76:1074–78. doi: 10.1073/pnas.76.3.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miles RW, Tyler PC, Furneaux RH, Bagdassarian CK, Schramm VL. Purine nucleoside phosphorylase. Transition state structure, transition state inhibitors and one-third-the-sites reactivity. In: Frey PA, Northrop DB, editors. Enzymatic Mechanisms. Washington, DC: IOS Press; 1999. pp. 32–47. [Google Scholar]
- 66.Lewandowicz A, Schramm VL. Transition state analysis for human and Plasmodium falciparum purine nucleoside phosphorylases. Biochemistry. 2004;43:1458–68. doi: 10.1021/bi0359123. [DOI] [PubMed] [Google Scholar]
- 67.Ho MC, Shi W, Rinaldo-Matthis A, Tyler PC, Evans GB, et al. Four generations of transition-state analogues for human purine nucleoside phosphorylase. Proc Natl Acad Sci USA. 2010;107:4805–12. doi: 10.1073/pnas.0913439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lewandowicz A, Tyler PC, Evans GB, Furneaux RH, Schramm VL. Achieving the ultimate physiological goal in transition state analogue inhibitors for purine nucleoside phosphorylase. J Biol Chem. 2003;278:31465–68. doi: 10.1074/jbc.C300259200. [DOI] [PubMed] [Google Scholar]
- 69.Evans GB, Furneaux RH, Hutchison TL, Kezar HS, Morris PE, Jr, et al. Addition of lithiated 9-deazapurine derivatives to a carbohydrate cyclic imine: convergent synthesis of the aza-C-nucleoside immucillins. J Org Chem. 2001;66:5723–30. doi: 10.1021/jo0155613. [DOI] [PubMed] [Google Scholar]
- 70.Schramm VL, Tyler PC. Imino-sugar based nucleosides. Curr Top Med Chem. 2003;3:525–40. doi: 10.2174/1568026033452465. [DOI] [PubMed] [Google Scholar]
- 71.Evans GB, Furneaux RH, Tyler PC, Schramm VL. Synthesis of a transition state analogue inhibitor of purine nucleoside phosphorylase via the Mannich reaction. Org Lett. 2003;5:3639–40. doi: 10.1021/ol035293q. [DOI] [PubMed] [Google Scholar]
- 72.Clinch K, Evans GB, Frohlich RF, Furneaux RH, Kelly PM, et al. Third-generation immucillins: syntheses and bioactivities of acyclic immucillin inhibitors of human purine nucleoside phosphorylase. J Med Chem. 2009;52:1126–43. doi: 10.1021/jm801421q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Taylor EA, Clinch K, Kelly PM, Li L, Evans GB, et al. Acyclic ribooxacarbenium ion mimics as transition state analogues of human and malarial purine nucleoside phosphorylases. J Am Chem Soc. 2007;129:6984–85. doi: 10.1021/ja071087s. [DOI] [PubMed] [Google Scholar]
- 74.Clinch K, Evans GB, Furneaux RH, Lenz DH, Mason JM, et al. A practical synthesis of (3R,4R)-N-tert-butoxycarbonyl-4-hydroxymethylpyrrolidin-3-ol. Org Biomol Chem. 2007;5:2800–2. doi: 10.1039/b708796a. [DOI] [PubMed] [Google Scholar]
- 75.Evans GB, Furneaux RH, Greatrex B, Murkin AS, Schramm VL, Tyler PC. Azetidine based transition state analogue inhibitors of N-ribosyl hydrolases and phosphorylases. J Med Chem. 2008;51:948–56. doi: 10.1021/jm701265n. [DOI] [PubMed] [Google Scholar]
- 76.Huang M, Wang Y, Gu J, Yang J, Noel K, et al. Determinants of sensitivity of human T-cell leukemia CCRF-CEM cells to immucillin-H. Leuk Res. 2008;32:1268–78. doi: 10.1016/j.leukres.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Balakrishnan K, Verma D, O’Brien S, Kilpatrick JM, Chen Y, et al. Phase 2 and pharmacodynamic study of oral forodesine in patients with advanced, fludarabine-treated chronic lymphocytic leukemia. Blood. 2010;116:886–92. doi: 10.1182/blood-2010-02-272039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bantia S, Parker C, Upshaw R, Cunningham A, Kotian P, et al. Potent orally bioavailable purine nucleoside phosphorylase inhibitor BCX-4208 induces apoptosis in B- and T-lymphocytes—a novel approach for autoimmune diseases, organ transplantation and hematologic malignancies. Int Immunopharmacol. 2010;10:784–90. doi: 10.1016/j.intimp.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 79.Albers E. Metabolic characteristics and importance of the universal methionine salvage pathway recycling methionine from 5′-methylthioadenosine. J Med Chem. 2009;61:1132–42. doi: 10.1002/iub.278. [DOI] [PubMed] [Google Scholar]
- 80.Grillo MA, Colombatto S. S-adenosylmethionine and its products. Amino Acids. 2008;34:187–93. doi: 10.1007/s00726-007-0500-9. [DOI] [PubMed] [Google Scholar]
- 81.Avila MA, Garcia-Trevijano ER, Lu SC, Corrales FJ, Mato JM. Methylthioadenosine. Int J Biochem Cell Biol. 2004;36:2125–30. doi: 10.1016/j.biocel.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 82.Pegg AE, Michael AJ. Spermine synthase. Cell Mol Life Sci. 2010;67:113–21. doi: 10.1007/s00018-009-0165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Singh V, Schramm VL. Transition-state structure of human 5′-methylthioadenosine phosphorylase. J Am Chem Soc. 2006;128:14691–96. doi: 10.1021/ja065419p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zappia V, Della Ragione F, Pontoni G, Gragnaniello V, Carteni-Farina M. Human 5′-deoxy-5′-methylthioadenosine phosphorylase: kinetic studies and catalytic mechanism. Adv Exp Med Biol. 1988;250:165–77. doi: 10.1007/978-1-4684-5637-0_15. [DOI] [PubMed] [Google Scholar]
- 85.Evans GB, Furneaux RH, Schramm VL, Singh V, Tyler PC. Targeting the polyamine pathway with transition-state analogue inhibitors of 5′-methylthioadenosine phosphorylase. J Med Chem. 2004;47:3275–81. doi: 10.1021/jm0306475. [DOI] [PubMed] [Google Scholar]
- 86.Evans GB, Furneaux RH, Lenz DH, Painter GF, Schramm VL, et al. Second generation transition state analogue inhibitors of human 5′-methylthioadenosine phosphorylase. J Med Chem. 2005;48:4679–89. doi: 10.1021/jm050269z. [DOI] [PubMed] [Google Scholar]
- 87.Basu I, Cordovano G, Das I, Belbin TJ, Guha C, Schramm VL. A transition state analogue of 5′-methylthioadenosine phosphorylase induces apoptosis in head and neck cancers. J Biol Chem. 2007;282:21477–86. doi: 10.1074/jbc.M702287200. [DOI] [PubMed] [Google Scholar]
- 88.Basu I, Locker J, Cassera MB, Belbin TJ, Merino EF, et al. Growth and metastases of human lung cancer are inhibited in mouse xenografts by a transition state analogue of 5′-methylthioadenosine phosphorylase. J Biol Chem. 2010;285 doi: 10.1074/jbc.M110.198374. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fuqua C, Greenberg EP. Listening in on bacteria: acyl-homoserine lactone signaling. Nat Rev Mol Cell Biol. 2002;3:685–95. doi: 10.1038/nrm907. [DOI] [PubMed] [Google Scholar]
- 90.Winzer K, Williams P. Quorum sensing and the regulation of virulence gene expression in pathogenic bacteria. Int J Med Microbiol. 2001;291:131–43. doi: 10.1078/1438-4221-00110. [DOI] [PubMed] [Google Scholar]
- 91.Vendeville A, Winzer K, Heurlier K, Tang CM, Hardie KR. Making ‘sense’ of metabolism: autoinducer-2, LuxS and pathogenic bacteria. Nat Rev Microbiol. 2005;3:383–96. doi: 10.1038/nrmicro1146. [DOI] [PubMed] [Google Scholar]
- 92.Sperandio V. Novel approaches to bacterial infection therapy by interfering with bacteria-to-bacteria signaling. Expert Rev Anti-Infect Ther. 2007;5:271–76. doi: 10.1586/14787210.5.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. The biology and future prospects of antivirulence therapies. Nat Rev Microbiol. 2008;6:17–27. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gutierrez JA, Crowder T, Rinaldo-Matthis A, Ho MC, Almo SC, Schramm VL. Transition state analogs of 5′-methylthioadenosine nucleosidase disrupt quorum sensing. Nat Chem Biol. 2009;5:251–57. doi: 10.1038/nchembio.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sing V, Lee JE, Nuńez S, Howell PL, Schramm VL. Transition state structure of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase from Escherichia coli and its similarity to transition state analogues. Biochemistry. 2005;44:11647–59. doi: 10.1021/bi050863a. [DOI] [PubMed] [Google Scholar]
- 96.Singh V, Schramm VL. Transition-state analysis of S. pneumoniae 5′-methylthioadenosine nucleosidase. J Am Chem Soc. 2007;129:2783–95. doi: 10.1021/ja065082r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Singh V, Luo M, Brown RL, Norris GE, Schramm VL. Transition-state structure of Neisseria meningitides 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. J Am Chem Soc. 2007;129:13881–83. doi: 10.1021/ja0754204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gutierrez JA, Luo M, Singh V, Li L, Brown RL, et al. Picomolar inhibitors as transition-state probes of 5′-methylthioadenosine nucleosidases. ACS Chem Biol. 2007;2:725–34. doi: 10.1021/cb700166z. [DOI] [PubMed] [Google Scholar]
- 99.Lord JM, Robertus LM, Robertus JD. Ricin: structure, mode of action and some current applications. FASEB J. 1994;8:201–8. [PubMed] [Google Scholar]
- 100.Glück A, Endo Y, Wool IG. The ribosomal RNA identity elements for ricin and for alpha-sarcin: mutations in the putative CG pair that closes a GAGA tetraloop. Nucleic Acids Res. 1994;22:321–24. doi: 10.1093/nar/22.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen X-Y, Berti PJ, Schramm VL. Ricin A-chain: kinetic isotope effects and transition state structures with stem-loop RNA. J Am Chem Soc. 2000;122:1609–17. [Google Scholar]
- 102.Chen X-Y, Berti PJ, Schramm VL. Transition state structure for depurination of DNA by ricin A-chain. J Am Chem Soc. 2000;122:6527–34. [Google Scholar]
- 103.Angelotti T, Krisko M, O’Connor T, Serianni AS. [1-13C]Aldono-1,4-lactones: conformational studies based on proton-proton, proton-carbon-13, and carbon-13-carbon-13 spin couplings and ab initio molecular orbital calculations. J Am Chem Soc. 1987;109:4464–72. [Google Scholar]
- 104.Roday S, Amukele T, Evans GB, Tyler PC, Furneaux RH, Schramm VL. Inhibition of ricin A-chain with pyrrolidine mimics of the oxacarbenium ion transition state. Biochemistry. 2004;43:4923–33. doi: 10.1021/bi0498499. [DOI] [PubMed] [Google Scholar]
- 105.Barbieri L, Valbonesi P, Gorini P, Pession A, Stirpe F. Polynucleotide: adenosine glycosidase activity of saporin-L1: effect on DNA, RNA and poly (A) Biochem J. 1996;319:507–13. doi: 10.1042/bj3190507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sturm MB, Tyler PC, Evans GB, Schramm VL. Transition state analogues rescue ribosomes from saporin-L1 ribosome inactivating protein. Biochemistry. 2009;48:9941–48. doi: 10.1021/bi901425h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ho MC, Sturm MB, Almo SC, Schramm VL. Transition state analogues in structures of ricin and saporin ribosome-inactivating proteins. Proc Natl Acad Sci USA. 2009;106:20276–81. doi: 10.1073/pnas.0911606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang Y, Schramm VL. Pyrophosphate interactions at the transition states of Plasmodium falciparum and human orotate phosphoribosyltransferases. J Am Chem Soc. 2010;132:8787–94. doi: 10.1021/ja102849w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang Y, Luo M, Schramm VL. Transition states of Plasmodium falciparum and orotate phosphoribosyltransferases. J Am Chem Soc. 2009;131:4685–94. doi: 10.1021/ja808346y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tao W, Grubmeyer C, Blanchard JS. Transition state structure of Salmonella typhimurium orotate phosphoribosyltransferase. Biochemistry. 1996;35:14–21. doi: 10.1021/bi951898l. [DOI] [PubMed] [Google Scholar]
- 111.Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–55. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 112.McCann JA, Berti PJ. Transition state analysis of acid-catalyzed dAMP hydrolysis. J Am Chem Soc. 2007;129:7055–64. doi: 10.1021/ja067371l. [DOI] [PubMed] [Google Scholar]
- 113.McCann JA, Berti PJ. Transition-state analysis of the DNA repair enzyme MutY. J Am Chem Soc. 2008;130:5789–97. doi: 10.1021/ja711363s. [DOI] [PubMed] [Google Scholar]
- 114.Bolhuis PG, Chandler D, Dellago C, Geissler PL. Transition path sampling: throwing ropes over rough mountain passes in the dark. Annu Rev Phys Chem. 2002;53:291–318. doi: 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- 115.Dellago C, Bolhuis PG, Chandler D. Efficient transition path sampling: application to Lennard-Jones cluster rearrangements. J Chem Phys. 1998;108:9236–45. [Google Scholar]
- 116.Basner JE, Schwartz SD. How enzyme dynamics helps catalyze a chemical reaction in atomic detail: a transition path sampling study. J Am Chem Soc. 2005;127:13822–31. doi: 10.1021/ja043320h. [DOI] [PubMed] [Google Scholar]
- 117.Ghanem M, Murkin AS, Schramm VL. Ribocation transition state capture and rebound in human purine nucleoside phosphorylase. Chem Biol. 2009;16:971–79. doi: 10.1016/j.chembiol.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wolfenden R. Transition state analog inhibitors and enzyme catalysis. Annu Rev Biophys Bioeng. 1976;5:271–306. doi: 10.1146/annurev.bb.05.060176.001415. [DOI] [PubMed] [Google Scholar]
- 119.Wolfenden R, Kati WM. Testing the limits of protein-ligand binding discrimination with transition-state analogue inhibitors. Acc Chem Res. 1991;24:209–15. [Google Scholar]
- 120.Stockridge RB, Schroeder GK, Wolfenden R. The rate of spontaneous cleavage of the glycoside bond of adenosine. Bioorg Chem. 2010;38:224–28. doi: 10.1016/j.bioorg.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Edwards AA, Mason JM, Clinch K, Tyler PC, Evans GB, Schramm VL. Altered enthalpy-entropy compensation in picomolar transition state analogues of human purine nucleoside phosphorylase. Biochemistry. 2009;48:5226–38. doi: 10.1021/bi9005896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ghanem M, Li L, Wing C, Schramm VL. Altered thermodynamics from remote mutations altering human toward bovine purine nucleoside phosphorylase. Biochemistry. 2008;47:2559–64. doi: 10.1021/bi702132e. [DOI] [PubMed] [Google Scholar]
- 123.Edwards AA, Tipton JD, Brenowitz MD, Emmett MR, Marshall AG, et al. Conformational states of human purine nucleoside phosphorylase at rest, at work, and with transition state analogues. Biochemistry. 2010;49:2058–67. doi: 10.1021/bi902041j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hirschi JS, Arora K, Brooks CL, Schramm VL. Conformational dynamics in human purine nucleoside phosphorylase with reactants and transition-state analogues. J Phys Chem B. 2010;114:16263–72. doi: 10.1021/jp108056s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Murkin AS, Tyler PC, Schramm VL. Transition-state interactions revealed in purine nucleoside phosphorylase by binding isotope effects. J Am Chem Soc. 2008;130:2166–67. doi: 10.1021/ja7104398. [DOI] [PubMed] [Google Scholar]
RELATED RESOURCES
- 1.Schramm VL. Enzymatic transition-state analysis and transition state analogs. Methods Enzymol. 1999;308:301–55. doi: 10.1016/s0076-6879(99)08015-5. [DOI] [PubMed] [Google Scholar]
- 2.Murkin AS, Schramm VL. Purine nucleoside phosphorylases as targets for transition-state analog design. In: Merz KM Jr, Ringe D, Reynolds CH, editors. Drug Design. New York: Cambridge Univ. Press; 2010. pp. 215–47. [Google Scholar]
- 3.Schramm VL. Binding isotope effects: boon and bane. Curr Opin Chem Biol. 2007;11:529–36. doi: 10.1016/j.cbpa.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Truhlar DG. Tunneling in enzymatic and nonenzymatic hydrogen transfer reactions. J Phys Org Chem. 2010;23:660–76. [Google Scholar]
- 5.Kamerlin SC, Warshel A. At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis? Proteins. 2010;78:1339–75. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu Rev Biochem. 2006;75:519–41. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]